


Abstract
Folding of group II introns is characterized by a first slow compaction of domain 1 (D1) followed by the rapid docking of other domains to this scaffold. D1 compaction initiates in a small subregion encompassing the κ and ζ elements. These two tertiary elements are also the major interaction sites with domain 5 to form the catalytic core. Here, we provide the first characterization of the structure adopted at an early folding step and show that the folding control element can be narrowed down to the three-way junction with the κ motif. In our nuclear magnetic resonance studies of this substructure derived from the yeast mitochondrial group II intron Sc.ai5γ, we show that a high affinity Mg(II) ion stabilizes the κ element and enables coaxial stacking between helices d′ and d′′, favoring a rigid duplex across the three-way junction. The κ-element folds into a stable GAAA-tetraloop motif and engages in A-minor interactions with helix d′. The addition of cobalt(III)hexammine reveals three distinct binding sites. The Mg(II)-promoted structural rearrangement and rigidification of the D1 core can be identified as the first micro-step of D1 folding.
INTRODUCTION
Self-splicing group II introns belong to the class of large ribozymes. Recognized as an independent class only in 1982 (1), they are found primarily in organellar genes of plants, fungi and lower eukaryotes, but also in many bacteria (2,3). It is thought that they were crucial in modeling the genome of almost all terrestrial life forms: a common ancestry has been proposed for group II introns and most nuclear introns, components of the telomerase and the abundant LINE–element form of transposons (4), as well as the eukaryotic spliceosome (5–9). Moreover, they have been found to be able to reinsert into RNA and DNA via a retro-homing mechanism (10–12), thus being mobile genetic elements responsible for the continuous diversification of different genomes.
Group II introns as all other RNA molecules are characterized by a secondary structure usually stabilized by monovalent metal ions, whereas the tertiary structure requires the presence of divalent metal ions (13). The latter are eventually also involved in catalysis (14–17). Despite a lack of primary structure conservation, all group II introns present a similar secondary structure, consisting of six domains originating from a central wheel (2,18,19). Domain 1 represents the largest domain. It is an independent folding unit (20,21) and provides a scaffold for the other domains to dock. Domain 2 and 3 stabilize the folding and enhance the catalytic activity of the whole intron, respectively. Domain 4 can contain an open reading frame encoding for a maturase protein useful for intron activity in vivo (10). Domain 5 is the genetically most conserved one and shares many features with the spliceosomal RNA U6 (22). It represents, together with domain 1, the minimal core to enable catalysis (3,5). Finally, domain 6 contains the bulged adenosine implicated as nucleophile in the first step of splicing (23). Several studies have been devoted to understanding and describing both the folding and the catalytic mechanism of group II introns. One of the best studied systems is the Saccharomyces cerevisiae intron Sc.ai5γ from baker’s yeast (24–29).
In particular, a shorter version of the whole intron containing domains 1, 3 and 5 (D135) has been extensively used for folding studies (28–32). The folding of large RNAs usually involves a rapid collapse on metal ion addition (33,34). The collapse can be unspecific or can be initiated by the formation of specific RNA substructures. Pyle et al. (34) showed that D135 folds in an apparent two state model: the first stage is slow (in the order of seconds) and requires the formation of specific structural features within D1. This stage is then followed by the fast docking of all the other domains. Both stages require above-physiological Mg(II) concentrations in vitro, the first one has a KD,Mg of ∼13 mM (21), the second of 20–40 mM (30).
A small subregion in the central core of D1 comprising the three-way junction around the κ motif (25) and an 11-nt tetraloop receptor (ζ) was proposed to act as folding control element (D1κζ, Figure 1) (24,25,28). In the presence of Mg(II), this region adopts a specific structure, which allows the whole intron to attain the active native state (29,34). Moreover, it was recently shown that Mss116p, a DEAD box protein, which promotes folding in vivo substituting in part for the Mg(II) (35,36) acts by stabilizing this small substructure (37). In a later stage of folding, D1κζ also offers the docking site for D5, as supported by biochemical studies and a recent crystal structure of a group IIC intron from Oceanobacillus iheyensis (38–40).
Figure 1.

(a) Predicted secondary structure of the group II intron Sc.ai5γ from S. cerevisiae (the κ-ζ region studied in this work is highlighted). (b) The wild-type κ-ζ region (top) and the 49 nucleotide construct used for NMR studies (bottom). Nucleotides that were added at the ends to stabilize helix d′′ and helix d are drawn as contours.
We have now investigated the structure of D1κζ by means of nuclear magnetic resonance (NMR) spectroscopy, and demonstrate the central role of Mg(II) in its stabilization. Our data show that the helices next to the three-way junction are destabilized in the absence of Mg(II). Coordination of Mg(II) leads to a rigidification of the central core of this substructure, resulting in the coaxial stacking of two helices as was suggested by Costa et al. (41) from comparative sequence analysis.
The bulged nucleotides of the D1κζ junction form a classical GAAA tetraloop motif interrupted between the third and the forth nucleotide by a short helix. In addition, adenosines in this motif are engaged in A-minor interactions with the adjacent helix. Interestingly, this A-minor motif is not present in the only available crystal structure of a group IIC intron (38–40), where the two adenines are interacting with the docked D5.
Coaxial stacking of helices and A-minor motifs have been recognized as among the most important interactions governing RNA tertiary structure (42) and are here combined together within this small subregion. Helices next to multibranch junctions tend to maximize coaxial stacking to reduce overall folding free energy (43), and the stacking has been shown to depend on di- or multivalent ions (44). Our detailed NMR data allow us to locate a possible binding pocket at the junction that explains the requirement of multivalent metal ions for stabilization of this region. Which helices in a junction stack and how they are oriented is strongly influenced by the bulged nucleotides. Geary et al. (45) have highlighted the versatility of A-minor interactions in determining the architecture of junctions and the relative orientation of the helices. To the best of our knowledge, the structural arrangement of the A-minor interactions in the three-way junction present in D1κζ has not been described previously.
This NMR solution structure of D1κζ adds to the scarce structural data on group II introns (23,27,38–40,46–48). Moreover, we reveal κ to be a flexible, bimodal structure, which keeps its characteristic fold both in the context of a minimal A-minor junction motif to the adjacent helix (as observed in the D1κζ construct), as well as in the tertiary interaction with D5 (in the full group II intron).
MATERIALS AND METHODS
All chemicals were purchased from either Fluka-Sigma-Aldrich (Buchs, Switzerland) or Brunschwig Chemie (Basel, Switzerland) at puriss p.a. or biograde. All solutions were prepared by using autoclaved and filtered ultrapure water. All DNA oligonucleotides were purchased from Microsynth (Balgach, Switzerland) and purified by polyacrylamide gel electrophoresis before usage. The nucleotide 5′-triphosphates (NTPs) were obtained from GE Healthcare (Glattbrugg, Switzerland), with the exception of uridine-5′-triphosphate (UTP) that was purchased from Acros-Organics (Geel, Belgium). 13C- and 15N-labeled NTPs were purchased from Silantes GmbH (München, Germany), whereas the partially deuterated NTPs were obtained from Cambridge Isotope Laboratories (Andover, USA). The RNA samples were prepared by in vitro transcription, using a home-made T7 polymerase (49) and purified through denaturing polyacrylamide gel electrophoresis (15–18%). The electroelution apparatus used to recover the RNA samples was a Whatman® Elutrap System with BT1 and BT2 membranes. For desalting, Vivaspin 2-mL ultrafiltration devices (Sartorius Stedim biotech, Aubagne, France) with 3-kDa cutoff membranes were used. RNA concentration was measured by UV-VIS. MgCl2 for the metal ion titration was obtained as ultrapure solution (1 M in H2O) from Fluka-Sigma-Aldrich. Cobalt(III)hexammine was synthesized according to literature procedure (50). The exact concentration of the MgCl2 stock solution in 100% D2O was determined by potentiometric pH titration with EDTA (51), whereas the one of Co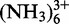 was measured by UV-VIS (52). Hundred percent D2O used for NMR sample preparation was purchased from Armar Chemicals (Döttingen, Switzerland).
was measured by UV-VIS (52). Hundred percent D2O used for NMR sample preparation was purchased from Armar Chemicals (Döttingen, Switzerland).
RNA transcription and sample preparation for NMR experiments
The RNA samples were transcribed and purified following standard procedures (49). The reaction mixture contained 5 mM of each NTP, 0.5 μM of the double-stranded DNA template, 0.1% Triton X-100, 40 mM TRIS-HCl [Tris(hydroxymethyl)-aminomethan-HCl, pH 7.5], 40 mM DTT, 2 mM spermidine and 30 mM MgCl2. The amount of polymerase depended on the effectiveness of the polymerase itself and was optimized for each polymerase batch. To avoid 3′-end inhomogeneity due to run-off transcription by the T7-RNA polymerase, we used a modified DNA template, in which the two nucleotides at the 5′-end contain a 2′-methoxy group (53). For the 49-nucleotide molecule used for this study, the best transcription yield was obtained by transcribing for 4 h at 37°C. Generally, of a 5 ml transcription, we obtained enough RNA to prepare 2–3 NMR samples at concentrations between 0.5 and 1 mM (in 300 μl). Before preparing the NMR sample, the RNA was desalted through several washing steps using first 1 M KCl, pH 8, then ultrapure water. Usage of KCl is critical, as all the TRIS must be removed from the solution, as it can compete with RNA for metal ion binding (54). The samples were lyophilized and re-dissolved in 300 μl 100% D2O or H2O/D2O (90:10) and transferred into Shigemi® tubes before the acquisition of the NMR spectra. All the samples contained 60 mM KCl, 10 µM EDTA and were of pH 6.5–6.9.
NMR spectroscopy
D1κζ was investigated by solution NMR methods generally used in RNA structural studies (55–57). The NMR spectra were recorded on a Bruker AV700 MHz spectrometer equipped with a CP-TXI z-axis pulsed field gradient CryoProbe®, on a Bruker AV600 MHz equipped with a TCI z-gradient CryoProbe® and on a Bruker AV500MHz, equipped with a QNP z-gradient CryoProbe®. In the context of the Bio-NMR project, additional spectra were acquired on a Bruker AV900MHz equipped with a TCI z-gradient CryoProbe® at CERM in Florence.
The spectra were acquired either on D1κζ constructs prepared with isotopes in natural abundance or constructs prepared by using partially deuterated nucleotides or 13C-, 15N-labeled nucleotides. The use of partially deuterated samples was crucial to avoid the massive overlap in the sugar regions: the sequential walk could be followed in the H6/H8-H1′ region and in the H6/H8-H2′ region. The sugar pucker of the nucleotides was determined by Total Correlation Spectroscopy (TOCSY) experiments with a sample with natural isotope abundance. Non-exchangeable protons were assigned from 2D [1H, 1H]-NOESY (Nuclear Overhauser Effect Spectroscopy) spectra in D2O, recorded at various temperatures (290, 295, 300, 305, 310 K) and with different mixing times (60, 150, 250, 350 ms); water suppression was either achieved by using a water presaturation pulse or an excitation sculpting pulse scheme. The same general spectral features were observed between 290 K and 310 K, but 300 K was found to be the optimum. The pH was kept between 6.5 and 6.9 without significant modification in the spectra. 2D [1H,1H]-TOCSY spectra (50 ms mixing time) were acquired to assess the sugar pucker. Exchangeable protons were assigned from [1H,1H]-NOESYs recorded in H2O/D2O at different temperatures (275, 280, 282, 285 K) with 150 ms mixing times, employing a 1-1 spin-echo pulse sequence or the excitation sculpting pulse scheme for water suppression. [1H,13C]-HSQC experiments were employed to attribute sugar and aromatic carbons. Two separate experiments were usually recorded for the aromatic and the aliphatic range of the 13C resonances. [1H,15N]-HSQC experiments were used to attribute the imino nitrogens. band-Selective Optimized Flip-Angle Short-Transient (SOFAST) sequences were used for these experiments (58). 3D-[1H,1H]-NOESY-[1H,13C]-HSQC were also performed (59).
The base-pairing pattern was established by recording a 2D JNN HNN-COSY. Such experiment correlates the imino nitrogens of uracil and guanine across the H-bond to the N1 of adenine or the N3 of cytosine on the other side of the double helix (60).
1H chemical shifts are directly referred to external DSS (0.2%, pH 7.5), whereas 13C and 15N are indirectly referred to 1H of DSS (61). All the spectra were recorded in the absence and in the presence of Mg(II).
For residual dipolar coupling (RDC) measurements, D1κζ was partially aligned using ∼8.0 mg/mL Pf1 filamentous bacteriophage (ASLA Biotech Ltd, Riga, Latvia). Before usage, the phages were spun down for 2 h at 50 000 rpm, at 4°C, and the phosphate buffer exchanged with a 60 mM KCl water solution, to avoid M2+-phosphate complexation. RDCs were measured using J-modulated [1H,13C]-HSQC experiments (62) with a 0.7 mM 13C-, 15N-labeled D1κζ in the presence of 7 mM Mg(II), run separately on aromatic and sugar regions. NOE peak volumes were integrated using Topspin 3.0 (Bruker) or CCPN (Collaborative Computing Project for NMR), www.ccpn.ac.uk/ccpn). The data were fitted using either Origin® or gnuplot (http://www.gnuplot.info/).
To assess the dimerization, an X-double-half-filtered experiment (63) was run on a sample containing 0.6 mM D1κζ in natural isotope abundance and 0.4 mM 13C-, 15N-labeled D1κζ in the presence of 12 mM Mg(II). This experiment yields four subspectra, f1/f2 edited, f1/f2 filtered and f1 filtered/f2 edited and vice versa. If intermolecular cross-peaks are present, they should be visible in the edited/filtered subspectra.
NMR data were processed using TopSpin 3.0 (Bruker) and analyzed using Sparky (http://www.cgl.ucsf.edu/home/sparky/). The 3D experiments were analyzed using CARA (http://cara.nmr.ch/doku.php). NOE peak volumes for the structure calculation were integrated with the Gaussian peak-fitting function in Sparky.
Titration with K+, Mg(II) and Co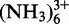
A 0.2 mM sample in H2O/D2O was titrated with increasing amounts of KCl (60, 100, 150, 200, 250, 300 and 400 mM). 1D 1H NMR experiments were recorded at each step to evaluate the changes of the imino protons. The solution became increasingly turbid on addition of large amounts of salt.
A 0.25 mM sample in H2O/D2O was titrated with increasing amounts of Mg(II) (0.5, 1, 1.5, 2, 2.5, 3 mM), and 1H-NMR was recorded at each step to follow the changes in the imino protons.
A 0.63 mM partially deuterated sample in D2O was titrated with increasing amount of Mg(II) (0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 7, 9 and 12 mM) at 300 K. [1H,1H]-NOESY experiments were recorded at each step. The chemical shift changes were plotted in bar diagrams using Origin® and gnuplot (http://www.gnuplot.info/). [1H,13C]-HSQC on the aromatic region and 2J-[1H,15N]-HSQC (in the region of N7 of guanosine and adenosine and N1 and N3 of adenosine) experiments were recorded with a 1.1 mM 13C-, 15N-labeled sample in the presence of increasing amounts of Mg(II) (0.5, 1, 1.5, 2, 3, 4, 6, 8 and 10 mM).
A 0.3 mM partially deuterated sample in D2O was titrated with increasing amounts of Co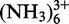 (0.6, 1.2, 1.8, 2.4, 3, 3.6, 4.2 mM) at 300 K. [1H,1H]-NOESY experiments were recorded at each step.
(0.6, 1.2, 1.8, 2.4, 3, 3.6, 4.2 mM) at 300 K. [1H,1H]-NOESY experiments were recorded at each step.
Structure calculation
Distance restraints were derived from [1H,1H]-NOESY spectra measured in D2O at 60 and 250 ms mixing time and from [1H,1H]-NOESY spectra measured in 90%H2O/10%D2O at 150 ms mixing time. Not or only moderately overlapping peaks were integrated in Sparky (http://www.cgl.ucsf.edu/home/sparky/), and distances were calibrated to pyrimidine H5H6 and H1′H2′ peak volumes using DYANA’s CALIBA macro (64). Based on these, all assignable peaks were categorized as strong (1.8–3 Å), medium (1.8–4.5 Å), weak (3–6 Å) and very weak (4–7 Å). In addition, dihedral angle restraints were used: For bases in stable base pairs and helical regions, chi was restrained to anti-conformation (−160° ± 20°). G32 was restrained to syn conformation (60° ± 20°) owing to its strong intranucleotide H8-H1′ peak, and U29, G15, A16, A17 and A18 were restrained to anti according to literature data (27,65). For all other bases, chi was left unrestrained. Sugar puckers were restrained to 3′-endo conformation (delta = 85°, nu1 = −25°, nu2 = 37.3°, ±10°) in helical regions, to 2′-endo conformation for residues showing strong H1′H2′ and medium H1′H3′ cross-peaks in 50 ms [1H,1H]-TOCSY spectra (delta = 145°, nu1 = 25°, nu2 = −35°, ±10°) (A6, A16, U30, C31 and A43) (27,66–68), and they were left unrestrained in all other cases. Backbone angles were restrained to standard A-RNA values in regions where the NOESY peak pattern and base-pairing strongly suggest standard helical conformation (alpha = –68°, beta = 178°, gamma = 54°, epsilon = −153°, zeta = −71°, ±20°), and left unrestrained in other regions. Hydrogen bonds in Watson–Crick base pairs were kept stable by short distance restraints between the hydrogen and the acceptor, as well as between the donor and the acceptor and by planarity restraints.
Starting from an extended conformation, an initial ensemble of 100 structures was calculated using CNS 1.21 (69,70) (40 ps torsion angle dynamics at 20 000 K, followed by slow cooling for 90 ps in torsion angle space and 30 ps in cartesian space). This was followed by 60 ps of refinement using XPLOR-NIH 2.24 (71,72), slowly cooling from 3000 to 50 K, during which 35 single-bond 1H-13C RDC restraints were introduced (with a force constant increasing from 0.01 to 1.0 kcal mol−1 Hz−2). Axial component and rhombicity of the alignment tensor were determined as −20.8/0.2 in a gridsearch using XPLOR-NIH (73). RDC error limits were set to generous 1.5 Hz, to allow for the uncertainty introduced by the facts that peaks are broadened in the presence of Mg(II) and the Pf1 phages.
The 20 lowest energy structures (none of them had NOE violations >0.2 Å, dihedral angle violations >5° or RDC violations >3 Hz) were selected as the final ensemble and analyzed using MolMol (74), Pymol (http://www.pymol.org) and APBS (http://www.poissonboltzmann.org/apbs) (75).
An additional set of structures was refined in XPLOR-NIH including distance restraints (6.0 Å to the central Co3+) to three cobalt(III)hexammine molecules.
RESULTS
Design of D1κζ
This work focuses on the solution structural characterization of the central core of domain 1, comprising the κ and ζ elements, of the mitochondrial group II intron Sc.ai5γ from baker’s yeast (Figure 1). To obtain a construct suitable for NMR measurements, a tetraloop needed to be inserted either at the A218-U374 or at the U386-A195 end. The resulting different constructs (Supplementary Figure S1) were transcribed and tested by NMR (see Supporting Information, Supplementary Figures S1–S5). The most promising construct was found to be a 49 nt long molecule (D1-49 in Supplementary Figure S1), to which we will refer as D1κζ throughout the text (Figure 1). Its NMR spectrum shows well-dispersed peaks, and the sequential walk could be followed all the way through, with only few exceptions (see later in the text). Moreover, good transcription yields were obtained, and all experiments were well reproducible (see Materials and Methods).
Proton attribution of D1κζ
D1κζ contains structural features common to many RNAs (76) like a three-way junction, an internal loop, as well as GAAA and UUCG tetraloops. Literature data from other RNA molecules are available on the chemical shifts of the tetraloop receptor region (ζ region) (66,68), as well as on the GAAA (27) and UUCG tetraloops (65,67). Comparison with literature data supports the assignment for the exchangeable and non-exchangeable protons. Moreover, a 27-nucleotide long construct (D1-27 in Supplementary Figure S1) was prepared and used to attribute the resonances belonging to the ζ region and the UUCG tetraloop in our experimental conditions.
D1κζ was prepared in good yields using wild-type nucleotides, partially deuterated nucleotides and 15N-, 13C-labeled nucleotides (77). In the absence of Mg(II), almost all non-exchangeable protons could be attributed except those between C11 and C14 and between A37 and A39 (located next to the junction, Figure 1), where the sequential walk is interrupted. Moreover, H1′ protons of G19 and G40 are missing (Figure 1). In line with the interruption of the sequential walk, hydrogen bonds of base pairs directly next to the κ region are not visible in a 2D JNN HNN COSY experiment (data not shown) in the absence of Mg(II). Furthermore, all neighboring base pairs (U10-A39, C14-G19, C25-G36, Figure 1) are characterized by broad imino resonances at 275 K. In total, only 9 of the 15 expected base pairs could be experimentally confirmed (Figure 2, left). The κ extension helix (Figure 1) is hardly formed at all, which is in line also with the absence of G19H1′.
Figure 2.

Imino proton region of NOESY spectra in the absence (left) and presence (right) of 10 mM Mg(II). The asterisks indicate G1 and G2 imino protons from n + 1 species (3′-end heterogeneity caused by T7 polymerase). The inset structures denote stable base-pairing that is observed in JNN HNN COSY spectra in green. Base pairs that are less stable, but still display observable imino-cross-peaks, are marked in orange. In the right panel, the NOE indicating coaxial stacking of helices d′ and d′′ is highlighted in a green box.
In summary, the resonances next to the three-way junction are either absent or very broad, and we do not observe base-pairing directly neighboring the three-way junction, indicating that no stable structure is achieved in this region in the absence of divalent ions.
Stabilization of D1κζ on Mg(II) addition
On addition of Mg(II) to D1κζ, the imino resonances that were previously missing appear (U20H3, U24H3 and G38H1, Figure 1), and their broad neighbors (U10H3, G19H1 and G36H1) sharpen. In contrast, no new signals are observed on addition of up to 300 mM KCl only (Supplementary Figure S6). The stabilization of H-bonds neighboring the three-way junction was confirmed by a 2D JNN HNN COSY experiment (data not shown). The newly observed sequential NOE cross-peak between U24H3 and G38H1 suggests that helix d′ and helix d′′ form a continuous helix (Figures 1 and 2, right).
Also, non-exchangeable proton resonances appear and increase in intensity, as observed in [1H,1H]-NOESY and can be identified with the help of [1H,13C]-HSQC spectra. At 3 mM Mg(II), the missing G19H1′ appears at 3.79 ppm, and the corresponding sugar C1′ carbon shows up in the [1H,13C]-HSQC spectrum (Figure 3). These strong upfield shifts are well-known for the nucleotides immediately following a GAAA tetraloop, due to the ring current of the preceding adenine (27). At 4 mM Mg(II), the gap in the sequential walk between C11 and C14, as well as between A37 and A39, is closed. C11H1′ is particularly strongly influenced by the addition of Mg(II). A broad peak in the absence of Mg(II), it broadens more [until 3 mM Mg(II)] and then gains intensity again, all the while moving downfield to an unusually deshielded position at 6.10 ppm (Figure 3). Interestingly, no matter the amount of added Mg(II), G40H1′ is never visible. No peaks for G40H8-H1′ and C41H6-G40H1′ could be identified under any of our experimental conditions, even if the equivalent signals in similar constructs like D1-27 and D1-45 (Supplementary Figures S1 and S2), containing the same region from helix d to d′, but no κ region, are visible and sharp. On the other hand, the missing G40H8-H1′ and C41H6-G40H1′ cross-peaks do become visible in D1κζ on the addition of cobalt(III)hexammine, suggesting a tighter binding of cobalt(III)hexammine with a stronger stabilizing effect. This is also reflected by generally sharper signals in [1H,1H]-NOESY spectra recorded in the presence of cobalt(III)hexammine (Supplementary Figure S7).
Figure 3.

[1H,1H]-NOESY (top) and [1H,13C]-HSQC (bottom) in the absence (left) and presence (right) of 9 mM Mg(II) and 12 mM Mg(II), respectively. Resonances that appear only in the presence of Mg(II) are highlighted in green. The same effect is observed with cobalt(III)hexammine (Supplementary Figure S7). All spectra were measured at 300 K, pH 6.6–6.8, 0.4–0.7 mM D1κζ, 600 or 700 MHz.
Figure 3 shows the appearance of crucial resonances at high Mg(II) concentration, both in the [1H,1H]-NOESY and in the [1H,13C]-HSQC experiments. Even if some of these signals are present also at lower Mg(II) concentration, they are sharper and better defined at 9–12 mM Mg(II) concentration.
In contrast to the other two linker regions of the junction, resonances of A22 and A23 in the third linker are observable both in the absence and in the presence of Mg(II).
In the presence of 4 mM Mg(II), a cross-peak between A18H2 and C9H1′ appears, together with a less-pronounced cross-peak between A18H2 and G8H2′. These cross-peaks would be expected for a cognate GAAA-tetraloop-receptor interaction, which would involve a base quadruple of G15-A18-G8-C41 and an AA platform involving A43 and A44 (78). Neither A18H2-C9H1′ nor A18H2-G8H2′ can be satisfied by an intramolecular interaction (Figure 4). The stronger A18H2-C9H1′ was confirmed to be an intermolecular peak in a double-half-filtered experiment (see Materials and Methods) on a sample containing both wild-type D1κζ and 13C-, 15N-enriched D1κζ and Mg(II). In addition, native gel assays showed that a small amount of the D1κζ dimer is already formed in the presence of 0.5 mM Mg(II) (data not shown).
Figure 4.

Left: Ensemble of the 20 lowest energy structures aligned over all heavy atoms, calculated including RDC restraints and in the presence of 9 mM Mg(II) (see also text). Right: Lowest energy structure. Orange: tetraloop receptor (ζ), light green: UUCG tetraloop, blue: GAAA tetraloop, dark green: κ.
Although a dimer could also be formed through an asymmetric interaction of one GAAA tetraloop of one molecule with the receptor of another, we have evidence that D1κζ forms a symmetric homodimer, with two symmetric tetraloop-tetraloop receptor (TL-TLR) interactions. In the classical TL-TLR interaction between the 11-nt TLR and a GAAA tetraloop (68), we would expect strong characteristic chemical shift changes in several tetraloop and receptor proton resonances (68), which we do not observe here. On the other hand, we see exchange cross-peaks between aromatic proton resonances of the GAAA tetraloop and resonances corresponding to the receptor-bound form of the tetraloop (68) at 9 mM Mg(II). This suggests that in contrast to the more stable contact between A18 and C9 (indicated by the sharp and pronounced A18H2-C9H1′ peak), interaction of the rest of the TL with the receptor is very transient.
One single TL-TLR interaction is not very strong (KD = 0.4 mM) so that already Davis et al. (68) followed the strategy of a homodimer to study this interaction by solution NMR. While the symmetric dimer of Davis et al. was designed for an optimal interaction, our construct obviously was not. Because the TL and the TLR receptor are not positioned exactly one helix turn apart, there is bound to be some steric hindrance for a symmetric interaction, which will prevent two stable classical TL-TLR receptor interactions. This explains why we see only exchange cross-peaks indicative of a transient interaction.
Nevertheless, the minor presence of the dimer is not expected to influence the main features of D1κζ, which are d′-d′′ coaxial stacking and the A-minor motif involving κ (see later in the text).
D1κζ forms a stable three-way junction in solution
An ensemble of D1κζ structures with an overall RMSD (Root Mean Square Deviation) of 8.21 Å was calculated using 848 NOE distance restraints collected from spectra recorded in the absence or presence of 9 mM Mg(II) [resonances peripheral from the junction are much sharper and allow for unequivocal assignment in the absence of Mg(II)] (Table 1) in restrained molecular dynamics simulations. In case of experimental evidence for Watson–Crick base pairing from sequential imino proton cross-peaks and JNN HNN COSY spectra (60), and if the peak pattern in [1H,1H]-NOESY spectra agreed with A-helical conformation, hydrogen bonds and standard backbone angles (±20°) were imposed. No backbone angles were restrained in the loop and bulge regions. Including a total of 35 RDC restraints collected in the presence of Mg(II) improved the overall RMSD to 4.3 ± 1.23 Å (Figure 4). Both UUCG and GAAA tetraloop as well as the helical regions converge well (RMSDs, see Table 1), with helix d′ and d′′ almost forming a continuous helix. The three adenines of the tetraloop receptor mostly stack into the helix, which is typical for the undocked tetraloop receptor (66) but also other conformations with protruding A6 or A44 are compatible with the NOE restraints that are relatively weak in this region due to peaks broadened by dynamics. The κ residues form a GAAA-like structure where A22, A23 and A12 stack (Figure 4) (79,80). Formation of a sheared GA base pair between G21 and A12 as is typical for a GAAA structure is compatible with our NOE restraints, but is not observed in all the structures. This is due to a scarceness of strong restraints in this region, owing to the fact that the sheared G-A base pair is not stable enough to allow the observation of its exchangeable protons. The H1′ of the nucleotide following the GAAA tetraloop at the 3′-side (here A13), that is generally strongly shielded owing to the ring current of the preceding adenine (see G19H1′ that resonates at 3.79 ppm), here is only moderately shielded. It resonates at 5.1 ppm, whereas adenosine H1′ are usually found at 5.8 ppm. This could be indicative of dynamics in this region where the tetraloop structure is only transiently formed or of a non-classical positioning of the nucleotide following the GAAA tetraloop.
Table 1.
NMR restraints and structural statistics for the D1κζ structure
| Restraint statistics | Without RDCs | With RDCs |
|---|---|---|
| NOE-derived distance restraints | 848 | 848 |
| Intranucleotide (i = j) | 256 | 256 |
| Sequential (|i–j| = 1) | 420 | 420 |
| Medium range (1 < |i–j| < 5) | 38 | 38 |
| Long range | 134 | 134 |
| Dihedral restraints | 276 | 276 |
| Hydrogen bond restraints | 83 | 83 |
| RDCs | 35 | |
| RMSDs (for all heavy atoms to mean structure) (Å) | ||
| Overall | 8.21 ± 2.69 | 4.30 ± 1.23 |
| κ extension (12–23) | 2.24 ± 0.70 | 1.82 ± 0.45 |
| Helix d′′ (24–37) | 1.56 ± 0.68 | 1.81 ± 0.66 |
| κ extension and helix d′′ (12–37) | 5.00 ± 2.10 | 3.24 ± 1.00 |
| Helix d, d′ and d′′ (1–11, 24–49) | 7.39 ± 2.82 | 4.11 ± 1.35 |
| κ, helix d′ and d′′ (8–12, 21–41) | 3.44 ± 1.20 | 2.76 ± 0.83 |
| NOE violations >0.2 Å | 0 | 0 |
| Dihedral violations >5° | 0 | 0 |
| Average NOE r.m.s. deviation (Å) | 0.007 | 0.008 |
| Average dihedral r.m.s. deviation (°) | 0.053 | 0.084 |
RMSDs are given for the 20 structures of 200 with lowest energies.
Several NOEs connect the adenines in κ to the minor groove of helix d′. The position of hydrogen bond donors at this interaction site in most of the calculated structures suggests the presence of hydrogen bonds from C11O2′ to A23N3 and A23O2′ and from U10O2′ to A22N1 (and possibly from A22O2′ to A39N3 and A39O2′) in the context of A-minor interactions (see Discussion) (81). However, the resolution especially around the A22-U10:A39 interaction is rather low, and all distances are usually longer than they should be in a hydrogen bond. Nonetheless, when the hydrogen bonds are included in the calculations as restraints, they are consistent with all experimental restraints.
Metal ion binding sites: Mg(II) and cobalt(III)hexammine
Metal ions are crucial for stabilizing RNA structure and promoting catalysis (82). By using NMR, it is possible to localize metal ions and also to quantify the strength of the interaction (83). To locate and characterize possible binding sites in D1κζ, we followed 1H, 13C and 15N chemical shift changes and line broadening in [1H,1H]-NOESY, 1J-[1H,13C]-HSQC and 2J-[1H,15N]-HSQC on Mg(II) addition (see ‘Materials and Methods’ section). Furthermore, cobalt(III)hexammine, which is often used as a mimic for the hexaqua complex of Mg(II), was added to observe NOE cross-peaks between the protons of NH3 and the protons of the RNA (84–86).
Proton resonances of a partially deuterated sample of D1κζ were attributed in [1H,1H]-NOESY spectra, and their chemical shifts plotted against increasing Mg(II) concentration. Some curves are clearly biphasic (Figure 5a), indicating a second effect, which could be structural rearrangement or a second metal ion binding close-by. This behavior mostly involves nucleotides located in the region of the GAAA loop, κ, the starting point but also in helices. As the two effects cannot be clearly separated, only a qualitative picture on metal ion binding sites within the molecule can be drawn.
Figure 5.

Chemical shift perturbations (Δδ) of G40H8 (at 300 K, 60 mM KCl) on titration with (a) MgCl2 and (b) cobalt(III)hexammine. (c) Chemical shift perturbations (at 300 K, 60 mM KCl, pD 6.85) of sugar H1′, H2′ and aromatic H6/8, H2 protons on addition of 9 mM MgCl2. Some values are missing because the resonances cannot be followed throughout the titration (H5 shifts are missing because partially deuterated samples were used).
Figure 5c shows the change in chemical shift experienced by sugar (H1′ and H2′) and aromatic protons (H6/H8/H2) on addition of 9 mM Mg(II). The nucleotides that are most strongly influenced by the presence of Mg(II) are C11 and A12 with resonances shifting by up to 0.4 ppm. C11 and A12 are in a critical position within the three-way junction, similar to A37 and G38, whose resonances also shift noticeably. In the GAAA tetraloop, we observe similar changes for all the sugar protons, without a clear pattern. Helix d′′ and the UUCG loop are remarkably unaffected by Mg(II), whereas the peaks of nucleotides located in the ζ region do shift considerably, get broader and eventually disappear when increasing the amount of metal ions. This could be due to metal ion binding, dimerization (see earlier in the text) or both.
We titrated an RNA sample with increasing amounts of cobalt(III)hexammine (from 0 to 3.6 mM, in six steps, see Materials and Methods) and evaluated both chemical shift variation and NOE cross-peaks. We observe the appearance of the same peaks around the three-way junction that also appear with Mg(II).
At 1 mM cobalt(III)hexammine concentration, it is possible to identify three binding sites through the NOE cross-peaks from the NH3 ligands to D1κζ protons: (i) the GAAA tetraloop; (ii) the GU wobble pair in the ζ region; and (iii) the CG-AU base pair step in helix d′′ (Figure 6a and b). Cross-peaks between cobalt(III)hexammine and U5H3 and U47H3 (Figure 6a) can be explained by the electrostatically favorable interaction of the complex with the major groove of the U5-G45 G-U wobble pair (87). Both the sign and magnitude of chemical shift variations induced by cobalt(III)hexammine are in general very similar to what we observe with Mg(II) (Supplementary Figure S8). However, the same chemical shift change is already reached at much lower concentration, suggesting that cobalt(III)hexammine binds at similar sites but with higher affinity (see, for example, G40H8, Figure 5a and b).
Figure 6.

(a) Sections of a [1H,1H]-NOESY spectrum recorded in 90% H2O (275 K, 0.4 mM D1κζ, pH 6.6, 60 mM KCl, 700 MHz) in the presence of 1 mM cobalt(III)hexammine indicating the cobalt(III)hexammine cross-peaks to imino protons. (b) D1κζ structure calculated including distance restraints to three cobalt(III)hexammine. Both the exchangeable and the non-exchangeable protons, which show cross-peaks to cobalt(III)hexammine are indicated as red spheres.
To evaluate possible inner sphere binding of Mg(II), 2J-[1H,15N]-HSQC spectra were recorded. Thus, direct coordination of the metal ion to the nitrogen of guanines and adenines can be detected (88), the N7 of guanines being generally the most favorable binding site (89). The chemical shift changes experienced by both N7/H8 in guanosines and adenosines as well as N1/N3/H2 in adenosines can be followed contemporaneously (88). Usually, an upfield shift of the N7 resonance on metal ion binding indicates a direct binding (88,90,91), and the corresponding H8 should be shifted downfield due to the electron-withdrawing effect of the coordinated metal ion. On the other hand, if a structural rearrangement takes place resulting in increased stacking, the H8 resonances would move upfield.
Figure 7 shows the overlay of [1H,15N]-HSQC spectra recorded with increasing amounts of Mg(II). Residues in the helices show a moderate upfield shift of N7 (<1 ppm) and almost no shift of N1 and N3, which is consistent with a general non-specific interaction with the N7 and bad accessibility of N1 and N3. In the ζ region, N1 of A6, A43, A44 and N7 of G45 broaden out and eventually disappear on addition of Mg(II). Such line broadening can be due to the ligand exchange rate in the first coordination shell of Mg(II), which is on the NMR timescale (88) and thus suggests inner-sphere coordination in the tetraloop receptor region. On the other hand, also 1H and 13 C resonances broaden out, and the cross-peaks observed in [1H,1H]-NOESY involving A43, A44 and G45 get broader more quickly in the presence of cobalt(III)hexammine than with Mg(II), and in the first case, this effect cannot be due to inner-sphere coordination. In addition, we have described earlier in the text that there are strong indications for a homodimer formation, which includes transient interactions of the GAAA tetraloop with the ζ region and is also expected to lead to a broadening of the signals. We observe a similar behavior for all N7 of the GAAA tetraloop nucleotides: the resonances move upfield more strongly than residues in the helices (>1 ppm) and without broadening. Similarly, also A22N7 and G21N7 in the κ tetraloop structure move upfield without broadening. A12N1 and A22N1 move by almost 2 ppm, whereas A17N1 and A18N1 turn around at ∼2 mM Mg(II) hinting at the interplay of structural rearrangements and metal ion interactions in this region (Figure 7).
Figure 7.

Overlay of 2J-[1H,15 N]-HSQC spectra in the region of purine N1, N3 and N7 resonances in the presence of increasing amounts of Mg(II). Resonances disappearing after the first addition of 0.5 mM Mg(II) are labeled in red. (300 K, 0.45 mM D1κζ, pD 6.74, 60 mM KCl, 10 μM EDTA, 700 MHz).
Mg(II) as a hard metal ion prefers oxygen over nitrogen binding. If there is nonetheless a strong direct interaction, it is expected to move the 15N resonance upfield by ∼6 ppm (90,92). None of our observable N1, N3 or N7 moves that much; only the G1N7 moves by 3.4 ppm, and this behavior is explained by the charge of the tri- or diphosphate present.
DISCUSSION
General structural features: the 11-nt tetraloop receptor and two classical tetraloops
The D1κζ construct contains several common structural motifs that have already been described in the literature. Both the GAAA and UUCG tetraloops as well as the tetraloop receptor region (ζ) agree reasonably well with published solution structures [2JYJ (68), 2KOC (65), 1TLR (66)]. The tetraloop receptor in its unbound form is characterized by a zipper-like stacking of A6 between A43 and A44, a U7-U43 mismatch and a G45-U5 wobble pair (66). Such an arrangement of the tetraloop receptor has been proposed also in the context of the complete D1 before docking of D5 (28) and is now confirmed by our observed NOE pattern in the absence of Mg(II).
κ forms an A-minor junction
In solution, the κ region (A12, G21-A23) of D1κζ folds into a GAAA tetraloop-like structure similar to the one in the published crystal structure of a group IIC intron from O. iheyensis (38–40) (Figures 8 and 9). The orientation of this motif differs from the one in the fully assembled group II intron (Figures 8 and 9). In the full-length group IIC intron structure, the κ region engages in H-bonds with the two G-C pairs of κ′ in D5 through the Hoogsteen face of A22 (N7), the WC face of A23 (N1) and the sugar face of A12 (N3) (numbering of D1κζ). In contrast, in D1κζ A22 and A23 form A-minor interactions to helix d′ (see Results, Figure 10a and b). The interactions can be classified as type 0 (A23N3 and O2′ to C11O2′, Figure 10a) and type I (A22N1 to U10O2′, A22O2′ to A39N3 and O2′, Figure 10b) according to Nissen et al. (81) and decrease in type order from 5′ to 3′ (81). An investigation of three-way junctions found in published crystal structures by Lescoute and Westhof (93) suggests that such an interaction as observed here, where adenines inside a junction engage in A-minor interactions to the adjacent helix, is common. Lescoute and Westhof classified the three-way junctions into three families. The D1κζ junction belongs to family C, which is the most abundant and appears in various structured RNAs. In this family, J31 is bigger than J23 and J12, and helix P3 (the κ extension in D1κζ) leans towards helix P1 (helix d′′) (Figure 10c and d). In addition, the J31 linker can contact the shallow minor groove of helix P2 (helix d′) (Figure 10d). Moreover, Lescoute and Westhof predict that the 0-nt linker will most likely be connecting helices that are coaxially stacking, which is also true in our three-way junction. Geary et al. (45) have even used the concept of A-minor interactions from a terminal GNRA-like loop to an adjacent helix to stabilize specific conformations of RNA four-way junctions. The three-way junction in D1κζ is completely homologous to the constructs they used, if the junction is moved by a 1–2 base pairs directly into the GNRA loop (Figure 10d and e). In D1κζ, the crossing of the strands takes place directly between A23 and A12, which are the third and the fourth nucleotide of the GAAA-motif (Figure 10d). Although it has been recognized long ago that the GNRA motif is a very favorable fold that can form even when the sequence is interrupted, database searches using FR3D (94) and RNA FRABASE (95) do not find this motif inside a three-way junction in any other structure except the O. iheyensis crystal structures (38–40).
Figure 8.

(a) Overlay of D1κζ (orange) with the corresponding region in the O. iheyensis group IIC intron crystal structure [dark grey; PDBID: 3G78, (12,40)]. The heavy atoms of the κ element were overlaid to visualize the different orientations of κ towards the rest of the molecule in the two structures. In green, the coordination loop adjacent to helix d′′ of the O. iheyensis group IIC intron interacting with the GAACA loop is shown. (b) Schematic representation of the structures depicted in (a). Tertiary interactions are indicated by asterisks. In D1κζ in solution (orange) κ interacts with helix d′, which probably disfavors the docking of D5 (shown in red). The GAAA tetraloop adjacent to κ does not interact with helix d′′. In the O. iheyensis crystal structure, the loop adjacent to κ interacts with the coordination loop, possibly disfavoring the inter-domain interaction observed in D1κζ and favoring docking of D5.
Figure 9.

(a) The three-way junction as found in the O. iheyensis group IIC intron. Interaction of κ (green) with κ′ in D5 (red) is indicated. Phosphate groups equivalent to those of A12 and U24 in D1κζ are colored. (b) A-minor interaction of κ residues A22 and A23 with helix d′ in D1κζ. Examples of observed NOE distances are indicated as blue dotted lines.
Figure 10.

(a) and (b) Detail of the A-minor interactions as observed in the ensemble of the 10 lowest energy D1κζ solution structures. (c) Schematic representation of a three-way junction indicating the nomenclature used to describe three-way junctions in the text, according to Lescoute and Westhof (93). (d) Schematic representation of the three-way junction within D1κζ. The color code of the junctions is in accordance to (c). The critical nucleotides within the junction are numbered; the red arrows indicate the A-minor interactions. (e) Schematic representation of an A-minor junction as described by Geary et al. (45).
The interaction of κ with helix d′ also helps to understand why the H1′ resonance of G40 is not observed in D1κζ. At first sight, this is unexpected considering that the residue is within a stable base pair in a helical region, and that the H1′ resonance is present and sharp in constructs lacking the κ region (see constructs D1-27 and D1-45 in Supplementary Figures S1 and S2). However, the final structures of D1κζ position G40H1′ just below the contact interface of κ and helix d′, where it transiently gets into contact with A22, inducing dynamics on a timescale that completely broadens the resonance.
Our spectra indicate that this A-minor interaction is strong and independent of the presence of Mg(II). In the crystal structure, comprising the whole intron in its native state, however, this A-motif is not present, but κ is positioned differently. There are two plausible reasons for that: (i) the docking of D5 itself switches the κ interaction or (ii) the contact between the capping loop of the κ extension (GAAA in our structure, GAACA in the O. iheyensis intron) and the lower part of the neighboring so-called coordination loop as observed in the crystal structure (Figure 8) is strong enough to disrupt the A-minor interactions. To prove the first hypothesis, we looked at D5 and D1κζ in solution, but could not detect inter-molecular NOEs or any other signs of interaction, e.g. in native gel studies (data not shown). The reason could be that the two constructs need to be embedded in the complex framework of the group II intron to adopt the global conformations that allow a favorable interaction. On the other hand, we can also not exclude that D5 in the full intron will be the factor that leads to the release of the A-minor interactions, as also the interactions to the coordination loop (second hypothesis) are not absolutely required for a functional group II intron. When the GAAA tetraloop is replaced by UUCG, full catalytic activity is retained (96). In the context of the full intron, a combination of the two factors might control the final orientation of the κ motif.
Mg(II) promotes coaxial stacking of helices d′ and d′′
On addition of Mg(II) and cobalt(III)hexammine, the appearance and sharpening of resonances neighboring the junction indicate that the three-way junction around κ is structurally stabilized in the presence of those metal ions. Effects of the stabilization are noticeable not only directly at the junction but extend also to the second base pairs in all three helices, i.e. d′, d′′ and the κ extension. The appearance of an NOE cross-peak between U24H3 and G38H1 is strong evidence for a stable coaxial stacking of helix d′ and d′′, only in the presence of Mg(II) or cobalt(III)hexammine. This coaxial stacking is observed also in the group IIC intron crystal structure (39) and thus is independent of the orientation of the κ extension, which bulges out between residue 11 and 24 (D1κζ numbering) in the crystal and in solution.
In our D1κζ structure, stacking between C11 and U24 as well as between A12 and A23 is observed. At the same time the two phosphate-sugar backbones approach each other very closely. Specifically, A12OP2 and U24OP1 are only ∼4 Å apart in most of the calculated structures (Figure 11), a configuration that is likely to require a charge-compensating metal ion. No Mg(II) ion was found at this location in the crystal structure of O. iheyensis intron, but A12 displayed a strong phosphorothioate effect in studies with the D135 ribozyme (28). The distance of the A12OP2 and U24OP1 phosphate oxygens is well within the range of a bridging Mg(II) ion. The fact that cobalt(III)hexammine has a stabilizing effect equivalent to Mg(II) indicates that direct inner-sphere coordination is not a requirement for a cation to be able to stabilize the region.
Figure 11.

Electrostatic surface potential map of D1κζ viewed from the top, looking at the three-way junction from between the κ extension and helix d′′. The highest negative density is observed at the phosphate groups of A12 and U24.
Obviously, di- or multivalent ions are a prerequisite for this close approach of the two backbones, and thus a stable coaxial stacking of helix d′ and helix d′′. Coaxial stacking is very common for helices in multibranch junctions in crystal structures (43). In solution, it has been shown for the four-way junction of the hairpin ribozyme that different conformations are sampled at low ionic strength, whereas one dominant coaxially stacked conformation is found under high salt conditions (97,98). We observe here a similar case: the presence of Mg(II) or cobalt(III)hexammine strongly stabilizes coaxial stacking of helix d′ and d′′.
High affinity metal ion binding within D1κζ
D1κζ was titrated with increasing amounts of Mg(II) or cobalt(III)hexammine. As described above, both ions behave similarly, although less cobalt(III)hexammine is needed for structural stabilization, well in line with the higher positive charge. The nucleotides most affected by the metal ion addition are near the tetraloop receptor and the three-way junction. For example, the strong chemical shift changes observed for G8H1′, C11H1′ and A12H2′ can be explained by the metal ion-dependent structural stabilization of the three-way junction (see earlier in the text). This clustering of residues around the three-way junction being affected by Mg(II) binding is well in line with earlier biochemical studies, where interferences of thiophosphate groups and purine N7 positions, both being primary Mg(II) binding sites, have been described (28).
The GAAA tetraloop resonances are strongly affected by both Mg(II) and cobalt(III)hexammine. Previous spectroscopic data proved the presence of a localized binding site in the GAAA tetraloop (99), with KD values in the low millimolar range (27,85,100). In our case, many chemical shift variations in this region show a biphasic behavior (see e.g. G40H8 in Figure 5a), which is likely due to the effects of both metal-ion interactions and metal-ion induced structural stabilization of the whole κ region. As the two effects cannot be distinguished spectroscopically at the various binding sites within D1κζ, a calculation of reliable KD value is so far not possible.
The triphosphate at the 5′-end is a strong Mg(II) binding site, and this is generally responsible for the disappearing of resonances from the first nucleotides at the 5′-end during the first steps of metal ion titration (101). Interestingly, here, these cross-peaks do not disappear. This behavior is rather unusual and can be explained by the presence of another high affinity site for Mg(II): our data imply a strong Mg(II) binding site within the three-way junction that competes with the interaction of Mg(II) at the 5′-end. The intrinsic dissociation constants for Mg(II) binding to 5′-triphosphates were found to be between 0.1 and 0.5 mM (100). As we observe structural changes in the three-way junction at already low mM concentration of Mg(II), this means that the dissociation constants must be in the same order of magnitude, i.e. in the micromolar range.
CONCLUSIONS
The structural stabilization of the three-way junction in the κ-ζ region within D1 of the group II intron Sc.ai5γ on Mg(II) binding was characterized in detail by NMR spectroscopy. The main feature of the stabilized structure is the coaxial stacking of helices d′ and d′′, which leads to a continuous helix across the three-way junction. Coaxial stacking of helices is also observed in the same region of D1 in the crystal structure of the group IIC intron O. iheyensis. We could show that also the GAAA-tetraloop motif of the κ-element observed in the crystal structure is already present in solution at low Mg(II) concentration. This demonstrates once more how very favorable this GAAA-fold is even if interrupted by a helix and in the absence of a tertiary interaction partner. In contrast to the crystal structure, in the isolated D1κζ in the absence of other parts of the intron, this motif is engaged in A-minor interactions to the adjacent d′ helix. A-minor motifs together with coaxial stacking are among the most significant interactions in shaping RNA architecture, and both the structural motifs are combined in our small D1κζ construct.
In conclusion, the structure of D1κζ in the presence of Mg(II) shows that essential structural features of the final catalytically prone group II intron core are already formed in the absence of any tertiary contacts, as present in the early steps of folding. This suggests that the metal ion-induced rigidification of the three-way junction may be one of the first steps in group II intron folding.
ACCESSION NUMBERS
Atomic coordinates for the refined structures have been deposited with the Protein Data Bank under accession code 2LU0. The NMR chemical shifts have been deposited at the Biological Magnetic Resonance Data Bank (BMRB) under the accession code 18503.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–8 and Supplementary Methods including references.
FUNDING
Marie Curie Intra-European Fellowship (FP7-MC-IEF) [236797 to D.D.]; Bio-NMR Research Infrastructure (FP7/2007-2013) [261863]; Swiss National Science Foundation [200021-124834 to R.K.O.S.]; Roche Research Foundation (to C.F. and R.K.O.S.); Novartis Stiftung (to C.F. and R.K.O.S.); University of Zurich. Funding for open access charge: University of Zurich.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
Financial support by a Marie Curie Intra European Fellowship (to D.D.) and in the form of access to the Bio-NMR Research Infrastructure within the 7th European Community Framework Programme, by the Roche Research Foundation, the Novartis Stiftung (both to C.F. and R.K.O.S.), the Swiss National Science Foundation (to R.K.O.S.) and the University of Zurich are gratefully acknowledged. R.K.O.S. is holder of an ERC starting grant 2010. The authors thank the Biochemistry Department at the University of Zurich for the use of the ultra-centrifuge.
REFERENCES
- 1.Michel F, Jacquier A, Dujon B. Comparison of fungal mitochondrial introns reveals extensive homologies in RNA secondary structure. Biochimie. 1982;64:867–881. doi: 10.1016/s0300-9084(82)80349-0. [DOI] [PubMed] [Google Scholar]
- 2.Michel F, Ferat JL. Structure and activities of group II introns. Annu. Rev. Biochem. 1995;64:435–461. doi: 10.1146/annurev.bi.64.070195.002251. [DOI] [PubMed] [Google Scholar]
- 3.Lehmann K, Schmidt U. Group II introns: structure and catalytic versatility of large natural ribozymes. Crit. Rev. Biochem. Mol. Biol. 2003;38:249–303. doi: 10.1080/713609236. [DOI] [PubMed] [Google Scholar]
- 4.Boeke JD. The unusual phylogenetic distribution of retrotransposons: a hypothesis. Genome Res. 2003;13:1975–1983. doi: 10.1101/gr.1392003. [DOI] [PubMed] [Google Scholar]
- 5.Keating KS, Toor N, Perlman PS, Pyle AM. A structural analysis of the group II intron active site and implications for the spliceosome. RNA. 2010;16:1–9. doi: 10.1261/rna.1791310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dayie KT, Padgett RA. A glimpse into the active site of a group II intron and maybe the spliceosome, too. RNA. 2008;14:1697–1703. doi: 10.1261/rna.1154408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michel F, Costa M, Westhof E. The ribozyme core of group II introns: a structure in want of partners. Trends Biochem. Sci. 2009;34:189–199. doi: 10.1016/j.tibs.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 8.Toor N, Keating KS, Pyle AM. Structural insights into RNA splicing. Curr. Opin. Struct. Biol. 2009;19:260–266. doi: 10.1016/j.sbi.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pyle AM. The tertiary structure of group II introns: implications for biological function and evolution. Crit. Rev. Biochem. Mol. Biol. 2010;45:215–232. doi: 10.3109/10409231003796523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambowitz AM, Belfort M. Introns as mobile genetic elements. Annu. Rev. Biochem. 1993;62:587–622. doi: 10.1146/annurev.bi.62.070193.003103. [DOI] [PubMed] [Google Scholar]
- 11.Lambowitz AM, Zimmerly S. Mobile group II introns. Annu. Rev. Genet. 2004;38:1–35. doi: 10.1146/annurev.genet.38.072902.091600. [DOI] [PubMed] [Google Scholar]
- 12.Dai L, Toor N, Olson R, Keeping A, Zimmerly S. Database for mobile group II introns. Nucleic Acids Res. 2003;31:424–426. doi: 10.1093/nar/gkg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sigel RKO. Group II intron ribozymes and metal ions - A delicate relationship. Eur. J. Inorg. Chem. 2005;2005:2281–2292. [Google Scholar]
- 14.DeRose VJ. Metal ion binding to catalytic RNA molecules. Curr. Opin. Struct. Biol. 2003;13:317–324. doi: 10.1016/s0959-440x(03)00077-0. [DOI] [PubMed] [Google Scholar]
- 15.Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc. Natl Acad. Sci. USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fedor MJ. The role of metal ions in RNA catalysis. Curr. Opin. Struct. Biol. 2002;12:289–295. doi: 10.1016/s0959-440x(02)00324-x. [DOI] [PubMed] [Google Scholar]
- 17.Sigel RKO, Pyle AM. Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem. Rev. 2007;107:97–113. doi: 10.1021/cr0502605. [DOI] [PubMed] [Google Scholar]
- 18.Qin PZ, Pyle AM. The architectural organization and mechanistic function of group II intron structural elements. Curr. Opin. Struct. Biol. 1998;8:301–308. doi: 10.1016/s0959-440x(98)80062-6. [DOI] [PubMed] [Google Scholar]
- 19.Toor N, Hausner G, Zimmerly S. Coevolution of group II intron RNA structures with their intron-encoded reverse transcriptases. RNA. 2001;7:1142–1152. doi: 10.1017/s1355838201010251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin PZ, Pyle AM. Stopped-flow fluorescence spectroscopy of a group II intron ribozyme reveals that domain 1 is an independent folding unit with a requirement for specific Mg2+ ions in the tertiary structure. Biochemistry. 1997;36:4718–4730. doi: 10.1021/bi962665c. [DOI] [PubMed] [Google Scholar]
- 21.Su LJ, Waldsich C, Pyle AM. An obligate intermediate along the slow folding pathway of a group II intron ribozyme. Nucleic Acids Res. 2005;33:6674–6687. doi: 10.1093/nar/gki973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shukla GC, Padgett RA. A catalytically active group II intron domain 5 can function in the U12-dependent spliceosome. Mol. Cell. 2002;9:1145–1150. doi: 10.1016/s1097-2765(02)00505-1. [DOI] [PubMed] [Google Scholar]
- 23.Erat MC, Zerbe O, Fox T, Sigel RKO. Solution structure of domain 6 from a self-splicing group II intron ribozyme: a Mg2+ binding site is located close to the stacked branch adenosine. ChemBioChem. 2007;8:306–314. doi: 10.1002/cbic.200600459. [DOI] [PubMed] [Google Scholar]
- 24.Costa M, Michel F. Frequent use of the same tertiary motif by self-folding RNAs. EMBO J. 1995;14:1276–1285. doi: 10.1002/j.1460-2075.1995.tb07111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boudvillain M, Pyle AM. Defining functional groups, core structural features and inter-domain tertiary contacts essential for group II intron self-splicing: a NAIM analysis. EMBO J. 1998;17:7091–7104. doi: 10.1093/emboj/17.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sigel RKO, Vaidya A, Pyle AM. Metal ion binding sites in a group II intron core. Nat. Struct. Biol. 2000;7:1111–1116. doi: 10.1038/81958. [DOI] [PubMed] [Google Scholar]
- 27.Sigel RKO, Sashital DG, Abramovitz DL, Palmer AG, Butcher SE, Pyle AM. Solution structure of domain 5 of a group II intron ribozyme reveals a new RNA motif. Nat. Struct. Mol. Biol. 2004;11:187–192. doi: 10.1038/nsmb717. [DOI] [PubMed] [Google Scholar]
- 28.Waldsich C, Pyle AM. A folding control element for tertiary collapse of a group II intron ribozyme. Nat. Struct. Mol. Biol. 2007;14:37–44. doi: 10.1038/nsmb1181. [DOI] [PubMed] [Google Scholar]
- 29.Waldsich C, Pyle AM. A kinetic intermediate that regulates proper folding of a group II intron RNA. J. Mol. Biol. 2008;375:572–580. doi: 10.1016/j.jmb.2007.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swisher JF, Su LJ, Brenowitz M, Anderson VE, Pyle AM. Productive folding to the native state by a group II intron ribozyme. J. Mol. Biol. 2002;315:297–310. doi: 10.1006/jmbi.2001.5233. [DOI] [PubMed] [Google Scholar]
- 31.Steiner M, Karunatilaka KS, Sigel RKO, Rueda D. Single-molecule studies of group II intron ribozymes. Proc. Natl Acad. Sci. USA. 2008;105:13853–13858. doi: 10.1073/pnas.0804034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steiner M, Rueda D, Sigel RKO. Ca2+ induces the formation of two distinct subpopulations of group II intron molecules. Angew. Chem. Int. Ed. 2009;48:9739–9742. doi: 10.1002/anie.200903809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sosnick TR, Pan T. RNA folding: models and perspectives. Curr. Opin. Struct. Biol. 2003;13:309–316. doi: 10.1016/s0959-440x(03)00066-6. [DOI] [PubMed] [Google Scholar]
- 34.Pyle AM, Fedorova O, Waldsich C. Folding of group II introns: a model system for large, multidomain RNAs? Trends Biochem. Sci. 2007;32:138–145. doi: 10.1016/j.tibs.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Fedorova O, Solem A, Pyle AM. Protein-facilitated folding of group II intron ribozymes. J. Mol. Biol. 2010;397:799–813. doi: 10.1016/j.jmb.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karunatilaka KS, Solem A, Pyle AM, Rueda D. Single-molecule analysis of Mss116-mediated group II intron folding. Nature. 2010;467:935–939. doi: 10.1038/nature09422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fedorova O, Pyle AM. The brace for a growing scaffold: Mss116 protein promotes RNA folding by stabilizing an early assembly intermediate. J. Mol. Biol. 2012;422:347–365. doi: 10.1016/j.jmb.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toor N, Rajashankar K, Keating KS, Pyle AM. Structural basis for exon recognition by a group II intron. Nat. Struct. Mol. Biol. 2008;15:1221–1222. doi: 10.1038/nsmb.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J. Inclusion of weak high-resolution X-ray data for improvement of a group II intron structure. Acta Crystallogr. D Biol. Crystallogr. 2010;66:988–1000. doi: 10.1107/S0907444910029938. [DOI] [PubMed] [Google Scholar]
- 41.Costa M, Michel F, Westhof E. A three-dimensional perspective on exon binding by a group II self-splicing intron. EMBO J. 2000;19:5007–5018. doi: 10.1093/emboj/19.18.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butcher SE, Pyle AM. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Acc. Chem. Res. 2012;44:1302–1311. doi: 10.1021/ar200098t. [DOI] [PubMed] [Google Scholar]
- 43.Tyagi R, Mathews DH. Predicting helical coaxial stacking in RNA multibranch loops. RNA. 2007;13:939–951. doi: 10.1261/rna.305307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lilley DMJ. Structures of helical junctions in nucleic acids. Q. Rev. Biophys. 2000;33:109–159. doi: 10.1017/s0033583500003590. [DOI] [PubMed] [Google Scholar]
- 45.Geary C, Chworos A, Jaeger L. Promoting RNA helical stacking via A-minor junctions. Nucleic Acids Res. 2011;39:1066–1080. doi: 10.1093/nar/gkq748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan RT, Robart AR, Rajashankar KR, Pyle AM, Toor N. Crystal structure of a group II intron in the pre-catalytic state. Nat. Struct. Mol. Biol. 2012;19:555–557. doi: 10.1038/nsmb.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seetharaman M, Eldho NV, Padgett RA, Dayie KT. Structure of a self-splicing group II intron catalytic effector domain 5: Parallels with spliceosomal U6 RNA. RNA. 2006;12:235–247. doi: 10.1261/rna.2237806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Doudna JA. Structural insights into group II intron catalysis and branch-site selection. Science. 2002;295:2084–2088. doi: 10.1126/science.1069268. [DOI] [PubMed] [Google Scholar]
- 49.Gallo S, Furler M, Sigel RKO. In vitro transcription and purification of RNAs of different size. Chimia. 2005;59:812–816. [Google Scholar]
- 50.Brauer G, Handa BK. Heterotype Mischkristalle bei Hexammincobaltjodiden. Z. Anorg. Allg. Chem. 1964;329:12–17. [Google Scholar]
- 51.Sigel H, Zuberbühler AD, Yamauchi O. Comments on potentiometric pH titrations and the relationship between pH-meter reading and hydrogen ion concentration. Anal. Chim. Acta. 1991;255:63–72. [Google Scholar]
- 52.Kankia BI, Buckin V, Bloomfield VA. Hexamminecobalt(III)-induced condensation of calf thymus DNA: circular dichroism and hydration measurements. Nucleic Acids Res. 2001;29:2795–2801. doi: 10.1093/nar/29.13.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kao C, Rüdisser S, Zheng M. A simple and efficient method to transcribe RNAs with reduced 3′ heterogeneity. Methods. 2001;23:201–205. doi: 10.1006/meth.2000.1131. [DOI] [PubMed] [Google Scholar]
- 54.Sigel H, Griesser R. Nucleoside 5′-triphosphates: self-association, acid-base, and metal ion-binding properties in solution. Chem. Soc. Rev. 2005;34:875–900. doi: 10.1039/b505986k. [DOI] [PubMed] [Google Scholar]
- 55.Flinders J, Dieckmann T. NMR spectroscopy of ribonucleic acids. Prog. Nucl. Magn. Reson. Spectrosc. 2006;48:137–159. [Google Scholar]
- 56.Fürtig B, Richter C, Wöhnert J, Schwalbe H. NMR spectroscopy of RNA. ChemBioChem. 2003;4:936–962. doi: 10.1002/cbic.200300700. [DOI] [PubMed] [Google Scholar]
- 57.Latham MP, Brown DJ, McCallum SA, Pardi A. NMR methods for studying the structure and dynamics of RNA. ChemBioChem. 2005;6:1492–1505. doi: 10.1002/cbic.200500123. [DOI] [PubMed] [Google Scholar]
- 58.Schanda P, Brutscher B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J. Am. Chem. Soc. 2005;127:8014–8015. doi: 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- 59.Davis AL, Keeler J, Laue ED, Moskau D. Experiments for recording pure-absorption heteronuclear correlation spectra using pulsed field gradients. J. Magn. Reson. 1992;98:207–216. [Google Scholar]
- 60.Dingley AJ, Grzesiek S. Direct observation of hydrogen bonds in nucleic acid base pairs by internucleotide 2JNN couplings. J. Am. Chem. Soc. 1998;120:8293–8297. [Google Scholar]
- 61.Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wüthrich K. Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Mol. Biol. 1998;280:933–952. doi: 10.1006/jmbi.1998.1852. [DOI] [PubMed] [Google Scholar]
- 62.Tjandra N, Bax A. Measurement of dipolar contributions to 1JCH splittings from magnetic-field dependence of J modulation in two-dimensional NMR spectra. J. Magn. Reson. 1997;124:512–515. doi: 10.1006/jmre.1996.1088. [DOI] [PubMed] [Google Scholar]
- 63.Otting G, Wüthrich K. Extended heteronuclear editing of 2D 1H NMR spectra of isotope-labeled proteins, using the X(w1, w2) double half filter. J. Magn. Reson. 1989;85:586–594. [Google Scholar]
- 64.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 65.Nozinovic S, Fürtig B, Jonker HRA, Richter C, Schwalbe H. High-resolution NMR structure of an RNA model system: the 14-mer cUUCGg tetraloop hairpin RNA. Nucleic Acids Res. 2010;38:683–694. doi: 10.1093/nar/gkp956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Butcher SE, Dieckmann T, Feigon J. Solution structure of a GAAA tetraloop receptor RNA. EMBO J. 1997;16:7490–7499. doi: 10.1093/emboj/16.24.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fürtig B, Richter C, Bermel W, Schwalbe H. New NMR experiments for RNA nucleobase resonance assignment and chemical shift analysis of an RNA UUCG tetraloop. J. Biomol. NMR. 2004;28:69–79. doi: 10.1023/B:JNMR.0000012863.63522.1f. [DOI] [PubMed] [Google Scholar]
- 68.Davis JH, Tonelli M, Scott LG, Jaeger L, Williamson JR, Butcher SE. RNA helical packing in solution: NMR structure of a 30 kDa GAAA tetraloop-receptor complex. J. Mol. Biol. 2005;351:371–382. doi: 10.1016/j.jmb.2005.05.069. [DOI] [PubMed] [Google Scholar]
- 69.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 70.Brünger AT. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 71.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 72.Schwieters CD, Kuszewski JJ, Clore GM. Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006;48:47–62. [Google Scholar]
- 73.Clore GM, Gronenborn AM, Tjandra N. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J. Magn. Reson. 1998;131:159–162. doi: 10.1006/jmre.1997.1345. [DOI] [PubMed] [Google Scholar]
- 74.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
- 75.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Batey RT, Rambo RP, Doudna JA. Tertiary motifs in RNA structure and folding. Angew. Chem., Int. Ed. 1999;38:2326–2343. doi: 10.1002/(sici)1521-3773(19990816)38:16<2326::aid-anie2326>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 77.Lu K, Miyazaki Y, Summers MF. Isotope labeling strategies for NMR studies of RNA. J. Biomol. NMR. 2010;46:113–125. doi: 10.1007/s10858-009-9375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Szewczak AA, Kundrot CE, Cech TR, Doudna JA. RNA tertiary structure mediation by adenosine platforms. Science. 1996;273:1696–1699. doi: 10.1126/science.273.5282.1696. [DOI] [PubMed] [Google Scholar]
- 79.Heus HA, Pardi A. Structural features that give rise to the unusual stability of RNA hairpins containing GNRA loops. Science. 1991;253:191–194. doi: 10.1126/science.1712983. [DOI] [PubMed] [Google Scholar]
- 80.Abramovitz DL, Pyle AM. Remarkable morphological variability of a common RNA folding motif: the GNRA tetraloop-receptor interaction. J. Mol. Biol. 1997;266:493–506. doi: 10.1006/jmbi.1996.0810. [DOI] [PubMed] [Google Scholar]
- 81.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc. Natl Acad. Sci. USA. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Donghi D, Schnabl J. Multiple roles of metal ions in large ribozymes. Met. Ions Life Sci. 2011;9:197–234. doi: 10.1039/9781849732512-00197. [DOI] [PubMed] [Google Scholar]
- 83.Donghi D, Sigel RKO. Metal ion-RNA interactions studied via multinuclear NMR. Methods Mol. Biol. 2012;848:253–273. doi: 10.1007/978-1-61779-545-9_16. [DOI] [PubMed] [Google Scholar]
- 84.Gonzalez RL, Tinoco I. Solution structure and thermodynamics of a divalent metal ion binding site in an RNA pseudoknot. J. Mol. Biol. 1999;289:1267–1282. doi: 10.1006/jmbi.1999.2841. [DOI] [PubMed] [Google Scholar]
- 85.Rüdisser S, Tinoco I. Solution structure of Cobalt(III)hexammine complexed to the GAAA tetraloop, and metal-ion binding to G.A mismatches. J. Mol. Biol. 2000;295:1211–1223. doi: 10.1006/jmbi.1999.3421. [DOI] [PubMed] [Google Scholar]
- 86.Gonzalez RL, Tinoco I. Identification and characterization of metal ion binding sites in RNA. Methods Enzymol. 2001;338:421–443. doi: 10.1016/s0076-6879(02)38231-4. [DOI] [PubMed] [Google Scholar]
- 87.Kieft JS, Tinoco I. Solution structure of a metal-binding site in the major groove of RNA complexed with cobalt(III)hexammine. Structure. 1997;5:713–721. doi: 10.1016/s0969-2126(97)00225-6. [DOI] [PubMed] [Google Scholar]
- 88.Erat MC, Kovacs H, Sigel RKO. Metal ion-N7 coordination in a ribozyme branch domain by NMR. J. Inorg. Biochem. 2010;104:611–613. doi: 10.1016/j.jinorgbio.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 89.Freisinger E, Sigel RKO. From nucleotides to ribozymes—a comparison of their metal ion binding properties. Coord. Chem. Rev. 2007;251:1834–1851. [Google Scholar]
- 90.Tanaka Y, Taira K. Detection of RNA nucleobase metalation by NMR spectroscopy. Chem. Commun. 2005;2005:2069–2079. doi: 10.1039/b415137m. [DOI] [PubMed] [Google Scholar]
- 91.Tanaka Y, Ono A. Nitrogen-15 NMR spectroscopy of N-metallated nucleic acids: insights into 15N NMR parameters and N-metal bonds. Dalton Trans. 2008:4965–4974. doi: 10.1039/b803510p. [DOI] [PubMed] [Google Scholar]
- 92.Wang G, Gaffney BL, Jones RA. Differential binding of Mg2+, Zn2+, and Cd2+ at two sites in a hammerhead ribozyme motif, determined by 15N NMR. J. Am. Chem. Soc. 2004;126:8908–8909. doi: 10.1021/ja049804v. [DOI] [PubMed] [Google Scholar]
- 93.Lescoute A, Westhof E. Topology of three-way junctions in folded RNAs. RNA. 2006;12:83–93. doi: 10.1261/rna.2208106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sarver M, Zirbel CL, Stombaugh J, Mokdad A, Leontis NB. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J. Math. Biol. 2008;56:215–252. doi: 10.1007/s00285-007-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Popenda M, Szachniuk M, Blazewicz M, Wasik S, Burke EK, Blazewicz J, Adamiak RW. RNA FRABASE 2.0: an advanced web-accessible database with the capacity to search the three-dimensional fragments within RNA structures. BMC Bioinformatics. 2010;11:231. doi: 10.1186/1471-2105-11-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hamill S, Pyle AM. The receptor for branch-site docking within a group II intron active site. Mol. Cell. 2006;23:831–840. doi: 10.1016/j.molcel.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 97.Jaeger L, Leontis NB. Tecto-RNA: one-dimensional self-assembly through tertiary interactions. Angew. Chem. Int. Ed. 2000;39:2521–2524. doi: 10.1002/1521-3773(20000717)39:14<2521::aid-anie2521>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 98.Nasalean L, Baudrey S, Leontis NB, Jaeger L. Controlling RNA self-assembly to form filaments. Nucleic Acids Res. 2006;34:1381–1392. doi: 10.1093/nar/gkl008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maderia M, Horton TE, DeRose VJ. Metal interactions with a GAAA RNA tetraloop characterized by 31P NMR and phosphorothioate substitutions. Biochemistry. 2000;39:8193–8200. doi: 10.1021/bi000140l. [DOI] [PubMed] [Google Scholar]
- 100.Erat MC, Coles J, Finazzo C, Knobloch B, Sigel RKO. Accurate analysis of Mg2+ binding to RNA: from classical methods to a novel iterative calculation procedure. Coord. Chem. Rev. 2012;256:279–288. [Google Scholar]
- 101.Erat MC, Sigel RKO. Determination of the intrinsic affinities of multiple site-specific Mg2+ ions coordinated to domain 6 of a group II intron ribozyme. Inorg. Chem. 2007;46:11224–11234. doi: 10.1021/ic701627t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.