

Abstract
Connective tissue growth factor (CTGF, CCN2) is a member of the CCN family of matricellular proteins. It interacts with many other proteins, including plasma membrane proteins, modulating cell function. It is expressed at low levels in normal adult kidney cells but is increased in kidney diseases, playing important roles in inflammation and in the development of glomerular and interstitial fibrosis in chronic disease. This review reports the evidence for its expression in human and animal models of chronic kidney disease and summarizes data showing that anti-CTGF therapy can successfully attenuate fibrotic changes in several such models, suggesting that therapies targeting CTGF and events downstream of it in renal cells may be useful for the treatment of human kidney fibrosis. Connective tissue growth factor stimulates the development of fibrosis in the kidney in many ways including activating cells to increase extracellular matrix synthesis, inducing cell cycle arrest and hypertrophy, and prolonging survival of activated cells. The relationship between CTGF and the pro-fibrotic factor TGFβ is examined and mechanisms by which CTGF promotes signalling by the latter are discussed. No specific cellular receptors for CTGF have been discovered but it interacts with and activates several plasma membrane proteins including low-density lipoprotein receptor-related protein (LRP)-1, LRP-6, tropomyosin-related kinase A, integrins and heparan sulphate proteoglycans. Intracellular signalling and downstream events triggered by such interactions are reviewed. Finally, the relationships between CTGF and several anti-fibrotic factors, such as bone morphogenetic factor-4 (BMP4), BMP7, hepatocyte growth factor, CCN3 and Oncostatin M, are discussed. These may determine whether injured tissue heals or progresses to fibrosis.
Keywords: CCN2, chronic kidney disease, connective tissue growth factor, connective tissue growth factor-receptors, fibrosis, TGFβ
Introduction
Chronic kidney disease (CKD) can develop in widely divergent disorders such as diabetes, auto-immune diseases and drug toxicity but it invariably leads to a single outcome, renal fibrosis. This has devastating effects on kidney function, and frequently long-term dialysis and renal transplantation are required to maintain the life of those affected by it. About 13% of the US population have CKD, a figure that is probably typical for western societies and that may be even higher in some other populations.
The kidney has a complex structure. In the adult human kidney, afferent arterioles carry blood into approximately 600,000 glomerular capillary tufts, across whose basement membranes it is filtered. Each glomerulus is connected to a tubular system through which the filtrate passes and in which it is modified, before entering collecting ducts as urine for transport to the ureter for passage to the bladder. The tubules are surrounded by a usually sparse interstitium which is permeated by blood capillaries and other small blood vessels. The many different cellular components of these structures are susceptible to injury, after which either repair occurs or progression to a state where cells lay down excessive amounts of extracellular matrix, forming fibrous scar tissue that either replaces normal tissue or impedes its function. In the glomeruli, mesangial cells become activated to a myofibroblast-like state and secrete abundant collagen and fibronectin, obstructing glomerular capillaries and filtration (Figure 1a). In the renal interstitium, excessive extracellular matrix is produced by a variety of cells including activated fibroblasts, pericytes and tubular epithelial cells, accompanied by tubular atrophy and peritubular capillary loss (Figure 1b). Many cellular and molecular processes that lead to this state have been identified (Liu 2011; Lόpez-Hernandez & Lόpez-Nova 2012). This review will focus on the role of connective tissue growth factor, a secreted 36–38 kDa protein that modulates inflammatory and fibrotic processes and that plays a key role in kidney fibrosis.
Figure 1.
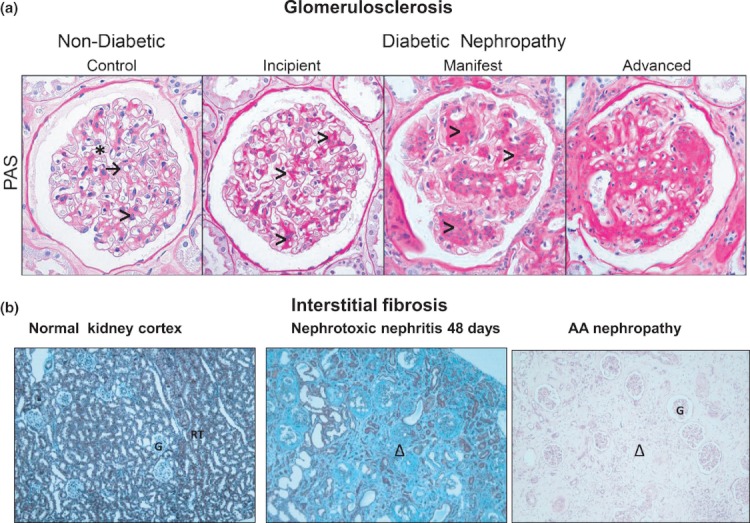
Normal and fibrotic kidney structure. (a) Glomerulosclerosis (glomerular fibrosis). The four frames show cross-sections of typical glomeruli from normal human kidney and from patients with incipient, manifest and advanced stages of diabetic nephropathy stained with periodic acid Schiff reagent (PAS; Wahab et al. 2005a, reproduced with permission from Diabetologia). Blood is filtered across the glomerular capillary basement membranes (→) into the urinary space (*) from where the filtrate drains into the tubular system. The capillary tuft is supported by the mesangium composed of mesangial cells and extracellular matrix (>). With incipient disease, there is diffuse expansion of mesangial matrix (>), strong pink staining), but otherwise normal cellularity and vasculature. At later stages (manifest and advanced), further mesangial expansion leads to increasing diffuse and nodular sclerosis with encroachment on and occlusion of capillary lumina and progressive loss of cells. ×400. (b) Renal interstitial fibrosis (pictures courtesy of Prof Terry Cook). Normal WKY rat kidney cortex, like human cortex, has an abundance of renal tubules (RT) with scattered glomeruli (G). Interstitial matrix between neighbouring tubules is barely detected. Forty-eight days after induction of nephrotoxic nephritis, the tubules have been lost, or have distorted morphology, and are replaced by abundant interstitial extracellular matrix. This fibrotic tissue is stained light blue with Masson's trichrome (Δ). Remnant glomeruli are also fibrotic. Human aristolochic acid nephropathy (AA nephropathy, syn Chinese herbal nephropathy) results in profound tubular loss and massive interstitial fibrosis (Δ) but glomeruli (G) show only mild ischaemic changes. Stain, heamatoxylin and eosin.
Ethical approval
Ethical approval was obtained for investigations in the authors laboratory using human tissue and animal experiments were carried out under the authority of a UK Home Office Licence.
CTGF is a CCN protein
Connective tissue growth factor (CTGF or CCN2) is a member of the CCN family of secreted matricellular proteins (Figure 2). The name CCN is derived from the first letter of three members of the family (underlined). The other family members are cysteine-rich angiogenic inducer 61 (CYR 61 or CCN1), nephroblastoma overexpressed gene (NOV or CCN3) and the Wnt-inducible signalling pathway proteins 1–3 (WISP 1–3 or CCN 4–6). The genes for CCN proteins encode a modular structure comprising a signal peptide followed by four domains, each named for its homology with other proteins. CCN5 lacks the C-terminal domain. Each domain is able to bind multiple ligands. In the case of CTGF, these include IGF-1, IGF-2 (IGF-BP domain); α5β3 integrin, TGFβ and bone morphogenetic factor-4 (BMP4; von Willebrand factor C domain); LRP-1, VEGF (TSP1 domain); and Wnt, heparan sulphate proteoglycan, integrins, LRP-5, LRP-6 (CT domain). With this large number of interactions, it is not surprising that CCN proteins, which are generally expressed at low levels in healthy adult tissue but increased in many disease settings, are able to affect many different biological functions. Alternative splicing of CCN mRNAs (Perbal 2009) and post-translational proteolytic cleavages of CCN proteins (De Winter et al. 2009) may result in yet further complexity in the biological roles of these molecules. These include effects on cell adhesion and migration, cell proliferation, survival and apoptosis, cellular transdifferentiation and extracellular matrix synthesis and turnover. Proteolytic processing of CTGF complexed with other proteins may also generate biological responses, for example, angiogenesis, when VEGF is released from an angiogenic-inhibitory complex with CTGF by matrix metalloproteinase 2 (MMP2) (Dean et al. 2007). Moreover, CCN proteins have been implicated as having key functions in pathologies ranging from wound healing to fibrosis to cancer and are potential therapeutic targets in these disorders (Jun & Lau 2011).
Figure 2.
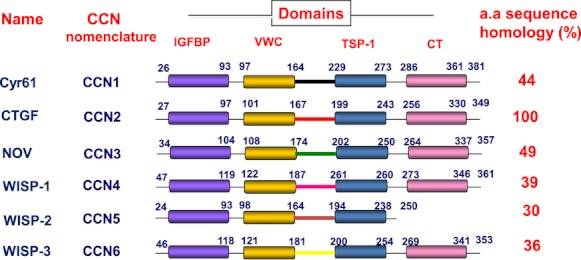
Domain structure and amino acid sequence homology of the CCN proteins. IGFBP, insulin-like growth factor binding protein domain; VWC, von Willebrand factor C domain; TSP-1, thrombospondin type 1 repeat domain; CT, C-terminal domain with cysteine knot motif; CTGF, connective tissue growth factor. Numbers refer to the amino acid (a.a.) sequence for each CCN protein. The per cent a.a homology shown is with reference to CTGF/CCN2.
Human CTGF and the mouse homologue, FISP12, were discovered independently in 1991 (Bradham et al. 1991; Ryseck et al. 1991). It was soon recognized that CTGF was induced by TGFβ1 in a sequential manner in wound healing and that it may be involved in tissue repair (Igarashi et al. 1993). However, it was found that there was a strong correlation between skin sclerosis and CTGF expression in dermal fibroblasts of patients with systemic sclerosis, implying that CTGF may also be involved in fibrosis (Igarashi et al. 1995). This was confirmed after injecting CTGF and TGFβ together into the skin of neonatal mice which, in contrast to either cytokine alone, induced persistent fibrosis (Mori et al. 1999). Subsequently, CTGF has been implicated in fibrosis in a variety of tissues and organs (Shi-Wen et al. 2008).
Connective tissue growth factor and inflammation
Chronic inflammation usually precedes and accompanies the development of fibrosis in organs. Tissue injury from infectious, immune, mechanical, chemical or other agent is followed by the acute release of inflammatory mediators, recruitment and activation of inflammatory cells (macrophages, leucocytes and T-lymphocytes), removal of damaged cells and pathogens, activation of fibroblasts to myofibroblasts and finally repair (Wynn 2007). Activation of inflammatory cells releases many factors including profibrotic cytokines such as TGFβ. With repeated or prolonged injury, myofibroblast activation persists, leading to fibrosis rather than repair. Activation of the transcription factor NF-κB, a key marker of inflammatory activity, occurs in tubular epithelial cells and in the glomerulus in human kidney diseases and in animal models, particularly when proteinuria is present (Mezzano et al. 2001, 2004). It seems likely that CTGF, which is expressed in tubular and other renal cells in kidney disease (Ito et al. 1998; Nonaka et al. 2008), promotes cellular infiltration by activating NF-κB (Sanchez-Lopez et al. 2009). Experimentally, 24 h after injecting normal mice i.p. with CTGF, inflammatory cells infiltrate the renal interstitium, with marked increases in CD68+ macrophages and CD3+ T-lymphocytes. NF-κB was activated in the kidney, and the cellular infiltration was attenuated by prior treatment with an NF-κB inhibitor. Expression of MCP-1 and RANTES, chemokines that promote inflammation, was increased in both tubular epithelial cells and infiltrating interstitial cells and was also dependent on NF-κB activation (Sanchez-Lopez et al. 2009). Connective tissue growth factor itself is chemotactic for monocytes in vitro (Cicha et al. 2005). These data indicate that CTGF provokes an inflammatory response in the kidney which is likely to contribute to the initiation of fibrosis in kidney disease.
Connective tissue growth factor and kidney fibrosis
Exploring potential mechanisms involved in fibrosis in diabetic nephropathy, we and others used suppression subtraction hybridization to reveal changes in gene expression in mesangial cells exposed to high glucose conditions (Mason et al. 1997; Murphy et al. 1999). Both studies showed that CTGF expression was upregulated in these conditions. Moreover, an earlier report implicating CTGF in the development of skin fibrosis (Igarashi et al. 1995) raised questions as to whether it is also involved in the development of glomerulosclerosis in diabetic nephropathy. A subsequent study confirmed that both mRNA and protein levels of glomerular CTGF increase progressively with advancing glomerulosclerosis in this disease (Wahab et al. 2005a). Moreover, a study on renal biopsies from patients with CKD of various origins indicated that those with glomerulosclerosis or interstitial fibrosis showed increased CTGF expression in cells at those sites, suggesting a role for CTGF in renal fibrosis generally (Ito et al. 1998). Subsequently, Suzuki et al. (2003) reported that CTGF expression was higher in glomerular mesangial and epithelial cells in diseases characterized by mesangial matrix expansion (diabetic nephropathy and IgA nephropathy) than it was in disorders associated with changes in the glomerular basement membrane (membranous nephropathy and minimal change nephrotic syndrome). Another study reported that CTGF was strongly expressed in cellular and fibrocellular crescents in human crescentic glomerulonephritis and proposed that it was involved in extracellular matrix production by parietal epithelial cells and fibrosis (Kanemoto et al. 2004). Animal models of kidney disease also showed changes in CTGF expression. For example, CTGF expression was markedly increased in interstitial fibroblasts during the development of tubular damage and interstitial fibrosis in the rat remnant kidney model (Frazier et al. 2000) and CTGF was expressed de novo in dilated proximal tubules of a rat model of diabetic nephropathy (Wang et al. 2001a,b). In a non-human primate (Papio hamadryas) model of type 1 diabetes, tubular CTGF protein scores after 5 years of disease predicted the degree of albuminuria at 10 years, suggesting CTGF as a contributory factor in the development of incipient nephropathy and an early marker of kidney disease (Thomson et al. 2008).
Whilst observational studies in man and animals are highly suggestive of a link between CTGF expression and the development of fibrosis in the kidney, they do not prove it. In contrast, interventional studies firmly establish this. Experimentally induced unilateral ureteric obstruction (UUO) in rodents leads to rapid development of renal interstitial fibrosis and is used as a model of CKD (Klahr & Morrissey 2002). Seven days after performing UUO, the number of myofibroblasts in the interstitium is markedly increased, accompanied by increased expression of type 1 collagen, fibronectin, EDA-fibronectin (a splice variant associated with fibrosis), CTGF and the development of fibrous tissue incorporating these molecules. Administration of CTGF antisense oligonucleotide (ASO) via the renal vein markedly attenuated these molecular events and the development of fibrosis in UUO (Yokoi et al. 2004). Interestingly, TGFβ levels that were much increased in UUO were not affected by the CTGF-ASO treatment. Another model of renal fibrosis, sub-total nephrectomy (5/6) in TGFβ-transgenic mice, leads to the development of glomerulosclerosis and interstitial fibrosis over the following 56 days (Okada et al. 2002). Intravenous administration of CTGF-ASO from days 29 to 56 after surgery blocked the expression of CTGF in proximal tubular epithelial cells and attenuated the development of interstitial fibrosis as shown by decreased peritubular collagen 1 deposition, decreased numbers of interstitial fibroblasts and reduced levels of α1COL1 and FN-IIIA mRNA in the remnant kidney. This occurred despite the continued expression of TGFβ (Okada et al. 2005). Connective tissue growth factor expression in the glomerulus and glomerulosclerosis was unaffected in the CTGF-ASO-treated mice, the authors attributing this to their earlier observation that intravenously injected oligonucleotides are not absorbed by glomerular cells. However, in another study, mice with streptozotocin-induced type 1 diabetes and mutant db/db mice with type 2 diabetes were given CTGF-ASO subcutaneously for 16 weeks. The mesangial matrix expanded in the type 1 mice, accompanied by an increase in kidney cortex mRNA for CTGF, collagen 1 (α1), fibronectin and TGFβ. The increase in glomerular size was attenuated, and the mRNA levels returned to those of controls in the ASO-treated animals. CTGF-ASO treatment had similar effects in the db/db mice, and improvements in kidney function tests (urinary protein loss, serum creatinine) were also noted (Guha et al. 2007). Collectively, these studies provide convincing evidence of a key role for CTGF in the development of both glomerulosclerosis and interstitial fibrosis following kidney injury. The experiments in mice provide proof of principle that anti-CTGF therapies are successful in attenuating the development of renal fibrosis. Moreover, a phase 1 trial of the effects of an anti-CTGF antibody in human diabetics with microalbuminuria (indicative of early renal changes) showed not only that the antibody was safe to use but also that it reduced microalbuminuria (Adler et al. 2010).
Investigation of mice in which CTGF expression has been genetically modified also supports the notion that CTGF is a key factor in the development of renal fibrosis. CTGF was overexpressed in fibroblasts in mice transgenic for the murine homologue, fisp12, under the control of the minimal promoter from the Col1a2 gene and a fibroblast-specific 6 kb upstream enhancer (Sonnylai et al. 2010). Mice that were homozygous for the transgene developed fibrosis in many tissues, including focal glomerulosclerosis and interstitial fibrosis in the kidney.
The promoter of the human nephrin gene was used to drive overexpression of murine CTGF specifically in podocytes. These transgenic mice had normal kidney histology but 12 weeks after induction of diabetes with streptozotocin they showed glomerular hypertrophy, mesangial expansion and proteinuria compared to diabetic non-transgenic mice. Glomerular CTGF mRNA levels were markedly increased in the diabetic transgenic mice compared to diabetic non-transgenic controls, but mRNAs for matrix encoding genes were not significantly increased. However, whilst expression of matrix metalloproteinase 2 (MMP) was increased in control diabetic mice, it was lowered in the diabetic transgenics. This difference in MMP2 expression was accompanied by a decline in MMP2 activity in the diabetic transgenics, so the mesangial matrix expansion associated with increased CTGF expression was likely due to decreased matrix degradation rather than to increased matrix protein expression. Interestingly although CTGF protein was increased in podocytes in diabetic transgenics, as expected, it was also increased in the mesangium. Thus, the podocyte transgene-CTGF likely had a paracrine inductive effect on mesangial cell CTGF expression (Yokoi et al. 2008).
Complementing these experiments, another group induced diabetes in CTGF+/− and CTGF+/+ mice and later subjected them to UUO to accelerate pathological changes in the kidney (Nguyen et al. 2008). After 17 weeks, renal cortex, plasma and urinary levels of CTGF were all increased in the CTGF+/+ mice, whilst cortex and plasma levels in CTGF+/− mice were similar to those in non-diabetic controls. Moreover, gelatinase activity (due to MMP2 and MMP9) was decreased in CTGF+/+ mice, but was maintained at control levels in the CTGF+/− mice, inferring again that higher CTGF levels decrease these metalloproteinases, which could slow matrix degradation.
It is noteworthy that the expression of CTGF in the renal cortex of transgenic mice does not always enhance the development of fibrosis in disease models. In contrast to homozygotes (see above), heterozygote Col1a2-CTGF mice have normal kidney structure, even though they have higher levels of cortical CTGF mRNA (Fragiadaki et al. 2011). When aristolochic acid (AA) nephropathy was induced in heterozygote Col1a2-CTGF mice, CTGF mRNA expression in the kidney cortex was markedly increased and there was a modest increase in CTGF protein compared to AA-treated wild-type litter mates. However, they only developed the same degree of interstitial fibrosis as that found in the AA-treated controls (Fragiadaki et al. 2011). This finding is similar to the observation that enhanced CTGF expression in cardiomyocytes did not evoke cardiac fibrosis in unchallenged transgenic mice, or increased cardiac damage when they were subjected to ischaemia-reperfusion (Panek et al. #b501). Thus, the relationship between CTGF and the development of fibrosis in a tissue appears to depend on context and may differ depending on factors such as the level of CTGF expressed, the cell types exposed to the factor, the receptors for CTGF expressed by them, the levels of the profibrotic factor TGFβ and the activity of its signalling pathway.
The relationship between CTGF and TGFβ in fibrosis
TGFβ has long been recognized as a key factor in the development of renal inflammation and fibrosis (Floege & Johnson 1993). The discovery that TGFβ stimulated expression of CTGF (Igarashi et al. 1993) raised questions about the relationship between the two factors in driving fibrosis. These have still not been completely resolved. Connective tissue growth factor binds to TGFβ [Kd 30 nM] promoting interaction with its receptor and increasing downstream signalling (Abreu et al. 2002). However, the observation that fibrosis could be ameliorated in some animal models by treatment with CTGF-ASO whilst TGFβ levels were unaffected (Okada et al. 2005) led to the idea that CTGF acted ‘downstream’ of TGFβ. Nevertheless, experimental data suggest that the two factors act in a cooperative rather than in a sequential manner to promote fibrosis. For example, repeated daily subcutaneous injections of TGFβ1, TGFβ2, TGFβ3 or CTGF into the skin of neonatal mice failed to induce fibrosis, but persistent fibrosis developed at the site when TGFβ and CTGF were injected together, or when TGFβ was injected for 3 days, followed by CTGF for 4 days (Mori et al. 1999). Using a similar protocol in transgenic mice harbouring a reporter gene for the COL1A2 gene promoter, the same group reported that injections of TGFβ3 or CTGF for 1 week failed to activate the promoter above control levels, but the sequential procedure stimulated a 12-fold increase in activity (Chujo et al. 2005). In another report, when CTGF and TGFβ2 were injected intraperitoneally together, neonatal mice developed severe fibrosis of the peritoneal membranes. However, injection of either TGFβ2 alone, or CTGF alone, did not induce fibrosis (Wang et al. 2011).
In vitro experiments have not yet fully explained the molecular basis of the cooperative effects of the two cytokines in driving fibrosis in vivo. In the TGFβ canonical signalling pathway, Smad2 and Smad3 are phosphorylated following TGFβ binding to its receptor (ALK5) and then translocate to the nucleus in a complex with Smad4, where they bind to the promoters of TGFβ-responsive genes, stimulating their transcription (Miyazawa et al. 2002). For example, the COL1A2 promoter contains a classical Smad-binding element (SBE) for Smad3 (Chen et al. 2000), whilst the COL1A1 promoter contains a CC(GG)-rich element to which Smad2 binds in association with Sp1 (Sysa et al. 2009). Smad2-deficient mice are embryonic lethal, whereas Smad3-deficient mice are viable and are protected from developing fibrosis in several different models (Flanders 2004). Mori et al. (2008) investigated whether responses to TGFβ differed in CTGF−/− and CTGF+/+ murine embryonic fibroblasts (MEF). Although there were differences in cell proliferation rate and fewer α-smooth muscle actin positive cells formed in response to TGFβ in CTGF−/− cells, activation of the TGFβ canonical signalling pathway was unaffected when compared to CTGF+/+cells. This was evidenced by normal Smad2 phosphorylation, Smad2/3 and Smad4 translocation to the nucleus, Smad3-dependent transcriptional activity, and synthesis of type 1 collagen and fibronectin in the null cells. However, in contrast to these results, in human mesangial cells, Smad2 and Smad3 phosphorylation was stimulated more when treated with TGFβ plus exogenous rCTGF than it was with TGFβ alone. Moreover, CTGF enhanced TGFβ-induced transcriptional activity of the SBE4-luc reporter gene in these cells, whilst treatment with an antisense CTGF-ASO abrogated much of the transcriptional response to TGFβ (Wahab et al. 2005b). The CTGF-ASO also markedly reduced TGFβ-stimulated transcription of TGFβ-responsive genes such as those for collagen III, PAI-1 and the cyclin-dependent kinase inhibitor, p15INK in mesangial cells. One factor that is likely to be relevant to the different results of these two studies is that mesangial cells exhibit basal expression of CTGF whilst the MEF line studied by Mori et al. (2008) does not (Wahab et al. 2005b). Liu et al. (2011) investigated TGFβ responses in adult dermal fibroblasts derived from wild-type mice and from mice in which CTGF expression in fibroblasts/smooth muscle cells was conditionally deleted. TGFβ induced the same level of expression of α-smooth muscle cell actin and Col1a1 mRNA in both cell types. They proposed that in cells in which CTGF is normally basally expressed, CTGF mediates induction of TGFβ-inducible mRNAs, whilst in cells lacking constitutive CTGF expression, responses to TGFβ are unchanged. Interestingly, in vivo, wild-type mice were susceptible to bleomycin-induced skin fibrosis, whereas mice in which fibroblast/smooth muscle cell CTGF expression was deleted were not susceptible. The authors concluded that lack of a fibrotic response to bleomycin in CTGF-knockout mice may relate to a failure to recruit myofibroblasts (derived from pericytes) to the site, possibly because cell migration requires CTGF/integrin interactions.
In another recent study, CTGF was depleted in foreskin fibroblasts with an adenoviral siRNA. These cells were unable to upregulate type 1 collagen mRNAs and protein in response to subsequent TGFβ treatment (Nakerakanti et al. 2011). Connective tissue growth factor depletion had no effect on Smad3 phosphorylation in response to TGFβ, but Smad1 phosphorylation was abolished and ERK1/2 phosphorylation much reduced. Smad1 was first reported as being activated by a BMP receptor (ALK1) in endothelial cells but subsequent studies showed that it is also activated by TGFβ in other cell types, including fibroblasts and epithelial cells, either via ALK1 or ALK5 (Miyazawa et al. 2002; Trojanowska 2009). Smad1 binds directly to the CTGF promoter activating it and increasing CTGF. It is proposed that secreted CTGF then binds to αvβ3 integrin activating the ERK1/2 MAP kinase pathway in a Src-dependent manner, this pathway being required for TGFβ-induced activation of Smad1 (Trojanowska 2009; Nakerakanti et al. 2011). Thus, the pathway has the potential for persistent cell activation. Importantly, Smad1 and p-Smad1 levels are elevated in systemic sclerosis (SSc) skin biopsies and siRNA depletion of Smad1 in SSc fibroblasts normalized their production of CTGF and collagen 1, indicating that the Smad1 pathway is involved in this fibrotic disease (Pannu et al. 2008). Collectively these reports highlight the importance of taking into account the possible involvement of the Smad1 pathway, as well as the Smad2/3 pathway, in future studies of the role of TGFβ and CTGF in fibrotic diseases.
It is possible that the concentrations of TGFβ and CTGF may affect the outcome of some experiments. Abreu et al. (2002) treated foetal mink lung cell cultures with TGFβ and CTGF. Whilst 6 nM CTGF strongly potentiated the phosphorylation of Smad2 in cells stimulated with 10 pM TGFβ, it had no effect with 50 pM TGFβ, suggesting the concentration ratio between the two factors is important in cooperative effects. Moreover, in cells in which TGFβ concentration was constant, even 3 nM CTGF stimulated 3TP-Lux activity, whilst maximum activity was achieved with 15 nM CTGF and 35 nM had no further effect.
There are also potential mechanisms for attenuating TGFβ responses by driving down CTGF expression. Both TGFβ1 and 2 upregulated the expression of sphingosine kinase-1 (SK-1) in an immortalized podocyte cell line. CTGF expression was increased in TGFβ-treated cells, but even more when they were transfected with an siRNA targeting SK-1. Conversely, CTGF expression decreased when SK-1 was over-expressed in TGFβ-treated cells and further up-regulated when such cells were exposed to an SK-1 pharmacological inhibitor (Ren et al. 2009). Collectively these results indicate that SK-1 exerts a braking effect on TGFβ-induced CTGF expression. Glomerular SK-1 expression was noted in human diabetic nephropathy biopsies but was barely detectable in controls, suggesting that the brake is applied in pathological situations where CTGF is upregulated. Similarly, glomerular CTGF was increased and albuminuria was worse in diabetic SK-1−/− mice than in SK-1+/+ mice (Ren et al. 2009). Further mechanisms that decrease CTGF actions, but which involve opposition by other cytokines, are described later.
Pro-fibrotic effects of CTGF on renal cells in vitro
Connective tissue growth factor has effects on many cell types in the kidney. Selected examples of CTGF-induced effects are discussed to illustrate the diversity of its potential roles in kidney fibrosis.
Stimulation of extracellular matrix deposition
In the glomerulus, mesangial cells lay down an extracellular matrix around themselves, providing support for the glomerular tuft and also contract in response to vasoconstrictors, helping to regulate blood flow through the glomerular capillaries. Glomerulosclerosis in diabetic nephropathy is due to the expansion of the mesangial extracellular matrix. The expression of both TGFβ and CTGF is increased in DN (Wahab et al. 2005a). Connective tissue growth factor stimulates the expression of fibronectin in mesangial cells in vitro, as does TGFβ. The stimulatory effect of the latter is mediated through CTGF (Wahab et al. 2001a,b). Both factors also increase the incorporation of the newly synthesized fibronectin into an insoluble extracellular matrix around the cells. This process is dependent on an increase in expression of cell surface α5β1 integrin that provides anchoring points for the formation of the extracellular fibronectin matrix (Weston et al. 2003). This TGFβ-driven effect was attenuated by blocking CTGF expression with antisense oligonucleotides, demonstrating a key role for the factor in matrix formation. The deposition of an insoluble fibronectin matrix in the mesangium provides a scaffold to which other matrix proteins such as collagens can attach to form a mature fibrotic tissue.
Induction of mesangial cell cycle arrest and hypertrophy
Mesangial cell hypertrophy occurs early in diabetic nephropathy and is due to the cells undergoing cell cycle arrest. The arrest, which occurs in the G1phase of the cycle, was attributed to TGFβ, itself induced by high glucose conditions (Wolf & Ziyadeh 1999). Our subsequent study showed that CTGF induces mesangial cells to enter the G1 phase from their quiescent G0 state and to express cyclin D1 and the cyclin-dependent kinase inhibitors (CDKIs), p15INK4, p21Cip1 and p27Kip1 which are known to bind and inactivate cyclinD/CDK4/6 and the cyclin E/CDK2 kinase complexes. This in turn leads to the maintenance of pRB protein in its hypophosphorylated state, thereby preventing cell cycle progression. TGFβ induces the same events but these are CTGF-dependent because they were attenuated by prior treatment with a CTGF-ASO (Wahab et al. 2002).
Prolonging survival of activated cells
It was reported that CTGF induces apoptosis in mesangial cells (Hishikawa et al. 2001), but we found no evidence of this. Indeed we reported that CTGF induces rapid synthesis of MAPK phosphatase-1 (MKP-1), an enzyme that dephosphorylates p38MAP kinase, inactivating it (Wahab et al. 2007). Inactive p38MAP kinase is unable to phosphorylate the anti-apoptotic protein Bcl-2, protecting it from degradation and allowing its dimerization with Bax, protecting the cell from apoptosis triggered by formation of Bax–Bax homodimers. siRNA depletion of MKP-1 in mesangial cells allowed p38 phosphorylation and under these conditions CTGF did promote apoptosis. In summary, arrest in G1 is likely to be important in the development of fibrosis because the cells are highly active in protein synthesis during this phase of the cycle, including extracellular matrix proteins. Continued production of matrix, as in fibrosis, depends on persistently activated cells, in contrast to wound healing and recovery from injury that requires apoptosis of previously active cells. Connective tissue growth factor is clearly implicated in cycle arrest and the accompanying elevated protein synthesis and in cell survival in diabetic glomerulosclerosis.
Transition of quiescent renal cells to a myofibroblastic phenotype
Connective tissue growth factor is able to stimulate transition of differentiated cells such as tubular epithelial cells, endothelial cells and interstitial fibroblasts to an activated myofibroblast cell phenotype in vitro. In this transition, proteins that are characteristically expressed by the differentiated cell are lost (e.g. E-Cadherin in epithelial cells), whereas proteins that are characteristically expressed by myofibroblasts, such as fibrillar collagens and α-smooth muscle actin, are expressed de novo (Fragiadaki & Mason 2011). Myofibroblasts occupy the intertubular spaces in renal interstitial fibrosis, generating the abundant collagenous extracellular matrix which is characteristic of this condition. They also express CTGF which is considered to be a marker of myofibroblasts and fibroproliferative disease (Leask et al. 2009). The transition of tubular epithelial cells to myofibroblasts (EMT) was thought to provide an important source of interstitial cells in in vivo models of renal interstitial fibrosis (Iwano et al. 2002). However, more recent cell lineage studies indicate that pericytes that support peritubular capillary walls are the major source of renal interstitial myofibroblasts in fibrosis disease models (Humphreys et al. 2010). Under stimulus of disease factors, pericytes detach from the capillary wall, migrate into the interstitium and undergo transition to myofibroblasts (Schrimpf & Duffield #b502). Liu et al. (2010) reported that about 85% of myofibroblasts are derived from CTGF-expressing pericytes in a bleomycin-induced model of skin fibrosis. Connective tissue growth factor stimulates the migration of retinal pericytes in vitro (Pi et al. 2011). We observed that CTGF can induce expression of Snail1 (Fragiadaki et al. 2012), a transcriptional regulator which is expressed in pericytes isolated from UUO kidney (Lin et al. 2008). Snail1 initiates the expression of fibronectin in epithelial cells and fibroblasts in association with NF-κB p65 subunit and PARP-1 (Stanisavljevic et al. 2011) and over-expression of Snail1 in transgenic mice induces interstitial fibrosis in the kidney (Boutet et al. 2006). Collectively these results strongly suggest that CTGF may be a key factor in promoting pericyte migration and transition to interstitial myofibroblasts in renal fibrosis. However, the debate concerning the role of tubular cell EMT as a source of myofibroblasts in renal fibrosis continues. We have reviewed the evidence for and against this recently (Fragiadaki & Mason 2011).
Recruitment of inflammatory cells
As described earlier, intraperitoneal injection of CTGF in mice activated NF-κB, induced an inflammatory cell infiltrate in the interstitium and stimulated the expression of cytokines in the kidneys. To define this pathway Sanchez-Lopez et al. (2009) investigated the effect of CTGF on murine proximal tubular epithelial cells (MCTcells) and on HK2 cells. NF-κB was rapidly activated in both cell types, as were the MAP kinases, ERK1/2, p38 kinase and JNK. NF-κB activation was dependent on the activation of all three kinases as it was blocked by specific kinase inhibitors. Connective tissue growth factor also upregulated expression of MCP-1, ICAM-1 and IL-6 in MCT and HK2 cells in an NF-κB-dependent manner. These results support the view that CTGF is likely to play an important role in the recruitment of inflammatory cells to the tubulointerstitial compartment and in the induction of chemokines, cytokines and extracellular matrix (ECM) proteins. We observed that CTGF also activates NF-κB in human mesangial cells, so this pathway is likely to be important in glomerular inflammation and subsequent sclerosis (Wahab and Mason, unpublished results).
Cellular receptors for CTGF and downstream signalling
Exposure of cells to CTGF activates numerous signalling molecules. For example, in mesangial cells ERK1/2, JNK, PKB, CamKII, PKCα and PKCδ are activated, suggesting that CTGF functions via one or more signalling receptors (Wahab et al. 2005c). Several CTGF receptors have been reported but none are unique for the cytokine. They include low-density lipoprotein receptor-related protein (LRP-1; Segarini et al. 2001), low-density lipoprotein receptor-related protein-6 (LRP-6; Mercurio et al. 2004), tropomyosin-related kinase A (TrkA; Wahab et al. 2005c), a 280 kDa chondrocyte receptor (Nishida et al. 1998) and several adhesion receptors (Jun & Lau 2011) such as β3 integrin (Crean et al. 2002), αVβ3 integrin (Gao & Brigstock 2004), α5β1 intergrin (Gao & Brigstock 2005) and αMβ2 integrin (Schober et al. 2002). The TGFβ type III receptor is also reported to bind CTGF (O'Donovan et al. 2012). Heparan sulphate proteoglycans function as co-receptors with integrin receptors (Gao & Brigstock 2004) and LRP-1 (Gao & Brigstock 2003; Jun & Lau 2011). To date, only some of these receptors have been reported in renal cells.
Low-density lipoprotein receptor-related protein-1
Low-density lipoprotein (LDL) receptor-related protein-1, a member of the LDL-receptor family of proteins can bind and endocytose many different proteins such as ECM proteins, growth factors and antigens, in addition to LDL. LRP-1 was proposed as a master regulator of the plasma membrane proteome, adapting it to the prevailing microenvironment by binding other plasma membrane proteins via bridging proteins, followed by endocytosis and lysosomal degradation. Binding of ligands, including extracellular matrix proteins, may also trigger phosphorylation of the cytoplasmaic tail of LRP-1, promoting its interaction with intracellular adaptor proteins and downstream cell signalling (Gonias et al. 2004).
Low-density lipoprotein receptor-related protein-1 was isolated as a CTGF-binding protein from the bone marrow stem cell line, BMS2. Radiolabelled CTGF was displaced from BMS2 cells by known ligands of LRP-1 such as the receptor-associated protein (RAP), and another cell line, deficient in LRP-1, was unable to bind CTGF, demonstrating the receptor–ligand relationship. Connective tissue growth factor endocytosis was blocked by RAP, indicating that cellular uptake was mediated by LRP-1 (Segarini et al. 2001). Renal fibrosis follows activation of cells to myofibroblasts in the interstitium. CTGF alone had no effect on normal rat kidney fibroblast cells (NRK-49F) but augmented the effect of TGFβ in stimulating fibronectin synthesis and de novo expression of αSMA, evidence of activation to myofibroblasts (Yang et al. 2004). Connective tissue growth factor, but not TGFβ, induced phosphorylation of LRP-1 in the NRK-49F cells. RAP inhibited this activation and reduced the synergistic action of CTGF on TGFβ-induced αSMA expression. The synergistic action of CTGF was dependent on its activation of the ERK1/2 signalling pathway, although whether this was downstream of LRP-1 phosphorylation or not was not investigated (Yang et al. 2004). Others showed subsequently that tissue-type plasminogen activator (tPA) also promoted TGFβ-stimulated transition of NRK-49F cells to myofibroblasts. This was dependant on tPA-induced phosphorylation of LRP-1 which led to recruitment of β1 integrin to form a complex with LRP1, activating integrin-linked kinase (ILK; Hu et al. 2007). This kinase can activate several downstream pathways including phophorylation of Akt. Hu et al. also showed that the tPA/LRP-1 pathway is active in vivo in the UUO model of renal fibrosis. Moreover in vivo inhibition of either ERK1/2 or Akt activation decreased myofibroblast markers in the UUO model, indicating a key role for both signalling pathways in myofibroblast proliferation in interstitial fibrosis (Rodriguez-Pena et al. 2008). Collectively these results indicate an important role for LRP-1 activation by different ligands, including CTGF, in triggering signalling involved in renal fibrosis.
Low-density lipoprotein receptor-related protein-6
Connective tissue growth factor-induced phosphorylation of glycogen synthase kinase 3β (GSK3β) and increased levels of β-catenin in mesangial cells, which could be blocked by DKK-1, a Wnt signalling pathway antagonist, observations consistent with activation of the canonical Wnt signalling pathway by CTGF (Rooney et al. 2011). The authors showed that mesangial cells expressed LRP-6, shown previously to be a Wnt co-receptor that interacts with CTGF (Mercurio et al. 2004). Connective tissue growth factor induced serine-phosphorylation of LRP-6 followed by nuclear accumulation of β-catenin and transcriptional activity as measured by a TCF/LEF-luciferase reporter. Rooney et al. (2011) identified several upregulated Wnt target genes in human nephropathy biopsies, in UUO, and in streptozotocin-induced diabetes in animal models. However, further research is needed to establish the precise role of the CTGF-activated LRP-6 pathway in mesangial cells. Chronic dysregulated Wnt signalling has been implicated in renal fibrosis, but Wnt pathways are also involved in healing processes following acute renal injury (Nelson et al. 2011).
Tropomyosin-related kinase A
Tropomyosin-related kinase A is a member of the neurotrophin receptor family (TrkA, TrkB and TrkC). Each forms a transmembrane homodimeric receptor tyrosine kinase on binding its ligand. Neurotrophins [nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3) and neurotrophin 4 (NT-4)] bind to one or more of these receptors, triggering phosphorylation in the cytosolic domain, binding of adaptor proteins and activation of downstream signalling pathways. Tropomyosin-related kinase A is the cognate receptor for NGF. All neurotrophins also bind to a common low-affinity receptor p75NTR that can associate with the Trk receptors, modifying their neurotrophin-binding selectivity (Allen & Dawbarn 2006). Other proteins have been reported to associate directly with Trk receptors, for example, the GPI-linked membrane protein Ephrin A5 to TrkB. In addition, Trk receptors can be transactivated by the Src-related kinase Fyn following the activation of some G-protein coupled receptors such as the A2a adenosine receptor and LRP-1 (Schecterson & Bothwell 2010).
Connective tissue growth factor binds and activates TrkA
We found that CTGF bound to the TrkA/p75NTR receptor in human mesangial cells, inducing phosphorylation at Y490 in the juxtamembrane region of TrkA (Wahab et al. 2005c). This is the docking site for the adaptor protein Shc that initiates signalling through the Ras-ERK1/2 and PI3 kinase/Akt (PKB) pathways in other cell types (Chao et al. 2006). We also reported that CTGF induced phosphorylation of Y674/675, key sites in TrkA activation for regulating its catalytic activity (Cunningham et al. 1997). The TrkA inhibitor K252a blocked CTGF-induced phosphorylation of ERK 1/2, Akt, JNK (Wahab et al. 2005c). In a subsequent investigation of the involvement of TrkA in diabetic nephropathy, we showed that high glucose conditions induce CTGF in mesangial cells which activates TrkA and downstream phosphorylation of ERK1/2. These steps were blocked by either siRNA knock-down of either CTGF or TrkA, confirming the specific involvement and importance of CTGF–TrkA-initiated signalling in the response to high glucose (Fragiadaki et al. 2012).
Connective tissue growth factor activation of TrkA induces TIEG-1, potentiating TGFβ signalling
Connective tissue growth factor induces TGFβ-inducible early-response gene-1 (TIEG-1) in mesangial cells, an action that was inhibited by K252a and thus likely mediated via TrkA activation (Wahab et al. 2005b). This transcriptional repressor binds to the promoter of Smad7, a negative regulator of the TGFβ/Smad signalling pathway, inhibiting its expression and so potentiating signalling (Johnsen et al. 2002). Connective tissue growth factor enhanced TGFβ-induced phosphorylation of Smad2 and 3, their nuclear translocation, increased activity of the SBE4-Luc reporter gene and the transcription of several TGFβ responsive gened in mesangial cells. Antisense oligonucleotide knock-down of TIEG-1 in mesangial cells showed that this CTGF potentiation of TGFβ-stimulated effects was indeed due to induction of TIEG-1 and downregulation of Smad7 expression. Thus, we proposed that under pathological conditions where CTGF expression is elevated the induction of TIEG-1 is a key step in promoting continued activation of the TGFβ signalling pathway (Figure 3).
Figure 3.
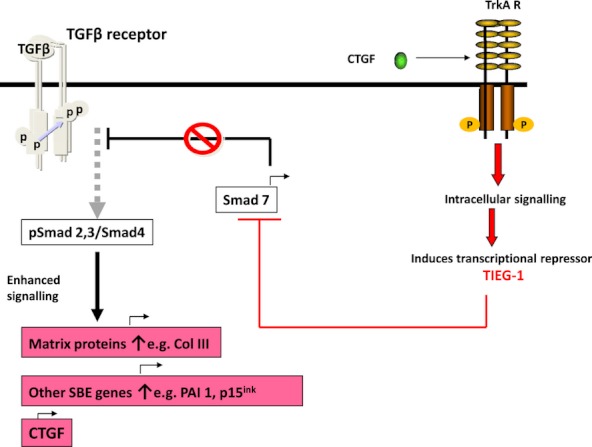
Connective tissue growth factor (CTGF) stimulates the TGFβ-Smad signalling pathway. Connective tissue growth factor activation of the tropomyosin-related kinase A (TrkA) receptor induces TIEG-1, a transcriptional repressor of Smad7. Decreased Smad7, a natural repressor of TGFβ signalling, promotes increased phospho-Smad2/3, promoting transcription of Smad-responsive genes. TIEG-1, TGFβ-inducible early-response gene 1; SBE, Smad binding element.
Tropomyosin-related kinase A expression is induced by hypoxia, a common feature of CKD
Although expressed on mesangial, tubular epithelial and podocyte cells in culture, TrkA expression in normal renal cells in vivo is very low. In contrast, it is expressed on both glomerular cells and tubular epithelial cells in diabetic nephropathy (Fragiadaki et al. 2012) and in other CKDs associated with glomerulosclerosis and interstitial fibrosis (Bonofiglio et al. 2007). We also showed TrkA activation in DN kidney biopsies. It seems likely that TrkA expression is induced in CKD by hypoxia which is commonly present. It has four canonical hypoxia responsive elements in its promoter and hypoxia inducible factor 1α (HIF1α), a transcription factor that binds these elements, accumulates in proximal tubular epithelial cells exposed to either dimethyloxaloylglycine (DMOG) or desferrioxamine (DFO), agents which are commonly used to mimic hypoxic changes in cells. Moreover, in vivo, TrkA is expressed de novo in renal tubular epithelial cells in the hypoxia/reperfusion model of kidney injury (Fragiadaki et al. 2012). It is noteworthy that CTGF is also induced by hypoxia (Higgins et al. 2004).
CTGF–TrkA signalling may require co-receptors
Mason (2009) speculated that CTGF–TrkA/p75 signalling may require ligation of other cell surface proteins such as integrins and heparan sulphate proteoglycans following clustering in the plasma membrane. Recently, Edwards et al. (2011) reported that CTGF binds to a β1integrin/TrkA plasma membrane complex in glioma tumour-initiating or tumour stem cells, activating NF-κB. In another setting, CTGF induced expression of the chemokines fractalkine, MCP-1 and RANTES in mesangial cells via an ERK1/2- and PI3-K/PKB/NF-κB-dependent pathway. It was proposed that this was initiated by CTGF interaction with TrkA and a heparan sulphate proteoglycan co-receptor as it was inhibited by K252a or heparin (Wu et al. 2008).
Connective tissue growth factor and TrkA are endocytosed and may participate in intracellular signalling
Connective tissue growth factor is internalized from the mesangial cell surface in endosomes and accumulates in a juxtanuclear organelle. Some CTGF then translocates to the cytosolic compartment where it is phosphorylated by protein kinase C before translocating to the nucleus (Wahab et al. 2001b). In unstimulated cells, TrkA is distributed in a punctate pattern over the whole cell and also in a ring-like structure around the nucleus. No p-TrkA is present. Following CTGF stimulation juxtanuclear TrkA becomes more prominent and punctate p-TrkA staining appears in the nucleus within 20 min (Fragiadaki and Mason unpublished results). Taken together these observations suggest that, as well as initiating signalling pathways at the plasma membrane, CTGF/TrkA may participate in complex intracellular signalling mechanisms, first in signalling endosomes (Miaczynska et al. 2004) and then possibly within the nucleus itself. The internalization of NGF/TrkA to form signalling endosomes is well established (Grimes et al. 1996). Treatment of isolated nuclei with CTGF markedly increased their RNA pool size, but no specific mRNAs encoding collagens, fibronectin, etc., were detected, leading us to conclude that the increased transcription may be attributed to ribosomal RNA (Wahab et al. 2001b).
Integrin adhesion receptors
All CCN family members interact with integrins. At least eight different integrins are involved, with Lau and colleagues proposing that CCN proteins mediate their diverse biological actions on many different cell types primarily through interactions with these cell adhesion receptors (Chen & Lau 2009; Jun & Lau 2011). Integrins are expressed at the sites of focal adhesions and are a family of heterodimeric membrane-spanning proteins that interact with a variety of proteins extracellularly. This initiates conformational changes that promote their assembly with protein complexes on their cytoplasmic domains. These both form bridges to the cytoskeleton of the cell and activate associated signalling kinases such as ILK and focal adhesion kinase (FAK). Integrins participate in both outside-in and inside-out signalling and are involved in many biological functions ranging from cell adhesion to sensing the extracellular environment and mediating cell responses to it. Ligand binding may promote clustering and cross-talk between integrin complexes and receptor tyrosine kinases (Campbell & Humphries 2011; McDonald et al. 2011; Millard et al. 2011).
Crean et al. (2002) reported that CTGF mediates signalling in mesangial cells at least in part through a β3-integrin-mediated process. Treatment with anti-β3 antibodies inhibited CTGF-induced activation of ERK1/2 (p42/44) and Akt (PKB) and blocked CTGF-stimulated fibronectin expression. Furthermore, CTGF-induced mesangial cell migration that was dependent on ERK1/2 and Akt signalling was blocked by antibodies against either α1, α2, β3 or αvβ3 integrins. We observed that K252a, a TrkA inhibitor, attenuated CTGF-induced ERK1/2 and Akt phosphorylation (Wahab et al. 2005c) and that siRNA depletion of either CTGF or TrkA blocked high glucose-induced ERK1/2 activation (Fragiadaki et al. 2012) in mesangial cells. Collectively these results suggest that CTGF activation of these signalling pathways in mesangial cells may involve crosstalk or interaction between integrins and the TrkA receptor, as discussed earlier. Heparan sulphate proteoglycans may be a further component of such a complex (Gao & Brigstock 2004).
As well as stimulating intracellular signalling, CTGF induces dissolution of focal adhesions in mesangial cells and actin filament disassembly, promoting cell migration in vitro and, perhaps more importantly, preventing a contractile response to vasoconstrictors. Hypocontractility is thought to occur in diabetic nephropathy. These changes involve dephosphorylation of FAK and paxillin, an intracellular protein interacting with both the integrin cytoplasmic domain and FAK (Crean et al. 2004). Src recruitment and activation follow exposure to CTGF, and the dephosphorylations appear to depend on increased activity of the Src-dependent tyrosine phosphatase, SHP-2. The detailed mechanism underlying this action of CTGF remains to be elucidated.
TGFβ receptor III
O'Donovan et al. (2012) proposed that CTGF switches TGFβ1 signalling from Smad-dependent to Smad-independent signalling, mediated at least in part by CTGF interaction with TGFβ receptor III (TGFβRIII). They reached this conclusion after finding that (i) CTGF co-immunoprecipitated with a V5-TGFβRIII fusion protein expressed in mesangial cells; (ii) treating TGFβ-stimulated mesangial cells with CTGF reduced pSmad2 and pSmad3 levels, whilst simultaneously inducing a positive shift toward non-canonical ERK1/2 MAPK signalling; (iii) CTGF also reduced TGFβ-transcriptional responses in HeLa cells transfected with the 3TP-Lux reporter gene; (iv) TGFβRIII knock-down with a shRNA reversed these signalling changes; (v) TGFβ1 binding to TGFβ1-receptor in HK2 cells was reduced in the presence of CTGF. These findings are, however, difficult to reconcile with previous reports. For example, CTGF treatment increased p-Smad2, p-Smad3 and transcriptional activity measured by the SBE4-Luc reporter gene and transcription of TGFβ-responsive genes in TGFβ-stimulated mesangial cells (Wahab et al. 2005c). In this study, p-Smad levels were measured in nuclear extracts since they modulate transcription in the nucleus, rather than in the whole cell lysates from which nuclear debris had been removed, used by O'Donovan et al. (2012). Secondly, Abreu et al. (2002) found that CTGF promoted TGFβ binding to its receptor, as discussed earlier. Thirdly, strong evidence to date indicates that CTGF promotes TGFβ-Smad signalling in vivo to induce fibrosis in models such as the UUO model (Yokoi et al. 2004) and the subtotal nephrectomy model (Okada et al. 2002), also discussed earlier. Thus, the findings by O'Donovan et al. (2012) and the role of the CTGF/TGFβRIII interaction require further clarification.
Interactions between CTGF and other cytokines may impact on renal fibrosis
Bone morphogenetic factor-4
Bone morphogenetic factor-4 (BMP4) is a member of the TGFβ superfamily. Connective tissue growth factor binds directly to BMP4 [Kd 5 nM] with a higher affinity than it does to TGFβ [Kd 30 nM]. Interaction of BMP4 with its receptor and downstream signalling via Smad1 phosphorylation were decreased in the presence of CTGF (Abreu et al. 2002), and this was attributed to direct binding between CTGF and BMP4. However, in vivo, although glomerular CTGF is increased in murine and human DN (Riser et al. 2000; Wahab et al. 2005a) diabetic mice with mesangial matrix expansion have increased glomerular BMP4 expression and p-Smad1 levels (Tominaga et al. 2011). Indeed, overexpression of BMP4 in transgenic mice leads to marked glomerulosclerosis and albuminuria, changes akin to those in diabetic nephropathy. Diabetic BMP4+/− mice have reduced glomerulosclerosis compared to BMP4+/+ mice, confirming a role for BMP4 in this disease (Tominaga et al. 2011). The interaction of CTGF with BMP4 in DN has yet to be explored but it seems unlikely that CTGF significantly suppresses the actions of BMP4, as might be predicted from the findings of Abreu et al. (2002).
Bone morphogenetic factor-7
Bone morphogenetic factor-7 is another member of the TGFβ superfamily. It also binds to CTGF (Kd 14 nM; Nguyen et al. 2008). Following reports that administration of BMP7 reduced glomerulosclerosis in several different animal models of renal fibrosis Wang and Hirschberg (2003) showed that this factor antagonizes TGFβ-induced fibrogenic responses in mesangial cells, including reducing fibronectin and type IV collagen accumulation, CTGF expression, PAI-1 promoter activity and MMP2 gelatinase activity. Bone morphogenetic factor-7 is expressed normally in tubular epithelial cells but this is lost during STZ-induced diabetes, as is expression of the BMP type II receptor and the ALK2 type I receptor. Gremlin, a BMP antagonist, increased. This and in vitro studies gave rise to the proposal that loss of BMP7 activity is profibrogenic (Wang et al. 2001b). Subsequently, Nguyen et al. (2008) found that nephropathy was attenuated in diabetic CTGF+/− mice in which glomerular CTGF levels are reduced compared to CTGF+/+ mice. Whilst BMP7 signalling was reduced in diabetic CTGF+/− mice as evidenced by decreased p-Smad1/5 levels and expression of the downstream target Id1, this was relatively preserved in the CTGF+/+ mice. Moreover, when CTGF was injected into non-diabetic mice, renal p-Smad 1/5 levels fell. In vitro studies on renal cells gave similar results (Nguyen et al. 2008). Thus, the higher levels of CTGF in DN, and probably in other CKDs, correlate with decreased BMP7-signalling and disease advancement.
Hepatocyte growth factor
Hepatocyte growth factor (HGF) is expressed widely in tissues, including in interstitial and endothelial cells of the kidney. Proximal tubular epithelial cells (PTECs) express the c-Met receptor for HGF. Mice that are transgenic for TGFβ1 under the control of the albumin promoter express TGFβ in the liver and have increased levels of plasma TGFβ compared to controls. When these transgenic mice were subjected to 5/6 nephrectomy, CTGF levels in tubular and glomerular epithelial cells in the remnant kidney increased markedly compared to controls, probably as a result of the higher plasma TGFβ levels, and the development of interstitial fibrosis was enhanced. Treatment with HGF significantly reduced the CTGF expression and attenuated the expression of collagen and development of fibrosis (Inoue et al. 2003). We have summarized previously the likely pathway, involving HGF induction of SnoN and TGIF (Wahab & Mason 2006). In vitro experiments with co-cultures of PTECs and a tubulointerstitial fibroblast cell line showed that HGF attenuated TGFβ-stimulated CTGF expression in the epithelial cells and reduced collagen expression in the fibroblasts which was dependent, at least in part, on CTGF secreted by the PTEC. Thus, HGF is able to attenuate TGFβ-stimulated fibrogenic responses by down-regulating CTGF (Inoue et al. 2003).
CCN3
Riser et al. (2009) discovered an intriguing relationship between CTGF (CCN2) and Nov (CCN3). CCN3 is expressed in unstimulated mesangial cells, whilst CTGF expression is negligible. Following stimulation with TGFβ, CCN3 expression declined and CTGF levels increased, followed by increase in collagen1 transcription and translation. However, addition of either conditioned medium enriched in CCN3 or purified recombinant CCN3 was able to block CTGF and collagen 1 expression in TGFβ-stimulated cells, suggesting a reciprocal or ‘ying–yang’ relationship between the two CCN proteins. The mechanism of CCN3 inhibition of CTGF expression is unknown but is not due to downregulation of Smad3 signalling. The authors hypothesize that CCN3 is an endogenous negative regulator of ECM and fibrosis. Thus, in normal wound healing, CCN3 may be expressed at the stage of resolution, switching off CTGF and extracellular matrix synthesis, whilst in fibrosis CCN3 levels remain low and CTGF continues to stimulate ECM production.
Oncostatin M
Oncostatin M (OSM) belongs to the IL-6 family of cytokines. We proposed that it may play a role in renal interstitial inflammation, reporting that it was expressed by activated peripheral blood mononuclear cells, that media conditioned by these cells induced the expression of the OSM-specific receptor β-subunit in human PTECs, and that OSM activated the Jak/Stat signalling pathway in these cells, which subsequently underwent EMT in vitro (Nightingale et al. 2004). Pollack et al. (2007) later speculated that OSM may be able to act as either a pro-fibrosis cytokine or, conversely, as a pro-healing cytokine, depending on the prevailing cellular microenvironment or nature of the cellular injury. They went on to report that OSM attenuated TGFβ-induced expression of CTGF and several other matricellular proteins in human PTEC. This effect was at least partly dependent on OSM-receptor-mediated Stat1/Stat3 signalling and independent of the Smad2/3 pathway (Sarkozi et al. 2011). Investigations into the relationship between OSM and CTGF in vivo would be of interest, especially whether OSM is able to suppress matricellular protein expression by PTECs in renal inflammation.
In summary, following tissue damage, the local concentrations of various cytokines acting on CTGF are likely to contribute importantly to whether injury is followed by healing and resolution, or by fibrosis.
Other factors that may impact on CTGF expression and renal fibrosis
MicroRNAs
MicroRNAs (miRNAs) are short non-coding RNAs which inhibit the expression of target genes. When dicer, an enzyme required to generate miRNAs, was selectively deleted in murine podocytes the mice developed proteinuria and glomerular lesions including glomerulosclerosis, demonstrating the importance of miRNAs in maintaining normal kidney structure and function (Shi et al. 2008). MicroRNAs are clearly involved in regulating TGFβ expression and fibrosis in the kidney (Patel & Noureddine 2012). Information about whether miRNAs are involved in regulating CTGF expression in the kidney is lacking. However, two miRNAs (miR-133 and miR-30c) that interact with the 3′ untranslated region of the CTGF transcript and are negative regulators of CTGF expression have been implicated in the pathophysiology of rodent and human left ventricular hypertrophy (LVH)/cardiac fibrosis (Duisters et al. 2009). Both miRNAs are down-regulated in LVH with concomitant increases in CTGF. Collectively these results suggest there may be an important role for miRNA regulation of CTGF expression in the development of kidney fibrosis.
Single nucleotide polymorphisms
A single nucleotide polymorphism (SNP) occurs in the CTGF promoter at G-945C. The GG genotype occurred more frequently in patients with systemic sclerosis than in controls in a large UK study (Fonseca et al. 2007). Moreover, the authors observed that the C allele has a high affinity for Sp3, a transcriptional regulator, and is associated with reduced transcriptional activity, repressing CTGF expression. However, a further large study on subjects of wide European origin did not find any significant association of this genetic variant with systemic sclerosis (Rueda et al. 2009). To date, there is little information concerning the possible effects of CTGF SNPs and susceptibility to renal fibrosis, although a study on the G-945C polymorphism in patients with diabetes with and without evidence of nephropathy did not find any correlation between the polymorphism and renal disease or the plasma CTGF level (Dendooven et al. 2011). Further studies on CTGF SNPs and the onset and progression of renal fibrosis in different kidney diseases are required to reveal whether such SNPs are important or not.
Concluding comments
A large body of evidence implicates CTGF in the development of renal fibrosis. This comes from both in vivo and in vitro experiments, from the investigation of naturally occurring diseases in which kidney fibrosis develops, from animal models and from experiments with renal cells. Connective tissue growth factor is involved in the early stages of inflammation before overt fibrosis and at later stages when it is well established. Connective tissue growth factor is a pleiotropic player in these processes, its actions ranging from receptor activation inducing cell signalling and de novo gene transcription, to participating in a delicate ‘yin-yang’ balance with other cytokines and growth factors, steering injured tissues towards fibrosis if CTGF predominates or healing if it does not. A recurrent but not exclusive theme is the role of CTGF acting in a cooperative manner with TGFβ to promote fibrotic responses. Overall its effects in adult tissues are pernicious. The evidence points to CTGF, its receptors, and the signalling pathways initiated by their activation, providing a range of targets for new therapeutic interventions in fibrotic kidney diseases. Current therapies for fibrotic diseases have very limited success. Reaching a complete understanding of mechanisms that control the yin–yang balance between CTGF and other factors, including miRNAs, in determining fibrosis or healing, the ways in which TGFβ and CTGF work together or not, and the relative importance of various receptors for CTGF and how they work cooperatively together, or independently, are likely to provide key challenges in developing successful new therapies to counter the development of renal fibrosis.
Acknowledgments
This review is based on the Fell-Muir Award lecture given at the Spring Meeting of the British Society for Matrix Biology, Oxford, 2012. I wish to acknowledge the excellent work of the members of my laboratory. I also acknowledge my early biochemistry teachers, the late Professors John Pryde and Ken Dodgson, whose influence directed me towards biochemical research in medicine. I thank Charles Pusey, David Abraham, George Bou-Gharios and Andrew Pitsillides for their comments on the draft manuscript.
Funding
This study was supported by the Medical Research Council UK and Diabetes UK.
Conflict of interest
None.
References
- Abreu JG, Ketpura NI, Reversade B, de Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGFβ. Nat. Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler SG, Schwartz S, Williams ME, et al. Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin. J. Am. Soc. Nephrol. 2010;5:1420–1428. doi: 10.2215/CJN.09321209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SJ, Dawbarn D. Clinical relevance of the neurotrophins and their receptors. Clin. Sci. 2006;110:175–191. doi: 10.1042/CS20050161. [DOI] [PubMed] [Google Scholar]
- Bonofiglio R, Antonucci MT, Papalia T, et al. Nerve growth factor (NGF) and NGF-receptor expression in diseased human kidneys. J. Nephrol. 2007;20:186–195. [PubMed] [Google Scholar]
- Boutet A, de Frutos CA, Maxwell PH, Mayol MJ, Romero J, Nieto MA. Snail1 activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006;25:5603–5613. doi: 10.1038/sj.emboj.7601421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradham DM, Igarashi A, Potter RL, Grotendorst GR. Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J. Cell Biol. 1991;114:1285–1294. doi: 10.1083/jcb.114.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell ID, Humphries MJ. Integrin structure, activation and interactions. Cold Spring Harb. Perspect. Biol. 2011;3:a004994. doi: 10.1101/cshperspect.a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV, Rajagopal R, Lee FS. Neurotrophin signalling in health and disease. Clin. Sci. 2006;110:167–173. doi: 10.1042/CS20050163. [DOI] [PubMed] [Google Scholar]
- Chen C-C, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int. J. Biochem. Cell Biol. 2009;41:771–783. doi: 10.1016/j.biocel.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SJ, Yuan W, Lo S, Trojanowska M, Varga J. Interaction of smad3 with a proximal smad-binding element of the human alpha2 (I) procollagen gene promoter required for transcriptional activation by TGF-beta. J. Cell. Physiol. 2000;183:381–392. doi: 10.1002/(SICI)1097-4652(200006)183:3<381::AID-JCP11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Chujo S, Shirasaki F, Kawara S, et al. Connective tissue growth factor causes persistent proa2 (I) collagen gene expression induced by transforming growth factor-b in a mouse fibrosis model. J. Cell Physiol. 2005;203:447–456. doi: 10.1002/jcp.20251. [DOI] [PubMed] [Google Scholar]
- Cicha I, Yilmaz A, Klien M, et al. Connective tissue growth factor is overexpressed in complicated atherosclerotic plaques and induces mononuclear cell chemotaxis in vitro. Artioscler. Thromb. Vasc. Biol. 2005;25:1008–1013. doi: 10.1161/01.ATV.0000162173.27682.7b. [DOI] [PubMed] [Google Scholar]
- Crean JKG, Findlay D, Murphy M, et al. The Role of p42/44 MAPK and protein kinase B in connective tissue growth factor induced extracellular matrix production, cell migration and actin cytoskeletal rearrangement in human mesangial cells. J. Biol. Chem. 2002;46:44187–44194. doi: 10.1074/jbc.M203715200. [DOI] [PubMed] [Google Scholar]
- Crean JK, Furlong F, Finlay D, et al. Connective tissue growth factor [CTGF]/CCN2 stimulates mesangial cell migration through integrated dissolution of focal adhesion complexes and activation of cell polarization. FASEB J. 2004;20:1712–1724. doi: 10.1096/fj.04-1546fje. [DOI] [PubMed] [Google Scholar]
- Cunningham ME, Stephens RM, Kaplan DR, Greene LA. Autophosphorylation of activation loop tyrosines regulates signalling by the TRK nerve growth factor receptor. J. Biol. Chem. 1997;272:10957–10967. doi: 10.1074/jbc.272.16.10957. [DOI] [PubMed] [Google Scholar]
- De Winter P, Leoni P, Abraham D. Connective tissue growth factor; Structure-function relationships of a mosaic, multifunctional protein. Growth Factors. 2009;26:80–91. doi: 10.1080/08977190802025602. [DOI] [PubMed] [Google Scholar]
- Dean RA, Butler GS, Hamma-Kourbali Y, et al. Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP-2) in cell-based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affin regulatory peptide (pleiotropin) and VEGF/connective tissue growth factor angiogenic inhibitory complexes by MMP-2 proteolysis. Mol. Cell Biol. 2007;27:8454–8465. doi: 10.1128/MCB.00821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dendooven A, Nguyen TQ, rosens L, et al. The CTGF-945GC polymorphism is not associated with plasma CTGF and does not predict nephropathy or outcome in type 1 diabetes. J. Negat. Results Biomed. 2011;10:4. doi: 10.1186/1477-5751-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duisters RF, Tijsen AJ, Schroen B, et al. MiR-133 and miR-30 regulate connective tissue growth factor. Implications for a role of microRNAs in myocardial matrix remodelling. Circ. Res. 2009;104:170–178. doi: 10.1161/CIRCRESAHA.108.182535. [DOI] [PubMed] [Google Scholar]
- Edwards LA, Woolard K, Son MJ, et al. Effect of brain- and tumor-derived connective tissue growth factor on glioma invasion. J. Natl Cancer Inst. 2011;103:1–17. doi: 10.1093/jnci/djr224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanders KC. Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floege J, Johnson RJ. Cytokines in renal inflammation. Curr. Opin. Nephrol. Hypertens. 1993;2:449–457. doi: 10.1097/00041552-199305000-00013. [DOI] [PubMed] [Google Scholar]
- Fonseca C, Lindahl GE, Ponticos M, et al. A polymorphism in the CTGF promoter region associated with systemic sclerosis. N. Engl. J. Med. 2007;357:1210–1220. doi: 10.1056/NEJMoa067655. [DOI] [PubMed] [Google Scholar]
- Fragiadaki M, Mason RM. Epithelial-mesenchymal transition in renal fibrosis – evidence for and against. Int. J. Exp. Pathol. 2011;92:143–150. doi: 10.1111/j.1365-2613.2011.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragiadaki M, Witherden AS, Kaneko T, et al. Interstitial fibrosis is associated with increased COL1A2 transcription in AA-injured renal tubular epithelial cells in vivo. Matrix Biol. 2011;30:396–403. doi: 10.1016/j.matbio.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Fragiadaki M, Hill N, Hewitt R, et al. Hyperglycaemia causes renal cell damage via CCN2-induced activation of the TrkA receptor: implications for diabetic nephropathy. Diabetes. 2012;61:2280–2288. doi: 10.2337/db11-1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier KS, Paredes A, Dube P, Styer E. Connective tissue growth factor expression in the rat remnant kidney model and association with tubular epithelial cells undergoing transdifferentiation. Vet. Pathol. 2000;37:328–335. doi: 10.1354/vp.37-4-328. [DOI] [PubMed] [Google Scholar]
- Gao R, Brigstock DR. Low density lipoprotein receptor-related protein (LRP) is a heparin dependant receptor for connective tissue growth factor (CTGF) in rat activated hepatic stellate cells. Hepatol. Res. 2003;27:214–220. doi: 10.1016/s1386-6346(03)00241-9. [DOI] [PubMed] [Google Scholar]
- Gao R, Brigstock DR. Connective tissue growth factor (CCN2) induces adhesion of rat hepatic stellate cells by binding of its C-terminal domain to integrin alpha (v) beta (3) and heparin sulphate proteoglycan. J. Biol. Chem. 2004;279:8848–8855. doi: 10.1074/jbc.M313204200. [DOI] [PubMed] [Google Scholar]
- Gao R, Brigstock DR. Connective tissue growth factor (CCN2) in rat pancreatic stellate cell function: integrin α5β1 as a novel CCN2 receptor. Gastroenterology. 2005;120:1019–1030. doi: 10.1053/j.gastro.2005.06.067. [DOI] [PubMed] [Google Scholar]
- Gonias SL, Wu L, Salicioni AM. Low density lipoprotein receptor-related protein: regulation of the plasma membrane proteome. Thromb. Haemost. 2004;91:1056–1064. doi: 10.1160/TH04-01-0023. [DOI] [PubMed] [Google Scholar]
- Grimes ML, Zhou J, Beattie EC, et al. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signalling endosomes. J. Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M, Xu Z-G, Tung D, Lanting L, Natarajan R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007;21:3355–3368. doi: 10.1096/fj.06-6713com. [DOI] [PubMed] [Google Scholar]
- Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH. Hypoxic induction of Ctgf is directly mediated by Hif-1. Am. J. Renal Physiol. 2004;287:F1223–F1232. doi: 10.1152/ajprenal.00245.2004. [DOI] [PubMed] [Google Scholar]
- Hishikawa K, Oemar BS, Nakaki T. Static pressure regulates connective tissue growth factor expression in human mesangial cells. J. Biol. Chem. 2001;276:16797–16803. doi: 10.1074/jbc.M010722200. [DOI] [PubMed] [Google Scholar]
- Hu K, Wu C, Mars WM, Liu Y. Tissue-type plasminogenactivator promotes murine myofibroblast activation through LDL receptor-related protein-1-mediated integrin signalling. J. Clin. Invest. 2007;117:3821–3832. doi: 10.1172/JCI32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor expression in human skin fibroblasts and during wound repair. Mol. Biol. Cell. 1993;4:637–645. doi: 10.1091/mbc.4.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi A, Nashiro K, Kikuchi K, Sato S, et al. Significant correlation between connective tissue growth factor gene expression and skin sclerosis in tissue sections from patients with systemic sclerosis. J. Invest. Dermatol. 1995;105:280–284. doi: 10.1111/1523-1747.ep12318465. [DOI] [PubMed] [Google Scholar]
- Inoue T, Okada H, Kobayashi T, et al. Hepatocyte growth factor counteracts transforming growth factor-beta1, through attenuation of connective tissue growth factor induction, and prevents renal fibrogenesis in 5/6 nephrectomized mice. FASEB J. 2003;17:268–270. doi: 10.1096/fj.02-0442fje. [DOI] [PubMed] [Google Scholar]
- Ito Y, Aten J, Bende RJ, et al. Expression of Connective tissue growth factor in human renal fibrosis. Kidney Int. 1998;53:853–861. doi: 10.1111/j.1523-1755.1998.00820.x. [DOI] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC. TGFbeta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene. 2002;21:5783–5790. doi: 10.1038/sj.onc.1205681. [DOI] [PubMed] [Google Scholar]
- Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011;10:945–963. doi: 10.1038/nrd3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemoto K, Usui J, Nitta K, Horita S, Koyama A, et al. In situ expression of connective tissue growth factor in human crescentric glomerulonephritis. Virchows Arch. 2004;444:257–263. doi: 10.1007/s00428-003-0959-z. [DOI] [PubMed] [Google Scholar]
- Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis. Am. J. Physiol. Renal Physiol. 2002;283:F861–F875. doi: 10.1152/ajprenal.00362.2001. [DOI] [PubMed] [Google Scholar]
- Leask A, Parapuram SK, Shi-wen X, Abraham DJ. Connective tissue growth factor (CTGF,CCN2) gene regulation: a potent clinical bio-marker of fibroproliferative disease? J. Cell Commun. Signal. 2009;3:89–94. doi: 10.1007/s12079-009-0037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-L, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008;173:1617–1727. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011;7:684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Taghavi R, Leask A. Connective tissue growth factor is induced in bleomycin-induced skin scleroderma. J. Cell Commun. Signal. 2010;4:25–30. doi: 10.1007/s12079-009-0081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Shi-wen X, Abraham DJ, Leask A. CCN2 is required for bleomycin-induced skin fibrosis in mice. Arth. Rheum. 2011;63:239–246. doi: 10.1002/art.30074. [DOI] [PubMed] [Google Scholar]
- Lόpez-Hernandez FJ, Lόpez-Nova JM. Role of TGF-β in chronic kidney disease: an integration of tubular, glomerular and vascular effects. Cell Tissue Res. 2012;347:141–154. doi: 10.1007/s00441-011-1275-6. [DOI] [PubMed] [Google Scholar]
- Mason RM. Connective tissue growth factor (CCN2), a pathogenic factor in diabetic nephropathy. What does it do? How does it do it? J. Cell Commun. Signal. 2009;3:95–104. doi: 10.1007/s12079-009-0038-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RM, Li XJ, Wahab NA. High glucose induces the expression of connective tissue growth factor in human mesangial cells. J. Am. Soc. Nephrol. 1997;8:642A. [Google Scholar]
- McDonald PC, Fielding AB, Dehar S. Integrin-linked kinase – essential roles in physiology and cancer biology. J. Cell Sci. 2011;121:3121–3132. doi: 10.1242/jcs.017996. [DOI] [PubMed] [Google Scholar]
- Mercurio S, Latinkic B, Itasaki N, Krumlauf R, Smith JC. Connective-tissue growth factor modulates WNT signalling and interacts with the WNT receptor complex. Development. 2004;131:2137–2147. doi: 10.1242/dev.01045. [DOI] [PubMed] [Google Scholar]
- Mezzano SA, Barria M, Droguett MA, et al. Tubular NF-kappaB and AP-1 activation in human proteinuric renal disease. Kidney Int. 2001;60:1366–1377. doi: 10.1046/j.1523-1755.2001.00941.x. [DOI] [PubMed] [Google Scholar]
- Mezzano S, Aros C, Droguett A, et al. NF-kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol. Dial. Transplant. 2004;19:2505–2512. doi: 10.1093/ndt/gfh207. [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr. Opin. Cell Biol. 2004;16:400–406. doi: 10.1016/j.ceb.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Millard M, Odde S, Neamati N. Integrin targeted therapeutics. Theranostics. 2011;1:154–188. doi: 10.7150/thno/v01p0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa K, Hara T, Furuya T, Miyazono K. Two major smad pathways in TGF-β superfamily signalling. Genes Cells. 2002;7:1191–1204. doi: 10.1046/j.1365-2443.2002.00599.x. [DOI] [PubMed] [Google Scholar]
- Mori T, Kawara S, Shinozaki M, et al. Role and interaction of connective tissue growth factor with transforming growth factor beta in persistent fibrosis: a mouse fibrosis model. J. Cell Physiol. 1999;181:153–159. doi: 10.1002/(SICI)1097-4652(199910)181:1<153::AID-JCP16>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Mori Y, Hinchcliff M, Wu M, Warner-Blankenship M, Lyons K, Varga J. Connective tissue growth factor. CCN2-null mouse embryonic fibroblasts retain intact transforming growth factor-β responsiveness. Exp. Cell Res. 2008;314:1094–1104. doi: 10.1016/j.yexcr.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Godson C, Cannon S, et al. Supression subtractive hybridization identifies high glucose level as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J. Biol. Chem. 1999;274:5830–5834. doi: 10.1074/jbc.274.9.5830. [DOI] [PubMed] [Google Scholar]
- Nakerakanti SS, Bujor AM, Trojanowska M. CCN2 is required for the TGF-β induced activation of smad1-Erk ½ signalling network. PLoS ONE. 2011;6:e21911. doi: 10.1371/journal.pone.0021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PJ, von Toerne C, Grone HJ. Wnt-signaling pathways in progressive renal fibrosis. Expert Opin. Ther. Targets. 2011;15:1073–1083. doi: 10.1517/14728222.2011.588210. [DOI] [PubMed] [Google Scholar]
- Nguyen TQ, Roestenberg P, van Nieuwenhoven FA, et al. CTGF inhibits BMP-7 signaling in diabetic nephropathy. J. Am. Soc. Nephrol. 2008;19:2098–2107. doi: 10.1681/ASN.2007111261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale J, Patel S, Suzuki N, et al. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J. Am. Soc. Nephrol. 2004;15:21–32. doi: 10.1097/01.asn.0000102479.92582.43. [DOI] [PubMed] [Google Scholar]
- Nishida T, Nakanishi T, Shimo T, et al. Demonstration of receptors specific for connective tissue growth factor on a human chondrocytic cell line (HCS-2/8) Biochem. Biophys. Res. Commun. 1998;247:905–909. doi: 10.1006/bbrc.1998.8895. [DOI] [PubMed] [Google Scholar]
- Nonaka TS, Fujita T, Takahashi T, et al. TGF-beta1 and CTGF mRNAs are correlated with urinary protein level in IgA nephropathy. J. Nephrol. 2008;21:53–63. [PubMed] [Google Scholar]
- O'Donovan HC, Hickey F, Brazil DP, et al. Connective tissue growth factor antagonizes transforming growth factor-β/Snad signalling in renal mesangial cells. Biochem. J. 2012;441:499–510. doi: 10.1042/BJ20110910. [DOI] [PubMed] [Google Scholar]
- Okada H, Inoue T, Kanno Y, et al. Interstitial fibroblast-like cells express renin-angiotensin system components in a fibrosing murine kidney. Am. J. Pathol. 2002;160:765–772. doi: 10.1016/S0002-9440(10)64898-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H, Kikuta T, Kobayashi T, et al. Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrogenesis. J. Am. Soc. Nephrol. 2005;16:133–143. doi: 10.1681/ASN.2004040339. [DOI] [PubMed] [Google Scholar]
- Panek AN, Posch MG, Alenina N, et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertropy and protection against pressure overload. PLoS One. 2009:e6743. doi: 10.1371/journal.pone.0006743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannu J, Asano Y, Nakerakanti S, et al. Smad1 pathway is activated in systemic sclerosis fibroblasts and is targeted by imatinib mesylate. Arthritis Rheum. 2008;58:2528–2537. doi: 10.1002/art.23698. [DOI] [PubMed] [Google Scholar]
- Patel V, Noureddine L. MicroRNAs and Fibrosis. Curr. Opin. Nephrol. Hypertens. 2012;21:410–416. doi: 10.1097/MNH.0b013e328354e559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perbal B. Alternative splicing of CCN mRNAs…it has been upon us. J. Cell. Commun. Signal. 2009;3:153–157. doi: 10.1007/s12079-009-0051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi L, Xia H, Liu J, Shenoy AK, Hauswirth WW, Scott EW. Role of connective tissue growth factor in retinal vasculature during development. Invest. Ophthalmol. Vis. Sci. 2011;52:8701–8710. doi: 10.1167/iovs.11-7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack V, Sarkozi R, Banki Z, et al. Oncostatin M-induced effects on EMT in human proximal tubular cells: differential role of ERK signalling. Am. J. Physiol. Renal Physiol. 2007;293:F1714–F1726. doi: 10.1152/ajprenal.00130.2007. [DOI] [PubMed] [Google Scholar]
- Ren S, Babelova A, Moreth K, et al. Transforming growth factor-beta2 upregulates sphingosine kinase-1activity, which in turn attenuates the fibrotic response to TGF-beta2 by impeding CTGF expression. Kidney Int. 2009;76:857–867. doi: 10.1038/ki.2009.297. [DOI] [PubMed] [Google Scholar]
- Riser BL, Denichilo M, Cortes P, et al. regulation of connective tissue growth factor activity in cultured rat mesangial cells and in experimental diabetic glomerulosclerosis. J. Am. Soc. Nephrol. 2000;11:25–38. doi: 10.1681/ASN.V11125. [DOI] [PubMed] [Google Scholar]
- Riser BL, Najmabadi F, Perbal B, et al. CCN3 (NOV) is a negative regulator of CCN2 (CTGF) and a novel endogenous inhibitor of the fibrotic pathway in an in vitro model of renal disease. Am. J. Pathol. 2009;174:1725–1734. doi: 10.2353/ajpath.2009.080241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Pena AB, Grande MT, Eleno N, et al. Activation of Erk1/2 and Akt following unilateral ureteral obstruction. Kidney Int. 2008;74:196–209. doi: 10.1038/ki.2008.160. [DOI] [PubMed] [Google Scholar]
- Rooney B, O'Donovan H, Gaffney A, et al. CTGF/CCN2 activates canonical Wnt signalling in mesangial cells through LRP6: implications for the pathogenesis of diabetic nephropathy. FEBS Lett. 2011;585:531–538. doi: 10.1016/j.febslet.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Rueda B, Simeon C, Hesselstrand R, et al. A large multicentre analysis of CTGF -945 promoter polymorphism does not confirm association with systemic sclerosis susceptibility or phenotype. Ann. Rheum. Dis. 2009;68:1618–1620. doi: 10.1136/ard.2008.100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryseck RP, Macdonald-Bravo H, Mattéi MG, Bravo R. Structure, mapping and expression of fisp-12, a growth factor-inducible gene encoding a secreted cysteine-rich protein. Cell Growth Differ. 1991;2:225–233. [PubMed] [Google Scholar]
- Sanchez-Lopez E, Rayego S, Rodrigues-Diez R, et al. CTGF promotes inflammatory cell infiltration of the renal interstitium by activating NF-κB. J. Am. Soc. Nephrol. 2009;20:1513–1526. doi: 10.1681/ASN.2008090999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkozi R, Hauser C, Noppert SJ, et al. Oncostatin M is a novel inhibitor of TGF-β1-induced matricellular protein expression. Am. J. Physiol. Renal Physiol. 2011;301:F1014–F1025. doi: 10.1152/ajprenal.00123.2011. [DOI] [PubMed] [Google Scholar]
- Schecterson LC, Bothwell M. Neurotrophin receptors: old friends with new partners. Dev. Neurobiol. 2010;70:332–338. doi: 10.1002/dneu.20767. [DOI] [PubMed] [Google Scholar]
- Schober JM, Chen N, Grzeskiewicz TM, et al. Identification of integrin αMβ2 as an adhesion receptor on peripheral blood monocytes for cyr 61 (CCN1) and connective tissue growth factor CTGF): immediate early gene products expressed in atherosclerosis. Blood. 2002;99:4457–4465. doi: 10.1182/blood.v99.12.4457. [DOI] [PubMed] [Google Scholar]
- Schrimpf C, Duffield JS. Mechanisms of fibrosis: the role of the pericyte. Curr. Opin. Nephrol. Hypertens. 2011;20:297–305. doi: 10.1097/MNH.0b013e328344c3d4. [DOI] [PubMed] [Google Scholar]
- Segarini PR, Nesbitt JE, Li D, Hays LG, Yates JR, III, Carmichael DF. The low density lipoprotein receptor-related protein/macroglobulin receptor is a receptor for connective tissue growth factor. J. Biol. Chem. 2001;44:40659–40667. doi: 10.1074/jbc.M105180200. [DOI] [PubMed] [Google Scholar]
- Shi S, Yu L, Chiu C, et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J. Am. Soc. Nephrol. 2008;19:2159–2169. doi: 10.1681/ASN.2008030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19:133–144. doi: 10.1016/j.cytogfr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Sonnylai S, Shi-wen X, Leoni P, et al. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic fibrosis. Arthritis Rheum. 2010;62:1523–1532. doi: 10.1002/art.27382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanisavljevic J, Porta-de-la Riva M, Batile R, de Herreros AG, Baulida J. The p65 subunit of NF-κB and PARP1 assist Snail1 in activating fibronectin transcription. J. Cell Sci. 2011;124:4161–4171. doi: 10.1242/jcs.078824. [DOI] [PubMed] [Google Scholar]
- Suzuki D, Toyoda M, Umezono T, Uehara G, Zhang SY, et al. Glomerular expression of connective tissue growth factor mRNA in various renal diseases. Nephrology (Carlton) 2003;8:92–97. doi: 10.1046/j.1440-1797.2003.00142.x. [DOI] [PubMed] [Google Scholar]
- Sysa P, Potter JJ, Liu X, Mezey E. Transforming growth factor-b1 up-regulation of human a1 (I) collagen is mediated by Sp1 and smad2 transacting factors. DNA Cell Biol. 2009;28:425–434. doi: 10.1089/dna.2009.0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson SE, McLennan SV, Kirwan PD, et al. Renal connective tissue growth factor correlates with glomerular basement membrane thickness and prospective albuminuria in a non-human primate model of diabetes: possible predictive marker for incipient diabetic nephropathy. J. Diabetes Complicat. 2008;22:284–294. doi: 10.1016/j.jdiacomp.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Tominaga T, Abe H, Ueda O, et al. Activation of bone morphogenetic protein 4 signaling leads to glomerulosclerosis that mimics diabetic nephropathy. J. Biol. Chem. 2011;286:20109–20116. doi: 10.1074/jbc.M110.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowska M. Noncanonical transforming growth factor β signalling in scleroderma fibrosis. Curr. Opin. Rheumatol. 2009;21:623–629. doi: 10.1097/BOR.0b013e32833038ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahab NA, Mason RM. A critical look at growth factors and epithelial to mesenchymal transition in the adult kidney. Nephron Exp. Nephrol. 2006;104:129–134. doi: 10.1159/000094963. [DOI] [PubMed] [Google Scholar]
- Wahab NA, Yevdokimova N, Weston BS, et al. Role of connective tissue growth factor in the pathogenesis of diabetic nephropathy. Biochem. J. 2001a;359:77–87. doi: 10.1042/0264-6021:3590077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahab NA, Brinkman H, Mason RM. Uptake and intracellular transport of the connective tissue growth factor: a potential mode of action. Biochem. J. 2001b;359:89–97. doi: 10.1042/0264-6021:3590089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahab NA, Weston BS, Roberts T, Mason RM. Connective tissue growth factor and regulation of the mesangial cell cycle: role in cellular hypertrophy. J. Am. Soc. Nephrol. 2002;13:2437–2445. doi: 10.1097/01.asn.0000031828.58276.02. [DOI] [PubMed] [Google Scholar]
- Wahab NA, Schaefer L, Weston BS, et al. Glomerular expression of thrombospondin -1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial responses to diabetic stimuli. Diabetologia. 2005a;48:2650–2660. doi: 10.1007/s00125-005-0006-5. [DOI] [PubMed] [Google Scholar]
- Wahab NA, Weston BS, Mason RM. Modulation of the TGFβ/Smad signalling pathway in mesangial cells by CTGF/CCN2. Exp. Cell Res. 2005b;307:305–315. doi: 10.1016/j.yexcr.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Wahab NA, Weston BS, Mason RM. Connective tissue growth factor CCN2 interacts with and activates the tryrosine kinase receptor TrkA. J. Am. Soc. Nephrol. 2005c;16:340–351. doi: 10.1681/ASN.2003100905. [DOI] [PubMed] [Google Scholar]
- Wahab N, Cox D, Witherden A, Mason RM. Connective tissue growth factor (CTGF)promotes activated mesangial cell survival via up-regulation of mitogen-activated protein kinase phphatase-1 (MKP-1) Biochem. J. 2007;406:131–138. doi: 10.1042/BJ20061817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Hirschberg R. BMP7 antagonizes TGF-β- dependent fibrogenesis in mesangial cells. Am. J. Physiol. Renal Physiol. 2003;284:1006–1013. doi: 10.1152/ajprenal.00382.2002. [DOI] [PubMed] [Google Scholar]
- Wang S, Denichilo M, Brubaker C, Hirschberg R. Connective tissue growth factor in tubulointerstitial injury of diabetic nephropathy. Kidney Int. 2001a;60:96–105. doi: 10.1046/j.1523-1755.2001.00776.x. [DOI] [PubMed] [Google Scholar]
- Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J. Am. Soc. Nephrol. 2001b;12:2392–2399. doi: 10.1681/ASN.V12112392. [DOI] [PubMed] [Google Scholar]
- Wang Q, Usinger W, Nichols B, et al. Cooperative interaction of CTGF and TGFβ in animal models of fibrotic disease. Fibrogenesis Tissue Repair. 2011;4:4–11. doi: 10.1186/1755-1536-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston BS, Wahab NA, Mason RM. CTGF mediates TGFβ induced fibronectin matrix deposition by upregulating active α5β1 integrin in human mesangial cells. J. Am. Soc. Nephrol. 2003;14:601–610. doi: 10.1097/01.asn.0000051600.53134.b9. [DOI] [PubMed] [Google Scholar]
- Wolf G, Ziyadeh FN. Molecular mechanisms of diabetic renal hypertrophy. Kidney Int. 1999;56:393–405. doi: 10.1046/j.1523-1755.1999.00590.x. [DOI] [PubMed] [Google Scholar]
- Wu SH, Lu C, Dong L, Chen ZQ. Signal transduction involved in CTGF-induced production of chemokines in mesangial cells. Growth Factors. 2008;26:192–200. doi: 10.1080/08977190802227828. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Huang H, Li J, Li D, Wang H. Tyrosine phosphorylation of the LDL receptor-related protein (LRP) and activation of the ERK pathway are required for connective tissue growth factor to potentiate myofibroblast differentiation. FASEB J. 2004;18:1920–1931. doi: 10.1096/fj.04-2357fje. [DOI] [PubMed] [Google Scholar]
- Yokoi H, Mukoyama M, Nagae T, et al. Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2004;15:1430–1440. doi: 10.1097/01.asn.0000130565.69170.85. [DOI] [PubMed] [Google Scholar]
- Yokoi H, Mukoyama M, Mori K, et al. Overexpression of connective tissue growth factor in podocytes worsens diabetic nephropathy in mice. Kidney Int. 2008;73:446–455. doi: 10.1038/sj.ki.5002722. [DOI] [PubMed] [Google Scholar]