

Abstract
Voltammetry of cytochrome P450 (cyt P450) enzymes in ultrathin films with polyions was related for the first time to electronic and secondary structure. Heterogeneous electron transfer (hET) rate constants for reduction of the cyt P450s depended on heme iron spin state, with low spin cyt P450cam giving a value 40-fold larger than high spin human cyt P450 1A2, with mixed spin human P450 cyt 2E1 at an intermediate value. Asymmetric reduction–oxidation peak separations with increasing scan rates were explained by simulations featuring faster oxidation than reduction. Results are consistent with a square scheme in which oxidized and reduced forms of cyt P450s each participate in rapid conformational equilibria. Rate constants for oxidation of ferric cyt P450s in films by t-butyl hydroperoxide to active ferryloxy cyt P450s from rotating disk voltammetry suggested a weaker dependence on spin state, but in the reverse order of the observed hET reduction rates. Oxidation and reduction rates of cyt P450s in the films are also likely to depend on protein secondary structure around the heme iron.
Introduction
Cytochrome P450s (cyt P450s) are metabolic enzymes containing a heme iron cofactor (Figure 1). They are the major enzymes for oxidative metabolism of lipophilic xenobiotic chemicals including drugs and environmental pollutants, and play a key role in metabolism-mediated toxicity.1–8 Cyt P450s have a cysteinyl residue coordinated to the heme iron and a distal water molecule bound above that can freely exchange with ligands and substrates. Lipophilic substrates bind in a hydrophobic pocket of the protein in the distal region, displacing the water.1,3 About 57 human cyt P450s had been identified by 2007,9 and their highest concentrations are in the liver, which is the major site of xenobiotic metabolism.4 Cyt P450s denoted as CYP1A2, CYP2E1, CYP3A4, CYP2D6, CYP2C9, and CYP2C19 are responsible for about 95% of human oxidative drug metabolism.1,10,11
Figure 1.
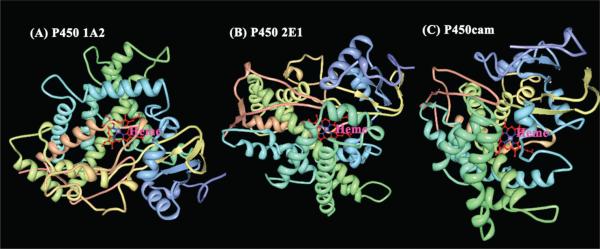
Crystal structures of (A) cyt P450 1A2 (PDB:2HI4),51 (B) cyt P450 2E1 (PDB:3E4E),52 and (C) cyt P450cam (PDB:2CPP)53 showing the heme iron in red. Structures were obtained from the protein data bank.54
Cyt P450s catalyze a wide variety of selective oxidations such as heteroatom oxygenation, carbon hydroxylation, epoxidation, dealkylation, and others.1–3 In the cyt P450 catalytic cycle,3,9 after substrate binding in the heme pocket, the cyt P450 NADPH-reductase reduces the substrate-bound ferric cyt P450 to ferrous cyt P450. This follows dioxygen binding to form a ferrous cyt P450–dioxygen complex. Then, a second electron transfer from NADPH-reductase and protonation yields a FeIII–hydroperoxo complex. In some cases, Cytochrome b5 has been shown to act as the second electron donor instead of the cyt P450 NADPH-reductase.3,9 Bacterial P450cam used in this study requires the iron–sulfur protein putidaredoxin as a redox partner to deliver electrons to cyt P450s.3 Protonation and heterolytic cleavage of the FeIII–hydroperoxo complex yields +•(P450—FeIV=O). This ferryloxy species is presumed to be the active oxidant that transfers oxygen to bound substrates.1,3 The presumed +•(P450—FeIV=O) form can also be generated directly by reaction with hydrogen peroxide or organic peroxides in the reverse of a process called the peroxide shunt.1,3 The use of the peroxide shunt pathway to hydroxylate the substrates bypasses complex electron delivery involving NADPH and cyt P450 reductases.3,9
Hill et al. first reported direct reversible voltammetry of purified, dissolved bacterial cyt P450cam at low temperatures.12 Cyt P450s have generated interest as highly regio- and stereo-selective synthetic catalysts for organic synthesis.13 Thus, electron transfer from doped tin oxide to putidaredoxin provided electrons to cyt P450cam to drive biocatalytic conversion of camphor to 5-exohydroxycamphor in solution.14 Electrochemically mediated α-hydroxylation of lauric acid by cyt P450 4A1 fusion protein with cyt P450 reductase or by purified P450 4A1 plus NADPH-reductase dissolved in solution was achieved.15 Electrocatalytic dehalogenation of haloalkanes by cyt P450cam in solution using ferredoxin as an electron mediator has been demonstrated.16 Synthetic applications have been reviewed recently.17,18
Over the past several decades, protein film voltammetry has emerged as a method of choice to investigate direct electron transfer and biocatalytic processes of enzymes.19–22 In this method, an electrode is coated with a thin film containing the protein in an environment that maintains native or near native structure and facilitates reversible electron exchange with the electrode. Advantages include avoidance of electron mediators and of diffusion of large protein molecules, minimizing or eliminating electrode fouling by denatured protein, and economy of enzyme use.
We reported the first direct reversible protein film voltammetry of cyt P450cam using thin surfactant films on pyrolytic graphite (PG) electrodes,23 as well as the first direct reversible voltammetry of this enzyme in protein–polyion films constructed layer-by-layer (LbL).24 We subsequently pursued direct cyclic voltammetry and qualitative mechanistic studies of electrochemical biocatalysis using bacterial and human cyt P450s in these films.25
A number of subsequent studies addressed other cyt P450s by direct film voltammetry. Farmer et al. reported electrocatalytic reduction of nitrite, nitric oxide, and nitrous oxide using thermophilic cyt P450 119 in didodecyldimethylammonium bromide (DDAB)-poly(styrene sulfonate) (DDAPSS) films.26 They also demonstrated electrocatalytic conversion of CCl4 to CH4 using this thermophilic P450 CYP119 in DDAPSS films at high temperatures.27 Other studies of cyt P450s on electrodes have been aimed at applications including drug metabolism sensors,28–30 drug sensors,31,32 monitors of cyt P450-drug interactions,33 and clinical uses.34
Scheller et al.35 used Laviron's method,36 to estimate heterogeneous electron transfer (hET) rate constants for cyt P450cam adsorbed to clay modified glassy carbon in the range 5–152 s−1 at scan rates 0.4–12 V s−1. This large variation of hET rate with scan rate suggests poor agreement of the Laviron model to the data. Electron transfer kinetics of cyt P450 2B4 at glassy carbon electrodes modified with nonionic detergent and colloidal clay37 yielded a rate constant of 80 s−1. Gilardi et al.38 reported hET rate constants for cyt P450 2E1 of 5 ± 0.5 s−1 immobilized on glassy carbon, and 2.0 ± 0.5 s−1 on glassy carbon/poly(diallyldimethyl ammonium chloride) (PDDA) or thiol-modified gold with PDDA as an underlayer. A ks of 10 ± 0.5 s−1 was obtained for cyt P450 2E1 covalently linked via cysteine to maleimide on cysteamine modified gold.38
Direct electrochemistry of human cyt P450s 2C9, 2C18, and 2C19 in DDAB films on an edge plane pyrolytic graphite was reported without hET rates.39 Martin et al. studied the influence of pH on the hET rate constant of human cyt P450 2C9 in DDAB films on edge-plane pyrolytic graphite.40 Using Laviron's method, they found ks of 139 s−1 at pH 6.0, 150 s−1 at pH 7.4, and 165 s−1 at pH 8.2. Gray et al.41 found a hET rate constant of 10 s−1 at 10 V s−1 for cyt P450 BM3 in sodium dodecyl sulfate films on basal plane pyrolytic graphite, and compared voltammetry of wild type cyt P450 BM3 with its mutant.42 Direct voltammetry of human cyt P450 2B6 on zirconium dioxide nanoparticles on glassy carbon was also reported.43
Considering these numerous prior reports, a major challenge remains to understand how the electronic structure of the heme iron and the secondary structure around it controls the dynamics of electron exchange of cyt P450s with electrodes and of forming the active ferryloxy form +•(P450—FeIV=O). Elucidating these relationships is the goal of the work described herein. Such information can provide insights into biological electron transfer processes of cyt P450 enzymes, and how they vary with enzyme structure, and are also important for electrochemical catalytic synthesis and sensing applications of cyt P450s.
In the present paper, we compare for the first time three different cyt P450s with regard to kinetics of direct electron transfer and formation of +•(P450—Fe(IV)=O) in ultrathin enzyme/polyion films constructed layer-by-layer (LbL) (Scheme 1). We chose the LbL method44,45 for enzyme immobilization because of its versatility, facilitation of direct electron transfer for enzymes,20,21 and ability to stabilize proteins in near native conformations.20,21,44,46 Kinetics of reduction and oxidation processes were measured by voltammetry for human cyt P450s 1A2 and 2E1, bacterial cyt P450cam, and the heme proteins myoglobin (Mb) and catalase. Results are explained by differences in FeIIIP450 spin states, secondary structures, and rapid conformational equilibria coupled to electron transfer.
Scheme 1.
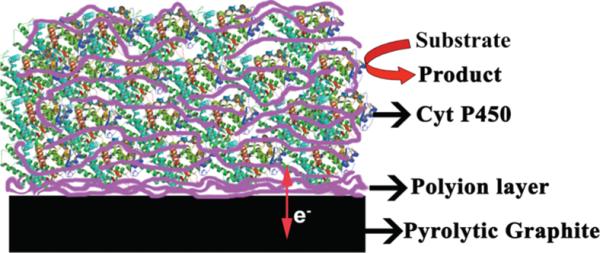
Representation of LbL Films of Cyt P450s and Polyions on PG Electrodesa
aPolyions are purple strands, and proteins are green/red ribbon structures.
Experimental Section
Bacterial Pseudomonas putida cyt P450 101 (cyt P450cam, MW 47 500),47 human cyt P450s 1A2 (MW 58 200),48 and 2E1 (MW 56 900)49 were expressed from DH5R Escherichia coli containing the relevant cDNA, then isolated and purified according to the referenced literature. Horse heart myoglobin (Mb, MW 16 900), poly(ethyleneimine) (PEI), poly(diallyldimethyl ammonium chloride) (PDDA), poly(sodium 4-styrene sulfonate) (PSS), catalase (Bovine liver, MW 247 500), and t-butyl hydroperoxide (t-BuOOH) were from Sigma.
A CHI 660A electrochemical analyzer was used for voltammetry at 25 °C. CHI software was used to simulate thin film voltammograms (see Supporting Information for full details). Thin film voltammetry was done in 3-electrode cell as described previously.50 Electrolyte was 50 mM pH 7.0 potassium phosphate buffer or appropriate buffers of other pH25b containing 0.1 M NaCl. Buffers were purged with purified nitrogen before voltammetry. Rotating disk voltammetry was done at 1000 rpm and scan rate 0.1 V s−1.50
Polyion/enzyme films were assembled layer-by-layer (LbL) on basal plane pyrolytic graphite (PG) electrodes as previously described (see Supporting Information for details).25 Assembly of LbL polyion-enzyme film was monitored at each step using a quartz crystal microbalance (QCM).25 Spectroscopy of P450/polyion LbL films was done with films fabricated on aminoalkylsilated fused silica slides as previously described.55
Results
LbL Assembly of Enzyme/Polyion Films
Table 1 describes LbL enzyme film architectures that were constructed for this work. QCM mass measurements on gold resonators (Supporting Information, Figure S1) having alkylthiol carboxylate groups to mimic the surface of pyrolytic graphite confirmed regular and reproducible assembly of polyion/enzyme films as described elsewhere.56 The nominal thickness of dry films and amount of enzyme in the film (Table 1) were estimated by QCM. While the films are constructed layer-by-layer, the final film structure features considerable mixing of neighboring layers (Scheme 1), which actually facilitates charge transport through the film.20,21,44
Table 1.
Average Characteristics of Enzyme Layer-by-Layer Assemblies from QCM Studies
| film architecture | thickness (nm) | amount of enzymea (nmol cm −2) | enzyme cone in film (μmol cm −3) |
|---|---|---|---|
| PEI(/PSS/1A2)4 | 24 ± 1 | 0.11 ± 0.01 | 46 ± 4 |
| PEI(/PSS/2E1)4 | 24 ± 3 | 0.12 ± 0.02 | 51 ± 8 |
| PSS(/PEI/cam)4 | 9 ± 1 | 0.05 ± 0.003 | 55 ± 4 |
| PEI(/PSS/Mb)2 | 9 ± 2 | 0.12 ± 0.01 | 130 ± 11 |
| PEI(/PSS/Mb)4 | 16 ± 3 | 0.21 ± 0.04 | 130 ± 24 |
| PEI(/PSS/Mb)6 | 24 ± 2 | 0.32 ± 0.03 | 140 ± 13 |
| PSS(/PEI/catalase)4 | 13 ± 1 | 0.015 ± 0.001 | 12 ± 1 |
| PSS(/PEI/hemin)4 | 19 ± 2 | 6.6 ± 0.82 | 3500 ± 430 |
Average ± SD for films on 3 resonators.
Cyt P450s 1A2 and 2E1 films had a similar nominal thickness of ~24 nm, whereas cyt P450cam films were about 9 nm thick. Though cyt P450 1A2, cyt P450 2E1, and cyt P450cam are of similar size, the bacterial cyt P450cam formed relatively thinner films than the human cyt P450s (Table 1). Dividing the amount of enzyme in each film in nmol cm−2 by film thickness reveals that the cyt P450 films have very similar enzyme concentrations (Table 1). The observed nominal film thickness of polyion and catalase films was 13 nm. With neutral buffer solutions from which to grow these enzyme films, the human cyt P450s (pI > 8) have a net positive charge, whereas bacterial cyt P450cam (pI 4.6) and catalase (pI 5.8) have a net negative charge. These observations are consistent with layer thickness for these enzymes controlled by relative surface charges of the film components.57
For myoglobin, 4-bilayer PSS/Mb films gave a thinner assembly of ~16 nm attributed to the smaller protein size (Table 1). In order to study the influence of LbL film thickness on the voltammetry, to be discussed below, we also assembled 2 and 6 bilayer films of PSS/Mb layers having the thicknesses 9 and 24 nm, respectively. Mb concentrations in these three films were similar (Table 1).
Spectroscopic Characterization
Spectra were taken of films similar to those used for voltammetry, with the first 2 layers of polyions (~1.5 nm) deposited first on aminoalkylsilated fused silica slides.46,55 As on the electrodes, this protects the protein from adsorbing on the bare surface and ensures that the spectra are characteristic of protein in the bulk of the film. Carbon monoxide binds to ferrous cyt P450s to form a cyt P450 FeII–CO complex that has a characteristic difference absorbance band near 450 nm that gave these enzymes their name.58 Denatured cyt P450s show the FeII–CO band near 420 nm. To verify that P450 enzymes in the LbL films retained structural integrity, CO-difference spectra were measured for human cyt P450s in LbL films grown on aminoalkylsilated glass slides. Figure 2A shows the CO difference spectrum for PEI(PSS/P450 1A2)6 film with the absorbance band near 450 nm consistent with that reported for native cyt P450 enzyme in solution, and for P450cam in LbL films.55
Figure 2.
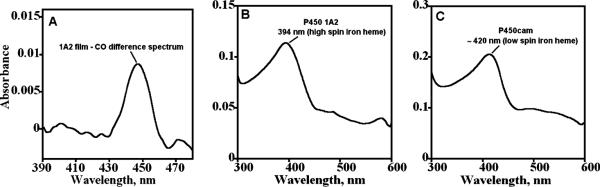
UV–vis spectra of films on aminosilane-functionalized fused silica slides: (A) CO difference spectrum for PEI(/PSS/cyt P450 1A2)6 film after reducing to the ferrous form and purging the pH 7 buffer with CO, (B) ferric form of enzyme in PEI(/PSS/cyt P450 1A2)6, and (C) ferric form of enzyme in PSS(/PEI/cyt P450cam)6 film.
Spectroscopic studies of ferric cyt P450 films were done to confirm heme iron spin states. Figure 2B shows the UV–vis spectrum of the PEI(/PSS/cyt P450 1A2)6 film, and Figure 2C is that of the PSS(/PEI/cyt P450cam)6 film. The absorbance maximum at 394 nm for the PEI(/PSS/cyt P450 1A2)6 film (Figure 2B) corresponds to the high spin heme iron in cyt P450 1A2,51,59 and that of 420 nm for the PSS(/PEI/cyt P450cam)6 film (Figure 2C) confirms the low spin electronic state of heme iron in cyt P450cam as reported previously.23,53,60,61
Voltammetry of Cyt P450 Films
While the LbL films are made one layer at a time, significant interlayer mixing in polyion–protein films made by LbL has been well documented.24,45 This mixing (cf. Scheme 1) facilitates charge transport through the film most likely by keeping the average distance between heme centers of neighboring protein molecules within a range in which electron hopping between them is relatively efficient.25 Efficient electron transport through LbL films of redox proteins and enzymes is reflected by nearly reversible CVs.20,21
Figure 3A,B represents the background subtracted cyclic voltammograms (CV) of PEI(/PSS/P450 2E1)4 and PSS(/PEI/P450cam)4 films on pyrolytic graphite electrodes with increasing scan rates in anaerobic pH 7.0 buffer. Well-defined quasireversible cyt P450 heme FeIII/FeII redox peaks can be observed from these CVs.
Figure 3.
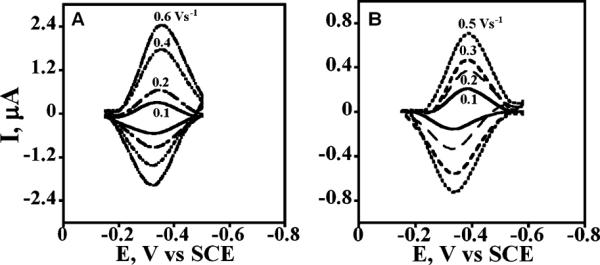
Background subtracted thin film voltammograms of cyt P450 enzymes in films on pyrolytic graphite electrodes in anaerobic 50 mM potassium phosphate buffer + 0.1 M NaCl, pH 7.0: (A) PEI(/PSS/cyt P450 2E1)4 and (B) PSS(/PEI/cyt P450cam)4.
Peak currents were proportional to scan rate and oxidation–reduction peak current ratios were close to 1. Table 2 gives formal potential () values, amount of electroactive enzymes in the films obtained from the integration of the reduction peak and using Faraday's law,21 and the percentage of electroactive enzyme.25
Table 2.
Electrochemical Parameters and Formal Potential Shift of Enzyme Films upon CO Binding in 50 mM Phosphate Buffer Plus 0.1 M NaCl at pH 7.0
| film assembly |
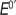 vs SCE (V) vs SCE (V) |
Γa (nmol cm−2) | % electroactive enzyme |
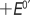 shift in CO (mV) shift in CO (mV) |
|---|---|---|---|---|
| PEI(/PSS/P450 1A2)4 | −0.330 ± 0.010 | 0.016 ± 0.005 | 15 ± 4 | 36 ± 4 |
| PEI(/PSS/P450 2E1)4 | −0.347 ± 0.009 | 0.030 ± 0.004 | 25 ± 3 | 50 ± 4 |
| PSS(/PEI/P450cam)4 | −0.363 ± 0.002 | 0.021 ± 0.004 | 42 ± 8 | 46 ± 3 |
| PSS(/PDDA/P450cam)4 | −0.366 ± 0.004 | 0.019 ± 0.003 | 38 ± 6 | 48 ± 5 |
| PEI(/PSS/Mb)4 | −0.337 ± 0.005 | 0.085 ± 0.007 | 40 ± 3 | 49 ± 4 |
| PSS(/PEI/catalase)4 | −0.467 ± 0.005 | 0.003 ± 0.001 | 20 ± 7 | |
| PSS(/PEI/hemin)4 | −0.370 ± 0.004 | 0.13 ± 0.02 | 2.0 ± 0.3 | 58 ± 3 |
Γ = surface concentration of enzyme in the film from CV; av ± SD for n = 4–6 electrodes.
Notably, the formal potential of the cyt P450cam film was similar when either PEI or PDDA was the polycation in the film, suggesting that the type of polycation has little effect on the voltammetry (Table 2). Furthermore, since the outer layer of each film is protein, the outer layer charge will be characteristic of the protein, while internal charge is completely compensated by protein-polyion interactions.44 The formal potential was most negative for the catalase film while that of the hemin film fell in the midrange of all the proteins examined (Supporting Information Figure S3). values for the enzymes are similar to those reported previously in LbL films.25b,c,38,62
The amounts of electroactive cyt P450s in the films were obtained from the ratio of electroactive surface concentration to total concentration from QCM and are in the range 15–40% (Table 2). For hemin alone, the electroactive fraction was very small (2%).
Table 2 also shows the shift in formal potential of cyt P450s and myoglobin films upon CO binding to form the cyt P450–FeII–CO complex. Complex formation following electron transfer results in a positive shift of midpoint potential that is characteristic of heme iron enzymes.20–24,35,40 For hemin, this shift was slightly higher than for protein films (Table 2).
Catalytic oxygen reduction from the CVs of cyt P450 films was observed from the increase in reduction peak current in the presence of oxygen over that of anaerobic buffer (Figure S5). Also, positive formal potential shifts in the CVs of protein films were observed with lowering pH in the pH range 4–9 (Figure S6) suggesting proton coupled electron transfer. The change in formal potential per pH unit was in the range 43–50 mV pH−1 for these films which is slightly less than the theoretical 59 mV pH−1 for a reversible one electron process coupled to proton transfer.
Heterogeneous Electron Transfer (hET) Rate Constants
For the estimation of hET rate constants, we used CVs of enzyme films at scan rates 0.005–1.4 V s−1 where good quality data were obtained. Following the suggestion of Hirst and Armstrong,63 we first subtracted the constant nonkinetic component from the oxidation–reduction peak separation (ΔEp) at lower scan rates from that at higher scan rates and fit the corrected peak separations to Butler–Volmer theoretical peak separation versus scan rate theory for surface confined voltammetry.36 Average standard hET rate constants (ks's) were estimated using the corrected ΔEp and the working curve for α = 0.5 in Figure 4, ref 36. Good fits of theory to experiment (Figure 4) indicated reliable estimates of ks.
Figure 4.
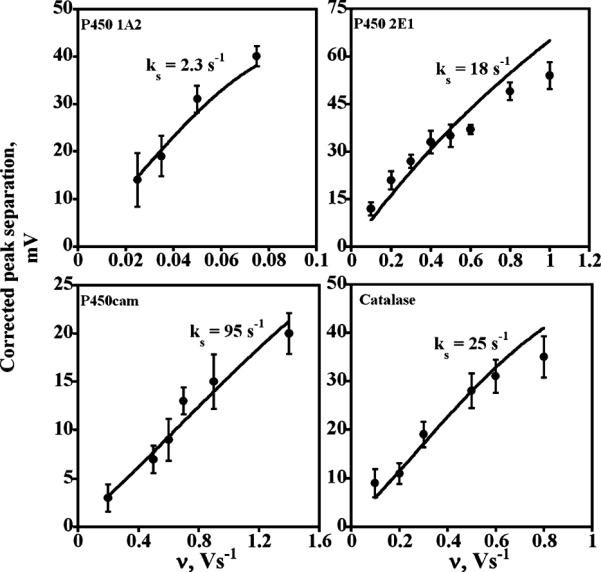
Influence of cyclic voltammetry scan rate for enzyme films on experimental (●) peak separation (ΔEp) corrected for scan rate independent nonkinetic contribution. The theoretical lines were computed for Butler–Volmer theory for the rate constant (ks) values shown and α = 0.5.
Figure 5 compares the ks values obtained at pH 7 for the enzyme films. Among the cyt P450s, cyt P450cam gave the largest hET rate constant, while cyt P450 2E1 was intermediate, and cyt P450 1A2 was the smallest. The electron transfer rate for the cyt P450 2E1 film is significantly larger than that reported for this enzyme on bare glassy carbon, a glassy carbon/polycation surface, or a gold electrode with a thiol monolayer and polycation surface.38 This suggests that faster electron transfer of cyt P450s can be achieved within the LbL architecture. Mb and catalase films gave hET rate values similar to cyt P450 2E1. Also, Mb films of thickness between 9 and 24 nm gave hET rate constants that were essentially the same, indicating that there is little effect of film thickness in this range on the measured ks values. This result confirms the validity of comparing hET rate constants for the cyt P450 films of different thicknesses.
Figure 5.
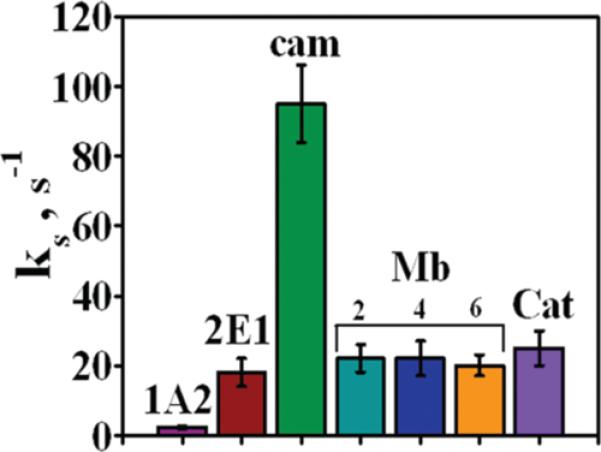
Apparent heterogeneous electron transfer rate constant (ks's) for films of cyt P450s, myoglobin (Mb), and catalase (Cat) from CVs in pH 7 buffer + 0.1 M NaCl. For Mb, the ks values for different films thicknesses utilizing 2, 4, or 6 bilayers are shown.
The linear relation of peak current to scan rate, similarity of oxidation and reduction peak heights, and good fits of ΔEp versus scan rate (Figure 4) are consistent with surface confined species, and suggest a nearly reversible thin film voltammetry model.20,21 However, the so-called trumpet plots of individual oxidation and reduction peak potentials versus scan rate should be symmetric for a simple quasireversible surface electron transfer process,19b,c but they were distinctly unsymmetrical for the cyt P450 films (Figure 6). Reduction peaks shifted more rapidly than oxidation peaks with increasing scan rate, which is also inconsistent with a simple electron transfer process.
Figure 6.

Trumpet plots showing the influence of scan rate on CV peak potentials in anaerobic pH 7.0 buffer. Experimental oxidation (red diamonds) and reduction (blue circles) peak potentials are shown for (A) PEI(/PSS/P450 1A2)4, (B) PEI(/PSS/P450 2E1)4, (C) PSS(/PEI/P450cam)4, (D) PSS(/PEI/catalase)4, and (E) PEI(/PSS/myoglobin)4 films. The lines were obtained by best fit simulations using an Ereduction/Eoxidation mechanism using the reduction and oxidation rate constants shown in each plot.
Thus, digital simulations of CVs were done to fit trumpet plots. The two simplest mechanisms likely to explain the data were considered: ECreduction/Eoxidation (where E = electron transfer, C = chemical step) and Ereduction/Eoxidation. While both mechanisms were capable of explaining the general trends observed in peak potentials, the Ereduction/Eoxidation simulation gave the best fits by far. These simulations embody the concept that the reduced species could be rapidly converted into a different form, e.g., by a conformation change, and so it could take on a different electrochemical rate constant for its oxidation compared to the reduction rate constant. Thus, taking the ks estimated from the Laviron model as the starting reduction rate constant (now denoted as ks,red) and evaluating multiples of this value for the oxidation rate (ks,ox), we arrived at the best fit illustrated by the theoretical lines that gave good fits to the experimental trumpet plots for all proteins (Figure 6, see Figure S7 for simulated CVs). As discussed later, these simulations are consistent with a square scheme in which oxidized and reduced forms of the proteins are involved in conformational equilibria that are fast with respect to the time scale of the voltammetry.
Kinetics of Ferryloxy Cyt P450 Formation
Rotating disk voltammetry was used to monitor the oxidation of cyt P450s to their active ferryloxy forms. A well studied example of this reaction is the oxidation of ferric Mb by t-BuOOH which produces the ferryloxy species ˙MbFeIV=O, a strong oxidant.64–67 Cyt P450s can be activated a similar way.1–3 On the basis of knowledge of these processes, a simplified Michaelis–Menten reaction pathway for RDV similar to that proposed for Mb in films can be represented by Scheme 2.50
Scheme 2.
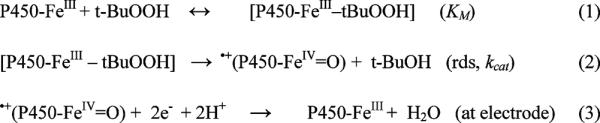
Oxidation Pathway of Cyt P450s by Organic Peroxides via RDV
Equation 1 in Scheme 2 describes binding of substrate (t-BuOOH) to ferric cyt P450 enzyme forming a cyt P450–substrate complex characterized by Michaelis dissociation constant KM, followed by formation of the ferryloxy radical cation and conversion of t-BuOOH into t-BuOH characterized by kcat, eq 2, Scheme 2. The formed ferryloxy species is subsequently reduced by the electrode regenerating ferric cyt P450 (eq 3, Scheme 2).
Figure 7 shows the representative steady state RDVs showing increasing limiting currents with increasing t-BuOOH concentration in pH 7.0 buffer due to this pathway. t-BuOOH is reduced by cyt P450s in the polyion films according to Scheme 2 to give the catalytic reduction limiting current that increases with increasing t-BuOOH concentration.
Figure 7.
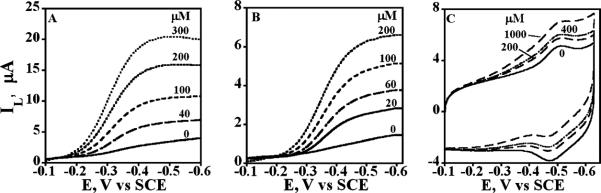
Representative steady state RDVs at 1000 rpm for protein films on PG electrodes for t-BuOOH reduction in anaerobic 50 mM potassium phosphate buffer + 0.1 M NaCl, pH 7.0 at 25 °C. RDVs for increasing t-BuOOH concentration (in μM) for (A) PEI(/PSS/P450 2E1)4, (B) PSS(/PEI/P450cam)4, and (C) PSS(/PEI/catalase)4 films.
The catalytic current Icat in rotating disk voltammetry is related to kinetic parameters (Scheme 2) by an electrochemical version of the Michaelis–Menten equation (eq 4):19a
| (4) |
where n is the number of electrons in the electrochemical reaction (n = 2 in our case, Scheme 2), F is Faraday's constant (96 485 C mol−1), A is electrode area (0.2 cm2), Γ is the electroactive surface concentration of enzyme in the film (in nmol cm−2), Cs is substrate concentration in solution (in mM), kcat is the catalytic rate constant (s−1) assumed to be rate limiting, and KM is the Michaelis–Menten dissociation constant. Icat is the catalytic current obtained by extrapolating limiting current (IL) to infinite rotation rate at each Cs. Here, to conserve enzyme, we used limiting currents at one large rotation rate to approximate Icat to obtain relative Michaelis parameters50 for comparisons between enzymes.
Figure 8 shows the turnover rates from RDV versus t-BuOOH concentration for all the films. Fitting these experimental turnover plots with the electrochemical Michaelis–Menten equation (eq 4), using nonlinear regression, provided apparent KM and kcat values. The ratio kcat/KM has units of a second order rate constant and is a relative measure of catalytic efficiency (Table 3). These data suggested the following order for rate of ferryloxy protein formation: P450 1A2 > P450 2E1 > myoglobin > P450cam ⪢ catalase.
Figure 8.
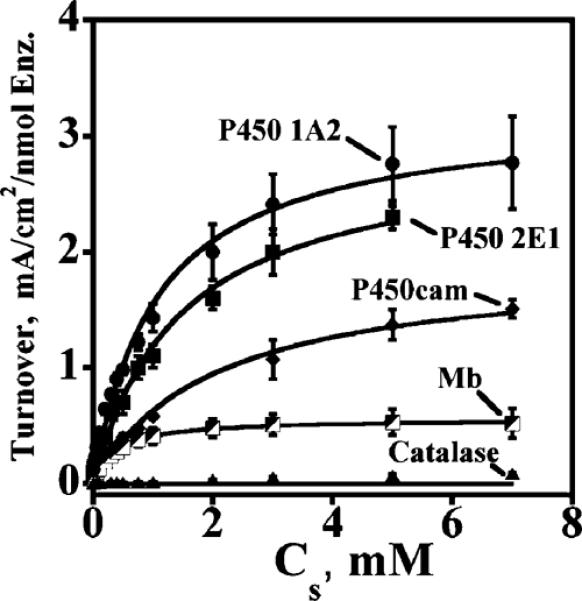
Michaelis–Menten fits (eq 4) of relative turnover rate (IL/Γ) vs t-BuOOH concentration for cyt P450, myoglobin, and catalase films. Points are experimental data, and solid lines represent the best fit of eq 4 by nonlinear regression. RDV parameters are as in Figure 7.
Table 3.
Apparent Relative kcat, KM, and kcat/KM Values Obtained from Michaelis–Menten Fitting of RDV Data for Reduction of t-BuOOH
| film assembly | kcat (s−1) | KM (mM) | 104 kcat/ KM (M−1 s−1) |
|---|---|---|---|
| PEI(/PSS/1A2)4 | 83 ± 7 | 1.05 ± 0.11 | 7.9 ± 0.8 |
| PEI(/PSS/2E1)4 | 74 ± 5 | 1.43 ± 0.1 | 5.2 ± 0.4 |
| PSS(/PEI/cam)4 | 49 ± 4 | 2.0 ± 0.24 | 2.5 ± 0.2 |
| PEI(/PSS/Mb)4 | 15 ± 2 | 0.36 ± 0.03 | 4.2 ± 0.5 |
| PSS(/PEI/catalase)4 | 7 ± 1 | 15.4 ± 3.7 | 0.045 ± 0.006 |
Discussion
Enzyme Characterization in Films
Near native enzyme structures in the films were confirmed by UV–vis spectroscopy reflecting known high and low spin ferric heme iron bands of cyt P450s and by characteristic 450 nm bands of reduced cyt P450–CO complexes (Figure 2). CO binding to reduced cyt P450s in the films was confirmed by positive midpoint potential shifts in CVs (Table 2).
While the spectroscopic data argue conclusively against it, in rare cases loss of heme from the protein in LbL films might be envisioned. Relevant to this issue, hemin films (Figure S3, Table 1) had peak potentials that changed very differently with increasing scan rate from the protein films (Figure S4), gave larger CO potential shifts than the proteins (Table 2), and had a fraction of electroactive hemin that was very low (Table 2). These data show that electrochemical properties of hemin LbL films are very different from those of protein films. Taken together with the spectroscopy and many studies showing that cytP450 enzymes retain enzyme activity in similar films,20,30,45,55,56 results strongly support the view that the cyt P450s retain near native structures in the films. This is also consistent with the many other enzymes demonstrated to maintain native structures and enzyme activities in LbL films.20,21,30,44–46
Electrochemical Dynamics
Well defined quasireversible CVs of cyt P450 films (Figure 3) enabled a comparative study of the effect of cyt P450 structure on the kinetics of direct electron transfer for the first time. Demonstration of good model fitting is an essential test confirming the estimation of reliable hET rate constants (ks).20,21 After a correction was made for nonkinetic peak separations that do not depend on scan rate,63 plots of peak separation versus scan rate agreed well with Butler–Volmer theory for surface confined electron transfer (Figure 4), and peak currents were proportional to scan rate. The values of ks obtained from this first level of analysis correlate with heme spin state. Cyt P450cam having predominantly low spin heme iron53,60 gave the largest electron transfer rate (95 ± 11 s−1, Figure 5), high spin cyt P450 1A2 (>90%)59 gave the smallest electron transfer rate (2.3 ± 0.4 s−1), and cyt P450 2E1 with mixed spin states (~80% high spin)49,52 gave an intermediate value (18 ± 4 s−1).
On the basis of biochemical studies, Guengerich et al.68–70 reported that low spin forms of cyt P450s 1A2 and 2E1 undergo significantly faster reduction than the corresponding high spin forms, although these correlations were difficult to uncouple from other structural factors. Cyt P450 2E1 is reduced by NADPH-cyt P450 reductase70 about 2.5 times faster than cyt P450 1A2 consistent with our finding that mixed spin cyt P450 2E1 is reduced about 7 times faster than high spin cyt P450 1A2 using the electrode as electron donor. Thus, qualitatively, the dependence of hET rates in the films on cyt P450 heme spin states seems consistent with literature precedents.
In addition, voltammetry studies by Feng and Schultz showed that free low spin heme iron complexes (not bound to proteins) gave faster electron transfer rates than high spin complexes by about 0.3–1.3 orders of magnitude.71 They explained that the reduction of high spin heme complexes involves greater activation energy than the reduction of low spin heme complexes. They also showed that the electron self-exchange rate is several-fold larger for low spin hemes compared to high spin.71 Unlike the natural reductase–P450 electron transfer involving a prior protein–protein interaction on a membrane, in thin film voltammetry an electron is transferred from the solid electrode to heme irons of the cyt P450 near the surface, and self-exchange within the film propagates charge transfer.20,21 Thus, correlation of hET rate constants to spin state the same as in model heme iron complexes is reasonable.
We also need to consider the structural factors (Figure 1) that may influence the electron transfer rates. Considering differences between the cyt P450s,72 human cyt P450s 1A2 and 2E1 share ~30% sequence identity, and cyt P450 1A2 has no sequence identity with cyt P450cam. The structure of cyt P450cam features flexible helices,73,74 while human cyt P450s have more compact secondary structures (Figure 1). This conformational flexibility of cyt P450cam structure may lead to subconformations with the result of entropically favored faster direct electron transfer rates between the electrode and cyt P450cam by facilitating a shorter average ET path. We are currently pursuing quantum mechanical/molecular mechanics (QM/MM) modeling of cyt P450s to better understand these structural influences on ET rates.
Asymmetry in the CV trumpet plots (Figure 6) features much smaller shifts in oxidation peak potentials compared to reduction peak potentials. This is most prominent for the cyt P450 2E1 film (Figure 6B) compared to cyt P450 1A2 and cyt P450cam (Figure 6A,C). Catalase and myoglobin films exhibited the smallest asymmetry in trumpet plots (Figure 6D,E).
This asymmetry was best fit by using digital simulations of the redox process (Figure 6) using an Ereduction/Eoxidation mechanism. As mentioned earlier, ECreduction/Eoxidation simulations were able to explain trends, but gave much worse fits to the data than Ereduction/Eoxidation simulations. Using the Occam's razor principle, we relied on Ereduction/Eoxidation simulations for a second level kinetic analysis, realizing that results need to be interpreted in a chemically logical manner. Data for cyt P450 2E1 films with the highest asymmetry (Figure 6B) were fit reasonably well by simulations using 18 s−1 for ks,red and a ≥100-fold larger ks,ox(≥1800 s−1). This simulation remained the same upon further increases of ks,ox to 18 × 106 s−1. On the other hand, when both the reduction and oxidation values were the same (18 s−1) or when ks,ox of only 10-fold (180 s−1) larger than the reduction rate (18 s−1), nearly symmetrical trumpet plots were found that gave poor fits to experimental data.
A simulation of cyt P450cam with ks,ox ≥ 950 s−1 and ks,red of 95 s−1 gave the best fit to experimental data (Figure 6C). Similarly, the less asymmetric cyt P450 1A2 (Figure 6A), catalase (Figure 6D), and Mb (Figure 6E) trumpet plots were best fit using a ks,ox 10 times larger than ks,red.
Simulations using the ECreduction/Eoxidation mechanism were tested for cyt P450 2E1 films featuring the same ks,red and ks,ox as used above, and with a reversible chemical (C) step using various values of forward (kf) and reverse (kb) chemical rate constants. Among all trials, the asymmetry was best reproduced using kf 3.6 s−1 and kb 0.018 s−1 (Figure S8) for the chemical equilibrium, but shifts in Ep,red with scan rate fit poorly compared to the Ereduction/Eoxidation best fit (Figure 6B).
Asymmetry in trumpet plots on the order of that for cyt P450cam and cyt P450 1A2 was found by Hirst and Armstrong for the S. acidocaldarius (Sa Fd) ferredoxin and A. Vinelandii ferredoxin-I mutant (D15N) adsorbed on edge plane pyrolytic graphite electrodes.63 They attributed this to the conformational or orientational changes coupled to electron transfer that gate electron transfer at scan rates well above those used in our work. A square scheme incorporating conformational equilibria coupled to the electron transfer was suggested,63 but this mechanism was not tested with simulations.
The Ereduction/Eoxidation simulation model used here requires an oxidized form of the enzyme reduced at a moderate rate, and a reduced form that is oxidized at a faster rate. This is an elaboration of the Butler–Volmer surface voltammetry model36 that assumes that a redox system can be characterized by a single ks that is the rate constant for ET at its formal potential.36 The Ereduction/Eoxidation model can be interpreted in terms of two separate redox couples, one for reduction and another for oxidation. In this way, different electrochemical rate constants for the reduction and oxidation can be rationalized. These two redox couples can arise if oxidized and reduced forms of the enzymes are involved in separate conformational equilibrium.75 Conformational equilibria and their influence on thin film voltammetry have been well documented for cytochrome c,76,77 myoglobin,78 and several ferredoxins.63 This concept implies a square scheme mechanism63,76 (Scheme 3). The horizontal reactions in Scheme 3 are electrochemical steps, and the vertical transitions are likely to be conformational equilibria. The fact that the Ereduction/Eoxidation simulations fit our data for the proteins implies that the conformational equilibria are fast relative to the CV time scale. This is also consistent with findings above for ferredoxins in which the proposed conformational-based asymmetry in trumpet plots63 was only manifested at much higher scan rates than was possible for the cyt P450 films.
Scheme 3.
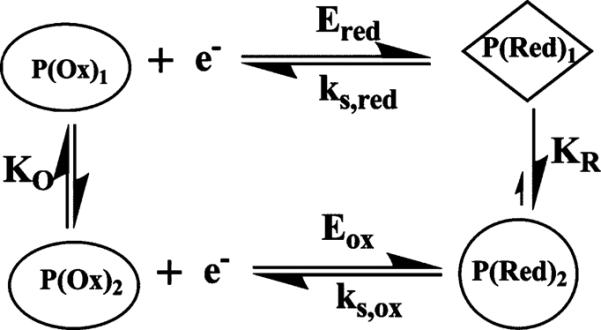
Square Scheme Explaining Cyt P450 Thin Film Voltammetry
The square scheme (Scheme 3) implies reduction of the cyt P450 enzyme with heme iron in FeIII state [P(Ox)1] to FeII heme [P(Red)1] by an electron supplied by the electrode. Conformational change produces P(Red)2 which is oxidized to P(Ox)2 with ks,ox ⪢ ks,red. This explains the smaller shifts in experimental Ep,ox with increasing scan rate compared to the shifts in Ep,red (Figure 6B). Since peak potentials are also pH dependent (Figure S6), the participation of protons in the conformational changes is also possible.
Certainly, the above electron transfer model remains an approximation as it does not consider electron self-exchange or “hopping”79 through the films of 9–24 nm. We roughly estimated the distance between adjacent redox centers of cyt P450s in the films. From the experimentally obtained amount of protein molecules per layer from QCM, protein diameter,80 and film thickness, we estimated an upper limit of protein heme group separation from its adjacent neighbor at ≤4 nm assuming a uniform protein distribution and absence of specific interactions. However, we know that strong electrostatic and other secondary interactions exist in LbL films as well as interlayer mixing of neighboring protein layers.25,44 This may further decrease the effective distance between adjacent protein redox centers in LbL films as would intermolecular electron transfer via water channels,81 all of which favor efficient electron self-exchange between protein redox centers. The role of electron hopping in direct electron transfer in protein LbL films has been documented in the literature. Lisdat et al.82 reported electron hopping in direct electron exchange of cytochrome c with electrodes in polyelectrolyte multilayer films. We realize that the hET rate constants reported in this paper should be considered characteristic of the charge transport properties of the entire film, and not only the proteins. However, since ks was independent of thickness of the films, internal charge is compensated, and outer layers are always protein and will have charge characteristic of the given protein, we can justify the use of kinetic constants for the relative comparisons for cyt P450 films we have discussed above. Clearly, the rate constant values should not be taken as absolute.
Oxidation to Ferryloxy Cyt P450s
Increasing RDV limiting currents with increasing t-BuOOH concentration are due to the enzyme-catalyzed reduction of t-BuOOH (Scheme 2, Figure 7A,B). Catalase films gave very small catalytic currents for the peroxide and serve as a negative control (Figure 7C).
The kcat for oxidation of human cyt P450s is about 5-fold greater than that of myoglobin (Table 3), and ~1.5 times larger than for cyt P450cam. The two human cyt P450s had comparable binding affinity for t-BuOOH as shown by KM, but cyt P450cam had (Table 3) less binding affinity. Myoglobin showed strong binding of t-BuOOH (lowest KM, 0.36 mM), and catalase showed the weakest binding (highest KM). The binding trends are most likely controlled by the nature of the protein secondary structure around the heme group, as discussed below.
The ratio kcat/KM, which can be considered relative catalytic efficiency,19 was larger for the human cyt P450s than for cyt P450cam and myoglobin. Again, this can be related to differences in the amino acids close to the active heme group, which are different in cyt P450cam than in human cyt P450s. Due to the stronger binding of peroxide to Mb, catalytic efficiency of Mb was slightly larger than that of cyt P450cam.
Overall, the trend of catalytic efficiency was cyt P450 1A2 > P450 2E1 > myoglobin > P450cam ⪢ catalase (Table 3). This order of +˙(P450—FeIV=O) formation rate for cyt P450s is the reverse of that obtained for hET rate, and reflects an ~3.5-fold faster oxidation of the high spin heme iron cyt P450 compared to low spin cyt P450cam. Thus, the dependence on spin state is weaker than for the hET rate constant.
Significantly, cyt C on surfaces having high spin heme iron gave higher activity for peroxide reduction than low spin cyt C.83–85 On the basis of quantum mechanical simulations, Boyes et al.86 reported stabilization of low spin cyt P450 that could lead to lower catalytic efficiency compared to high spin P450s. Thus, kcat/KM in the order for cyt P450 1A2 films seems consistent with the influence of spin state on peroxidase activity of other heme enzymes.
The influence of amino acid residues in the distal and proximal regions of the heme in enhancing the formation rate of +˙(P450—FeIV=O) must also be considered. Cyt P450s with their active site residues around the heme distal region that participate in peroxide reduction are shown in Figure 9.51–53 Peroxidases and Mb have proximal histidine coordinated to the heme, whereas in cyt P450s the proximal iron-binding ligand is cysteine thiolate (Figure 9).3b
Figure 9.
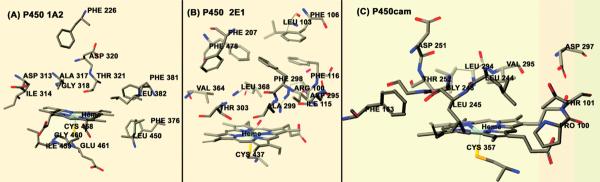
Heme structures with distal and proximal region amino acid residues for (A) cyt P450 1A2 (PDB:2HI4),51(B) cyt P450 2E1 (PDB:3E4E),52and (C) cyt P450cam (PDB:2CPP).53
Recently, Roth and Cramer87 proposed alternate mechanisms involving reversible O—O heterolysis/homolysis for peroxide activation of heme peroxidases. A recent study on cyt P450cam using QM/MM showed that proton coupled electron transfer played a role in formation of ferryloxy radical cation (Scheme 2).88 As we can presently only speculate on the influence of enzyme structure, we plan future QM/MM studies to address the involvement of specific residues in the distal/proximal heme regions of cyt P450 films in formation of +˙(P450—Fe(IV)=O).
Conclusions
We have shown for the first time that rates of electrochemical reduction of cyt P450s in thin LbL polyion films depend significantly on spin state, with low spin cyt P450cam giving the largest rate. Observed asymmetry in CV trumpet plots for the enzyme films was explained by a square scheme supported by simulations featuring faster oxidation than reduction. To a lesser extent and in the opposite order, the rate of oxidation of cyt P450s by an organic peroxide also correlated with spin state. However, spin state does not control all aspects of these reactions, and oxidation and reduction rates of cyt P450s in the films are also likely to depend on protein secondary structure around the heme group.
Supplementary Material
Acknowledgment
This work was supported financially by U.S. PHS Grant ES03154 from the National Institute of Environmental Health Sciences (NIEHS), NIH. We thank Dr. Eli Hvastkovs for providing purified cyt P450s and Dr. Dominic Hull for assistance in using CHI simulation software. We thank Professor Jose Gascon and L. Menikarachchi of the University of Connecticut for their help in representing protein structures. J.F.R. thanks Science Foundation Ireland for a Walton Research Fellowship in 2009.
Footnotes
Supporting Information Available: Detailed procedures used for simulations of voltammetry and making layer-by-layer films. Eight additional figures giving data for QCM, CVs of cyt P450 films without background subtraction, CVs and trumpet plot for hemin, CVs of cyt P450 films upon catalytic oxygen reduction and at various pH values, and simulation results with an alternative mechanism. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Guengerich FP. Chem. Res. Toxicol. 2008;21:70–83. doi: 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- (2).Liebler DC, Guengerich FP. Nat. ReV. Drug DiscoVery. 2005;4:410–420. doi: 10.1038/nrd1720. [DOI] [PubMed] [Google Scholar]
- (3).(a) Ortiz de Montellano PR. Cytochrome P450. 3rd ed. Kluwer/Plenum; New York: 2005. [Google Scholar]; (b) Schenkman JB, Greim H, editors. Cytochrome P450. Springer-Verlag; Berlin: 1993. [Google Scholar]
- (4).Ioannidis C, Lewis DFV. Curr. Top. Med. Chem. 2004;4:1767–1788. doi: 10.2174/1568026043387188. [DOI] [PubMed] [Google Scholar]
- (5).Parikh A, Gillam EMJ, Guengerich FP. Nat. Biotechnol. 1997;15:784–788. doi: 10.1038/nbt0897-784. [DOI] [PubMed] [Google Scholar]
- (6).Estabrook RW. Drug Metab. Dispos. 2003;31:1461–1473. doi: 10.1124/dmd.31.12.1461. [DOI] [PubMed] [Google Scholar]
- (7).Rusling JF, Hvastkovs EG, Schenkman JB. In: Drug Metabolism Handbook. Nassar A, Hollenburg PF, Scatina J, editors. Wiley; New York: 2009. pp. 307–340. [Google Scholar]
- (8).(a) Williams DP, Kitteringham NR, Naisbitt DJ, Pirmohamed M, Smith DA, Park BK. Curr. Drug Metab. 2002;3:351–366. doi: 10.2174/1389200023337423. [DOI] [PubMed] [Google Scholar]; (b) Hecht SS. Chem. Res. Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- (9).Guengerich FP. J. Biochem. Mol. Toxicol. 2007;21:163–168. doi: 10.1002/jbt.20174. [DOI] [PubMed] [Google Scholar]
- (10).Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, Ball SE. Drug Metab. Dispos. 2004;32:1201–1208. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- (11).Hodgson J. Nat. Biotechnol. 2001;19:722–726. doi: 10.1038/90761. [DOI] [PubMed] [Google Scholar]
- (12).Kazlauskaite J, Westlake ACG, Wong L-L, Hill HAO. J. Chem. Soc., Chem. Commun. 1996:2189–2190. [Google Scholar]
- (13).Schoemaker HE, Mink D, Wubbolts MG. Science. 2003;299:1694–1697. doi: 10.1126/science.1079237. [DOI] [PubMed] [Google Scholar]
- (14).Reipa V, Mayhew MP, Vilker VL. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13554–13558. doi: 10.1073/pnas.94.25.13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Faulkner KM, Shet MS, Fisher CW, Estabrook RW. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7705–7709. doi: 10.1073/pnas.92.17.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wirtz M, Klucik J, Rivera M. J. Am. Chem. Soc. 2000;122:1047–1056. [Google Scholar]
- (17).Coon MJ. Annu. Rev. Pharmacol. Toxicol. 2005;45:1–25. doi: 10.1146/annurev.pharmtox.45.120403.100030. [DOI] [PubMed] [Google Scholar]
- (18).Urlacher VB, Eiben S. Trends Biotechnol. 2006;24:324–330. doi: 10.1016/j.tibtech.2006.05.002. [DOI] [PubMed] [Google Scholar]
- (19).(a) Sucheta A, Cammack R, Weiner J, Armstrong FA. Biochemistry. 1993;32:5455–5465. doi: 10.1021/bi00071a023. [DOI] [PubMed] [Google Scholar]; (b) Armstrong FA, Heering HA, Hirst J. J. Chem. Soc. Rev. 1997;26:169–179. [Google Scholar]; (c) Armstrong FA, Wilson GS. Electrochim. Acta. 2000;45:2623–2645. [Google Scholar]; (d) Heering HA, Hirst J, Armstrong FA. J. Phys. Chem. B. 1998;102:6889–6902. [Google Scholar]; (e) Leger C, Elliott SJ, Hoke KR, Jeuken LJC, Jones AK, Armstrong FA. Biochemistry. 2003;42:8653–8662. doi: 10.1021/bi034789c. [DOI] [PubMed] [Google Scholar]; (f) Vincent KA, Parkin A, Armstrong FA. Chem. Rev. 2007;107:4366–4413. doi: 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- (20).(a) Rusling JF, Wang B, Yun SE. In: Bioelectrochemistry. Bartlett PN, editor. John Wiley; New York: 2008. pp. 39–86. [Google Scholar]; (b) Rusling JF, Zhang Z. In: Electroanalytical Methods for Biological Materials. Chambers JQ, Brajter-Toth A, editors. Marcel Dekker; New York: 2002. pp. 195–231. [Google Scholar]
- (21).(a) Rusling JF, Zhang Z. In: Handbook of Surfaces and Interfaces of Materials, Vol. 5. Biomolecules, Biointerfaces, and Applications. Nalwa RW, editor. Academic Press; New York: 2001. pp. 33–71. [Google Scholar]; (b) Rusling JF, Zhang Z. In: Biomolecular Films. Rusling JF, editor. Marcel Dekker; New York: 2003. pp. 1–64. [Google Scholar]
- (22).Udit AK, Gray HB. Biochem. Biophys. Res. Commun. 2005;338:470–476. doi: 10.1016/j.bbrc.2005.08.087. [DOI] [PubMed] [Google Scholar]
- (23).Zhang Z, Nassar A-EF, Lu Z, Schenkman JB, Rusling JF. J. Chem. Soc., Faraday Trans. 1997;93:1769–1774. [Google Scholar]
- (24).Lvov YM, Lu Z, Schenkman JB, Zu X, Rusling JF. J. Am. Chem. Soc. 1998;120:4073–4080. [Google Scholar]
- (25).(a) Zu X, Lu Z, Zhang Z, Schenkman JB, Rusling JF. Langmuir. 1999;15:7372–7377. [Google Scholar]; (b) Munge B, Estavillo C, Schenkman JB, Rusling JF. ChemBioChem. 2003;4:82–89. doi: 10.1002/cbic.200390018. [DOI] [PubMed] [Google Scholar]; (c) Estavillo C, Lu Z, Jansson I, Schenkman JB, Rusling JF. Biophys. Chem. 2003;104:291–296. doi: 10.1016/s0301-4622(02)00383-6. [DOI] [PubMed] [Google Scholar]
- (26).Immoos CE, Chou J, Bayachou M, Blair E, Greaves J, Farmer PJ. J. Am. Chem. Soc. 2004;126:4934–4942. doi: 10.1021/ja038925c. [DOI] [PubMed] [Google Scholar]
- (27).Blair E, Greaves J, Farmer PJ. J. Am. Chem. Soc. 2004;126:8632–8633. doi: 10.1021/ja0488333. [DOI] [PubMed] [Google Scholar]
- (28).Bistolas N, Wollenberger U, Jung C, Scheller FW. Biosens. Bioelectron. 2005;20:2408–2423. doi: 10.1016/j.bios.2004.11.023. [DOI] [PubMed] [Google Scholar]
- (29).Iwuoha EI, Joseph S, Zhang Z, Smyth MR, Fuhr U, Ortiz de Montellano PR. J. Pharm. Biomed. Anal. 1998;17:1101–1110. doi: 10.1016/s0731-7085(98)00076-4. [DOI] [PubMed] [Google Scholar]
- (30).Joseph S, Rusling JF, Lvov YM, Friedberg T, Fuhr U. Biochem. Pharmacol. 2003;65:1817–1826. doi: 10.1016/s0006-2952(03)00186-2. [DOI] [PubMed] [Google Scholar]
- (31).Das A, Grinkova YV, Sligar SG. J. Am. Chem. Soc. 2007;129:13778–13779. doi: 10.1021/ja074864x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liu S, Peng L, Yang X, Wu Y, He L. Anal. Biochem. 2008;375:209–216. doi: 10.1016/j.ab.2007.12.001. [DOI] [PubMed] [Google Scholar]
- (33).Antonini M, Ghisellini P, Pastorino L, Paternolli C, Nicolini C. IEE Proc.: Nanobiotechnol. 2003;150:31–34. doi: 10.1049/ip-nbt:20030534. [DOI] [PubMed] [Google Scholar]
- (34).Shumyantseva VV, Bulko TV, Archakov AI. J. Inorg. Biochem. 2005;99:1051–1063. doi: 10.1016/j.jinorgbio.2005.01.014. [DOI] [PubMed] [Google Scholar]
- (35).Lei C, Wollenberger U, Jung C, Scheller FW. Biochem. Biophys. Res. Commun. 2000;268:740–744. doi: 10.1006/bbrc.2000.2200. [DOI] [PubMed] [Google Scholar]
- (36).Laviron E. J. Electroanal. Chem. 1979;101:19–28. [Google Scholar]
- (37).Shumyantseva VV, Ivanov YD, Bistolas N, Scheller FW, Archakov AI, Wollenberger U. Anal. Chem. 2004;76:6046–6052. doi: 10.1021/ac049927y. [DOI] [PubMed] [Google Scholar]
- (38).Fantuzzi A, Fairhead M, Gilardi G. J. Am. Chem. Soc. 2004;126:5040–5041. doi: 10.1021/ja049855s. [DOI] [PubMed] [Google Scholar]
- (39).Shukla A, Gillam EM, Mitchell DJ, Bernhardt PV. Electro-chem. Commun. 2005;7:437–442. [Google Scholar]
- (40).Johnson DL, Lewis BC, Elliot DJ, Miners JO, Martin LL. Biochem. Pharmacol. 2005;69:1533–1541. doi: 10.1016/j.bcp.2005.02.020. [DOI] [PubMed] [Google Scholar]
- (41).Udit AK, Hill MG, Gray HB. Langmuir. 2006;22:10854–10857. doi: 10.1021/la061162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Udit AK, Hindoyan N, Hill MG, Arnold FH, Gray HB. Inorg. Chem. 2005;44:4109–4111. doi: 10.1021/ic0483747. [DOI] [PubMed] [Google Scholar]
- (43).Peng L, Yang X, Zhang Q, Liu S. Electroanalysis. 2008;20:803–807. [Google Scholar]
- (44).Lvov Y. In: Handbook of Surfaces and Interfaces Of Materials, Vol. 3. Nanostructured Materials, Micelles and Colloids. Nalwa RW, editor. Academic Press; San Diego: 2001. pp. 170–189. [Google Scholar]
- (45).Rusling JF, Hvastkovs EG, Hull DO, Schenkman JB. Chem. Commun. 2008:141–154. doi: 10.1039/b709121b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Panchagnula V, Kumar CV, Rusling JF. J. Am. Chem. Soc. 2002;124:12515–12525. doi: 10.1021/ja020683d. [DOI] [PubMed] [Google Scholar]
- (47).O'Keefe DH, Ebel RE, Petersen JA. Methods Enzymol. 1978;52:151–157. doi: 10.1016/s0076-6879(78)52017-x. [DOI] [PubMed] [Google Scholar]
- (48).Fisher CW, Caudle DL, Martin-Wixtrom C, Quattrochi LC, Turkey RH, Waterman MR, Estabrook RW. FASEB J. 1992;6:759–764. doi: 10.1096/fasebj.6.2.1537466. [DOI] [PubMed] [Google Scholar]
- (49).Gillam EMJ, Guo ZY, Guengerich FP. Arch. Biochem. Biophys. 1994;312:59–66. doi: 10.1006/abbi.1994.1280. [DOI] [PubMed] [Google Scholar]
- (50).Guto PM, Rusling JF. J. Phys. Chem. B. 2005;109:24457–24464. doi: 10.1021/jp054621w. [DOI] [PubMed] [Google Scholar]
- (51).Sansen S, Yano JK, Reynald RL, Schoch GA, Griffin KJ, Stout CD, Johnson EF. J. Biol. Chem. 2007;282:14348–14355. doi: 10.1074/jbc.M611692200. [DOI] [PubMed] [Google Scholar]
- (52).Porubsky PR, Meneely KM, Scott EE. J. Biol. Chem. 2008;283:33698–33707. doi: 10.1074/jbc.M805999200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Poulos TL, Finzel BC, Howard AJ. J. Mol. Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- (54).Moreland JL, Gramada A, Buzko OV, Zhang Q, Bourne PE. BMC Bioinf. 2005;6:21. doi: 10.1186/1471-2105-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Hvastkovs EG, So M, Krishnan S, Bajrami B, Tarun M, Jansson I, Schenkman JB, Rusling JF. Anal. Chem. 2007;79:1897–1906. doi: 10.1021/ac061975q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zhou L, Yang J, Estavillo C, Stuart JD, Schenkman JB, Rusling JF. J. Am. Chem. Soc. 2003;125:1431–1436. doi: 10.1021/ja0290274. [DOI] [PubMed] [Google Scholar]
- (57).Schenkman JB, Jansson I, Lvov Y, Rusling JF, Boussaad S, Tao NJ. Arch. Biochem. Biophys. 2001;385:78–87. doi: 10.1006/abbi.2000.2140. [DOI] [PubMed] [Google Scholar]
- (58).Omura T, Sato R. J. Biol. Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- (59).Sandhu P, Guo Z, Baba T, Martin MV, Tukey RH, Guengerich FP. Arch. Biochem. Biophys. 1994;309:168–177. doi: 10.1006/abbi.1994.1099. [DOI] [PubMed] [Google Scholar]
- (60).Poulos TL, Finzel BC, Howard AJ. Biochemistry. 1986;25:5314–5322. doi: 10.1021/bi00366a049. [DOI] [PubMed] [Google Scholar]
- (61).(a) Peterson JA. Arch. Biochem. Biophys. 1971;144:678–693. [Google Scholar]; (b) Harris D, Loew G. J. Am. Chem. Soc. 1993;115:8775–8779. [Google Scholar]
- (62).Shen L, Hu N. Biomacromolecules. 2005;6:1475–1483. doi: 10.1021/bm049248x. [DOI] [PubMed] [Google Scholar]
- (63).Hirst J, Armstrong FA. Anal. Chem. 1998;70:5062–5071. doi: 10.1021/ac980557l. [DOI] [PubMed] [Google Scholar]
- (64).Allentoff AJ, Bolton JL, Wilks A, Thompson JA, Ortiz de Montellano PR. J. Am. Chem. Soc. 1992;114:9744–9749. [Google Scholar]
- (65).Reeder BJ, Svistunenko DA, Cooper CE, Wilson MT. Antioxidants Redox Signaling. 2004;6:954–966. doi: 10.1089/ars.2004.6.954. [DOI] [PubMed] [Google Scholar]
- (66).Chen Y-R, Mason RP. Biochem. J. 2002;365:461–469. doi: 10.1042/BJ20020170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Van der Zee J. Biochem. J. 1997;322:633–639. doi: 10.1042/bj3220633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Yamazaki H, Ueng Y-F, Shimada T, Guengerich FP. Biochemistry. 1995;34:8380–8389. doi: 10.1021/bi00026a020. [DOI] [PubMed] [Google Scholar]
- (69).Yamazaki H, Johnson WW, Ueng Y-F, Shimada T, Guengerich FP. J. Biol. Chem. 1996;271:27438–27444. doi: 10.1074/jbc.271.44.27438. [DOI] [PubMed] [Google Scholar]
- (70).Guengerich FP, Johnson WW. Biochemistry. 1997;36:14741–14750. doi: 10.1021/bi9719399. [DOI] [PubMed] [Google Scholar]
- (71).Feng D, Schultz FA. Inorg. Chem. 1988;27:2144–2149. [Google Scholar]
- (72). Sequence identity between P450s was determined using the BLAST program from the following website: http://blast.ncbi.nlm.nih.gov/Blast.cgi.
- (73).Dunn AR, Dmochowski IJ, Bilwes AM, Gray HB, Crane BR. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12420–12425. doi: 10.1073/pnas.221297998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Prasad S, Mazumdar S, Mitra S. FEBS Lett. 2000;477:157–160. doi: 10.1016/s0014-5793(00)01745-2. [DOI] [PubMed] [Google Scholar]
- (75).Hoffman BM, Ratner MA. J. Am. Chem. Soc. 1987;109:6237–6243. and references therein. [Google Scholar]
- (76).Kasmi AE, Leopold MC, Galligan R, Robertson RT, Saavedra SS, Kacemi KE, Bowden EF. Electrochem. Commun. 2002;4:177–181. [Google Scholar]
- (77).Ikeshoji T, Taniguchi I, Hawkridge FM. J. Electroanal. Chem. 1989;270:297–308. [Google Scholar]
- (78).Nassar A-EF, Zhang Z, Hu N, Rusling JF, Kumosinski TF. J. Phys. Chem. B. 1997;101:2224–2231. and references therein. [Google Scholar]
- (79).Andrieux CP, Saveant JM. Molecular Design of Electrode Surfaces. In: Murray RW, editor. Techniques of Chemistry. Vol. 22. Wiley-Interscience; New York: 1992. pp. 207–270. [Google Scholar]
- (80).Bayburt TH, Sligar SG. Proc. Natl. Acad. Sci. U.S.A. 2002;99:6725–6730. doi: 10.1073/pnas.062565599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Lin J, Balabin IA, Beratan DN. Science. 2005;310:1311–1313. doi: 10.1126/science.1118316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Beissenhirtz MK, Scheller FW, Stocklein WFM, Kurth DG, Mohwald H, Lisdat F. Angew. Chem., Int. Ed. 2004;43:4357–4360. doi: 10.1002/anie.200352804. [DOI] [PubMed] [Google Scholar]
- (83).Lee CH, Mou CY, Ke SC, Lin TS. Mol. Phys. 2006;104:1635–1641. [Google Scholar]
- (84).Deere J, Magner E, Wall JG, Hodnett BK. J. Phys. Chem. B. 2002;106:7340–7347. [Google Scholar]
- (85).Busi E, Howes BD, Pogni R, Basosi R, Tinoco R, Vazquez-Duhalt R. J. Mol. Catal. B: Enzym. 2000;9:39–48. [Google Scholar]
- (86).Segall MD, Payne MC, Ellis W, Tucker GT, Boyes N. Chem. Res. Toxicol. 1998;11:962–966. doi: 10.1021/tx980017j. [DOI] [PubMed] [Google Scholar]
- (87).Roth JP, Cramer CJ. J. Am. Chem. Soc. 2008;130:7802–7803. doi: 10.1021/ja802098c. [DOI] [PubMed] [Google Scholar]
- (88).Chen H, Hirao H, Derat E, Schlichting I, Shaik S. J. Phys. Chem. B. 2008;112:9490–9500. doi: 10.1021/jp803010f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.