

Background: Alleles Prnpa and Prnpb of mouse prion protein (PrP) influence the incubation period of prion disease.
Results: PrPa and PrPb, products of these alleles, aggregate differently in vitro.
Conclusion: The polymorphism at 108/189 influences the oligomeric stages of PrP polymerization.
Significance: Elucidating the mechanism of PrP aggregation is relevant to understanding prion disease susceptibility, prion strains, and species barriers.
Keywords: Amyloid, Circular Dichroism (CD), Kinetics, Prions, Protein Aggregation, Allele, Electron Microscopy, Polymorphism, Seeding, Thioflavin T
Abstract
Prion diseases are fatal neurodegenerative disorders associated with the polymerization of the cellular form of prion protein (PrPC) into an amyloidogenic β-sheet infectious form (PrPSc). The sequence of host PrP is the major determinant of host prion disease susceptibility. In mice, the presence of allele a (Prnpa, encoding the polymorphism Leu-108/Thr-189) or b (Prnpb, Phe-108/Val-189) is associated with short or long incubation times, respectively, following infection with PrPSc. The molecular bases linking PrP sequence, infection susceptibility, and convertibility of PrPC into PrPSc remain unclear. Here we show that recombinant PrPa and PrPb aggregate and respond to seeding differently in vitro. Our kinetic studies reveal differences during the nucleation phase of the aggregation process, where PrPb exhibits a longer lag phase that cannot be completely eliminated by seeding the reaction with preformed fibrils. Additionally, PrPb is more prone to propagate features of the seeds, as demonstrated by conformational stability and electron microscopy studies of the formed fibrils. We propose a model of polymerization to explain how the polymorphisms at positions 108 and 189 produce the phenotypes seen in vivo. This model also provides insight into phenomena such as species barrier and prion strain generation, two phenomena also influenced by the primary structure of PrP.
Introduction
Prion diseases are protein folding disorders that include Creutzfeldt-Jakob disease in humans, bovine spongiform encephalopathy in cattle, chronic wasting disease in cervids, and scrapie in sheep. These diseases have several similarities to other protein folding neurodegenerative disorders, such as Alzheimer disease, but the hallmark of prion diseases is the infectious nature of the protein responsible for the neurodegeneration (1). The protein in question is the prion protein (PrP),2 and its infectious form is generated when the monomeric, predominantly α-helical, and soluble form (PrPC) is structurally converted into an amyloid structure, oligomeric in nature, high in β sheet, and partially protease-resistant (PrPSc). Many different forms of PrPSc can be generated, and this structural promiscuity is thought to be the molecular basis for prion “strains,” defined by distinct incubation time, phenotype, and/or pathology (2–4). Although certain structures of PrPSc may be better able to convert PrPC from select species, once the process has begun, it is relentless. More PrPC is converted into PrPSc, PrPSc accumulates, and neuronal death follows.
The primary structure of the host PrPC is a major determinant of the host prion disease susceptibility. For example, subtle differences in PrPC sequence are sufficient to render some mammals, such as rabbits and horses, immune to prion disease (5–7). Sometimes this species barrier can be crossed, although generally it is an inefficient process, with newly infected species having prolonged incubation periods (7–9). Interestingly, with sequential passages through the new host species, incubation periods become shorter and stabilize at a new and constant incubation period for that host (9). During these passages, it is believed that a process of conformational adaptation or a selection of one subtype of PrPSc conformation is taking place (10). This adaptation is influenced primarily by the sequence of host PrP (7).
The PrP sequence can also vary within a species; these polymorphisms too can influence disease susceptibility or even phenotype. In humans, methionine homozygosity at codon 129 of the prion protein gene (Prnp) increases susceptibility to prion disease. Codon 129 also determines the phenotype of a genetic form of Creutzfeldt-Jakob disease caused by the D178N mutation (11). The effect of Prnp polymorphism on disease susceptibility can be found in other species too, including codon 132 in cervids (12) and codons 136/154/171 in sheep (13–17).
Given the clear influence of PrPC sequence on prion disease, the next question is by what mechanism is this influence conferred? The PrPC sequence may dictate which PrPSc conformations are permissive or preferred, or it may influence how readily the conversion process can occur under the guidance a given PrPSc structure. Although the mechanism of prion conversion into amyloids remains obscure, the canonical model proposed for amyloid formation is nucleated polymerization (18) consisting of two phases: (i) a nucleation phase where monomers undergo conformational change and self-associate to form oligomeric nuclei; and (ii) an elongation phase, in which nuclei rapidly grow by the further addition of monomers, forming larger fibrils until saturation (19). PrPC sequence may regulate either or both of these phases.
In mice, the presence of Prnp allele a (Prnpa, Leu-108/Thr-189) or allele b (Prnpb, Phe-108/Val-189) dramatically influences the incubation period of prion infection (20, 21). Typically, Prnpa mice exhibit shorter incubation times (∼100–200 days) when compared with Prnpb mice (∼255–300 days) (22–24), although the opposite trend was seen in one study (21). The reason for this discrepancy is not clear, but different prion strains were used; in the latter study, the strain used for inoculation was first passaged in Prnpb mice, whereas Prnpa mice were used as the source of strains for the other studies (25, 26).
As the differences in the primary structure of PrPa and PrPb are the main distinguishing features in these studies, it follows that this polymorphism is the major factor determining the incubation period in vivo. Both residues have been implicated in prion disease, either by playing a role in the initial stages of PrPC conversion (27–29) or by influencing the susceptibility to prion infection (13–16).
We hypothesized that different polymerization kinetics could explain the different incubation periods of these two alleles and that we would be able to detect these kinetic differences in vitro by putting recombinant mouse PrPa or PrPb into fibril-forming assays. Having found this to be the case, we then proceeded to use this in vitro model of conversion to explore the potential mechanisms of fibril formation process in each allele type.
In this work, we compare for the first time the kinetics of amyloid fibril formation of recombinant mouse PrPa and PrPb. From these results, together with the conformational and structural analysis of the formed fibrils, we propose different mechanisms of polymerization for these two isoforms of mouse PrP.
EXPERIMENTAL PROCEDURES
Expression and Purification of Mouse PrP (89–230)
The codon-optimized synthetic genes corresponding to the C-terminal domain of mouse prion protein (89–230) for allele A and B were expressed in Escherichia coli BL21 (DE3) as described earlier (30). The E. coli BL21 (DE3) cells were grown in 25 ml of LB plus 100 μg/ml ampicillin overnight. 10 ml was used to inoculate 500 ml of LB plus 100 μg/ml ampicillin and set to shake at 225 rpm at 37 °C until it reached an A600 of about 1.0. Cells were pelleted down and resuspended in fresh Terrific Broth plus 100 μg/ml ampicillin medium and set to shake at 225 rpm at 37 °C for 1 h before induction with 1 mm isopropyl-1-thio-β-d-galactopyranoside. Induced cells were grown for a further 12–18 h to reach an A600 of about 2.0. These cells were harvested by centrifugation at 4,500 × g for 25 min at 4 °C. The cell pellet was resuspended in 100 ml of lysis buffer (8 m urea, 10 mm Tris, 100 mm Na2PO4, pH 8.0) using vortex and incubated at room temperature for 1 h before sonication using a microtip at 30% amplitude for 10 cycles of 30 s. The cell debris was removed by centrifugation at 15,000 × g for 1 h. The supernatant was incubated with 30 ml of nickel-nitrilotriacetic acid resin with continuous stirring for 30–45 min and transferred to a column. On-column refolding was done by slow gradual change from 8 to 0 m urea (10 mm Tris, 100 mm Na2PO4, pH 6.3) (31, 32). Contaminants were removed using 5 column volumes of 10 mm Tris, 100 mm Na2PO4, 50 mm imidazole, pH 6.3. The His-tagged prion proteins were eluted using 10 mm Tris, 100 mm Na2PO4, 500 mm imidazole, pH 6.5. The sample was dialyzed against 10 mm Tris, 2 mm CaCl2, pH 6.3, and the His tag was removed with thrombin by incubation for 24 h at 4 °C. The samples were dialyzed against 10 mm ammonium carbonate, lyophilized, and kept at −80 °C until use.
PrP Fibril Formation
Lyophilized samples of recombinant PrPa and PrPb were dissolved in 6 m guanidinium hydrochloride (GdnHCl) at a protein concentration of 5 mg/ml. Stock solutions were diluted in 50 mm sodium phosphate buffer, pH 7.0, to reach a final concentration of 2 m GdnHCl. The final protein concentration was 0.5 mg/ml unless otherwise indicated. Fibril formation reactions were carried out in 96-well plates (white plate, clear bottom, Costar 3610) covered with thermal adhesive sealing film (08-408-240; Fisher Scientific) at reaction volumes of 200 μl/well. The samples were incubated at 37 °C with continuous shaking at 500 rpm in the presence of 10 μm ThT. Fluorescence measurements were taken at 445/482 nm excitation/emission and 475 nm cutoff on a M5 SpectraMax fluorescence plate reader (Molecular Devices). At least three replicates were measured. For seeding experiments, the reaction was carried out in the presence of sonicated preformed fibrils (1 min in a cup sonicator). Estimation of lag phase was done as reported previously (33). In brief, the data were fitted to the limiting forms of the hyperbolic cosine solution of the equation developed by Bishop and Ferrone (34). The data corresponding to the nucleation stage were fitted to the quadratic equation ½B2At2, whereas those corresponding to the elongation stage were fitted to the exponential function ½AeBt, where t is time and A and B are fitting coefficients. The lag phase was defined as the time point where the fitting curves for quadratic and exponential equation intersect. If the time value for the intersection point is equal to or less than zero, there is an absence of lag phase.
Prefibrillar Oligomer Formation
To monitor the formation of ThT-negative oligomers, aggregation reactions were carried out in 1.5-ml test tubes (protein LoBind, Eppendorf) at reaction volumes of 1.2 ml. The samples were incubated at 37 °C with continuous shaking at 500 rpm, and 50 μl of sample was withdrawn at different times and incubated for 15 min at room temperature with 1 μl of 15 μm p-FTAA or 1 μl of 2 mm ThT. Spectra for p-FTAA were recorded between 480 and 700 nm with excitation at 450 nm. The ratio 530/630 nm was plotted.
Circular Dichroism
1 ml of 0.5 mg/ml PrPa or PrPb was put under aggregation conditions in a 1.5-ml test tube, and at indicated time points, aliquots of 40 μl were removed for CD measurements in a 0.1-mm cuvette. The spectra were recorded on a Chirascan CD spectrometer (Applied Photophysics) instrument between 200 and 260 nm, with sampling points every 1 nm. For each sample, 10 scans were averaged, and base-line spectra were subtracted. Thermal denaturation and renaturation were monitored by heating the sample from 15 to 70 °C and cooling from 70 to 15 °C at 1 °C/min in a 1-mm cuvette. The ellipticity was recorded at 222 nm, and the percentage of unfolded protein was plotted. For GdnHCl-induced denaturation, separate samples were prepared by diluting a concentrated stock of PrP to 0.5 mg/ml in 50 mm phosphate buffer, pH 7.0, and varying the GdnHCl concentration. The samples were incubated for 2 h at room temperature, and spectra were recorded in a 1-mm cuvette. Data were processed using Applied Photophysics Chirascan Viewer and Microsoft Excel.
Conformational Stability Assay
Aliquots of 0.1 mg of PrPa and PrPb fibrils were incubated with various concentrations of GdnHCl for 2 h at 25 °C. The samples were diluted with conformational stability assay buffer (10 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.5% Nonidet P-40, and 0.5% deoxycholate) to reach a final concentration of 0.4 m GdnHCl and digested with 10 μg/ml proteinase K for 1 h at 37 °C. The reaction was stopped with a 5-min incubation with 1.5 mm PMSF at 37 °C. Samples were centrifuged 1 h at 100,000 × g, and the pellet was resuspended in 20 μl of sample buffer, boiled for 10 min, and loaded onto NuPAGE 4–12% BisTris gels. Protein was transferred to PVDF membrane and detected with SAF83 antibody.
Transmission Electron Microscopy (TEM)
Once the aggregation reaction reached the plateau phase, aliquots of PrPa and PrPb samples were adsorbed onto 400 mesh carbon-coated copper grids (Ted Pella, Inc., Redding CA), negatively stained with 2% (w/v) uranyl acetate, and subsequently imaged in a Philips EM-420 transmission electron microscope (FEI Ltd., Eindhoven, The Netherlands) operated at 100 kV. Images were recorded on films (Kodak SO-163) at a nominal magnification of 13,500× to 31,000×. The films were developed and then scanned at 21.16 μm/pixel for a final resolution range of 6.8–15.7 Å/pixel. Fibril dimensions were determined as reported previously (35). Between 15 and 30 randomly chosen fibrils were measured per sample; widths and lengths are reported as means ± the S.E.
RESULTS
Aggregation Kinetics Reveal Differences in the Lag Phases of PrPa and PrPb
We compared the aggregation process of recombinant PrPa (89–230) and PrPb (89–230) in phosphate buffer, 2 m GdnHCl, pH 7.0 under shaking conditions as described previously (36). The presence of chaotropic agent GdnHCl at this concentration has been demonstrated to improve aggregation efficiency (36). Sigmoidal kinetic profiles typical for amyloid fibril formation were obtained for both PrPa and PrPb when the process was followed by measuring changes in ThT fluorescence intensity (Fig. 1A). A clear difference in lag phase was observed, with PrPb taking much longer to develop ThT signal than PrPa. Even with variability between preparations, PrPb always had longer lag phase (supplemental Fig. S1). Quantitative analysis of the lag phases was performed as described previously (34) (see “Experimental Procedures” and supplemental Fig. S2). Lag phases were 36 (± 4.3) h for PrPa and 140 (± 23.0) h for PrPb (Fig. 1C). Of note, these lag phase differences were not specific to the denaturing GdnHCl conditions as PrPa also aggregated more quickly than PrPb in acetate buffer, pH 4.0, under the same temperature and shaking conditions (supplemental Fig. S3).
FIGURE 1.

Time courses of PrPa and PrPb fibril formation monitored by ThT fluorescence. PrP (22 μm) was incubated in 50 mm phosphate buffer, pH 7.0, 2 m GdnHCl at 37 °C and 500 rpm in the absence or in the presence of seeds. A and B, kinetics of unseeded (A) and seeded (B) polymerization reactions of PrPa (empty circles) and PrPb (filled circles). C, lag phases for the kinetics of PrPa (empty bars) and PrPb (filled bars) plotted in A and B. D, lag phase of PrPa (left) and PrPb (right) aggregation process in the presence of 0.01, 0.1, 0.5, or 2% preformed homologous fibrils. The fibril formation process was followed by ThT fluorescence, and the lag phase was estimated as the intersection of quadratic and exponential fitting curves as described under ”Experimental Procedures.“ Three replicates were plotted for each reaction. Error bars indicate ± S.E.
PrPa and PrPb Lag Phases Differ in Response to Seeding
In nucleated polymerization, the rate-limiting step is nucleus formation. Therefore, the addition of preformed nuclei should eliminate the lag phase and push the process into the elongation phase. To test whether lag phase could be eliminated in our reactions, we added preformed fibrils of homologous PrP (PrPa seeds to PrPa monomers; PrPb seeds to PrPb monomers). When the reaction was seeded with 0.5% (v/v) of homologous fibrils (Fig. 1B), the variability between replicates was lower than in unseeded reaction. As expected, the lag phase was eliminated for PrPa; however, although the lag phase for PrPb was substantially reduced from 140 to 20 h, it was not eliminated (Fig. 1C). Using separate preparations and different concentrations of seed, these differences were still observed. As little as 0.01% (v/v) of seed was sufficient to eliminate the lag phase for PrPa (Fig. 1D). For PrPb, increasing seed from 0.01 to 0.1% (v/v) did shorten the lag phase from 23.0 (± 0.8) to 17.3 (± 1.1) h, but further increase of seed concentration to 0.5 and 2% (v/v) had no effect.
Cross-seeding Experiments Reveal Similar Efficiency of Seeding by PrPa and PrPb Fibrils
To test whether PrPb fibrils are simply less efficient at seeding than PrPa fibrils, we added preformed fibrils of heterologous PrP to our reactions (PrPb seeds were added to PrPa monomers; PrPa seeds were added to PrPb monomers) (Fig. 2). The lag phase was eliminated when PrPa was seeded with PrPb fibrils, indicating that PrPb fibrils can indeed seed efficiently. Surprisingly, an identical residual lag phase was observed for PrPb regardless of whether reactions were seeded with heterologous PrPa or homologous PrPb fibrils.
FIGURE 2.

Time courses of PrPa and PrPb fibril formation in the presence of homologous and heterologous seeds. Monomeric PrP (22 μm) was seeded with 0.5% (v/v) of fibrils, and the kinetics were followed by ThT fluorescence. A, monomeric PrPa (empty circles) and PrPb (filled circles) were aggregated in the presence of preformed fibrils of homologous PrP (PrPa with PrPa fibrils; PrPb with PrPb fibrils). B, monomeric PrPa (empty circles) and PrPb (filled circles) were aggregated in the presence of preformed fibrils of heterologous PrP (PrPa with PrPb fibrils; PrPb with PrPa fibrils). C, the lag phases for PrPa and PrPb in the presence of homologous (Homo.) and heterologous (Hete.) seeds were calculated as described under ”Experimental Procedures.“ The average of three replicates was plotted for each reaction. Error bars indicate ± S.E.
Indications for a Receptive Substrate in the PrPb Aggregation Mechanism
The residual lag phase in seeded PrPb reactions indicated that the starting PrPb monomers were not the immediate substrate being incorporated into fibrils during the elongation phase. Rather, a secondary conformation must have formed first, and it was only this receptive substrate that could be polymerized by the seeds. The generation of this receptive substrate could be (i) a “seed-independent” process, where a set amount of time is required for the starting monomers to form this substrate; or (ii) a “seed-influenced” process, where the fibrils actually facilitate receptive substrate formation. To distinguish between these possibilities, we used a delayed seeding reaction in which PrPb was put under aggregation conditions and preformed PrPb fibrils were added at different times during the lag phase. We predicted that in a seed-independent process, a set amount of time would be required to form the receptive substrate followed by the same onset of exponential growth phase regardless of when the seeds were added. For a seed-influenced process, we predicted that the onset of exponential growth would correlate with the timing of seed addition. As shown in Fig. 3A, the latter phenomenon was demonstrated. Later seed addition produced longer lag phases (seed time 0, lag phase 9.4 ± 0.3 h; seed time 5 h, lag phase 12.2 ± 0.5 h; seed time 10 h, lag phase 16.2 ± 0.5 h). Also of note was that the later the seed addition, the less time required between seed addition and onset of exponential phase (time 0, lag phase 9.4 h; time 5 h, lag phase 7.2 h; time 10 h, lag phase 6.2 h).
FIGURE 3.

Kinetics of PrPb aggregation seeded at different times. A, monomeric PrPb (22 μm) was aggregated in the absence (filled circles) or in the presence (empty squares, triangles, and circles) of 0.5% (v/v) homologous seeds. The seeds were added at the beginning of the reaction (squares), after 5 h (triangles), or after 10 h (empty circles). The kinetics were normalized, and the percentage of ThT fluorescence was graphed. B, the same data were plotted as absolute fluorescence using a different y scale to focus on the processes happening before the exponential phase. The slopes were calculated by fitting the data with linear regression. The arrows in A and the dotted lines in B indicate the times at which the seeds were added. Three replicates were averaged for each reaction. Error bars indicate ± S.E.
PrPb Seeding Induces an Initial Linear Increase in ThT Fluorescence
Looking at the ThT fluorescence levels immediately after seed addition in the delayed seeding experiment, we observed a small but steady linear increase over time prior to the exponential growth phase (Fig. 3B). The immediate jump in ThT can be attributed to the addition of seed as the seed mixture contained some ThT. However, the subsequent rate of increase of ThT was higher when seed was added later. The increase (in ThT a.u./hour) was 6.8 ± 0.6, 14.4 ± 1.7, and 20.7 ± 2.7, for seeding times of 0, 5, and 10 h, respectively.
ThT-negative Oligomers Are Formed Prior to the Exponential Growth of PrPb Fibrils
To determine whether oligomeric species that do not bind ThT (ThT-negative oligomers) might be contributing to the early fibrillization process, we examined seeded and unseeded reactions with the anionic oligothiophene derivative p-FTAA, which has been reported to bind prefibrillar oligomers of recombinant PrP (37). In the absence of seeds, an increase in p-FTAA signal was observed ∼20 h before the increase in ThT signal, demonstrating the early formation of ThT-negative oligomeric species (Fig. 4). When the reaction was carried out in the presence of seeds, p-FTAA-positive oligomers appeared immediately, whereas ThT-positive fibril formation began 6 h later (Fig. 4).
FIGURE 4.

PrPb prefibrillar (ThT-negative) oligomer formation monitored by p-FTAA. Monomeric PrPb (20 μm) was put into aggregation assays in 1.5-ml test tubes. 50 μl was withdrawn at different times and incubated with p-FTAA (squares) or ThT (circles) to monitor the formation of prefibrillar oligomers and fibril formation, respectively. The assay was carried out in the absence (filled symbols) or in the presence (empty symbols) of 1% (v/v) of seeds.
PrPb Aggregation Is Not Predictably Influenced by Protein Concentration
In unseeded reactions undergoing nucleation polymerization, a higher monomer concentration should produce a shorter lag phase, a higher slope at elongation phase, and a higher plateau (19). Therefore, we followed the kinetics of unseeded aggregation as a function of concentration of PrP monomer (Fig. 5). PrPa kinetics were proportional to monomer concentration (Fig. 5, A and C). For 11, 16, and 27 μm PrPa, respectively, the lag phases were 36.7 (± 4.3), 26.3 (± 1.2), and 18.0 (± 1.7) h, the slopes for elongation phase were 6.1 (± 0.8), 88.8 (± 7.9), and 191.0 (± 45.2) ThT a.u./h, and the plateaus were 1180 (± 32), 2790 (± 99), and 4800 (± 111) ThT a.u. Importantly, these kinetic parameters changed linearly with monomer concentration, whereas an exponential correlation is expected from a pure nucleated polymerization model. For PrPb, no clear correlation was observed between these parameters and protein concentration for PrPb (Fig. 5, B and C). In fact, the highest concentration (27 μm) PrPb reactions consistently had the longest lag phases, opposite to what the nucleated polymerization model predicts. There was also significantly more kinetic variability among PrPb replicates as shown in the non-averaged replicate curves in Fig. 5B.
FIGURE 5.

PrP aggregation at different monomeric concentrations. A and B, PrPa (A) and PrPb (B) were put into aggregation assays at three different concentrations: 27 μm (squares), 16 μm (circles), and 11 μm (triangles). Three replicates were averaged for PrPa (A), but individual replicates were plotted for PrPb (B) to better display the high heterogeneity observed for these reactions. C, the lag phases for PrPa (empty bars) and PrPb (filled bars) under the different PrP monomer concentrations were calculated from the intersection of quadratic and exponential fitting curves, as described under ”Experimental Procedures.“ Error bars indicate ± S.E.
Monomeric PrPa and PrPb Have Similar Secondary Structure
Given the apparent aggregation differences between starting monomers for PrPa and PrPb, we compared their secondary structure by far-ultraviolet circular dichroism (supplemental Fig. S4). Identical, predominantly α-helical signals with minima at 208 and 222 nm were obtained for both PrPa and PrPb in Tris-HCl, pH 7.0 (supplemental Fig. S4A). The spectra were also similar under the conditions used for aggregation experiments (2 m GdnHCl and 37 °C) (supplemental Fig. S4B).
Monomeric PrPa and PrPb Share Denaturation/Renaturation Characteristics
A correlation between the thermal lability of mouse PrP and conversion efficiency has been reported (38), and distinct unfolding properties of PrPa and PrPb monomers might explain their different kinetic profiles. We therefore compared the denaturation profiles of PrPa and PrPb by following the 222 nm intensity as a function of temperature or GdnHCl concentration. Loss of the 222 nm minimum correlates with loss of α-helix structure. Thermal denaturation and renaturation of PrP produced identical curves for both PrP isoforms (supplemental Fig. S4C, inset); GdnHCl denaturation curves for PrPa and PrPb were also identical (supplemental Fig. S4D).
Seed Type Influences the Conformational Stability of PrPb Fibrils More than PrPa Fibrils
We used a conformational stability assay to study differences between PrPa and PrPb fibrils generated under unseeded and seeded conditions. In this assay, increasing concentrations of GdnHCl are used to gradually denature the fibrils, rendering them more susceptible to proteolysis with proteinase K. Structural differences in fibrils can affect the denaturation process and generate distinct proteinase K digestion profiles.
All proteinase K-treated PrPa fibrils yielded prominent bands of ∼13 and 18 kDa (Fig. 6, A–C). In unseeded reactions, the 18-kDa band was more intense and more sensitive to protease action, disappearing in the range of 5.4–5.8 m GdnHCl. The 13-kDa band was still visible at 6 m GdnHCl (Fig. 6A). Similar results were obtained for PrPa fibrils generated in the presence of either homologous or heterologous seeds (Fig. 6, B and C). The pattern was not exactly replicated; the relative band intensities were reversed in the homologous seeded reaction, the 13-kDa band being more intense than the 18-kDa band, and in the heterologous seeded reaction, the intensities of the two bands were comparable. Also, two other faint bands were only seen in the unseeded reactions.
FIGURE 6.
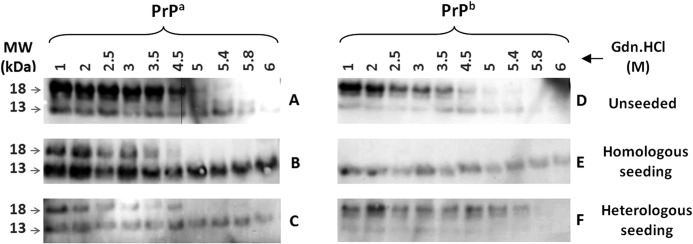
Conformational stability assays of PrP fibrils formed under seeded and unseeded conditions. A–F, Western blot of PrPa (A–C) and PrPb (D–F) fibrils formed in the absence (A and D) or in the presence of homologous (B and E) or heterologous (C and F) seeds and subjected to GdnHCl-induced denaturation and proteinase K digestion. 12 μl of proteinase K-treated samples was analyzed by Western blotting using SAF83 antibody, as described under “Experimental Procedures.” MW, molecular weight markers.
In contrast, differences in PrPb fibril stability were more apparent and highly dependent on the seeding conditions. Unseeded PrPb fibrils had a pattern similar to unseeded PrPa fibrils, with a resistant 13-kDa band and a more intense 18-kDa band that disappeared after 5.4–5.8 m GdnHCl treatment (Fig. 6D). However, under homologous seeding conditions (Fig. 6E), the 13-kDa band remained resistant and the 18-kDa band was not present at all, indicating a dramatically different structural state in these PrPb fibrils. Under heterologous seeding conditions (Fig. 6F), the 18-kDa band was present, was more intense, and disappeared with 5.8 m GdnHCl, as in the unseeded reactions. The usually resistant 13-kDa band was more sensitive, disappearing by 5.4 m GdnHCl.
Seed Type Influences the Kinetics of Fibril Formation for PrPb but Not for PrPa
Based on our conformational stability assay results, we speculated that if different seeds could strongly influence the stability of PrPb fibrils generated, different seeds might also affect PrPb aggregation kinetics more strongly than PrPa kinetics. Because of the variability of unseeded PrPb fibril reactions, we were able to generate two PrPb seeds with distinct kinetic profiles. The “slow” (S) fibrils had an elongation phase slope of 4.1 ThT a.u./h, whereas the “fast” (F) fibrils had one of 13.6 ThT a.u./h (Fig. 7, dashed and dotted curves). The lag phases for S and F fibril formation were 15 and 19 h, respectively. S and F seeds were used to seed PrPa and PrPb reactions.
FIGURE 7.
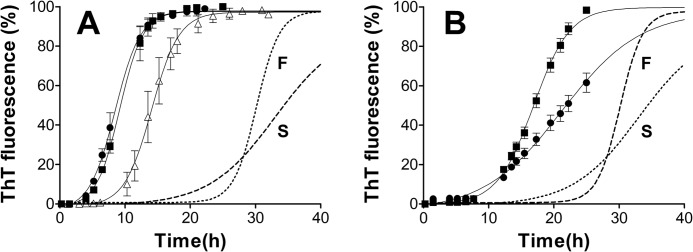
Influence of slow and fast seeds on the kinetics of PrP fibril formation. A and B, 22 μm PrPa (A) or PrPb (B) was incubated in the absence (A, open triangles) or the presence of 0.5% (v/v) slow (filled circles) or fast (filled squares) seeds. The original kinetics of the reactions that produced the S and F seeds are shown for comparison, as a dotted line (F) and dashed line (S). n = 3 for unseeded reactions; n = 2 for seeded reactions. Error bars indicate ± S.E.
As expected, PrPa lag phase was eliminated regardless of the type of seed used (Fig. 7A), whereas for PrPb, residual lag phases between 7 and 9 h occurred with seeding (Fig. 7B). For PrPa, the kinetics of reactions seeded with S or F were almost identical, with slopes of 12.5 and 13.5 ThT a.u./h, respectively (Fig. 7A). These slopes were also similar to those obtained for the elongation phase of unseeded reactions of PrPa (Fig. 7A, open triangles). In contrast, two distinct kinetic profiles were obtained for PrPb seeded with S or F fibrils, the slopes being 4.1 and 8.7 ThT a.u./h, respectively (Fig. 7B). The slope of the S-seeded reaction is identical to that of the original S fibril formation, whereas that for the F-seeded reaction is intermediate between the original S and F fibril formation kinetics.
TEM of PrP Fibrils
Fibril production was confirmed using TEM, and differences between PrPa and PrPb samples were also noted. All PrPa samples contained straight fibrils of similar length (≅600–800 nm) and width (≅10 and 20 nm in narrow and wide regions) regardless of whether the fibrils were produced under seeded or unseeded conditions and regardless of whether S or F seeds were used (Fig. 8, A–C, and Table 1). However, although unseeded PrPb fibrils (Fig. 8D) had grossly similar structures to PrPa fibrils (Table 1), different features occurred with seeding. PrPb fibrils formed in the presence of F seeds (Fig. 8E and Table 1) were shorter (≅100 nm) and uniform in width (≅15 nm); those seeded with S seeds (Fig. 8F and Table 1) were of intermediate length (≅300 nm) and sometimes curved, in contrast to the straight fibrils of PrPa.
FIGURE 8.
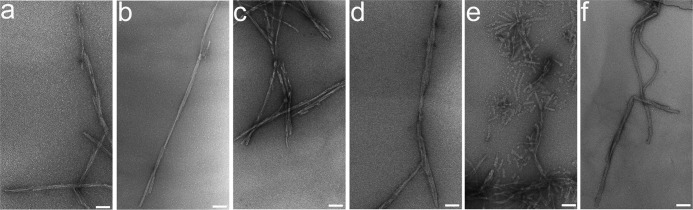
Influence of different seeds on the structure of PrP fibrils. a–f, images of PrPa (a–c) and PrPb (d–f) fibrils generated in the absence of seeds (a and d) or in the presence of fast (b and e) or slow (c and f) seeds were obtained by TEM. Scale bars, 100 nm.
TABLE 1.
Quantitative analysis of PrPa and PrPb fibrils generated under unseeded and seeded conditions
The width and length (expressed in nm ± S.E.) of fibrils from the six samples depicted in Fig. 8 were determined.
| PrPa fibrils |
PrPb fibrils |
|||||
|---|---|---|---|---|---|---|
| Unseeded | Fast seeds | Slow seeds | Unseeded | Fast seeds | Slow seeds | |
| No. of fibrils analyzed | 21 | 27 | 23 | 15 | 18 | 20 |
| Fibril width narrow region | 10.7 ± 0.8 | 9.6 ± 0.6 | 8.8 ± 0.6 | 10.2 ± 0.6 | 14.5 ± 0.7 (uniform width) | 8.4 ± 0.4 |
| Fibril width wide region | 23.3 ± 0.8 | 20.7 ± 1.1 | 19.5 ± 1.1 | 19.2 ± 1.1 | 14.5 ± 0.7 (uniform width) | 20.0 ± 1.1 |
| Fibril length | 821 ± 126 | 681 ± 82 | 602 ± 67 | 616 ± 86 | 102 ± 13 | 332 ± 58 |
DISCUSSION
In mice, the polymorphisms at positions 108 and 189 are major determinants of prion disease incubation time (20, 21), but the molecular basis for this phenomenon is unclear. We hypothesized that the effect of host PrP sequence on incubation time was related to the mechanism of prion conversion, involving the oligomerization of PrPC into PrPSc. In this study, we provide evidence that PrP aggregation does not follow the canonical nucleation polymerization and that in vivo findings can be explained by different polymerization kinetics and seeding responses of the two mouse PrP isoforms.
PrPa and PrPb differ only at residues 108 and 189, and we did not detect differences in α-helical content or thermal/chemical stability between the two monomers. Instead, the primary differences were found between their kinetic and seeding behaviors.
Based on our kinetic data, neither PrP isoform follows true nucleation polymerization. This model predicts that fibril mass is proportional to t2 during nucleation, meaning that there is never a flat lag phase (39, 40). Both PrPa and PrPb had prolonged flat lag phases in unseeded conditions. This better fits a double-nucleation mechanism (39). Also, PrPa concentration had a linear effect on elongation rate and lag phase, as opposed to the exponential effect predicted by nucleation polymerization. PrPb concentration did not correlate at all with lag phase, rate of elongation, or maximum ThT fluorescence. In fact, at highest PrPb concentration, a paradoxical increase in lag phase was observed. This is possibly explained by an accumulation of off-pathway oligomers that interfered with the aggregation process (41), although we were not able to detect these by TEM or in supernatants of centrifuged samples after aggregation.
Other aspects of PrPb kinetics also support an alternate model. Our data indicate that the PrPb starting monomers, in 2 m GdnHCl, cannot be directly incorporated into fibrils. This is why the lag phase persists despite the addition of seed. Therefore, it is a different component, which we call a receptive substrate, that is incorporated into the fibrils during exponential growth. How then do the starting monomers become receptive substrates? When unseeded, the starting monomers do eventually form receptive substrates and become fibrils, but the process is extremely long with greater variation in kinetic profile and fibril structure. This variability suggests that a number of pathways to receptive substrate formation are possible and that several different receptive substrates can be produced, each of which then produces a different kinetic profile and fibril product. Alvarez-Martinez et al. (42) have explained variation in aggregation kinetics by proposing the existence of “conformationally active monomers,” which randomly form different nuclei. Whichever nucleus forms first is the one that dictates the dynamics of the aggregation process and the resulting fibril structure. This model could explain the variability seen in our data, but not why we cannot eliminate the lag phase with seeding nor why we do not see a correlation between PrPb kinetics and monomer concentration. Rather than monomers, our data suggest that conformationally active oligomers may be the necessary precursors to the formation of receptive substrates.
If the production of conformationally active oligomers is the rate-limiting step, dependent on structural change from the monomer state, it follows that changes in monomer concentration will not facilitate the process of fibril growth, as found in our study. Also, the addition of seed to starting monomers does not immediately lead to exponential growth in PrPb because starting monomers must first form conformationally active oligomers and then receptive substrates; only the receptive substrates are incorporated into fibril growth. The presence of these early oligomers (ThT-negative and ThT-low oligomers) was confirmed by p-FTAA fluorescence. A similar mechanism, “nucleated conformational conversion,” has been described for the yeast prion element [PSI+] where nuclei are formed by conformational rearrangement of less structured oligomeric intermediates that are in equilibrium with monomers (43).
Of note, although seeding does not eliminate PrPb lag phase, seed addition does greatly accelerate the time to exponential growth phase, meaning that the seed must be affecting the process in some manner. Seed addition produces an immediate and rapid increase in p-FTAA fluorescence and a slower linear increase in ThT fluorescence (prior to the exponential growth phase), with later addition yielding both a higher rate of ThT increase and a shorter time to onset of exponential growth phase. Given this, we propose the following mechanism (Fig. 9). Starting monomers gradually aggregate into larger and larger oligomers (which are ThT-negative but p-FTAA-positive). When these ThT-negative oligomers bind seeds, they become conformationally active and acquire some low ThT fluorescence (ThT-low, p-FTAA-positive), but do not yet become fully receptive substrates (and therefore do not undergo exponential growth). As larger ThT-negative oligomers are expected to accumulate over time, seeds that are added to the process later will bind larger ThT-negative oligomers that in turn become conformationally active, thus producing higher rates of ThT increase. The larger oligomers may also be closer to adopting a receptive substrate conformation, which would explain why there is an apparent acceleration to exponential phase after delayed seed addition.
FIGURE 9.

Proposed mechanism of PrPb fibril formation in unseeded and delayed seeding conditions. A, the unseeded reaction is shown, where monomers (dark triangles) first form ThT-negative (p-FTAA-positive) oligomers of increasing size and ultimately form conformationally active oligomers (light triangles, ThT-low, p-FTAA-positive) followed many hours later by receptive substrates (white squares, ThT-high), which go on to form fibrils. B, a typical seeded reaction is shown, with seed added at time 0. When monomers bind seed, they are immediately converted into a conformationally active state (ThT-low, p-FTAA-positive). As more monomers bind and are converted, a sufficient number or size is reached such that receptive substrates form. C and D, with delayed seeding, larger ThT-negative (p-FTAA-positive) oligomers are already present when seed is added, so immediate conversion of the larger oligomers occurs (becoming ThT-low, p-FTAA-positive), giving a greater linear increase in ThT fluorescence signal over time and allowing receptive substrates to form in a shorter time period after seed addition.
It should be noted that PrPa could fundamentally share the mechanism that we propose for PrPb and still display the kinetic and seeding tendencies we observed. If PrPa converts to conformationally active oligomers and then to receptive substrates more efficiently, and is not rate-limiting, the conformationally active oligomers would not be detectable in our seeded kinetic studies. The efficient formation of receptive substrates might correlate with less off-pathway aggregation, explaining the shorter lag phases and reduced variability seen in unseeded PrPa reactions. It would also explain why PrPa is not overly influenced by seed type; there is less time for seed and oligomer to interact.
We are proposing that the seed induces structural change without immediately triggering polymerization. Such a process has been described in “surface-catalyzed nucleation,” where monomers nonspecifically bind the lateral aspects of fibrils with subsequent conformational rearrangement to form on pathway oligomers, and ultimately nuclei, which can bind the ends of fibrils and proceed with exponential growth (44, 45). Such lateral association is a recognized process in recombinant PrP fibril formation (46).
Somewhat unexpectedly, our conformational stability assay and TEM experiments revealed that PrPb may be structurally influenced by seeds to a greater extent than PrPa; the morphology and stability of PrPa fibrils did not vary as much as PrPb fibrils. Also, PrPb more closely mimicked the kinetics of the seeds used.
The purpose of this study was to gain insight into how polymorphisms of PrP translate into phenotypic differences in prion disease by studying kinetic profiles of fibril formation in vitro. The long lag phases for PrPb correlate with long incubation times in Prnpb mice (21–25). In addition, if conformationally active oligomers are part of the PrP aggregation mechanisms, they may present a new therapeutic target, one that lies structurally in between monomeric and fibrillar states. Finally, our data may also inform the study where a shorter incubation time was seen in Prnpb mice. Here, strain properties may be key, and we have found that PrPb appears to be more strongly influenced by the type of seed used. Importantly, this seed influence could have risk implications as hosts presumed to be less susceptible based on PrP sequence may simply need to be exposed to the appropriate strain. A real life example of this can be found in the rise of chronic wasting disease. At present, chronic wasting disease does not appear to cross the species barriers into humans, but there is growing evidence for distinct strains of chronic wasting disease, and the influence of human PrP polymorphism on their transmission characteristics is unknown.
Acknowledgments
We thank Byron Caughey, David Westaway, and Holger Wille for the critical reading of this manuscript, David Wishart for providing the DNA constructs for PrP expression, and Peter Nilsson for the provision of p-FTAA dye.
This work was supported by Alberta Innovates Health Solutions (formerly Alberta Heritage Foundation for Medical Research).

This article contains supplemental Figs. S1–S4.
- PrP
- prion protein
- PrPC
- cellular form of prion protein
- PrPSc
- disease-associated form of prion protein
- ThT
- thioflavin T
- p-FTAA
- 4′,3‴-bis(carboxymethyl)[2,2′;5′,2″,5″,2‴;5‴,2⁗]quinquethiophene-5,5⁗-dicarboxylic acid
- TEM
- transmission electron microscopy
- GdnHCl
- guanidinium hydrochloride
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- a.u.
- arbitrary units
- S
- slow
- F
- fast.
REFERENCES
- 1. Prusiner S. B. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 [DOI] [PubMed] [Google Scholar]
- 2. Bruce M. E., Fraser H. (1991) Scrapie strain variation and its implications. Curr. Top. Microbiol. Immunol. 172, 125–138 [DOI] [PubMed] [Google Scholar]
- 3. Fraser H., Dickinson A. G. (1973) Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. J. Comp. Pathol. 83, 29–40 [DOI] [PubMed] [Google Scholar]
- 4. Bruce M. E. (1993) Scrapie strain variation and mutation. Br. Med. Bull. 49, 822–838 [DOI] [PubMed] [Google Scholar]
- 5. Gibbs C. J., Jr., Gajdusek D. C. (1973) Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science 182, 67–68 [DOI] [PubMed] [Google Scholar]
- 6. Barlow R. M., Rennie J. C. (1976) The fate of ME7 scrapie infection in rats, guinea-pigs, and rabbits. Res. Vet. Sci. 21, 110–111 [PubMed] [Google Scholar]
- 7. Moore R. A., Vorberg I., Priola S. A. (2005) Species barriers in prion diseases–brief review. Arch. Virol. Suppl. 187–202 [DOI] [PubMed] [Google Scholar]
- 8. Chen S. G., Gambetti P. (2002) A journey through the species barrier. Neuron 34, 854–856 [DOI] [PubMed] [Google Scholar]
- 9. Hill A. F., Collinge J. (2004) Prion strains and species barriers. Contrib. Microbiol. 11, 33–49 [DOI] [PubMed] [Google Scholar]
- 10. Collinge J., Clarke A. R. (2007) A general model of prion strains and their pathogenicity. Science 318, 930–936 [DOI] [PubMed] [Google Scholar]
- 11. Goldfarb L. G., Petersen R. B., Tabaton M., Brown P., LeBlanc A. C., Montagna P., Cortelli P., Julien J., Vital C., Pendelbury W. W., et al. (1992) Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science 258, 806–808 [DOI] [PubMed] [Google Scholar]
- 12. Green K. M., Browning S. R., Seward T. S., Jewell J. E., Ross D. L., Green M. A., Williams E. S., Hoover E. A., Telling G. C. (2008) The elk PRNP codon 132 polymorphism controls cervid and scrapie prion propagation. J. Gen. Virol. 89, 598–608 [DOI] [PubMed] [Google Scholar]
- 13. Foster J. D., Parnham D. W., Hunter N., Bruce M. (2001) Distribution of the prion protein in sheep terminally affected with BSE following experimental oral transmission. J. Gen. Virol. 82, 2319–2326 [DOI] [PubMed] [Google Scholar]
- 14. Goldmann W., Hunter N., Smith G., Foster J., Hope J. (1994) PrP genotype and agent effects in scrapie: change in allelic interaction with different isolates of agent in sheep, a natural host of scrapie. J. Gen. Virol. 75, 989–995 [DOI] [PubMed] [Google Scholar]
- 15. Houston F., Goldmann W., Chong A., Jeffrey M., González L., Foster J., Parnham D., Hunter N. (2003) Prion diseases: BSE in sheep bred for resistance to infection. Nature 423, 498. [DOI] [PubMed] [Google Scholar]
- 16. van Keulen L. J., Vromans M. E., Dolstra C. H., Bossers A., van Zijderveld F. G. (2008) Pathogenesis of bovine spongiform encephalopathy in sheep. Arch. Virol. 153, 445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Westaway D., Zuliani V., Cooper C. M., Da Costa M., Neuman S., Jenny A. L., Detwiler L., Prusiner S. B. (1994) Homozygosity for prion protein alleles encoding glutamine-171 renders sheep susceptible to natural scrapie. Genes Dev. 8, 959–969 [DOI] [PubMed] [Google Scholar]
- 18. Jarrett J. T., Lansbury P. T., Jr. (1993) Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73, 1055–1058 [DOI] [PubMed] [Google Scholar]
- 19. Harper J. D., Lansbury P. T., Jr. (1997) Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66, 385–407 [DOI] [PubMed] [Google Scholar]
- 20. Westaway D., Goodman P. A., Mirenda C. A., McKinley M. P., Carlson G. A., Prusiner S. B. (1987) Distinct prion proteins in short and long scrapie incubation period mice. Cell 51, 651–662 [DOI] [PubMed] [Google Scholar]
- 21. Moore R. C., Hope J., McBride P. A., McConnell I., Selfridge J., Melton D. W., Manson J. C. (1998) Mice with gene targetted prion protein alterations show that Prnp, Sinc, and Prni are congruent. Nat. Genet. 18, 118–125 [DOI] [PubMed] [Google Scholar]
- 22. Lloyd S. E., Thompson S. R., Beck J. A., Linehan J. M., Wadsworth J. D., Brandner S., Collinge J., Fisher E. M. (2004) Identification and characterization of a novel mouse prion gene allele. Mamm. Genome 15, 383–389 [DOI] [PubMed] [Google Scholar]
- 23. Carlson G. A., Goodman P. A., Lovett M., Taylor B. A., Marshall S. T., Peterson-Torchia M., Westaway D., Prusiner S. B. (1988) Genetics and polymorphism of the mouse prion gene complex: control of scrapie incubation time. Mol. Cell. Biol. 8, 5528–5540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Akhtar S., Wenborn A., Brandner S., Collinge J., Lloyd S. E. (2011) Sex effects in mouse prion disease incubation time. PloS One 6, e28741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lloyd S. E., Onwuazor O. N., Beck J. A., Mallinson G., Farrall M., Targonski P., Collinge J., Fisher E. M. (2001) Identification of multiple quantitative trait loci linked to prion disease incubation period in mice. Proc. Natl. Acad. Sci. U.S.A. 98, 6279–6283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lloyd S. E., Linehan J. M., Desbruslais M., Joiner S., Buckell J., Brandner S., Wadsworth J. D., Collinge J. (2004) Characterization of two distinct prion strains derived from bovine spongiform encephalopathy transmissions to inbred mice. J. Gen. Virol. 85, 2471–2478 [DOI] [PubMed] [Google Scholar]
- 27. Dima R. I., Thirumalai D. (2004) Probing the instabilities in the dynamics of helical fragments from mouse PrPC. Proc. Natl. Acad. Sci. U.S.A. 101, 15335–15340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tizzano B., Palladino P., De Capua A., Marasco D., Rossi F., Benedetti E., Pedone C., Ragone R., Ruvo M. (2005) The human prion protein α2 helix: a thermodynamic study of its conformational preferences. Proteins 59, 72–79 [DOI] [PubMed] [Google Scholar]
- 29. Knaus K. J., Morillas M., Swietnicki W., Malone M., Surewicz W. K., Yee V. C. (2001) Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat. Struct. Biol. 8, 770–774 [DOI] [PubMed] [Google Scholar]
- 30. Bjorndahl T. C., Zhou G. P., Liu X., Perez-Pineiro R., Semenchenko V., Saleem F., Acharya S., Bujold A., Sobsey C. A., Wishart D. S. (2011) Detailed biophysical characterization of the acid-induced PrPc to PrPβ conversion process. Biochemistry 50, 1162–1173 [DOI] [PubMed] [Google Scholar]
- 31. Julien O., Chatterjee S., Bjorndahl T. C., Sweeting B., Acharya S., Semenchenko V., Chakrabartty A., Pai E. F., Wishart D. S., Sykes B. D., Cashman N. R. (2011) Relative and regional stabilities of the hamster, mouse, rabbit, and bovine prion proteins toward urea unfolding assessed by nuclear magnetic resonance and circular dichroism spectroscopies. Biochemistry 50, 7536–7545 [DOI] [PubMed] [Google Scholar]
- 32. Zahn R., von Schroetter C., Wüthrich K. (1997) Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 417, 400–404 [DOI] [PubMed] [Google Scholar]
- 33. Makarava N., Lee C. I., Ostapchenko V. G., Baskakov I. V. (2007) Highly promiscuous nature of prion polymerization. J. Biol. Chem. 282, 36704–36713 [DOI] [PubMed] [Google Scholar]
- 34. Bishop M. F., Ferrone F. A. (1984) Kinetics of nucleation-controlled polymerization. A perturbation treatment for use with a secondary pathway. Biophys. J. 46, 631–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qi X., Moore R. A., McGuirl M. A. (2012) Dissociation of recombinant prion protein fibrils into short protofilaments: implications for the endocytic pathway and involvement of the N-terminal domain. Biochemistry 51, 4600–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Polano M., Bek A., Benetti F., Lazzarino M., Legname G. (2009) Structural insights into alternate aggregated prion protein forms. J. Mol. Biol. 393, 1033–1042 [DOI] [PubMed] [Google Scholar]
- 37. Hammarström P., Simon R., Nyström S., Konradsson P., Aslund A., Nilsson K. P. (2010) A fluorescent pentameric thiophene derivative detects in vitro-formed prefibrillar protein aggregates. Biochemistry 49, 6838–6845 [DOI] [PubMed] [Google Scholar]
- 38. Kirby L., Agarwal S., Graham J. F., Goldmann W., Gill A. C. (2010) Inverse correlation of thermal lability and conversion efficiency for five prion protein polymorphic variants. Biochemistry 49, 1448–1459 [DOI] [PubMed] [Google Scholar]
- 39. Padrick S. B., Miranker A. D. (2002) Islet amyloid: phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry 41, 4694–4703 [DOI] [PubMed] [Google Scholar]
- 40. Ferrone F. (1999) Analysis of protein aggregation kinetics. Methods Enzymol. 309, 256–274 [DOI] [PubMed] [Google Scholar]
- 41. Baskakov I. V., Bocharova O. V. (2005) In vitro conversion of mammalian prion protein into amyloid fibrils displays unusual features. Biochemistry 44, 2339–2348 [DOI] [PubMed] [Google Scholar]
- 42. Alvarez-Martinez M. T., Fontes P., Zomosa-Signoret V., Arnaud J. D., Hingant E., Pujo-Menjouet L., Liautard J. P. (2011) Dynamics of polymerization shed light on the mechanisms that lead to multiple amyloid structures of the prion protein. Biochim. Biophys. Acta 1814, 1305–1317 [DOI] [PubMed] [Google Scholar]
- 43. Serio T. R., Cashikar A. G., Kowal A. S., Sawicki G. J., Moslehi J. J., Serpell L., Arnsdorf M. F., Lindquist S. L. (2000) Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289, 1317–1321 [DOI] [PubMed] [Google Scholar]
- 44. Ferrone F. A., Hofrichter J., Eaton W. A. (1985) Kinetics of sickle hemoglobin polymerization. II. A double nucleation mechanism. J. Mol. Biol. 183, 611–631 [DOI] [PubMed] [Google Scholar]
- 45. Ruschak A. M., Miranker A. D. (2007) Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 12341–12346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Anderson M., Bocharova O. V., Makarava N., Breydo L., Salnikov V. V., Baskakov I. V. (2006) Polymorphism and ultrastructural organization of prion protein amyloid fibrils: an insight from high resolution atomic force microscopy. J. Mol. Biol. 358, 580–596 [DOI] [PubMed] [Google Scholar]