

Abstract
Cell aggregation in unicellular organisms, induced by either cell non-sexual adhesion to yield flocs and biofilm, or pheromone-driving sexual conjugation is of great significance in cellular stress response, medicine, and brewing industries. Most current literatures have focused on one form of cell aggregation termed flocculation and its major molecular determinants, the flocculation (FLO) family genes. Here, we implemented a map-based approach for dissecting the molecular basis of non-sexual cell aggregation in Saccharomyces cerevisiae. Genome-wide mapping has identified four major quantitative trait loci (QTL) underlying nature variation in the cell aggregation phenotype. High-resolution mapping following up with knockout and allele replacement experiments resolved the QTL into the underlying genes (AMN1, RGA1, FLO1, and FLO8) or even into the causative nucleotide. Genetic variation in the QTL genes can explain up to 46% of phenotypic variation of this trait. Of these genes, AMN1 plays the leading role, differing from the FLO family members, in regulating expression of cell clumping phenotype through inducing cell segregation defect. These findings provide novel insights into the molecular mechanism of how cell aggregation is regulated in budding yeast, and the data will be directly implicated to understand the molecular basis and evolutionary implications of cell aggregation in other fungus species.
Keywords: cell aggregation, map-based cloning, QTL analysis, Saccharomyces cerevisiae
1. Introduction
Eukaryotic cells possess a remarkable capacity to aggregate together and form into a clump, which is crucial for the assembly of tissues and nervous systems, embryonic development, and cellular communication. Saccharomyces cerevisiae, the single-cell eukaryote, is also able to agglomerate cells into organized structures such as flocs, biofilm, and pseudohyphae. These non-sexual clumps create protective shields for cells in inner parts to escape from abiotic/biotic attacks,1 respond to nutrient starvation,2,3 and have been widely used in industrial productions.4 Thus, understanding the underlying mechanisms of the cellular behaviour has significant medical and evolutionary implications. However, most studies in the literature have focused on flocculation, a non-sexual type of cell clumping, induced by interaction between cell wall glycoproteins encoded by highly homologous flocculation (FLO) genes (FLO1, FLO5, FLO9, and FLO10).5,6 In particular, FLO1 was reported to be a key gene whose expression led to strong flocculation1 and was transcriptionally activated by FLO8.7,8 So far, these studies followed a reverse genetic approach to screen for the flocculins that bound specific sugar residues on the surface of other cells and led to formation of cell flocs.9,10 However, the reverse approach has been debated for its requirement of prior information on the underlying pathways leading to phenotypic expression of the traits,11 ignorance of the interactions between a gene of interest and the genetic background, and its incapacity to detect crucial polygenic interactions on phenotype.12–14
We present here a forward genetic approach to uncover the molecular basis of genetic variation in non-sexual cell aggregation in a natural population of budding yeast (S. cerevisiae). The approach integrates mapping of quantitative trait loci (QTL) with major effects on the cell aggregation trait, resolving the major QTL into the QTL genes, identifying causative nucleotides within the QTL genes, and characterizing molecular functions of these QTL genes and their interactive effects on the trait.
2. Materials and methods
2.1. Yeast strains and growth conditions
All yeast strains used in the present study are listed in Supplementary Table S1. YH1A, an isogenic haploid strain of the standard reference strain S288C, was crossed with YL1C, also a haploid strain described elsewhere.15 F1 hybrids from crossing the parental lines were sporulated to generate 292 segregants that were equivalent to gametes, forming an F2 generation. We created two other isogenic haploid strains from the parental lines with changed mating types through modifying their HO genes. We used the standard yeast media and growth conditions in mating, sporulation, and tetrad dissection experiments in the present study.16
2.2. Scoring phenotype of cell aggregation
The method used to score phenotype of cell aggregation in this study was modified from that proposed by Guo et al.6 In detail, any strain preserved under –70°C was recovered by plate streaking, and three single clones for the strain were obtained. Each clone was cultured in a 5-ml YPD liquid medium at 30°C at 280 rpm for 22–24 h to make the cells enter the stationary phase of growth. The cultures were swirled briefly with a vortex shaker before a sediment test. Phenotype of the tested strain was recorded as a sedimentary time in hours (T), i.e. the time taken for the tested cells to sink completely in the culture buffer in a test tube. On the basis of the sedimentary time records, we grouped phenotype of all tested strains into four categories when contrasted with the sedimentary records of the parental strains, TYH1A and TYL1C, which are illustrated in Fig. 1a. Phenotype of a tested strain was scored as one if T ≥ TYH1A, two if TYL1C < T < TYH1A, three if 0 < T ≤ TYL1C, or four if T = 0. In other words, phenotype of cell aggregation was quantified as a categorical character with multiple thresholds in the present study.
Figure 1.

QTL fine mapping. (a) Cell aggregation phenotype of two parental strains YH1A and YL1C and their representative offspring segregants labelled 1–4. The phenotype was assayed as sediment time in hours (left) and by microscope viewing (right) at stationary phase of cell growth. Red horizontal line in the left panel marks the top boundary of cell sediment. (b) Mapping of major cell aggregation QTLs on the yeast chromosome II and XV and candidate genes in the QTL regions. The physical map information of the candidate genes was from Saccharomyces Genome Database (www.yeastgenome.org).
In order for clear visualization of the cell aggregation phenotype, we present here microscope images for each of the four categorical phenotypes. To generate these images, the tested cells were first stained with Gram's iodine and then observed and photomicrographed using a Leica fluorescence microscope. All images illustrated in this paper were printed at the same magnification.
2.3. Marker development and genotyping
We first searched the genome sequence of yeast S. cerevisiae (http://www.yeastgenome.org/) for short tandem repeats (STR) within the genome by making use of the computer software Tandem Repeats Finder 3.21.17 We tested 561 evenly distributed STR sequences from the search as candidate markers and confirmed 264 STR loci that exhibited polymorphism between the two parental strains. These polymorphic markers were used as the basic marker set in the primary mapping experiment. Primer sequences for amplifying these STR markers were detailed in our previous study together with the experimental protocols for collecting and analysing STR genotype data.15 Beyond these STR markers, we developed a second set of single nucleotide polymorphism (SNP) markers to be used in the second stage of fine scale mapping of cell aggregation QTL by directly sequencing the relevant DNA regions. This added other 25 SNP markers in the QTL regions inferred from the primary QTL mapping (see marker locations and primer sequences in Supplementary Table S2).
2.4. Gene knockout, gene replacement, and nucleotide-specific mutagenesis
We implemented the standard polymerase chain reaction (PCR)-based gene disruption techniques to knock out the candidate genes.18,19 Single- and dual-knockout strains were constructed by replacing the target genes with the anti-hygromycin and anti-nourseothricin genes. The triple knockout strain was selected from offspring segregants from crossing of the dual-and single-knockout parental strains. All knockout strains obtained in the present study were confirmed by PCR experiments. The primer sequences for these PCRs are available upon request from the corresponding authors.
To perform the allelic exchange experiments and nucleotide-directed mutagenesis, we replaced the allele in situ with the replacement modules comprising the counterpart alleles and the Zeocin resistance gene as a dominant marker (detailed in Supplementary Figs S7 and S8).
2.5. Preparation of RNA samples and RT-qPCR analysis
Cells from a tested strain were cultured in 50 ml YPD liquid medium until reaching OD600 = 0.8. Total RNA was isolated using the hot phenol protocol,20 purified with RNase-free DNase (Promega), and subjected to first-strand cDNA synthesis with SuperScriptTM III Reverse transcriptase (Invitrogen). One microlitre of the single-strand cDNA after 10-fold dilution was used as template for real-time quantitative PCR (RT-qPCR) with SYBR-green (Toyobo) and ACT1 as the internal control. Every tested strain was independently cultured three times to gain three independent samples, and each of the samples was assayed for three repeated times of qPCR. The oligonucleotide primers for the three genes assayed were RGA1f: 5′-CGGTACAGTCACTGGAATCGAA-3′, RGA1r: 5′-ACGAATCAAACCTGGAGCAAA-3′;AMN1f: 5′-AGGCAAATACACACAGGTGGAA-3′, AMN1r: 5′-TTCCCAAATACCGGCATCA-3′; and ACTINf: 5′-TCGTTCCAATTTACGCTGGTT-3′, ACTINr: 5′-CGGCCAAATCGATTCTCAA-3′) for RGA1, AMN1, and ACT1, respectively.
We first sequenced the FLO1 gene of the two parental strains, YL1C and YH1A, and found that there were only 44 bp in the FLO1 gene, which distinguish FLO1 from the paralogous genes FLO5 and FLO9 (Supplementary Fig. S6). The sense primer of qPCR was designed across the 44-bp specific region, whereas the antisense primer on a non-specific region in 87-bp downstream. The respective specific sequences were ‘GCCTCATCGCTATATGTTTTTGG’ and ‘GCTGGTAAGCACGCCTCTGT’. The PCR product was sequenced and its specificity was confirmed.
2.6. Preparation of protein samples and western blotting assay
We followed the PCR-based epitope tagging method to insert 6His into the C-terminal of the target protein as an epitope.21 Proteins were extracted using the protocol described by Adams et al.16 Western blotting was performed using the standard laboratory procedures with monoclonal antibody specific for 6His (Univ. Co, China).
2.7. Yeast two-hybrid analysis
Yeast two-hybrid assay in the present study was carried out according to the manufacturer's manual (Clontech, CA USA). Three alleles of AMN1 (i.e. AMN1YH1A, AMN1YL1C, and AMN1V368D) were cloned independently into the vector pGADT7 (GAD), whereas TEM1 was inserted into the vector pGBKT7 (GBK). These vector constructs were confirmed by sequencing before being introduced into the reporter strains AH109 and Y187. These strains were then screened for positive co-transformation of the paired plasmids through growing them on the SC-Trp-Leu plates, synthetic complete medium lacking tryptophan and leucine. Positive interactions between AMN1 and TEM1 were detected as successful growth of the co-transformed strains on high-stringency SC-Trp-Leu-His plates (synthetic complete medium lacking tryptophan, leucine, and histidine) in the AH109 system.22 The positive interaction was also confirmed independently through the quantitative β-galactosidase assay in the Y187 system.23
2.8. Data analyses
We implemented a Bayesian approach based on Gibbs sampling as proposed in Sorenson et al.24 to estimate additive genetic variance component and, in turn, heritability of the multiple categorical phenotype of cell aggregation. In the statistical analysis, yi, phenotype of the ith segregant, is determined by a simple liability model 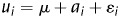 , i.e. yi = j (1 ≤ j ≤ 4), if
, i.e. yi = j (1 ≤ j ≤ 4), if 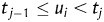 . In the liability model, μ is the population mean,
. In the liability model, μ is the population mean, 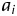 is a random variable of additive genetic effect of the segregant on its phenotype, and
is a random variable of additive genetic effect of the segregant on its phenotype, and 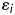 is the residual or environmental random variable. We assumed that the random variables,
is the residual or environmental random variable. We assumed that the random variables, 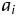 and
and 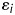 , follow independently a normal distribution with mean zero and variance
, follow independently a normal distribution with mean zero and variance 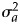 and a standard normal distribution, respectively. The threshold parameters satisfy
and a standard normal distribution, respectively. The threshold parameters satisfy 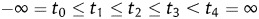 . We implemented the algorithm developed by Dr Zhaobang Zeng at North Carolina State University for carrying out multiple-interval mapping of QTL affecting the multiple categorical phenotype of cell aggregation.25
. We implemented the algorithm developed by Dr Zhaobang Zeng at North Carolina State University for carrying out multiple-interval mapping of QTL affecting the multiple categorical phenotype of cell aggregation.25
3. Results
3.1. Characterizing cell aggregation of yeast
We first established a reliable and feasible method to quantify the aggregation phenotype, which was essential for a quantitative genetic analysis and further functional analysis with the cellular behaviour character. The left panel in Fig. 1a shows sediment status of cells from the clumpy and non-clumpy parental strains YL1C and YH1A and their four representative segregants at three sediment time points, 0, 1.0, and 2.5 h. Thus, cell aggregation was, here, scored as a categorical quantitative trait, with the two parental strains, YH1A and YL1C, having phenotype categories 1 and 3, respectively and their offspring being scored as one of the four possible categories. In particular, cells of the segregant four sunk immediately in the clumping buffer (0 h) and the segregant was scored as category 4, a phenotype outside of the range of its two parents, showing transgressive segregation of the complex cell behaviour character. This method of categorizing the clumping character coordinates very well with that of the microscope views of the cell flocs, indicating the feasibility and reliability of the phenotyping assay.
3.2. Mapping and dissection of cell aggregation QTL
From crossing the parental strains, YH1A and YL1C, that are highly divergent in cell aggregation phenotype, we created a full-sibling population comprising 292 segregants. We scored the segregants for their clumping phenotype and genotyped them at 264 segregating simple sequence repeat (SSR) markers. Based on the phenotype and marker genotype data, we implemented the multiple-interval mapping method25 for a preliminary genome-wide scan for QTL affecting the multiple categorical phenotype of cell aggregation. The analysis detected four significant QTLs that mapped on the yeast chromosomes I, II, V, and XV, respectively. To narrow down these QTL regions, we genotyped the segregants at 1, 9, 4, and 11 further SNP markers in the 3 QTL regions, respectively and reran the multiple-interval mapping analysis. Because the chromosome I and V QTLs contain FLO1 and FLO8, respectively (Supplementary Fig. S1), two previously known genes involved in flocculation in S. cerevisiae,5,26,27 our focus here was on the other two QTLs on the yeast chromosomes II and XV. Figure 1b shows the likelihood profiles for the two major QTLs together with physical maps of candidate genes within the QTL regions. It can be seen that the fine mapping enables to resolve the chromosome II QTL into six candidate genes and the chromosome XV QTL into seven candidates.
3.3. Major-effect genes for cell aggregation and causative nucleotide in chromosome II QTL
Among the six candidate genes within the chromosome II QTL (Fig. 1b), three (RIB7, RPB5, and CNS1) are essential genes. We first knocked out each of the remaining three (SLI15, ICS2, and AMN1) in both the parental strains, YH1A and YL1C, and then evaluated the knockout strains for their clumping phenotype. We found that phenotype of the clumpy parental strain, YL1C, was dramatically altered after its AMN1 gene was knocked out (Fig. 2a). To further consolidate the candidacy of AMN1 as the QTL gene, we carried out an allelic exchange experiment, in which the AMN1 allele of the YL1C strain was replaced with that of the YH1A strain and vice versa. The aggregation phenotype of YL1C was clearly reduced after its AMN1 allele was replaced by that of YH1A (Fig. 2b). Moreover, the non-clumpy strain, YH1A, showed cell aggregation when its AMN1 allele was replaced by that from YL1C (Fig. 2b). These findings provide the strong evidence supporting that AMN1 is indeed the major clumping QTL gene. It should be noted that effects of replacing AMN1 alleles are not reciprocally constant between the two parental strains.
Figure 2.

Cell aggregation phenotype of engineered parent strains across QTL genes. Cell aggregation phenotype that was scored by the sedimentary assay (the left panel) and microscope view (the right panel) for the two parental strains and their genetically modified strains through knock out or allele replacement. Red horizontal line in the left panel marks the top boundary of cell sediment. (a) The three QTL genes (AMN1, FLO8, and RGA1) were knocked out in the clumpy parent YL1C. ‘Δ’ represents deletion, i.g. ‘amn1Δ’ for deletion AMN1. (b) The alleles of three QTL genes were replaced in situ by their counterpart alleles between the parents. The superscripts ‘A’ and ‘C’ refer to the YH1A-and YL1C-derived allele, respectively, i.g. AMN1A represents the allele of AMN1 derived from YH1A.
We sequenced the AMN1 alleles from the parental and several other laboratory strains and found three polymorphic sites within the Amn1 (Fig. 3a). Meanwhile, we assayed their expression at both transcriptional and translational levels through RT-qPCR and western blotting assays. Although the AMN1 gene is expressed significantly differentially between the two parental strains at both the mRNA and protein levels (Fig. 3c), the polymorphisms in the open reading frame (ORF) are sufficient to lead to phenotypic variation according to the results of allelic exchange (Fig. 2b).
Figure 3.

Genetic variation of QTL genes between two parental strains. Partial amino acid sequences of the cell aggregation QTL genes AMN1 (a) and RGA1 (b) with amino acid substitutions highlighted. The amino acid sequence in other yeast species is shown under the polymorphic residues when available: Saccharomyces paradoxus, Saccharomyces mikatae, Saccharomyces bayanus, Saccharomyces kudriavzevii, Saccharomyces kluyveri, Candida glabrata, Pichia angusta, Yarrowia lipolytica, and Kluyveromyces lactis. (c) Comparison in mRNA and protein expression of AMN1 between the two parental strains, YH1A and YL1C.
We further used site-directed mutagenesis to identify the causative quantitative trait nucleotide(s) in the QTL gene AMN1 by replacing the AMN1 gene of the YL1C strain with three different DNA segments of 2150 bp in length from the strain YH1A, accordingly. The segments covered the whole coding sequence of the AMN1 gene with the YH1A origin, but differed only at each of the three mutant nucleotides. We assayed these genetically modified strains for their clumping phenotype and found that only the strain that carried the V368D substitution in the AMN1 gene could recover the phenotype of YL1C (Supplementary Fig. S2a), strongly supporting the mutation as the causative quantitative trait nucleotide.
3.4. V368D variation determined the cell aggregation phenotype through the MEN pathway
It has been reported that AMN1 plays a key role as a daughter-specific switch that helps cells exit from the mitotic exit and reset the yeast cell cycle through binding to Tem1, a Ras-related GTPase. The TEM1 gene activates a signal transduction pathway, mitotic exit network (MEN) to allow mitotic exit.28 Based on this observation, we performed the yeast two-hybrid experiment in a standard strain AH109 and demonstrated that only Amn1YL1C and Amn1V368D can bind to Tem1 effectively (Fig. 4a). The quantitative β-galactosidase analysis yielded the results that were highly consistent with the two-hybrid analysis (Supplementary Fig. S3). Furthermore, we compared expression of the downstream genes DSE1, DSE2, and SCW11 between YL1C and YH1A, using the Affymetrix microarray hybridization data described previously in Wang et al.29 These genes were reported to underlie the cell wall degradation and daughter cell separation,30 and the expressions could be activated by Ace2, the transcriptional factor regulated by the MEN pathway.31 Our study shows a significantly lowered level of expression of these genes in YL1C than in YH1A (Fig. 4b), suggesting that the transcription of DSE1, DSE2, and SCW11 would be inhibited in the clumpy parent strain, YL1C. Then, we constructed two strains both derived from YL1C, which differed only at the 368th amino acid residue of Amn1. The expression of these downstream genes DSE1, DSE2, and SCW11 exhibited significant difference between the strains (Fig. 4c). These observations support the hypothesis that the V368D substitution in the Amn1 of the clumpy parent YL1C enhanced its binding to Tem1 and, in turn, blocked the MEN pathway, reduced transcription of the downstream genes DSE1, DSE2, and SCW11, and finally resulted in inhibition of daughter cells' separation and formation of clumps (Fig. 4d).
Figure 4.

Functional verification of AMN1 underlying yeast cell aggregation. (a) Yeast two-hybrid assay on interaction between the AMN1 and TEM1 genes. (b) Expressional assay of the three downstream genes, DSE1, DSE2, and SCW11, from the two parental strains. (c) The microscopic view and the expression assay of DSE1, DSE2, and SCW11 between two strains derived from YL1C, only differing in the 368th amino acid residue of Amn1. (d) A hypothetical model on how the single nucleotide substitution (V368D) of Amn1 affects the MEN signal transduction pathway, which, in turn, determines daughter cell separation and formation of cell clumps.
3.5. RGA1 as the chromosome XV clumping QTL gene
To dissect the chromosome XV QTL, we screened for the candidate(s) that responded to knockout and gene replacement treatments. Of the seven ORFs within the QTL, only when the RGA1 gene of the clumpy strain YL1C was silenced through knockout, its clumping phenotype was visibly altered (Fig. 2a). In addition, the phenotype was also changed significantly when its RGA1 allele was replaced with that from YH1A (Fig. 2b). These observations strongly support the candidacy of RGA1 as another major QTL gene. We compared the Rga1 sequence between the two parental strains and detected four polymorphic sites (Fig. 3b). We failed to find a single nucleotide substitution that could explain the observed change in the phenotype, although we found that the third polymorphic site in the gene had a significant causative effect on invasive growth of the yeast cells (Supplementary Fig. S4). Furthermore, we compared, but did not detect any significant change in expression of the RGA1 alleles between the two parental strains at either the transcriptional or translational level (Supplementary Fig. S5).
Rga1, a member of the Cdc42 GTPase-activating protein family, has been reported as an apical and isotropic switch in controlling the budding pattern of yeast cells by influencing hydrolytic status.32,33 In the presence of normally functional RGA1, Cdc42-GTP hydrolyzes and becomes Cdc42-GDP, leading to axial budding of cells that shows round cell morphology. The cells show bipolar budding and elongated morphology when their RGA1 is silenced. However, it should be stressed that effect of the RGA1 on cell budding and morphology would be only partial because there exist other family members such as RGA2 and BEM3. Their presence may compensate for defects of the silent RGA1.34 In light of the observation, we examined joint effects of the two verified QTL genes, RGA1 and AMN1, on cell aggregation. Figure 5 illustrates sequential influences of the two major clumping QTL genes, RGA1 and AMN1, on budding pattern, cellular morphology, and cell separation of the clumpy strain YL1C. When the AMN1 gene alone was knocked out whereas RGA1 remained normal, the strain showed cell separation and largely round shape. When only RGA1 was silenced whereas AMN1 functioned normally, daughter cells of the strain were inhibited to separate from mother cells. Some of the cells then displayed bipolar budding and apical attachment between mother–daughter cells in elongated shape, resulting in smaller clumps, and the presence of strings comprises a few apically linked cells. Whenever both the genes were knocked out in the clumpy strain, cells were promoted to bud in bipolarization, and inhibition of cell separation was released, leading to formation of bar-like cell clumps. These explain why single-knockout strain of AMN1 has a more defected phenotype than dual-knockout strain.
Figure 5.

Diagrammatic illustration of sequential effects of the two underlying QTL genes, AMN1 and RGA1. The two genes affect yeast cell aggregation through controlling cell separation and budding pattern, respectively. The black line indicates the effect of AMN1 on the aggregation trait and the blue the effect of RGA1 on the trait. The solid (or dotted) lines specify the genetic effects of the QTL genes in a normal (or dysfunctional) form.
3.6. FLO1, FLO8, and transgressive segregation
Our QTL analysis detected two significant QTL regions containing the two genes relevant to flocculation, FLO1 and FLO8. To test their candidacy as major cell aggregation genes, we first compared sequence of the two genes between the two parental strains, YH1A and YL1C, and found that the FLO1 allele in the clumpy strain YL1C had eight fewer tandem repeats of an intragenic domain than those in the non-clumpy parent YH1A. This disagrees with the previous observations that the degree of cell flocculation was found to be proportional to the number of tandem repeats of the intragenic domain structure.1,35 Comparison of genic sequence of FLO8 between the two parental strains revealed a nonsense substitution at the 425th nucleotide in YH1A, consistent with the previous study.26 The mutation creates a stop codon (TAG) and results in a truncated protein. The defective FLO8 in YH1A cannot activate expression of its FLO1, thus explaining why the FLO1 gene did not lead to stronger flocculation as previously expected, although the non-clumpy strain carried the allele enhancing flocculation phenotype.
We knocked out the FLO1 and FLO8 genes individually in YH1A and YL1C and observed that the clumping phenotype of YL1C was markedly altered after either FLO1 or FLO8 gene was silenced (Supplementary Fig. S2b). Moreover, we replaced the FLO8 allele in YL1C with the YH1A-derived counterpart and observed a similar phenotype to the FLO1 or FLO8 deletion strain (Supplementary Fig. S2b). The data showed that genetic variation of both the FLO genes contributed significantly to the clumpy phenotype. We attempted but failed to replace the FLO1 gene in the same way as reported above. This can be attributed to the difficulties arising from the location of the FLO1 gene at the chromosomal end with rich heterochromatin, thus keeping the gene in a protected status from genetic modification. Secondly, the FLO1 gene has an extremely high level of homology with its family members FLO5 and FLO9, making it extremely difficult to ensure specificity in replacing the target gene.
Although it was highly technically challenging to profile expression of the FLO1 gene due to its extremely high level of sequence homology with other FLO family members FLO5 and FLO9, we succeeded in assaying expression of the FLO gene in the two parental strains through an FLO1-specific RT-qPCR experiment (see Supplementary Fig. S6 for detail). We analysed the FLO1 and FLO8 expression levels of the parental strains and their eight offspring individuals with two different genotypes at the QTL genes and varying phenotypes. FLO8 expression showed no difference in the tested strains. Moreover, a highly expressed FLO1 gene was common for the strong cell clumping phenotype (the phenotype categories 3 and 4) but was certainly not an indicator of clumpiness because the gene was also observed to be highly expressed in the non-clumpy strains (those with phenotype category 1) (Fig. 6a).
Figure 6.
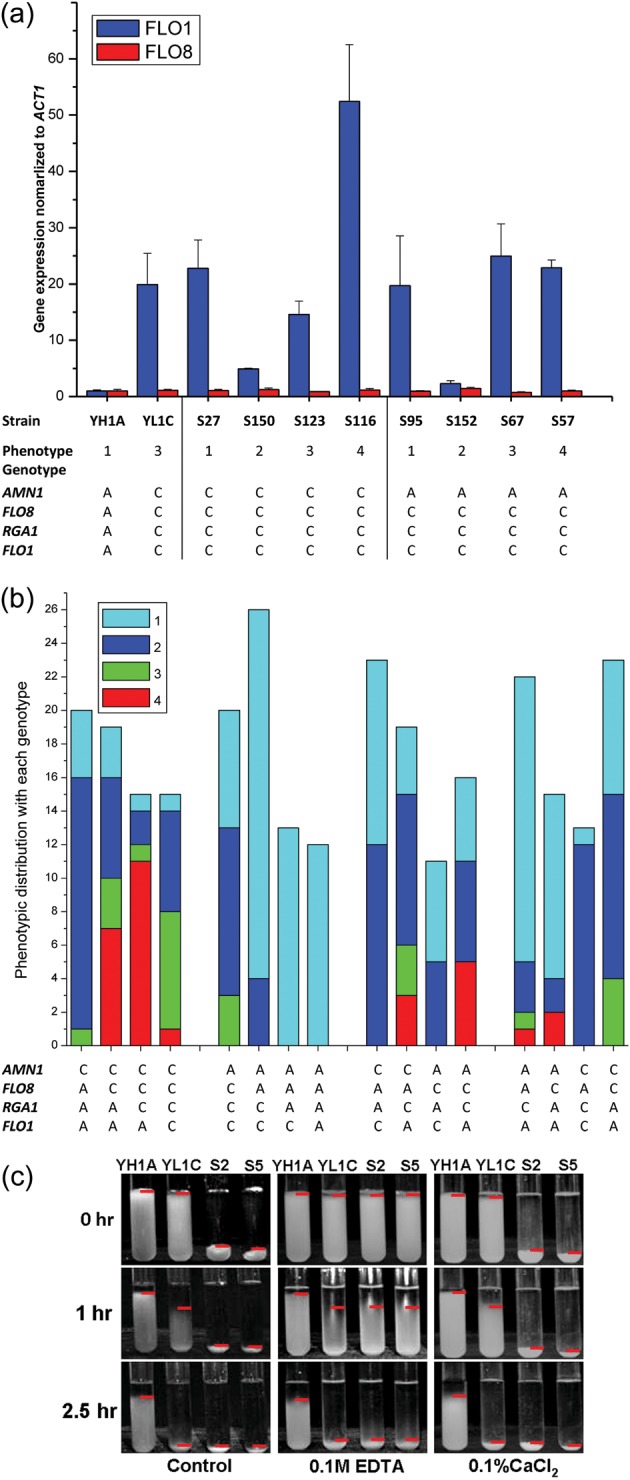
Genetic interactions of the QTL genes. ‘A’ and ‘C’ in (a) and (b) refer to as the YH1A- and YL1C-derived alleles, respectively. (a) Expression of FLO1 and FLO8 in two parental strains and their two groups of offspring segregants. The genotype of each strain at the four QTL genes is shown below, accordingly. (b) Distribution of segregants with different cell aggregation phenotypes (1–4) within each of genotypes at the four QTL genes. Each column represents a group of individuals shared with the same genotype at the four QTL genes. (c) Cell aggregation phenotype of the two parental strains and their two selected offspring segregants (S2 and S5) after different treatments of ethylenediaminetetraacetic acid and CaCl2. Panel 1(control) corresponds to where cells of the tested strains were scored without being treated, panel 2 (0.1 M ethylenediaminetetraacetic acid) to where the cells were washed twice with 0.1 M ethylenediaminetetraacetic acid and other two times with sterile water before being tested, and panel 3 (0.1% CaCl2) to where the cells were treated in the same way as those in panel 2, but tested in the flocculation buffer with 0.1% CaCl2 added. Red horizontal line represents the top boundary of cell sediment.
Intriguingly, 30 extremely clumpy segregants (T = 0) exhibited more strongly clumpy than parental strains, which is termed as transgressive segregation. To explore how the QTL genes affect the transgressive segregation, we first grouped the 292 segregants into 4 panels of 16 groups according to their allelic origin or joint genotype at the 4 QTL genes. Figure 6b illustrates distributions of the segregants with different phenotypes (1–4) within each of the four-gene genotypes and shows that the segregants receiving the four QTL genes from the clumpy parent, YL1C, have a much lower probability of expressing extreme clumpy phenotype (phenotypic category 4) than those with a YH1A-derived allele of FLO1, indicating a dispersion distribution of cell aggregation causing alleles at the QTL genes between the two yeast parental strains and thus explaining the transgressive segregation observed in the segregant population. Moreover, the phenotypic effect of the FLO1 allele was highly background dependent. This FLO1 allele would not cause the clumping phenotype in the absence of the other three QTL genes from the clumpy parent YL1C (group 7 vs. group 3 in Fig. 6b), but plays the role of clumping enhancer.
Given the fact that the flocculins leading to cell flocculation share a common three-domain structure and require Ca2+ ions to stabilize the structure,9 we explored the effect of Ca2+ ions on clumping phenotypes of the two parental strains and their two segregants (S2 and S5) that showed the transgressive phenotype. Figure 6c shows that the cell aggregation phenotype of the strains S2 and S5 was reduced to the extent of YL1C when Ca2+ ions were chelated with the ethylenediaminetetraacetic acid treatment (panel 2), but recovered when the ions were added as CaCl2 in the clumping buffer, suggesting that the transgressive phenotype can be attributed to FLO gene family. This assumption was confirmed by the further knockout experiments in which the transgressive phenotype was disrupted when either FLO1 or FLO8 was silenced in S2 and S5 strains (Supplementary Fig. S2b). Strikingly, the treatment did not result in altered phenotype of the clumpy parental strain (YL1C). This together with the fact that this parent carried the non-clumpy FLO1 allele indicates that the phenotypic response to the Ca2+ treatment actually reflects response of the Ca2+-dependent cell aggregation induced by the cell wall protein, which differs from that mediated by AMN1.
We assessed relative contribution of the QTL genes to phenotypic variation in cell aggregation and found that genetic variation in AMN1 explained 25% of the phenotypic variation, whereas the other three QTL genes FLO8, RGA1, and FLO1, respectively, explained 17, 5, and 11% of the variation. Variation between aggregative genotypes at all the four genes explained 46% of the phenotypic variation. To further explore the joint and interactive effects of the genes, we generated three dual-knockout strains (YL1C-amn1Δ&flo8Δ, YL1C-amn1Δ&rga1Δ, and YL1C-flo8Δ&rga1Δ) and one triple-deletion strain (YL1C-amn1Δ&flo8Δ&rga1Δ). Phenotypes of these engineered strains are shown together with other single QTL gene knockout strains and the wild-type strain in Fig. 2. Although the degree of cell aggregation of the single-gene knockout strains was further reduced when they were knocked out for additional one or two of the QTL genes, it is clear that different configurations of the knockout QTL genes have led to different degrees of clumpiness. This, together with the observation illustrated in Fig. 6b, reveals that the QTL genes show clearly epistatic effects on cell aggregation.
4. Discussion
The genetic architecture underlying a quantitative trait is determined by the number of genes that contribute to their additive and interactive effects on variation of the trait phenotype.36 Although many genomes have been fully sequenced and the number of genes with annotated function increases rapidly in the era of functional genomics, it still remains a great challenge to reconstruct the architecture with a full insight of the genetic parameters. Budding yeast, the single-cell eukaryote, but with probably the richest structural and functional information of the genome, provides an ideal model for dissecting the molecular basis of genetic variation in typical quantitative traits such as high-temperature growth12 and sporulation efficiency37 and in genetic control of transcriptome abundance.38 The present study extends these successful examples by dissecting multiple-threshold phenotypic variation of yeast cell aggregation into a genic level.
Several genes have been reported previously in the literature to control clumping phenotype variation in budding yeast. Of these, the most prominent are the FLO gene family responsible for many cellular adhesion phenotypes, including flocculation.10 In sharp contrast, the present study reveals the two novel genes, AMN1 and RGA1, in addition to previously identified two genes, FLO1 and FLO8, that are significant determinants for the phenotype. Success of the present study in resolving the mapped QTLs into the underlying QTL genes can be attributed to that the major QTLs have been mapped at the high resolution by which direct gene targeting becomes possible. We noted that the simulation study by Li et al. predicted a much larger confidence interval for mapping categorical traits from a simulated backcross population with 200 individuals genotyped at genetic markers of every 10 cM. When compared with the simulation study, the present analysis has employed a substantially larger sample size (292 segregants) and higher density of markers in the QTL regions. These together with the fact that the budding yeast has a highly recombiogenic and compact genome paved the basis for the mapping resolution we have achieved.
The combined effect of these QTL genes explains 46% of the overall phenotypic variation, leaving the remaining 54% of the variation to be residual. The residual variation can be explained as two parts as well as their interaction, the genetic component whose underlying genetic effects are too small to be detected by the experiment, and the component contributed by environmental modification to the quantitative trait. In addition, we demonstrated that the QTL genes exhibited clear epistatic effects on the aggregation trait. These show that the underlying mechanism of yeast cell aggregation is more complicated than expected in the previous literature.
Of these QTL genes, AMN1 plays the leading role in regulating cell aggregation. We provide strong and direct experimental evidence that supports this is through the genes modulating the mitotic exit network in S. cerevisiae.28 We have identified the AMN1 as a major QTL gene of cell aggregation through a genome-wide QTL scan in a natural population and provided here the direct experimental evidence for the V368D substitution as the causative genetic variant. Our results also strongly indicate the putative domain of AMN1, and the functional significance of V368D, which has been recently predicted as a superfamily being highly conserved (e-value of 6.38e-96) across species.39 On the other hand, the RGA1 gene, a member of the GTPase-activating family, is first identified in the present study as a major gene affecting cellular clumping in budding yeast. This gene is required for yeast cells to switch from the apical to isotropic growth phase.40 The present study creates an opportunity to explore the joint effect of this cell aggregation QTL gene, a budding pattern controller, and another QTL gene AMN1, also a mitotic exit regulator. Genetic modification to these genes through gene knockouts disperses the well-known grape-like strings of cells (Fig. 5).
In addition to the above two major genetic factors, FLO1 and FLO8 were also detected as the QTL genes through the present forward genetic analysis. Highly expressed FLO1, a cell surface adhesion regulator, has been widely recognized as a biomarker for cell flocculation in many candidate-based analysis. However, we present here the role of the gene, as merely one of the building elements in the genetic architecture underlying the complex phenotype, and show that FLO1 is highly background dependent in determining the phenotype of its carrier. Unlike what has been widely accepted in the literature of yeast cell flocculation, expressed FLO1 does not necessarily confer a clumping phenotype to its carriers. We demonstrate that the FLO1 allele from a non-clumpy parental strain, in the presence of the other three QTL genes from a clumpy parental strain, creates the genotype for the clumping phenotype that substantially surpasses performance of both the parents, i.e. transgressive segregation of the QTL genes. Taking all these together, the QTL genes unveiled in the present study have been shown to be involved in multiple pathways that govern the cellular development (RGA1), the behaviour of daughter cells (AMN1), and the cell-surface adherence property (FLO1 and FLO8). In contrast to the studies in which biological functions and their phenotypic significance of these QTL genes were examined either independently or jointly, the present study unveils the QTL genes as major determinants of the clumping phenotype in a systematic way. This clearly indicates the power and appropriateness of the forward genetic approach employed in the present study in uncovering the genetic basis of an aggregate polygenic trait such as cellular clumping that involves multiple constituent components.
The evolutionary significance of cellular clumping has recently been highlighted in the single-cell species because the clumping phenotype can protect inner individuals in cell clumps from biotic or/and abiotic stresses such as alcohol, anti-microbials, and physical attacks. The FLO1 gene is recognized as a ‘green beard gene’ because its induced expression in a non-flocculent laboratory strain, S288C, can restore its cells to flocculate, and the cellular flocs, thus, generated provide a cooperative shield to protect from stresses in the living environment.1 Our data demonstrate that expression of FLO1 may not be the biomarker for a clumping phenotype, although an unexpressed FLO1 allele from the non-clumpy strain, which is isogenic to S228C, may enhance clumping of its offspring strains carrying other major clumping genes. In the polygenic system uncovered here, AMN1 is found to be a more important genetic determinant than FLO1 in controlling expression of the clumping phenotype, and this endogenously expressed gene may make a more appropriate target for investigating the molecular mechanism underlying the ‘green beard’ effect.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by the National Basic Research Program of China (2012CB316505) and National Natural Science Foundation of China (91231114, 31271335).
Supplementary Material
Acknowledgements
We thank Prof. M.J. Kearsey and Dr Julia Lodge for their comments on an earlier version of this paper. In particular, we thank two anonymous reviewers for their constructively critical comments and suggestions that have helped improve clarity and presentation of this paper.
Footnotes
Edited by Dr Katsumi Isono
References
- 1.Smukalla S., Caldara M., Pochet N., et al. FLO1 is a variable green beard gene that drives biofilm-like cooperation in budding yeast. Cell. 2008;135:726–37. doi: 10.1016/j.cell.2008.09.037. doi:10.1016/S0083-6729(03)01002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gimeno C.J., Ljungdahl P.O., Styles C.A., Fink G.R. Unipolar cell divisions in the yeast S. cerevisiae lead to flamentous growth: regulation by starvation and RAS. Cell. 1992;68:1077–90. doi: 10.1016/0092-8674(92)90079-r. doi:10.1681/ASN.2006090975. [DOI] [PubMed] [Google Scholar]
- 3.Cullen P.J., Sprague G.F. Glucose depletion causes haploid invasive growth in yeast. Proc. Natl. Acad. Sci. USA. 2000;97:13461–3. doi: 10.1073/pnas.240345197. doi:10.1210/jc.2003-032149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pretorius I.S. Tailoring wine yeast for the new millennium: novel approaches to the ancient art of winemaking. Yeast. 2000;16:675–729. doi: 10.1002/1097-0061(20000615)16:8<675::AID-YEA585>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 5.Teunissen A.W., van den Berg J.A., Steensma H.Y. Localization of the dominant flocculation genes FLO5 and FLO8 of Saccharomyces cerevisiae. Yeast. 1995;30:735–45. doi: 10.1002/yea.320110805. doi:10.1159/000318776. [DOI] [PubMed] [Google Scholar]
- 6.Guo B., Styles C.A., Feng Q.H., Fink G.R. A Saccharomyces gene family involved in invasive growth, cell-cell adhesion, and mating. Proc. Natl. Acad. Sci. USA. 2000;97:12158–63. doi: 10.1073/pnas.220420397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi O., Suda H., Ohtani T., Sone H. Molecular cloning and analysis of the dominant flocculation gene FLO8 from Saccharomyces cerevisiae. Mol. Gen. Genet. 1996;251:707–15. doi: 10.1007/BF02174120. [DOI] [PubMed] [Google Scholar]
- 8.Rupp S., Summers E., Lo H.J., Madhani H., Fink G. MAP kinase and cAMP filamentation signaling pathways converge on the unusually large promoter of the yeast FLO11 gene. EMBO J. 1999;18:1257–69. doi: 10.1093/emboj/18.5.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verstrepen K.J., Reynolds T.B., Fink G.R. Origins of variation in the fungal cell surface. Nat. Rev. Microbiol. 2004;2:533–40. doi: 10.1038/nrmicro927. [DOI] [PubMed] [Google Scholar]
- 10.Verstrepen K.J., Klis F.M. Flocculation, adhesion and biofilm formation in yeasts. Mol. Microbiol. 2006;60:5–15. doi: 10.1111/j.1365-2958.2006.05072.x. [DOI] [PubMed] [Google Scholar]
- 11.Mackay T.F. The genetic architecture of quantitative traits. Annu. Rev. Genet. 2001;35:303–39. doi: 10.1146/annurev.genet.35.102401.090633. [DOI] [PubMed] [Google Scholar]
- 12.Steinmetz L.M., Sinha H., Richards D.R., et al. Dissecting the architecture of a quantitative trait locus in yeast. Nature. 2002;416:326–30. doi: 10.1038/416326a. [DOI] [PubMed] [Google Scholar]
- 13.Tabor H.K., Risch N.J., Myers R.M. Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat. Rev. Genet. 2002;3:391–7. doi: 10.1038/nrg796. [DOI] [PubMed] [Google Scholar]
- 14.Mehrabian M., Allayee H., Stockton J., et al. Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat. Genet. 2005;37:1224–33. doi: 10.1038/ng1619. [DOI] [PubMed] [Google Scholar]
- 15.Hu X.H., Wang M.H., Tan T., et al. Genetic dissection of ethanol tolerance in the budding yeast Saccharomyces cerevisiae. Genetics. 2007;175:1479–87. doi: 10.1534/genetics.106.065292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams A., Gottschling D.E., Kaiser C.A., Stearns T. Methods in Yeast Genetics. A Cold Spring Harbor Laboratory Course Manual. New York: Cold Spring Harbor Laboratory Press:; 1998. [Google Scholar]
- 17.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baudin A., Ozier-Kalogeropoulos O., Denouel A., Lacroute F., Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:3329–30. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorenz M.C., Muir R.S., Lim E., McElver J., Weber S.C., Heitman J. Gene disruption with PCR products in Saccharomyces cerevisiae. Gene. 1995;158:113–7. doi: 10.1016/0378-1119(95)00144-u. [DOI] [PubMed] [Google Scholar]
- 20.Schmitt M.E., Brown T.A., Trumpower B.L. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 1990;18:3091–2. doi: 10.1093/nar/18.10.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knop M., Siegers K., Pereira G., et al. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15:963–72. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 22.James P., Halladay J., Craig E.A. Genomic Libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–36. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harper J.W., Adami G.R., Wei N., Keyomarsi K., Elledge S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 24.Sorensen D.A., Anderson S., Gianola D., Korsgaard I. Bayesian inference in threshold models using Gibbs sampling. Genet. Sel. Evol. 1995;27:229–49. [Google Scholar]
- 25.Li J., Wang S., Zeng Z.B. Multiple-interval mapping for ordinal traits. Genetics. 2006;173:1649–63. doi: 10.1534/genetics.105.054619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu H., Styles C.A., Fink G.R. Saccharomyces cerevisiae S288C has a mutation in FLO8, a gene required for filamentous growth. Genetics. 1996;144:967–78. doi: 10.1093/genetics/144.3.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi O., Yoshimoto H., Sone H. Analysis of the genes activated by the FLO8 gene in Saccharomyces cerevisiae. Curr. Genet. 1999;36:256–61. doi: 10.1007/s002940050498. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y., Shirogane T., Liu D., Harper J.W., Elledge S.J. Exit from exit: resetting the cell cycle through Amn1 inhibition of G protein signaling. Cell. 2003;112:697–709. doi: 10.1016/s0092-8674(03)00121-1. [DOI] [PubMed] [Google Scholar]
- 29.Wang M., Hu X., Li G., et al. Robust detection and genotyping of single feature polymorphisms from gene expression data. PLoS Comput. Biol. 2009;5:e1000317. doi: 10.1371/journal.pcbi.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voth W.P., Yu Y., Takahata S., Kretschmann K.L., Lieb J.D. Forkhead proteins control the outcome of transcription factor binding by antiactivation. EMBO J. 2007;26:4324–34. doi: 10.1038/sj.emboj.7601859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lerner A., Chin T.E., Brent R. Yeast Cbk1 and Mob2 activate daughter specific genetic programs to induce asymmetric cell fates. Cell. 2001;107:739–50. doi: 10.1016/s0092-8674(01)00596-7. [DOI] [PubMed] [Google Scholar]
- 32.Caviston J.P., Longtine M., Pringle J.R., Bi E. The role of Cdc42p GTPase-activating proteins in assembly of the septin ring in yeast. Mol. Biol. Cell. 2003;14:4051–66. doi: 10.1091/mbc.E03-04-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tong Z., Gao X.D., Howell A.S., Bose I., Lew D.J., Bi E. Adjacent positioning of cellular structures enabled by a Cdc42 GTPase-activating protein–mediated zone of inhibition. J. Cell Biol. 2007;179:1375–84. doi: 10.1083/jcb.200705160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith G.R., Givan S.A., Cullen P., Sprague G.F., Jr GTPase-activating proteins for Cdc42. Eukaryot. Cell. 2002;1:469–80. doi: 10.1128/EC.1.3.469-480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verstrepen K.J., Jansen A., Lewitter F., Fink G.R. Intragenic tandem repeats generate functional variability. Nat. Genet. 2005;37:986–90. doi: 10.1038/ng1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lynch M., Walsh B. Genetics and Analysis of Quantitative Traits. Sunderland, MA, USA: Sinauer Associates; 1998. [Google Scholar]
- 37.Deutschbauer A.M., Davis R.W. Quantitative trait loci mapped to single-nucleotide resolution in yeast. Nat. Genet. 2005;37:1333–40. doi: 10.1038/ng1674. [DOI] [PubMed] [Google Scholar]
- 38.Brem R.B., Yvert G., Clinton R., Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–5. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- 39.Marchler-Bauer A., Lu S., Anderson J.B., et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:225–9. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saito K., Fujimura-Kamada K., Hanamatsu H., et al. Transbilayer phospholipid flipping regulates Cdc42p signaling during poparized cell growth via Rga GTPase-activating proteins. Dev. Cell. 2007;13:743–51. doi: 10.1016/j.devcel.2007.09.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.