

Abstract
Epoxy-fatty acids have been recognized as important cell signaling molecules with multiple biological effects including anti-nociception. The main degradation pathway of these signaling molecules is via the soluble epoxide hydrolase (sEH) enzyme. Inhibitors of sEH extend the anti-nociceptive effects of fatty acid epoxides. In this study two models of pain with different etiology, streptozocin induced type I diabetic neuropathic pain and lipopolysaccharide induced inflammatory pain were employed to test sEH inhibitors. A dose range of three sEH inhibitors with the same central pharmacophore but varying substituent moieties was used to investigate maximal anti-allodynic effects in these two models of pain. Inhibiting the sEH enzyme in these models successfully blocked pain related behavior in both models. The sEH inhibitors were more potent and more efficacious than celecoxib in reducing both diabetic neuropathic pain and lipopolysaccharide induced inflammatory pain. Because of their ability to block diabetic neuropathic pain sEH inhibition is a promising new approach to treat chronic pain conditions.
1. Introduction
Epoxy-fatty acids are endogenous lipid metabolites with important roles in cellular signaling which is underscored by their tight regulation (Bernstrom et al., 1992; Spector and Norris, 2007). These epoxy-metabolites are formed by cytochrome P450 enzymes acting on parent fatty acids released from cellular membranes by lipases including phospholipase A2 (Imig, 2012; Spector, 2009; Tomita-Yamaguchi et al., 1990). Epoxy-fatty acids undergo rapid enzymatic degradation by the soluble epoxide hydrolase (sEH, EPHX2 EC:3.3.2.10). The specific epoxy-fatty acids most frequently investigated are the epoxyeicosatrienoic acids. The epoxyeicosatrienoic acids are arachidonic acid metabolites with multiple biological activities including lowering blood pressure and attenuating inflammation and inflammatory pain (Imig et al., 2002; Inceoglu et al., 2006; Schmelzer et al., 2005). Epoxyeicosatrienoic acids are degraded by sEH into their corresponding diols the dihydroxyeicosatrienoic acids. Inhibitors of sEH have been used to stabilize epoxy-fatty acids, increasing their residence time and lowering dihydroxyeicosatrienoic acid levels both in vitro and in vivo (Spector, 2009). Early sEH inhibitors were successful in vitro but their formulation was problematic for use in vivo. The physical and chemical properties of the inhibitors have been systematically optimized (Morisseau et al., 1999; Morisseau et al., 2006; Shen and Hammock, 2012) and demonstrate improvements in bioefficacy (Hwang et al., 2007; Tsai et al., 2010). Direct application of epoxy-fatty acids including epoxyeicosatrienoic acids also mediates pain relief in rats (Inceoglu et al., 2006; Morisseau et al., 2010). Recently, results using a PGE2 induced pain model suggest a mode of action independent of their anti-inflammatory activity (Inceoglu et al., 2011). In addition, results of sEH inhibition in a model of diabetic neuropathic pain support the hypothesis that there is a mode of action independent of anti-inflammation (Inceoglu et al., 2008). These experiments revealed sEH inhibition was anti-hyperalgesic and equipotent to low dose morphine on thermal hyperalgesia in diabetic rats (Inceoglu et al., 2008). However, the extent of sEH inhibitor mediated anti-hyperalgesia and the efficacy of the inhibitors on neuropathic mechanical pain were both unknown. Here, we probed for maximum anti-nociceptive efficacy in two in vivo rat models. The nociceptive assays quantified mechanical allodynia, a pain associated with a stimulus that is normally innocuous and present in both models. Special attention is given to APAU which has investigational new drug status and has the possibility of being used in additional human clinical trials in the near future (Shen and Hammock, 2012). APAU is compared to the selective COX-2 inhibitor celecoxib in both a chronic diabetic neuropathic pain and an acute lipopolysaccharide induced inflammatory pain model. Then a dose range of three sEH inhibitors including APAU were compared in both models to test the hypothesis that sEH inhibitors dose dependently reduce both inflammatory and neuropathic allodynia. The sEH inhibitor mediated pain relief was tested with up to a 10 fold increase in dose compared to previous published data and evaluated for time dependent effects. In addition to examining maximum efficacy, these experiments add information about the possible mechanism of action of sEH inhibitors in induced pain states.
2. Materials and Methods
All experiments used groups of Sprague–Dawley male rats (250–300 g) purchased from Charles River Laboratories. The rats were allowed to habituate 3 days before the beginning of each experiment and housed under standard conditions (25°C) in a fixed 12-h light/dark cycle with ad libitum food and water. These experiments were performed in accordance with protocols approved by the University of California Davis Animal Use and Care Committee and with great care to minimize suffering of the animals.
2.1 Chemicals
The sEH inhibitors APAU: 1-(1-acetypiperidin-4-yl)-3-adamantanylurea; t-AUCB: trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid; and t-TUCB: trans-4-[4-(3-trifluoromethoxyphenyl-l-ureido)-cyclohexyloxy]-benzoic acid used for the experiments were synthesized and characterized in house as previously described (Hwang et al., 2007; Jones et al., 2006). The sEH inhibitor APAU is also referred to as AR9281 in published literature (Anandan et al., 2011; Imig and Hammock, 2009). The IC50 values for t–TUCB on recombinant rat and mouse sEH were determined per previously described methods (Wolf et al., 2006). Doses of sEH inhibitors were formulated in the polyethylene glycol PEG400 for the experiments. The highest doses of the inhibitors were first dissolved in a small amount of DMSO (10 % v/v final) to which was added PEG400. Both vehicle concentrations were tested in the nociceptive assay and oxylipin analysis and showed no statistically significant differences (data not shown). Celecoxib was purchased from (Fisher Scientific, USA) and formulated in PEG400 vehicle. Morphine sulfate was purchased (Fisher Scientific, USA) and diluted in saline.
2.2 Diabetic neuropathic pain model
Diabetic neuropathic pain was modeled using the antibiotic drug streptozocin which targets and kills the pancreatic beta islet cells rendering the animals with type I diabetes. The ensuing decrease in nociceptive thresholds develops within five days, persists the lifetime of the animal and was used as the model of diabetic neuropathic pain. Before induction of diabetes rats were acclimated for one hour and tested for baseline thresholds. Baseline mechanical withdrawal thresholds were established using the von Frey mechanical nociceptive test with an electronic anesthesiometer (IITC, Woodland Hills, CA). Subsequently, streptozocin (55 mg/kg) in saline was injected via tail vein per previously reported methods (Aley and Levine, 2001). After five days the allodynia of diabetic rats was confirmed. Rats that scored 65% or lower of the original pain free baseline were considered allodynic and included the presented groups.
2.3 Lipopolysaccharide induced inflammation model
On each test day the rats were acclimated for one hour and baseline mechanical withdrawal thresholds were measured with the von Frey anesthesiometer. One hour after pretreatment 10μg of lipopolysaccharide (Sigma, St. Louis MO) in 50μl saline was injected intraplantar in one hind paw of the rats (Kanaan et al., 1996). The mechanical withdrawal threshold of this hind paw was monitored for 6 h following the injection. For oral gavage administration, 3 mg/kg APAU in PEG400 was administered one hour prior to intraplantar injection of lipopolysaccharide.
2.4 Nociceptive and motor skill bioassays
An electronic von Frey aesthesiometer was used to quantify allodynia baselines on all test days. Rats were placed in clear acrylic chambers on a steel mesh floor. The hind paw of the rat was probed through the mesh with a rigid tip probe connected to an electronic readout pressure meter set to the maximum hold setting. The withdrawal thresholds per rat were measured 3–5 times at 1 minute intervals for each time point. For the diabetic neuropathy model, rats were injected s.c. with vehicle, celecoxib or sEH inhibitor and tested at 15min, 30min, 1, 2, 3, 4, 5, and 6 h post injection for mechanical withdrawal thresholds. For the inflammatory pain model rats were administered vehicle, celecoxib or sEH inhibitor 60 min prior to intraplantar lipopolysaccharide injection. These procedures were followed for 0.1, 0.3, 1, 3, 10, 30 and 100 mg/kg of APAU and t-TUCB and 1, 3, 10, 30 and 100 mg/kg of t-AUCB given this inhibitor was inactive at low dose in the assay. The presence of the disease state precluded pretreatment in the diabetic neuropathy model; therefore rats were tested at equivalent time intervals to the inflammatory pain model but immediately after sEH inhibitor administration. The reported scores are the grams of force required to elicit a hind paw withdrawal averaged with standard error of the mean (S.E.M.) per a group of rats tested on the same day under the same conditions. For the diabetic neuropathy model, the baseline scores were normalized to 100 percent to reflect the response to treatments which are reported as % of post diabetic neuropathic baseline. Therefore the diabetic neuropathy model scores can improve to over 100%. For the inflammatory pain model, the baseline scores are considered pain free and assigned 100%. The scores are group averages ± S.E.M. reported as percent of the baseline. Therefore the inflammatory pain model graphs reflect more painful responses as low percent scores with the object of returning toward 100% or more. For the rotorod test the rats were not pre-trained but tested for their baseline scores prior to vehicle or compound. Scores for each rat consisted of three consecutive trials per time point at an accelerating speed from 2–20 RPM on a 7.0 cm shaft diameter Rota-rod Treadmill (MedAssociates Inc., St. Albans, VT). A cutoff endpoint of 180 seconds was used for successful trials. The scores are the group average ± S.E.M. per time point.
2.5 Oxylipin and blood inhibitor analysis
The inflammatory pain model was used to investigate changes in plasma lipid metabolites after APAU treatment. 3 and 100 mg/kg of APAU were used to analyze sEH substrate and product levels as well as key prostaglandins compared to vehicle controls. For the oxylipin analysis, blood was sampled two hours post lipopolysaccharide or control intraplantar injection and centrifuged to obtain plasma. The plasma was extracted via solid phase extraction followed by LC/MS analysis as described previously (Yang et al., 2009). APAU concentration was measured using 10μl of whole blood sampled via tail vein puncture added to 50 μl distilled water. The samples were flash frozen, extracted and analyzed per previous methods (Liu et al., 2009).
2.6 Statistical Analysis
All results are expressed as mean ± S.E.M. Data were analyzed using SigmaPlot 11.0 for windows (Systat Software Inc., San Jose, CA). The applied statistical methods are specified in the figure legends with P values 0.05 considered significant.
3. Results
3.1 sEH inhibitors outperform efficacy of celecoxib in both pain models
The pain models used in these experiments differ, however, both models effectively induce allodynia which is quantifiable and of similar magnitude when measured with the von Frey assay. The diabetic neuropathy model is a chronic pain model designed to emulate the effects of nerve injury following prolonged hyperglycemia. The inflammatory pain model is an acute model that relies on the induction of the inflammatory cascade involving Toll-like receptors and the associated pain. In the diabetic neuropathy model the mean of the mechanical withdrawal thresholds for all groups (n=139) measured five days post streptozocin was 46.0 ± 1.0 S.E.M. grams of force. This decrease was 57% of the pre-diabetic average mechanical withdrawal threshold and was consistent with earlier findings (Aley and Levine, 2001). The PEG400 vehicle did not significantly change diabetic neuropathy thresholds over the entire time course (Fig. 1A). Lipopolysaccharide administration in vehicle control rats decreased the mean mechanical withdrawal thresholds from 85.0 grams to an average of 41.5 ±2.8 S.E.M. grams of force over the time course with the lowest average equaling 36.6% at 6 h. This average decrease was 48.8% of naïve baseline scores (Fig. 1B).
Fig. 1. The sEH inhibitor APAU is superior to celecoxib in both diabetic neuropathic and inflammatory pain models.

(A) A s.c. administration of celecoxib (10mg/kg) was statistically ineffective, APAU (10 mg/kg) significantly blocked the pain related behavior (Kruskal-Wallis One Way ANOVA on Ranks, P ≤0.001, n=6–11 /group). The converted pain free pre-diabetic baseline average for groups included in 1A was 182±20 % S.E.M. of the post diabetic neuropathic baseline. (B) Inflammatory pain was induced with a 10μg intraplantar injection of lipopolysaccharide into rat hind paw. Celecoxib (10 mg/kg) 1h prior to lipopolysaccharide while having a marginal effect at 3 h, was not significantly antihyperalgesic over the time course compared to vehicle controls. APAU (10 mg/kg) was highly significant against lipopolysaccharide induced allodynia over the time course (Kruskal-Wallis One Way ANOVA on Ranks, Dunn's method, P ≤0.001, n=6 /group).
While celecoxib has been shown to be ineffective in diabetic neuropathic pain, reports indicate that it is still used for this condition (Gore et al., 2007). We therefore examined it compared to sEH inhibitors in the diabetic neuropathy model. As expected, 10 mg/kg of celecoxib had no significant effect on the neuropathic allodynia when compared to the vehicle (Fig. 1A). However, 10 mg/kg of APAU significantly decreased allodynia when compared to both the celecoxib dose and the vehicle. These results of sEH inhibitors in the diabetic neuropathy model were in keeping with previous findings (Inceoglu et al., 2008). In the inflammatory pain model 10 mg/kg of celecoxib had a marginal effect at the 3 hour time point (Fig.1B). In contrast, 10 mg/kg of APAU significantly blocked the lipopolysaccharide induced allodynia outperforming celecoxib.
3.2 Evaluation of sEH inhibitor administration for dose response relationships and time dependent effects
Given the results of APAU in both models, a dose range of 0.1–100 mg/kg was used to investigate the optimal dose per model and the maximal possible effects of sEH inhibitors in the assay. The lower doses overlap with doses used in previous studies. In order to test whether the efficacy of APAU was independent of compound structure we compared APAU to two potent sEH inhibitors, t-TUCB and t-AUCB, in the same models. The nociceptive assay and administration route of all three sEH inhibitors were held constant for both models over a standard time course and responses compared for their dose response relationships (Fig. 2). This analysis revealed that the three sEH inhibitors performed differently between these two models. In addition there were differences in time dependence among the inhibitors with t-TUCB exhibiting a dose response relationship later in the time course (3 h) than APAU or t-AUCB (2 h). This may be related to differences in specific physical and chemical properties of the inhibitors such as water solubility (Table 1). In the diabetic neuropathy model APAU at 2 h significantly blocked allodynia at moderate doses with 3 mg/kg appearing to be the most effective for this compound (Fig. 2A). Reponses to t-TUCB in this model at 3 h were significant only at 10 mg/kg the maximal response in the dose range (Fig. 2B). The range of t-TUCB doses revealed a failure to increase response levels with increasing high doses in the diabetic neuropathy model which was observed for APAU as well. The sEH inhibitor t-AUCB did not follow this relationship and lacked significant effect except at 100 mg/kg (Fig. 2C). The effects of t-AUCB at low doses are not significant in this assay and therefore the lowest doses were not included in the figure for this inhibitor. Interestingly, there seemed to be a dose threshold for increased effects with t-AUCB in these nociceptive assays. An evaluation of the time dependent effects using 3mg/kg of each sEH inhibitor revealed that APAU had an effect early in the diabetic neuropathy model (Fig. 2D). APAU blocked allodynia by the first measurement at 15 minutes and this anti-nociception was sustained up to 5 h before declining (Fig. 2D). This rapid onset of efficacy is possibly associated with its high water solubility (Table 1). At 3 mg/kg t-TUCB was significantly effective versus vehicle control over the time course but was delayed and less active when compared to APAU. The third compound t-AUCB, was significant versus vehicle control over the time course but overall the effect was brief diminishing by 2 h and was less effective than APAU and t-TUCB in the diabetic neuropathy model (Fig. 2D). Thus, while the sEH inhibitors seemed to exhibit different optimal doses, all of them blocked allodynia in the diabetic neuropathy model.
Fig. 2. The efficacy of sEH inhibitor is dose dependent and time related in a neuropathic pain model.

(A) APAU at 2 h significantly attenuated allodynia when compared to vehicle controls (Kruskal-Wallis One Way ANOVA on Ranks, Dunn's method, P ≤0.001, n=6–16/dose). (B) t-TUCB at 3 h exhibited a dose dependent anti-allodynic response which reached significance at the maximal response of 10 mg/kg One Way ANOVA Holm-Sidak method P=0.014, n=6/dose). (C) t-AUCB on the other hand was significantly effective at the highest dose while lower doses (1–30 mg/kg) were not as anti-allodynic (Kruskal-Wallis One Way ANOVA on Ranks, Dunn's method, P =0.041, n=6/dose). (D) At 3 mg/kg all three sEH inhibitors significantly blocked pain related behavior over the time course. (One way ANOVA, Dunnett's method, P≤0.001, n=6/sEH inhibitor). The converted pain free pre-diabetic baseline average for groups included in 2D was 179±15 % S.E.M. of the post diabetic neuropathic baseline.
Table 1.
Physical and Biological Properties of Soluble Epoxide Hydrolase Inhibitors
| Name | APAU | t-TUCB | t-AVCB |
|---|---|---|---|
| Structure |
|
|
|
| Mass | 319.4 | 438.3 | 412.5 |
| Melting Point | 205-6 °Ca | 212.2 °Ca | 250-55 °Ca |
| eLogP (cLogP) * | 2.1 (0.77) | 1.6 (5.04) | 2.0 (3.05) |
| Water Solubility | 310 ±60 mg/Lb | 5 mg/Lc | 160 ±20mg/Lb |
| IC50(nM) | |||
| Rat | 6a | 16d | 8a |
| Mouse | 9a | 1.3d | 8a |
| Human | 15a | 0.9a | 2a |
The physical and chemical properties of the sEH inhibitors used in these experiments were previously described or were determined per published methods.
The IC50 values were determined with a fluorescent assay as described and presented in the following:
values from Hwang 2007,
values from Liu 2009,
values from Ulu 2011,
values determined per Hwang 2011.
The elogP was determined experimentally as described by Tsai et al. 2010 and clogP was calculated with Crippen's method using ChemDraw Ultra version 9.0. The clogP was not modeified for the compound class.
In the inflammatory pain model APAU was significantly effective versus vehicle control over a broader range of doses compared to the results in the diabetic neuropathy model (Fig. 3A). The effect of t-TUCB in the inflammatory pain model was similar to the diabetic neuropathy model with the highest effect at 10 mg/kg and with a dose response at 3 h (Fig. 3B). The results for t-AUCB were similar to the diabetic neuropathy model with lower doses being less active but higher doses of 30 and 100 mg/kg being significantly effective at 2 h (Fig. 3C). Assessing the time dependent effects of the inhibitors at 3 mg/kg in the inflammatory pain model revealed that APAU blocked pain at the earliest time point of 15 minutes and was significant versus vehicle control over the time course (Fig. 3D). t-TUCB was also highly significant, however t-AUCB at this dose lacked a significance versus vehicle control over the time course.
Fig. 3. The efficacy of sEH inhibition is dose dependent and time related in an inflammatory pain model.

(A) APAU at 2 h appears more potent in relieving pain in the inflammatory pain than the diabetic neuropathy model. All doses of APAU except 0.1 mg/kg significantly blocked the lipopolysaccharide induced allodynia (One way ANOVA, Holm-Sidak method, P ≤0.001, n=6/dose). t-TUCB at 3 h was significant at all doses (One way ANOVA, Holm-Sidak method, P =0.001, n=6/dose). (C) t-AUCB significantly blocked pain related behavior only at the two highest doses 30 and 100 mg/kg (One way ANOVA, Holm-Sidak method, P ≤0.001, n=6/dose). (D) At 3 mg/kg both APAU and t-TUCB are significant versus the control over the time course. In addition APAU is significant compared to both t-TUCB and t-AUCB at this dose in the inflammatory pain model (Two ANOVA Holm-Sidak method, P ≤0.001, n=6/sEH inhibitor).
3.3 APAU in Absence of Pain State
The dose response analysis revealed that higher doses of two of the inhibitors were associated with a decline in improved threshold responses. Therefore, the highest dose of APAU was tested in the assay in the absence of a pain state to ensure that this dose was not eliciting painful responses itself. 100 mg/kg APAU did not significantly alter the nociceptive thresholds compared to vehicle control in the von Frey assay (Fig. 4A). This is consistent with earlier observations that sEH inhibitors do not alter nociceptive thresholds in the absence of pain (Inceoglu et al., 2006; Inceoglu et al., 2008; Inceoglu et al., 2011). Morphine at 3 mg/kg was tested as a positive control in naive rats. It showed a characteristic and significant increase in mechanical withdrawal thresholds above the 100% baseline for several hours indicating analgesia. 3mg/kg APAU, an equal dose to morphine also showed no effect in the assay (Fig. 4A). These comparisons reveal that this sEH inhibitor has a mode of action distinct from opioids because APAU is not analgesic in naive rats. Thus, these data support the argument that a pain state is required for the anti-nociceptive activity of the sEH inhibitors and epoxyeicosatrienoic acids. In addition to testing high dose APAU in the nociceptive assay the rats were tested on a rotorod to ensure motor integrity during the treatment. The rats were not trained on the apparatus prior to the test day and therefore show improvement over time from their baseline scores as anticipated. There was no decline in motor skill for either the 3 or 100 mg/kg APAU doses compared to the vehicle (Fig. 4B). Thus this assay fails to demonstrate a negative effect on either coordination or learning.
Fig. 4. Unlike morphine APAU reduces the perception of pain only in a state of enhanced pain perception and without motor skill impairment.
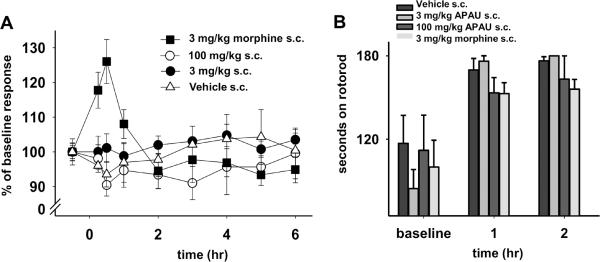
High dose APAU does not significantly change the nociceptive responses in naive animals compared to vehicle controls. (A) APAU at high or moderate dose did not significantly alter mechanical withdrawal thresholds. Unlike APAU, 3 mg/kg of morphine increased the withdrawal thresholds of naive rats. This highlights a difference between sEH inhibitors that lack this effect and narcotics such as morphine (Kruskal-Wallis One Way Analysis of Variance on Ranks, Dunn's Method, P =0.007, n=4–6). (B) Naive rats treated with high and moderate doses of APAU do not show reduced ambulation on a rotorod. No standard error bars appear for 3 mg/kg APAU at 2 h because all trials reached the cut off. Rotorod scores increase from baseline due to a training effect over the time course. While 3 mg/kg of morphine had no significant effect on rotorod performance compared to vehicle controls, it exhibited analgesia in the nociceptive assay. None of the treatments showed significant change from each other at 1 or 2 h including vehicle control (Kruskal-Wallis One Way ANOVA on Ranks, n=4–6/group).
3.4 APAU administration route and activity versus blood concentration
Given the rapid initiation of effects observed in both models, a dose via oral gavage was compared to s.c. administration. The effects of the 3 mg/kg APAU oral gavage dose equaled the maximal effect of the s.c. dose of APAU. However as expected; there were differences over the time course. The analysis revealed APAU is rapidly available at high concentration with the s.c. administration (Fig. 5A). The s.c. APAU rapidly increased mechanical withdrawal thresholds by >20% post lipopolysaccharide injection compared to controls. The oral gavage dose increased responses to the same level but much later (Fig. 5B). The blood concentration of the same dose of APAU is lower at early time points with oral gavage administration, and the mechanical withdrawal thresholds in the nociception assay reflect this.
Fig. 5. sEH inhibitors reach effective blood concentrations independent of administration route.
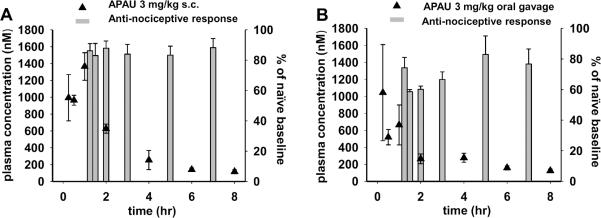
The blood concentrations in naïve rat (black triangles) are compared to the efficacy in the inflammatory pain model (grey bars) which shows 3 mg/kg APAU via both routes is effective in reducing pain perception. (A) 3mg/kg APAU was administered to rats via s.c. injection 60 min prior to lipopolysaccharide injection which significantly raised mechanical withdrawal thresholds compared to vehicle controls (shown in 3D). APAU s.c. at 15min reaches near 1000 nM which is approximately 166× the IC50 on the recombinant rat enzyme. As expected the onset of efficacy in the nociceptive assay is much faster with s.c. than oral administration. APAU maintained an effective concentration measuring 116 nM at 8 h well above 10× the IC50 for the rat sEH enzyme. (B) 3 mg/kg APAU oral gavage administration 1 h prior to lipopolysaccharide injection increased thresholds compared to vehicle. However this occurred later in the time course than the s.c. administration (Kruskal-Wallis One Way ANOVA on Ranks, P ≤0.001). Oral gavage of 3 mg/kg APAU led to lower blood concentrations at 2 h than the s.c. dose remained at an effective concentration well above the 10× of the IC50 for the rat sEH enzyme measuring 124 nM at 8 h.
Interestingly, with both administration routes, robust responses were observed at a time when blood concentrations were diminishing in rats. APAU has a relatively short plasma half-life based on previously published results (Liu et al., 2009), it is therefore surprising that significant responses to APAU were of long duration in the nociceptive assay. Inhibition of sEH has been shown to both down regulate the induction of COX-2 as well as block PGE2 pain (Inceoglu et al., 2011), effects which may limit the inflammation cascade and result in anti-nociception lasting beyond peak blood concentration of the inhibitors. APAU is believed to be rapidly metabolized primarily via hydroxylation of the adamantyl moiety (Tsai et al., 2010). However, the in vitro IC50 value of APAU on the recombinant rat sEH enzyme with a fluorescent substrate is 6nM (Hwang et al., 2007). These blood concentrations for both administrations are well above 10 fold higher than the IC50 values for the entire time course despite the steep decline over time.
3.5 Oxylipin Analysis
Using the 3 mg/kg dose that had an effective plasma concentration and significant results in both models APAU was analyzed for evidence of target engagement. Inhibition of sEH halts the degradation of epoxyeicosatrienoic acids and thus leads to decreased levels of the dihydroxyeicosatrienoic acid products. The results of the PEG400 vehicle group were in line with previously published results for the vehicle epoxyeicosatrienoic acid and dihydroxyeicosatrienoic acid levels (Inceoglu et al., 2006). The APAU treatments did not significantly alter sum epoxyeicosatrienoic acids levels compared to vehicle controls. Importantly however, APAU at both doses significantly lowered dihydroxyeicosatrienoic acids compared to vehicle controls. Thus, while the epoxyeicosatrienoic acids were not dramatically increased in plasma, both doses of the inhibitor were able to significantly lower the dihydroxyeicosatrienoic acid levels (Fig. 6A, Table S1). Because sEH inhibitors attenuate inflammation, we expected lower levels of key inflammatory pain mediating prostaglandins, specifically PGE2 and PGD2, when compared to vehicle. Lipopolysaccharide treatment did not significantly increase circulating PGE2 and PGD2 levels in plasma compared to vehicle (Fig. 6B, Table S2). However, APAU at 3 mg/kg reduced both prostaglandins significantly compared to lipopolysaccharide +vehicle and PGE2 compared to vehicle. A comparison of the two dose levels yielded an unexpected result. The 100 mg/kg dose of APAU did not significantly decrease PGE2 and PGD2 levels compared to lipopolysaccharide +vehicle.
Fig. 6. APAU reduces dihydroxyeicosatrienoic acids and abolishes pro-inflammatory prostaglandins.

Plasma of rats treated with APAU or vehicle was analyzed for oxylipin metabolites. (A) Changes in sum of epoxyeicosatrienoic acids [(8(9), 11(12), and 14(15) isomers] across treatments were not significant. Inhibition of sEH lowered sum dihydroxyeicosatrienoic acid plasma concentrations, the product of the sEH enzymatic activity in the inflammatory pain model. Sum dihydroxyeicosatrienoic acids were significantly reduced by APAU at 3 and 100 mg/kg compared to vehicle and lipopolysaccharide control groups (Kruskal-Wallis One Way ANOVA on Ranks, Dunn's Method, P ≤0.001, n=5–10/group). (B) Key inflammatory pain mediators PGE2 and PGD2 were greatly decreased after APAU administration. The 3 mg/kg dose significantly reduced PGE2 and PGD2 compared to lipopolysaccharide controls but the 100 mg/kg dose lacked significance (Kruskal-Wallis One Way ANOVA on Ranks, Dunn's Method, P ≤0.001, n=5–10/group).
4. Discussion
The newly discovered antihyperalgesic effects resulting from inhibiting sEH are compelling and may form the basis of a translational effort to discover novel drugs acting on the CYP450 branch of the arachidonic acid cascade. The sEH inhibitors have been described in several models with a variety of assays. Here a direct comparison using the same assay was used to evaluate the efficacy of three sEH inhibitors on two pain models in the rat.
4.1 Comparison of pain models
Although the models differ in etiology, it is useful to compare the efficacy of the sEH inhibitors against allodynia in two independent models. The results from both models illustrate that the sEH inhibitors usually are more effective over a broader dose range in the inflammatory pain model than the diabetic neuropathy model. It is also evident that the effects of sEH inhibitors can be saturated or even decreased at high dose. Additionally, the effective doses for pain relief may be different between these models. This is not surprising given the anti-inflammatory efficacy of the sEH inhibitors and the multi-faceted etiology of type I diabetic neuropathy. However, sEH inhibitors block allodynia in this diabetic neuropathy model. Other drugs targeting inflammation such as non-steroidal anti-inflammatory drugs (NSAIDs) and selective COX-2 inhibitors have no proven efficacy against diabetic neuropathy in man or rodents but are often used for this chronic pain (Gore et al., 2007; Inoue et al., 2009). Importantly, the long term use of NSAIDs and selective COX-2 inhibitors is counter indicated for patients with nephropathy and hypertension which are typical comorbidities of diabetes. Thus, NSAIDs may offer some relief of mixed etiology pain (Cohen and Harris, 1987; Gore et al., 2007), but due to their side effects they are not appropriate therapies for patients suffering moderate or severe diabetic neuropathy. There is currently no clear evidence of a single mechanism for diabetic neuropathic pain. The ability of sEH inhibitors to block allodynia in this model while other anti-inflammatory drugs have limited efficacy indicates that sEH inhibitor activity is distinct and not solely related to reducing inflammation. Several studies have addressed the role of inflammation in streptozocin induced pancreatic beta cell injury (Eizirik et al., 2009; Fukuda et al., 2008). It is possible that sEH inhibition could decrease the inflammation that is implicated in the streptozocin induced progressive damage to pancreatic beta cells of this diabetic neuropathy model, but this is a subject for further experiments. Here the diabetic neuropathy model was used to address the effect of sEH inhibitors on allodynia after neuropathy developed. The effectiveness of sEH inhibitors in the diabetic neuropathy model argues that the inhibitors and elevated epoxyeicosatrienoic acids have multiple modes of action. Due to this, sEH inhibitor administration may be advantageous over available pain therapeutics. For instance, tricyclic anti-depressants, a current front line therapy for chronic pain conditions including diabetic neuropathy, have no additional anti-inflammatory action. Celecoxib had limited or no effect on allodynia in these experiments. Notably, the sEH inhibitors were far superior to celecoxib in reducing allodynia in both the neuropathic and inflammatory pain assays. Additionally, the inhibitor stabilized epoxyeicosatrienoic acids have anti-hypertensive and cardio-protective activities (Imig and Hammock, 2009; Nithipatikom et al., 2006). Unlike NSAIDs and selective COX-2 inhibitors, sEH inhibitors do not disturb prostacyclin production or the prostacyclin to thromboxane ratio (Schmelzer et al., 2006). This property eliminates the thrombotic events associated with COX-2 selective drugs (Liu et al., 2010). Finally, sEH knockout mice were found resistant to gastrointestinal lesions associated with COX-1 inhibition (Zhang et al., 2012). Thus, sEH inhibitors may be an attractive pain relief alternative to NSAIDs and selective COX-2 inhibitors with known risks of adverse cardiovascular effects (Trelle et al., 2011).
4.2 Comparison of sEH inhibitors
The sEH inhibitors were effective in reducing pain in both the diabetic neuropathy and inflammatory pain models. However, there were also a number of observable differences among these inhibitors within the models which most likely relates to their physical and chemical properties. Despite differences in chemical structure APAU, t-AUCB and t-TUCB have experimental log P values from 1.6 to 2.1 and low nM potency (in vitro IC50) for sEH enzyme inhibition with the recombinant rat enzyme (Table 1). APAU was effective at lower doses in both models while t-TUCB was effective at a slightly higher dose level. Notably in the rat, 10 mg/kg of t-TUCB increased mechanical withdrawal thresholds to pre-diabetic pain free scores at early time points in the diabetic neuropathy model (Fig. S1, Table S3). In the inflammatory pain model 10 mg/kg of t-TUCB was unique compared to other sEH inhibitors as it had a later onset but higher efficacy reaching 100% of baseline mechanical withdrawal thresholds by 6 h (Fig. S1). In contrast to the other sEH inhibitors, t-AUCB was significantly active in both models only at the highest dose. This relationship was observed only for t-AUCB and therefore may be related to the pharmacokinetics, tissue levels or off-rate of the inhibitor from the enzyme. The effects of sEH inhibition are dependent on the presence of epoxy-fatty acids, therefore sEH inhibitors do not produce a greater effect than stabilizing the endogenous levels of these bioactive lipids. Thus, a response rate that slows at higher doses of APAU and t-TUCB is not surprising because they only inhibit the enzymatic degradation of epoxy-fatty acids. Other routes of epoxy-fatty acid elimination exist, some of which may dominate in the absence of sEH mediated degradation (Spector et al., 2004). t-AUCB was previously found to have superior pharmacokinetics to APAU based on the larger plasma concentration measured as area under the concentration-time curve (AUC) (Liu et al., 2009). When evaluated in dog there is a massive difference among the three compounds with c-TUCB (400) >> t-AUCB (160) >> APAU (2.5) when data are expressed as a function of exposure to potency (plasma AUC / IC50) on the target sEH enzyme (Tsai et al., 2010). The mean plasma AUCs for these three sEH inhibitors in rat reveals a rank of these inhibitors similar to the results in dog (S2). In rats t-TUCB showed the best overall pharmacokinetics at low dose which may explain its higher efficacy compared to t-AUCB. The success of APAU is surprising given it has a short half-life. Despite this, APAU reaches a high maximum concentration and it has a more potent IC50 on recombinant rat enzyme compared to t-TUCB and t-AUCB. In a pharmacokinetics study for the human clinical trial of APAU, this compound displayed remarkable absorption in multiple species and notably an oral bioavailability of 100% in the rat compared to 25% in cynomolgus monkey (Anandan et al., 2011). Additionally, the plasma protein binding of APAU in cynomolgus monkey was 35.5% versus 98% for t-AUCB and 99% for t-TUCB (Ulu et al., 2011). Thus, the success of APAU in these nociceptive assays could be related to its comparative bioavailability and particularly to its higher potency on the recombinant rat sEH. It, for example, has a much slower off rate from the rat recombinant sEH than corresponding mouse or human enzyme. There are a number of advantages of APAU over the other sEH inhibitors as a candidate compound to treat pain. The major advantage is that it has investigational new drug status with the U.S. FDA (Shen, 2010). APAU is smaller in molecular weight making it more attractive as a drug. As expected for ureas all three compounds have high melting points and have similar experimental logP values. Yet their potencies are such that all three compounds are relatively easy to formulate for oral administration. The physical properties of APAU are somewhat unique because the calculated logP value is much lower than the others and the water solubility is far higher. This allows it unlike t-AUCB and t-TUCB to be formulated as a dry powder. Its solubility in water also makes it more attractive for alternative routes of administration. However, APAU also has limitations. In this study all pain assays were carried out for only 6 hours. With longer assays, the short half-life of APAU might lead to a shorter duration of action. APAU is more active on the murine and rat sEH enzymes than the human, thus testing the rat model in these studies might over estimate its potency. Therefore there are cautions regarding the use of APAU for treating pain in man. It has been previously noted that the fluorescent assay cited here over estimates the potency of APAU compared to other sEH inhibitors, particularly on the human enzyme (Tsai et al., 2010). This could limit its proper evaluation in alternate pain models and potential studies in man.
5. Conclusion
Pain control is a major clinical challenge specifically for patients suffering from chronic pain conditions. Despite decades of research into understanding mechanisms of pain signaling and developing therapies there is currently no effective treatment for these chronic pain conditions. Thus, there is a strong need for new therapeutics to treat pain and to further understand the pathobiology of chronic pain conditions. While these assays in rats have limitations in their predictive capacities they do show a robust and reproducible antihyperalgesic response to sEH inhibition in whole animal. The sEH inhibition is effective in both the lipopolysaccharide induced inflammatory pain model and the streptozocin induced type 1 diabetic neuropathy model. The efficacy of sEH inhibitors against diabetic neuropathic pain is noteworthy because of its potential to address the unmet therapeutic need of relieving this chronic pain condition.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of Environmental Health Sciences (NIEHS) Grant R01 ES002710 (to B.D.H.), NIEHS Superfund Research Program P42 ES004699, and Grants NIEHS T32ES007059 and NIH 5T32DC008072-05 (to K.W.). B.D.H. is a George and Judy Marcus Senior Fellow of the American Asthma Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aley KO, Levine JD. Rapid onset pain induced by intravenous streptozotocin in the rat. J Pain. 2001;2:146–150. doi: 10.1054/jpai.2001.21592. [DOI] [PubMed] [Google Scholar]
- Anandan S-K, Webb HK, Chen D, Wang Y-X, Aavula BR, Cases S, Cheng Y, Do ZN, Mehra U, Tran V, Vincelette J, Waszczuk J, White K, Wong KR, Zhang L-N, Jones PD, Hammock BD, Patel DV, Whitcomb R, MacIntyre DE, Sabry J, Gless R. 1-(1-Acetylpiperidin-4-yl)-3-adamantan-1-yl-urea (AR9281) as a potent, selective, and orally available soluble epoxide hydrolase inhibitor with efficacy in rodent models of hypertension and dysglycemia. Bioorganic & Medicinal Chemistry Letters. 2011;21:983–988. doi: 10.1016/j.bmcl.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstrom K, Kayganich K, Murphy RC, Fitzpatrick FA. Incorporation and distribution of epoxyeicosatrienoic acids into cellular phospholipids. J Biol Chem. 1992;267:3686–3690. [PubMed] [Google Scholar]
- Cohen KL, Harris S. Efficacy and safety of nonsteroidal anti-inflammatory drugs in the therapy of diabetic neuropathy. Arch Intern Med. 1987;147:1442–1444. [PubMed] [Google Scholar]
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Tesch GH, Yap FY, Forbes JM, Flavell RA, Davis RJ, Nikolic-Paterson DJ. MKK3 signalling plays an essential role in leukocyte-mediated pancreatic injury in the multiple low-dose streptozotocin model. Lab Invest. 2008;88:398–407. doi: 10.1038/labinvest.2008.10. [DOI] [PubMed] [Google Scholar]
- Gore M, Dukes E, Rowbotham DJ, Tai KS, Leslie D. Clinical characteristics and pain management among patients with painful peripheral neuropathic disorders in general practice settings. Eur J Pain. 2007;11:652–664. doi: 10.1016/j.ejpain.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92:101–130. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006;79:2311–2319. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Jinks SL, Ulu A, Hegedus CM, Georgi K, Schmelzer KR, Wagner K, Jones PD, Morisseau C, Hammock BD. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A. 2008;105:18901–18906. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Wagner K, Schebb NH, Morisseau C, Jinks SL, Ulu A, Hegedus C, Rose T, Brosnan R, Hammock BD. Analgesia mediated by soluble epoxide hydrolase inhibitors is dependent on cAMP. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5093–5097. doi: 10.1073/pnas.1101073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue N, Ito S, Tajima K, Nogawa M, Takahashi Y, Sasagawa T, Nakamura A, Kyoi T. Etodolac attenuates mechanical allodynia in a mouse model of neuropathic pain. J Pharmacol Sci. 2009;109:600–605. doi: 10.1254/jphs.08287fp. [DOI] [PubMed] [Google Scholar]
- Jones PD, Tsai HJ, Do ZN, Morisseau C, Hammock BD. Synthesis and SAR of conformationally restricted inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett. 2006;16:5212–5216. doi: 10.1016/j.bmcl.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan SA, Saade NE, Haddad JJ, Abdelnoor AM, Atweh SF, Jabbur SJ, Safieh-Garabedian B. Endotoxin-induced local inflammation and hyperalgesia in rats and mice: a new model for inflammatory pain. Pain. 1996;66:373–379. doi: 10.1016/0304-3959(96)03068-0. [DOI] [PubMed] [Google Scholar]
- Liu JY, Li N, Yang J, Qiu H, Ai D, Chiamvimonvat N, Zhu Y, Hammock BD. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17017–17022. doi: 10.1073/pnas.1011278107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Tsai HJ, Hwang SH, Jones PD, Morisseau C, Hammock BD. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Br J Pharmacol. 2009;156:284–296. doi: 10.1111/j.1476-5381.2008.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci U S A. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Inceoglu B, Schmelzer K, Tsai HJ, Jinks SL, Hegedus CM, Hammock BD. Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res. 2010;51:3481–3490. doi: 10.1194/jlr.M006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Newman JW, Tsai HJ, Baecker PA, Hammock BD. Peptidyl-urea based inhibitors of soluble epoxide hydrolases. Bioorg Med Chem Lett. 2006;16:5439–5444. doi: 10.1016/j.bmcl.2006.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithipatikom K, Moore JM, Isbell MA, Falck JR, Gross GJ. Epoxyeicosatrienoic acids in cardioprotection: ischemic versus reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;291:H537–542. doi: 10.1152/ajpheart.00071.2006. [DOI] [PubMed] [Google Scholar]
- Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:13646–13651. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HC. Soluble epoxide hydrolase inhibitors: a patent review. Expert Opin Ther Pat. 2010;20:941–956. doi: 10.1517/13543776.2010.484804. [DOI] [PubMed] [Google Scholar]
- Shen HC, Hammock BD. Discovery of Inhibitors of Soluble Epoxide Hydrolase: A Target with Multiple Potential Therapeutic Indications. J Med Chem. 2012 doi: 10.1021/jm201468j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res. 2009;50(Suppl):S52–56. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- Tomita-Yamaguchi M, Babich JF, Baker RC, Santoro TJ. Incorporation, distribution, and turnover of arachidonic acid within membrane phospholipids of B220+ T cells from autoimmune-prone MRL-lpr/lpr mice. J Exp Med. 1990;171:787–800. doi: 10.1084/jem.171.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trelle S, Reichenbach S, Wandel S, Hildebrand P, Tschannen B, Villiger PM, Egger M, Juni P. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011;342:c7086. doi: 10.1136/bmj.c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HJ, Hwang SH, Morisseau C, Yang J, Jones PD, Kasagami T, Kim IH, Hammock BD. Pharmacokinetic screening of soluble epoxide hydrolase inhibitors in dogs. Eur J Pharm Sci. 2010;40:222–238. doi: 10.1016/j.ejps.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulu A, Appt S, Morisseau C, Hwang S, Jones P, Rose T, Dong H, Lango J, Yang J, Tsai H, Miyabe C, Fortenbach C, Adams M, Hammock B. Pharmacokinetics and in vivo potency of soluble epoxide hydrolase inhibitors in cynomolgus monkeys. Br J Pharmacol. 2011 doi: 10.1111/j.1476-5381.2011.01641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf NM, Morisseau C, Jones PD, Hock B, Hammock BD. Development of a high-throughput screen for soluble epoxide hydrolase inhibition. Anal Biochem. 2006;355:71–80. doi: 10.1016/j.ab.2006.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Schmelzer K, Georgi K, Hammock BD. Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Anal Chem. 2009;81:8085–8093. doi: 10.1021/ac901282n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yang AL, Liao J, Li H, Dong H, Chung YT, Bai H, Matkowskyj KA, Hammock BD, Yang GY. Soluble Epoxide Hydrolase Gene Deficiency or Inhibition Attenuates Chronic Active Inflammatory Bowel Disease in IL-10(−/−) Mice. Digestive diseases and sciences. 2012;57:2580–2591. doi: 10.1007/s10620-012-2217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.