

Abstract
Oxidative stress plays a significant role in the development of insulin resistance; however, the cellular targets of oxidation that cause insulin resistance have yet to be fully elucidated. Methionine sulfoxide reductases (Msr) reduce oxidized methionine residues, thereby repairing and protecting proteins from oxidation. Recently, several genome-wide analyses have found human obesity to be strongly correlated with polymorphisms near the methionine sulfoxide reductase A (MsrA) locus. In this study, we tested whether modulation of MsrA expression significantly alters the development of obesity and/or insulin resistance in mice. We show that mice lacking MsrA (MsrA−/−) are prone to the development of high fat diet-induced insulin resistance and a reduced physiological insulin response compared to high fat-fed wild type mice. We also show that oxidative stress in C2C12 cell cultures reduces both insulin-stimulated phosphorylation and autophosphorylation of the insulin receptor. Tissues from high fat-fed mice show similar reduction in insulin receptor function and the lack of MsrA further diminishes these functions. Together, these data demonstrate for the first time that MsrA plays a role in the regulation of glucose homeostasis. In addition, these data support a novel hypothesis that obesity-induced insulin resistance is caused in part by reduced function of insulin signaling proteins arising from protein oxidation.
Keywords: oxidative stress, methionine sulfoxide, diabetes, obesity, glucose homeostasis
Introduction
Excessive fat accumulation is well-established as a primary risk factor for the development of insulin resistance, one of the earliest defects in Type 2 diabetes mellitus (T2DM) [1,2]. However, the precise mechanisms by which increases in adiposity reduce insulin sensitivity remain largely unclear. Recent findings suggest that obesity-induced insulin resistance might be caused in part by dysregulation of oxidative stress. Accumulation of fat has been shown to increase oxidative stress and the accumulation of oxidative damage in clinical studies and experimental models. For example, markers of DNA, lipid and protein oxidation are relatively high in plasma and tissue (skeletal muscle and adipose) samples from obese human subjects [3,4,5]. Similarly, plasma and tissue lipid and protein oxidation markers and levels of hydrogen peroxide (H2O2) are elevated in both genetically obese mice and mice fed a high fat diet [4,6]. Obesity has also been shown to increase the production of reactive oxygen species (ROS) from both the mitochondria and the activation of NADPH oxidases [4,7,8,9]. The most direct evidence that oxidative stress is a critical factor in the development of insulin resistance is primarily from cell culture studies. For example, insulin sensitivity of cell lines (3T3L1 and L6 myotubes) is dramatically reduced by treatment with oxidative stress [9,10,11]. Further, H2O2 treatment significantly decreases insulin signaling in ex vivo rat soleus muscle cultures [12]. In addition, cellular insulin resistance induced by treatment with either TNFα or glucocortocoids is oxidative stress-dependent and is prevented by antioxidants [13].
The direct mechanism by which oxidative stress causes insulin resistance has yet to be fully elucidated. Oxidative stress has been commonly thought to reduce insulin signaling by activating the mitogen-activated protein kinase c-jun N terminal kinase (JNK). Activation of JNK can directly phosporylate insulin substrate receptor 1 (IRS-1) at inhibitory sites that then disrupt the propagation of signals through the insulin signaling pathway [14,15]. However, several intriguing findings suggest that oxidative stress may negatively affect the cellular insulin response by directly causing oxidative damage to proteins. Oxidation of proteins can cause significant alterations to their structure or damage catalytic sites, both of which can lead to decreased function of the oxidized protein [16]. For example, treatment of the enzyme glutamine synthetase with H2O2 in vitro dramatically reduces the function of this enzyme [17] and increased protein oxidation associated with disease states in vivo is associated with changes in protein conformation and reduced activity [18]. In vitro studies have also shown that oxidative stress impairs the ability of insulin receptor (InR) to correctly bind with insulin which then diminishes the ability of InR to internalize insulin in cells [19]. Oxidative stress has also been shown to reduce phosphatidylinositol 3-kinase (PI3K) activity and prevent PI3K from mobilizing to the correct subcellular locations necessary for transduction of insulin signals [20].
Oxidized and damaged proteins are dealt with by the cell either through removal of the protein (i.e., proteasomal degradation or autophagy) or through repair of the oxidation adducts. The methionine sulfoxide reductases (Msr) represent one of the few classes of enzymes capable of the reductive repair of protein oxidation. Msr help maintain proteins in the reduced state by repairing oxidative damage to methionine (Met). Due to the sulfur atom in their structure, Met residues are exquisitely sensitive to oxidation damage that results in the formation of methionine sulfoxide (MetO) residues [21]. The MetO residue is reduced in a reaction catalyzed by methionine sulfoxide reductase (Msr) using thioredoxin as a cofactor. In mammals, Msr exists as two general enzymes that repair specific diastereomers of MetO; MsrA is thought to reduce the S form of MetO while the isoforms of MsrB reduce the R form [21,22]. The bulk of Msr activity has generally been thought to be due to MsrA in part because this enzyme is found in both the cytosol and mitochondria in most mammalian tissues [23,24]. While the activity of Msr is specific for repairing Met oxidation, Msr activity may also, in part, regulate overall levels of protein oxidation. Because Met residues are highly prone to oxidation and repairable by Msr, they may act as a free radical sink within proteins by reducing the likelihood that other amino acids will be damaged by oxygen free radicals [17,25]. Altering either the content of Met or the presence of Msr would then be predicted to alter the susceptibility of proteins to oxidation damage beyond MetO.
Recent studies have indicated that polymorphisms in loci near the MsrA gene are associated with obesity and metabolic disease in humans [26,27,28]. To determine whether these associations are due to alterations in MsrA function, we tested whether reduction of MsrA levels in mice could significantly affect the development of obesity-induced insulin resistance. We show here that MsrA−/− mice fed a high fat diet develop a more severe insulin resistant phenotype and reduced insulin signaling than do high fatfed wild type mice. We also show that oxidative stress can directly reduce insulin receptor function. Tissues from high fat fed mice have a clear decline in the ability to undergo either insulin-stimulated phosphorylation or autophosphorylation; furthermore, this decline is exacerbated in tissues from MsrA−/− mice as is the increase in protein oxidation associated with high fat feeding. These data are the first to indicate that MsrA expression can significantly alter the development of obesity-induced insulin resistance and provide direct evidence for a role of MsrA in the regulation of glucose metabolism.
Materials and Methods
Animals
The generation and breeding scheme for MsrA−/− and wild type animals has been published elsewhere [29]. The breeding lines for MsrA−/− mice have been backcrossed into the C57BL/6 genetic background for >10 generations. Mice were genotyped using primers for the neocassette in the targeted construct; in addition, we checked for the presence of the MsrA protein by Western blot analysis at sacrifice. For the MsrA−/− study, male mice of 12–16 weeks male mice were used; wild type mice for all studies were of the MsrA+/+ genotype. All procedures involving the mice were approved by the Subcommittee for Animal Studies at the Audie L. Murphy Veterans Administration Hospital at San Antonio and the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio.
Diets
From weaning until initiation of experimental diet protocol, all mice were maintained at 3–4 mice per cage on standard NIH-31 chow diet (Harlan Teklad, Madison WI) provided ad libitum. Cages were randomly assigned to either a low fat defined diet (10% kCal from fat, D12450B, Test Diets, Richmond IN) or to a high fat defined diet (45% kCal from fat, D12451, Test Diets) when mice were approximately 10–16 weeks of age. Diets were provided ad libitum and food consumption and body weight were monitored bi-weekly. Body composition of non-anesthetized mice was analyzed by Quantitative Magnetic Resonance imaging (QMRi) using an EchoMRI 3-in-1 composition analyzer (Echo Medical Systems, Houston TX).
Glucose and insulin tolerance tests
For glucose tolerance tests, mice were fasted 6 hours and given 1.5 g glucose (Sigma, St. Louis MO)/kg of body weight by intraperitoneal (IP) injection. Blood glucose levels were measured at 0, 15, 30, 60, and 120 minutes following injection. For insulin tolerance tests, mice were fasted 5 hours and given 1 U insulin (Novalin; Novo Nordisk, Princeton NJ)/kg of body weight by IP injection. Blood glucose levels were measured at 0, 15, 30, and 60 minutes following injection. All blood glucose measurements were made using a ONE Touch Ultra handheld glucometer. Levels of insulin in serum were measured using the Ultra Sensitive Mouse Insulin ELISA (Crystal Chem, Downer's Grove IL) following the manufacturer's instructions.
Immunoblots
Total protein extracts were isolated from skeletal muscle, and visceral fat tissue that had been collected from mice, snap-frozen in liquid nitrogen, and stored at −80° C until use. Mice were sacrificed 15 minutes after either 1) overnight fast followed by intraperitonial (IP) injection of insulin (1 U/kg body weight) or 2) overnight fast followed by IP injection of saline. Protein extracts were made in RIPA buffer with added protease and phosphatase inhibitors (Thermo Scientific, Rockford IL), centrifuged at 13,000 g and 4° C for 15 min, then stored at −80° C until use. Total protein content was measured by the Pierce BCA assay (Bio-Rad, Hercules, CA, USA). For blots, total cellular protein extract was prepared in 6× SDS sample buffer, subjected to SDS-polyacrylamide electrophoresis, and transferred to PVDF membrane (Millipore, Billerica, MA, USA). Primary antibodies and dilutions used in this study include phospo-Akt Ser473 (1:1000), Akt (1:1000), phospho-SAPK/JNK (Thr183/Tyr185) (1:1000), SAPK/JNK (1:1000), phospho-IGF1 receptor β/Insulin receptor β (1:1000), insulin receptor (1:1000), phospho-GSK3 (1:1000), GSK3(1:1000), phospho-AS160 (1:1000) and AS160 (1:1000), (Cell Signaling, Beverly MA). β-tubulin and actin (1:5000, Sigma) were used as loading controls. Anti-MsrA was shared by the laboratory of Rodney Levine at NIH/NHLBI. All alkaline phosphatase-conjugated secondary antibodies (anti-mouse and anti-rabbit) were purchased from Santa Cruz Biotechnologies (Santa Cruz CA). Protein bands on immunoblots were detected using ECL+ reagent (GE Healthcare, Piscataway, NJ, USA).
Cell culture experiments
C2C12 cell lines (ATCC; Manassas, VA) were maintained under 5% CO2 at 37° C in DMEM (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin and Streptomycin. Cells were differentiated in low serum media containing 2% horse serum for several days, exchanging fresh media daily. Myotube formation can be visualized after 24 hours in differentiation medium. Differentiated myocytes were treated with different concentrations of H2O2 in low serum culture media for 48 hours with fresh media containing H2O2 exchanged at 24 hours. Cells were then incubated with serum free DMEM for 3 hours and either 1) treated with DMEM only or 2) treated with media containing 100 nM insulin for 15 minutes. Cells were then washed with cold PBS and lysed with RIPA buffer while on ice.
Insulin receptor autophosphorylation assay
Protein samples from C2C12 experiments were homogenized immediately after H2O2 treatment. Skeletal muscle and white adipose tissue samples were isolated from MsrA−/− and wild type mice that had been fasted overnight; these samples were then flash frozen in liquid nitrogen and stored at −80° C until analyzed. To prepare protein samples, cells or tissue sections were homogenized in Cell Extraction Buffer (Invitrogen, Carlsbad CA) containing Halt Protease and Phosphatase Inhibitor (Thermo Scientific). Homogenates were centrifuged for 30 minutes at 15,000 g and supernatant was used for protein assays. InR tyrosine 1158 autophosphorylation was then analyzed using a modified Insulin Receptor [pY1158] ELISA Kit (Invitrogen) protocol as described previously by Wang and Brodey (Invitrogen). In short, whole protein homogenates were incubated in 96 well plates coated with a monoclonal antibody specific for the InR β subunit. This immobilized the InR protein allowing unbound proteins to be washed away using phosphate buffered saline. Autophosphorylation of bound InR was then stimulated through protein incubation in InR Autophosphorylation Substrate Solution containing ATP, insulin, MnCl2, HEPES, NaCl and MgCl2. After incubation, the ELISA plate was washed, terminating InR autophosphorylation. InR tyrosine 1158 phosphorylation was then monitored using anti-InR [pY1158] antibody and additional substrates as directed in the phosphoELISA protocol (Invitrogen). The reaction was quantified by measuring well absorbance at 450 nm with intensity being directly proportional to pY1158 InR concentration.
Protein carbonyl levels
Protein carbonyls were labeled using a modified protocol of [49]. Snap frozen tissues were homogenized in 50 mM phosphate buffer, pH 6.0; 0.5 mM MgCl2; and 10 mM EDTA containing cocktail of protease inhibitors (0.5 mM PMSF, 1 mg/ml leupeptin, and 1 mg/ml aprotinin) then centrifuged at 4°C, 100,000 g for 1 hour. Cell extracts were incubated with 1% streptomycin sulfate at 37°C for 30 min then centrifuged at 16,000 g for 10 min. Soluble proteins from extract (1 mg/ml) were incubated with 1 mM fluorescein-5-thiosemicarbazide (FTC) in deaerated phosphate buffer, pH 6.0, with 0.3 M guanidine for 2 h at 37°C in the dark. Labeled proteins were precipitated with trichloroacetic acid (TCA) then centrifuged at 16,000 g at room temperature for 10 minutes. To measure carbonyls in total cellular protein homogenates, pellets were washed with ethanol/ethyl acetate, resuspended in 20 mM phosphate buffer, pH 8.0, containing 8 M urea, then boiled in Laemmli buffer, samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). After electrophoresis, fluorescent labeled proteins were imaged by Typhoon 9400 (excitation wavelength of 532 nm, emission filter at 526 nm, 40 nm bandpass; GE Healthcare Life Sciences, Piscataway, NJ, USA). The gel was then stained with Coomassie R250 (Sigma-Aldrich Corp., St. Louis, MO, USA), destained and imaged by AlphaImage 3400 (Alpha Innotech Corp., San Leandro, CA, USA). Intensity of fluorescence and Coomasie staining were calculated using ImageQuant 5.0 software (Molecular Dynamics, Amersham Biosciences Corp., Piscataway, NJ, USA) and carbonyl content was calculated as ratio of intensities (Flour/Coom). To measure carbonyls in isolated InR, protein pellets were washed, resuspended, and incubated with Protein A Dynabeads (Invitrogen) prepared for immunoprecipitation using antibody to InR (Cell Signaling) and the included Dynabeads Antibody Binding and Wash mixture provided with the kit. The protein-Dynabead mixture was incubated with rotation for 1 hour and then transferred to Dynamag for washing. After removing supernatant, proteins were eluted using the provided Buffer and resuspended in Dynabeads Western Running buffer. After separation by SDS-PAGE, samples were analyzed as above for total cellular protein homogenates.
Statistical analysis
Animal and tissue weights, insulin levels, and blood glucose at each time interval during glucose and insulin tolerance tests were analyzed for statistical significance by t-test comparing wild type to MsrA−/− animals within each diet group. Area under the curve for glucose tolerance tests was calculated using the trapezoidal rule. For immunoblots, autophosphorylation and carbonyl assays perfomed using tissue lysates, signal intensity was measured using ImageJ software (NIH) and were analyzed for statistical significance by t-test comparing either 1) diet groups within a genotype or 2) genotypes within a diet group. For immunoblots and autophosphorylation assays perfomed using cell culture lysates, data were analyzed for statistical significance by 2-Way ANOVA. For all, statistical significance was determined by p < 0.05. All data are presented as the mean ± SEM and sample sizes for each experiment are presented in figure legends.
Results
Lack of MsrA exacerbates insulin resistance caused by high fat diet
Prior to the initiation of feeding experiments, all mice were maintained on standard chow-based diet (NIH-31); we found no difference between MsrA−/− and wild type mice in their body weight prior to the switch to defined diets (data not shown). Further, we have previously shown that these mice do not differ from wild type mice in body weight or lifespan when maintained on this standard chow throughout life [29]. At 10–16 weeks of age, male mice were randomly assigned to be fed either a diet with 10% kCal from fat (low fat) or 45% kCal from fat (high fat); both diets were specifically defined in their nutrient sources (see Experimental Procedures) and utilized a single fat source (lard). Mice were maintained on these diets for a period of 16 weeks. Mice fed the high fat diet were significantly heavier and had increased adipose content; however, within each diet treatment, MsrA−/− and wild type mice did not differ in body weight, fat mass or lean mass (Figure 1A, 1B). Food consumption was similar for both genotypes of mice on both diets throughout the course of this experiment (data not shown).
Figure 1. Body weight and adipose content are unaffected by lack of MsrA on either low fat or high fat diets.

A) Mean body weight of wild type (open bars) or MsrA−/− mice (closed bars) after feeding either low fat or high fat diets. B) Mean fat mass (left) and lean mass (right) of wild type (open bars) or MsrA−/− mice (closed bars) after feeding either low fat or high fat diets. Sample sizes for each group are: low fat wild type = 14, low fat MsrA−/− = 13, high fat wild type = 21, high fat MsrA−/− = 18. Error bars represent ± SEM and p values are for student t-test between the groups connected by horizontal bar.
We then measured whether the absence of MsrA in mice significantly affected glucose metabolism on either diet. We found no significant difference between low fat-fed MsrA−/− and wild type in their responses to glucose tolerance tests (Figure 2A). Furthermore, fasting blood glucose levels of low fat-fed MsrA−/− and wild type mice did not differ (Figure 1A, Low fat time point 0). All mice fed high fat diet became glucose intolerant and blood glucose levels in high fat-fed MsrA−/− mice were significantly elevated relative to 45% fat-fed wild type mice at 120 minutes following glucose injection (Figure 2A). Area under the curve analysis of these data showed no significant difference between wild type and MsrA−/− on either diet (Figure 2B). However, we found that serum levels of insulin were higher in MsrA−/− mice following glucose injection suggesting a potential compensation for a reduced cellular response to insulin (Figure 2C). To address this possibility, we then performed insulin tolerance tests on these mice. In low fat-fed mice, we found no difference in insulin tolerance tests between wild type and MsrA−/− mice (Figure 2D). In high fat-fed mice, however, blood glucose levels in MsrA−/− mice remained significantly higher than wild type mice at 15 and 30 minutes following insulin injection suggesting that the lack of MsrA reduced insulin sensitivity under these conditions. Together, these data suggest that the lack of MsrA promotes the development of peripheral insulin resistance in response to high fat diet/obesity.
Figure 2. Lack of MsrA in mice promotes insulin resistance when fed a high fat diet.

A) Glucose tolerance tests for wild type (open circles) and MsrA−/− mice (closed circles) fed either low fat (left) or high fat diet (right). B) Average area under curve values for presented in A). Bars represent mean value ± SEM. C) Serum insulin levels for high fat fed mice following IP glucose injection. D) Insulin tolerance tests for wild type (open circles) and MsrA−/− mice (closed circles) fed either low fat (left) or high fat diet (right). For A, C and D, circles represent the mean value measured at each time point after IP injection. Error bars represent ± SEM. Asterisks represent p < 0.05 by Student t-test between genotypes at each given time point, p value given represents same. For glucose tolerance tests, sample sizes are: low fat wild type = 13, low fat MsrA−/− = 11, high fat wild type = 21, high fat MsrA−/− = 18. Serum insulin levels were measured for 3 mice/group. For insulin tolerance tests, sample sizes are low fat wild type = 7, low fat MsrA−/− = 6, high fat wild type = 12, high fat MsrA−/− = 9.
Reduced insulin signaling in high fat fed MsrA−/− mice
To understand the molecular mechanism(s) responsible for the increased insulin resistance in the absence of MsrA, we assessed whether MsrA−/− mice exhibited defects in insulin signaling in skeletal muscle and adipose tissue. Insulin resistance caused by obesity is commonly associated with reduction in insulin-stimulated signaling through the insulin signaling pathway with one of the most common defects a reduction in insulin-stimulated phosphorylation of Akt [30]. We measured Ser473 phosphorylation of Akt in skeletal muscle and white adipose tissue isolated from fasted mice given an IP injection of either saline or insulin (1 U/kg body weight). In low fat-fed animals, we found no difference between MsrA−/− and wild type mice in Ser473 phosphorylation of Akt in skeletal muscle or white adipose tissue in either saline-treated or insulin-treated mice (Figure 3A). This finding is consistent with the lack of difference between low fat-fed MsrA−/− and wild type mice in their response to insulin tolerance tests. In contrast, high fat feeding caused a significant reduction in the insulin-stimulated Ser473 phosphorylation of Akt in skeletal muscle and white adipose tissue from wild type mice; this reduction was exacerbated further in mice lacking MsrA (Figure 3A). We then characterized further the reduction in insulin signaling in high fat-fed mice lacking MsrA. In both skeletal muscle and adipose tissue from high fat fed mice, we found that tissues from MsrA−/− mice showed significant reductions in insulin-stimulated phosphorylation of GSK3α, GSK3β, and AS160 (Figure 3B). Together, these data are consistent with the results of insulin tolerance tests of high fat-fed mice wherein MsrA−/− mice showed a significantly blunted insulin response.
Figure 3. Reduction in insulin signaling in high fat fed MsrA−/− mice.

A) Representative blots (left) and quantification (right) for insulin-stimulated Ser473 phosphorylation of Akt in skeletal muscle and adipose tissue (WAT). The diet fed to each animal is indicated at the top, saline (0) or insulin (+) treatment is indicated by the “Insulin” line and genotype is indicated by “MsrA” line. For quantifications, mean values for p-Akt/Akt ratio are presented for wild type (open bars) and MsrA−/− mice (closed bars) with error bars representing ± SEM. Sample size = 3–7 for each group. p value is given for student t-test comparing groups indicated by horizontal line. B) Representative blot (left, WAT) and quantifications (right) for insulin-stimulated GSK3β phosphorylation (left) and AS160 phosphorylation in WAT and skeletal muscle from high fat fed mice. For quantifications, mean values for p-GSK3/GSK3 or p-AS160/AS160 ratio are presented for wild type (open bars) and MsrA−/− mice (closed bars) with error bars representing ± SEM. Sample size = 3–4 for each group. Asterisk represents p <0.05 between groups for given treatment.
Akt, GSK3 and AS160 are all downstream mediators of the insulin response. Because phosphorylation of all was reduced in MsrA−/− mice, we tested whether this could be explained by defects in upstream members of the insulin signaling pathway. To address this, we measured whether there were specific defects in the insulin stimulated phosphorylation of the InR in skeletal muscle and adipose tissue. Again, in low fat-fed mice we found no difference between MsrA−/− and wild type in the levels of InR phosphorylation; these groups did not differ in either the fasted or the insulin-stimulated state (Figure 4). However, we found animals fed the high fat diet showed a reduced phosphorylation of InR in response to insulin and that phosphorylation of InR was even further reduced in MsrA−/− mice compared to wild type mice. These data suggest that the reduced insulin response of MsrA−/− mice is caused in part by reduced ability to propagate signals from the InR.
Figure 4. Reduction in insulin stimulated phosphorylation of InR in high fat fed MsrA−/− mice.
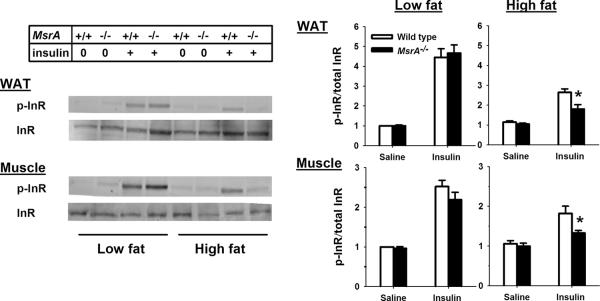
Representative blot (left) and quantification (right) for insulin-stimulated phosphorylation of InR in skeletal muscle and adipose tissue (WAT). The diet fed to each animal is indicated at the top, saline (0) or insulin (+) treatment is indicated by the “Insulin” line and genotype is indicated by “MsrA” line. For quantifications, mean values for p-InR/InR ratio are presented for wild type (open bars) and MsrA−/− mice (closed bars) with error bars representing ± SEM. Sample size = 3–7/group. P value is given for student t-test comparing groups indicated by horizontal line.
Oxidative stress reduces insulin receptor function
Because obesity affects many factors in vivo other than oxidative stress, we tested directly the effects of oxidative stress on InR function using C2C12 skeletal muscle cell lines treated with H2O2. Following treatment with H2O2, insulin stimulated phosphorylation of InR was reduced in a H2O2 dose-dependent manner (Figure 5A). These data were significant by 2 Way ANOVA (H2O2 dose and insulin treatment) and post-hoc analysis by the Holm-Sidak method determined that there were no differences in InR phosporylation among the samples not treated with insulin, whereas among samples treated with insulin, treatment with 10 μM or 30 μM H2O2 showed significant reduction in p-InR among the insulin-treated samples.
Figure 5. Oxidative stress reduces InR function.

A) Relative saline (open gray bars) or insulin-stimulated (gray hatched bars) InR phosophorylation of H2O2-treated C2C12 myotubes. B) Mean in vitro autophosphorylation of InR from H2O2-treated C2C12 myotubes. For both A and B, treatment dose of H2O2 given on horizontal axis. Bars represent mean of 4 independent experiments ± SEM. For A and B, letters shared by treatments indicate no difference between indicated groups by Holm-Sidak post-hoc test following 2 Way ANOVA. C) Mean in vitro autophosphorylation of InR from skeletal muscle (left) and adipose tissue (right) of wild type mice (open bars) and MsrA−/− mice (closed bars). Diet is indicated by X axis. Bars represent mean values; error bars represent ± SEM and p value is given for student t-test comparing groups indicated by horizontal line. Sample size = 3–4/group.
Binding of insulin to InR stimulates autophosphorylation of tyrosine residues in the InR β-subunit that increases the kinase activity of InR on downstream substrates [31]. Reduction of this function could directly diminish insulin response and induce insulin resistance. We determined then whether oxidative stress can alter the function of InR in terms of its autophosphorlation ability. Using an ELISA-based test of InR autophosphorylation, we found that InR isolated from cultures treated with H2O2 again showed a dose-dependent reduction in their ability to undergo autophosphorylation (Figure 5B). There was a significant effect of H2O2 dose on autophosphorylation of InR (F=7.1, p=0.012; 2 Way ANOVA comparing H2O2 and day of testing), with post-hoc analysis by Holm-Sidak showing that 30 μM H2O2 treatment significantly reduced autophosphorylation relative to control and 1 μM H2O2 treatment. Together, these data show that oxidative stress can directly diminish InR function, and suggest that oxidative damage of InR may be a proximate cause of exacerbated insulin resistance in MsrA−/− mice.
To address this idea, we tested whether high fat feeding and/or the lack of MsrA modified InR autophosphorylation in mice. Using the same ELISA-based assay as above, we found that InR isolated from the skeletal muscle of high fat-fed wild type mice had a significantly lower ability to undergo autophosporylation relative to InR isolated from low fat-fed wild type mice (Figure 5C). When compared to high fat-fed wild type, InR isolated from high fat-fed MsrA−/− skeletal muscle showed even further reduced autophosphorylation (Figure 5C). The effect of high fat feeding was even more dramatic when we analyzed InR isolated from the adipose tissue; high fat feeding causes a significant reduction in autophosphorylation that is further exacerbated by the lack of MsrA (Figure 5C).
To address whether these findings could be correlated with increased levels of oxidative damage, we measured levels of protein carbonylation, the most widely-used marker of protein oxidation [16]. High fat feeding caused a significant increase in carbonyl levels among total proteins isolated from both skeletal muscle and adipose tissue (Figure 6A). In addition, the lack of MsrA significantly exacerbated this effect in adipose tissue. To address whether InR was oxidatively damaged with high fat feeding, we used immunoprecipitation to isolate InR from carbonyl-labeled total cellular protein homogenates from skeletal muscle. Interestingly, we found that InR isolated from the tissues of high fat-fed mice was heavily carbonylated compared to low fat-fed mice (Figure 6B). Furthermore, InR isolated from MsrA−/− mice showed significantly higher levels of carbonyls than their wild type counterparts on each respective diet. These data show a clear correlation between InR oxidative damage and autophosphorylation function; that is, the highest levels of InR oxidative damage (high fat MsrA−/− mice) corresponded with the lowest ability of InR to undergo autophosphorylation. High fat feeding did not alter the levels of MsrA protein expression in either muscle or adipose from wild type mice suggesting that the increase in protein oxidation was likely due to increased oxidative stress and not diminished oxidative repair (Figure 6C). Together, these data suggest that MsrA plays an significant role in regulating the decline in InR function associated with obesity and high fat feeding. Furthermore, these data suggest that one factor in the development of insulin resistance with obesity may be oxidation of proteins associated with the insulin signaling pathway.
Figure 6. Increased protein oxidation directly affects InR.

A) Total cellular protein carbonyl levels in muscle and WAT. Representative carbonyl gels (left) and quantification (right) are presented and tissue type, genotype, and diet are indicated. B) Representative experiment (top) and quantification (bottom) for relative carbonyl levels in isolated InR. For A) and B), bars represent mean values from wild type mice (open bars) and MsrA−/− mice (closed bars) ± SEM with a sample size of 4–5/group. Values are presented as relative to those from low fat wild type mice for each tissue. Asterisk represents p <0.05 between groups for given treatment. C) Representative blot (top, adipose) and quantification (bottom) of relative MsrA expression for muscle and WAT isolated from wild type mice fed either low fat (gray) or high fat (hatched gray) diets. Bars represent mean MsrA/Actin ratio ± SEM for a sample size of 4/group
Discussion
The results of this study suggest a novel hypothesis that the protein repair enzyme MsrA can play a significant role in the development of obesity-induced insulin by preserving the function of InR. The function of MsrA is specific to the protection of proteins from oxidative stress, and thus these results hint that oxidative damage to insulin signaling proteins like InR might lead to defects in their function that could contribute in part to the etiology of insulin resistance. It is known that excessive fat accumulation and high blood glucose levels are associated with increased oxidative stress [4], but prior to this study, there has been limited evidence that oxidative damage to proteins in the insulin signaling pathway could lead to general reductions in their function. There had been some previous suggestion in endothelial cell lines that oxidative stress can cause oxidation of InR that reduces the internalization of insulin in cells, thereby diminishing insulin sensitivity [19]. Oxidative stress also reduces the ability of PI3K to redistribute to correct subcellular locations necessary for efficient transduction of signals following insulin stimulation [20]. GLUT4 function is also hindered by oxidative stress, in part due to oxidant effects on PI3K and in part due to oxidative damage to nuclear proteins that form protein-DNA complexes to regulate GLUT4 expression [32,33]. Our results are in line with these studies and suggest that protection of InR from oxidation could potentially prevent insulin resistance caused by obesity. To better understand the novel relationship between protein oxidation and insulin resistance, it will be important to more fully characterize the oxidation status of these proteins, and others, under different conditions including oxidative stress and obesity. Within the context of these studies, MsrA mutant mice, which have altered levels of a primary component of protein oxidation repair, will be an interesting and important model to further explore this paradigm.
The role of InR in the development of insulin resistance is still controversial, but there are reports that the early etiology of reduced insulin response can be attributed to reduced function of InR. The binding of insulin to the α-subunit of InR causes a conformational change of the β-subunits of InR that then undergo autophosphorylation of multiple tyrosine residues in an ATP-dependent process. Our findings are similar to those of Youngren et al. who found that InR autophosphorylation in skeletal muscle was significantly reduced after feeding rats a high fat/high sucrose diet for just 2 weeks [34], though others have been able to confirm this [35]. Skeletal muscles samples from obese patients show general reduced insulin action due to impaired insulin-induced InR autophosphorylation [36,37]. Because autophosphorylation of InR is required for subsequent InR tyrosine phosphorylation and activation of IRS-1, this could be a proximate cause of insulin resistance in these models. Oxidative stress (H2O2) in acute doses can stimulate InR phosphorylation [38,39]; however, the data here suggest that chronic oxidative stress both in vivo and in vitro can decrease this process. Our data are in line with findings by Zhou et al. and Van den Dobbelsteen et al. that show high doses of peroxinitrite or redox-cycling agents can inhibit InR autophosphorylation [40,41]. It is interesting to note that calorie restriction, which is known to reduce oxidative stress and oxidative damage, has also been shown to increase insulin-induced InR phosphorylation [42,43].
Future studies to determine the therapeutic potential of MsrA or other means to reduce protein oxidation will require fine-scale analysis of the potential sites of oxidation on InR that may contribute to reductions in insulin-stimulated phosphorylation and autophosphorylaion. It is conceivable that Met residues within the autophosphoylation domain of InR might be prone to oxidation that thereby reduces their function. Leconte and Clauser studied two sequences flanking the InR autophosphorylation site, one of which contains a Met residue [44]. Replacement of this Met residue with Leucine ablated tyrosine kinase activity required for InR autophosphorylation [44]. Presumably, oxidation of this Met residue might also reduce InR function and insulin sensitivity. Cama et al. found a Met located near the tyrosine phosphorylation sites of InR (Met-1153) that, when substituted with Isoleucine, impaired InR autophosphorylation in 3T3L1 cell lines [45]. Furthermore, this same substitution in InR was found to be associated with human case of insulin resistance with obesity [45]. It is also possible that MsrA could protect other amino acid residues in InR and other proteins. It has been previously proposed that because Met residues can be repaired by Msr, these residues may act as a “free radical sink” to protect other amino acids from oxidation [17]. There are 34 Met residues among the 1372 amino acids of InR [NCBI Accession Number P15208]. Under normal conditions, these Met residues may be present to protect the other amino acids. When MsrA is lacking, this might then promote the oxidation of neighboring amino acids and lead to the reduction of InR function. It will be of great interest in future studies to characterize the potential sites of oxidation on InR that may lead to insulin resistance.
There is growing evidence that the MsrA gene may significantly alter adiposity and glucose metabolism in human populations. Lindgren et al. found that a single nucleotide polymorphism (SNP) in a locus near the MsrA gene was significantly correlated with central adiposity and fat distribution in a meta-analysis of genome-wide association studies (GWAS) data in individuals of European origin [26]. A second study found that this locus is also significantly correlated with childhood and adolescent extreme obesity and adult waist circumference [28]. A similar association between this locus and body mass index and central obesity was found in a group made up of individuals of Singaporean Chinese, Malay and Asian-Indian populations and a group of adult Danes respectively [27,46]. Because GWAS can only confirm associations and not causality, it is not clear whether these associations are due to alterations in MsrA activity. Clearly, our data support the notion that genetic reduction of MsrA activity promotes insulin resistance associated with obesity and thus raise the possibility that Msr activity and MsrA might be viewed as a potential treatment option or preventative for insulin resistance and metabolic disease. It is interesting to note that in obese rats, MsrA activity has been shown to be reduced in adipose tissue [47]. Identification of putative activators of Msr will be a critical step in the design of future translational studies.
In this study, we report three significant findings: 1) reduction of MsrA in mice has a significant effect on susceptibility to obesity-induced insulin resistance, 2) the function of InR is reduced with both oxidative stress and obesity and can be correlated to increased oxidative damage to InR itself, and 3) the lack of MsrA exacerbates the effects on InR associated with obesity. Whereas previous studies have shown that enhancement of the antioxidant defense system in mice (either through pharmaceutical treatment or through genetic mutation) can prevent the development of insulin resistance when fed a diet high in fat [7,48], MsrA does not generally reduce oxidative stress, but rather acts to protect proteins from oxidation. Thus, the findings presented here hint at the importance of protein oxidation, as regulated by MsrA, in the development of insulin resistance. Supporting this idea, oxidative damage caused a significant reduction in InR function in cells, and reduction of MsrA expression exacerbates the decline in InR function with obesity. These findings point to oxidation of InR as a significant cause of reduced insulin signaling with obesity, and suggest that MsrA and Msr activity may be a novel future target for treatment of obesity-associated metabolic diseases.
Highlights
Mice lacking MsrA are susceptible to obesity-induced insulin resistance
Fat accumulation promotes protein oxidation in muscle and adipose
Oxidative stress significantly reduces insulin receptor function
MsrA protects insulin receptor function to preserve glucose metabolism in mice
Acknowledgments
The authors would like to thank Rodney Levine, Geumsoo Kim, and Hang Zhao from the NIH/NHLBI Laboratory of Biochemistry for sharing MsrA−/− mice to generate our breeding colonies. This study was supported by National Institutes of Health (NIH) training grant T32 AG021890-05, NIH MERIT grant R37AG026557, and the Mitochondrial Function and Oxidative Damage Core Facility of the San Antonio Nathan Shock Center of Excellence in the Basic Biology of Aging.
Abbreviations
- Akt
protein kinase B
- InR
insulin receptor
- IP
intraperitonial
- IRS-1
insulin receptor substrate 1
- Met
methionine
- MetO
methionine sulfoxide
- Msr
methionine sulfoxide reductase
- MsrA
methionine sulfoxide reductase A
- PI3K
phosphatidylinositol 3-kinase
- T2DM
Type 2 diabetes mellitus
- QMRi
quantitative magnetic resonance imaging
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM. A balanced overview. Diabetes Care. 1992;15:318–368. doi: 10.2337/diacare.15.3.318. [DOI] [PubMed] [Google Scholar]
- 2.Kahn CR. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes. 1994;43:1066–1084. doi: 10.2337/diab.43.8.1066. [DOI] [PubMed] [Google Scholar]
- 3.Frohnert BI, Sinaiko AR, Serrot FJ, Foncea RE, Moran A, et al. Increased Adipose Protein Carbonylation in Human Obesity. Obesity. 2011;19:1735–1741. doi: 10.1038/oby.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keaney JF, Jr., Larson MG, Vasan RS, Wilson PW, Lipinska I, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol. 2003;23:434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 6.Grimsrud PA, Picklo MJ, Griffin TJ, Bernlohr DA. Carbonylation of Adipose Proteins in Obesity and Insulin Resistance. Molecular & Cellular Proteomics. 2007;6:624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. The Journal of Clinical Investigation. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruggiero C, Ehrenshaft M, Cleland E, Stadler K. High-fat diet induces an initial adaptation of mitochondrial bioenergetics in the kidney despite evident oxidative stress and mitochondrial ROS production. Am J Physiol Endocrinol Metab. 2011;300:E1047–1058. doi: 10.1152/ajpendo.00666.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gardner CD, Eguchi S, Reynolds CM, Eguchi K, Frank GD, et al. Hydrogen peroxide inhibits insulin signaling in vascular smooth muscle cells. Exp Biol Med. 2003;228:836–842. doi: 10.1177/15353702-0322807-09. [DOI] [PubMed] [Google Scholar]
- 10.Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem. 1999;274:27905–27913. doi: 10.1074/jbc.274.39.27905. [DOI] [PubMed] [Google Scholar]
- 11.Dokken BB, Saengsirisuwan V, Kim JS, Teachey MK, Henriksen EJ. Oxidative stress-induced insulin resistance in rat skeletal muscle: role of glycogen synthase kinase-3. Am J Physiol Endocrinol Metab. 2008;294:E615–621. doi: 10.1152/ajpendo.00578.2007. [DOI] [PubMed] [Google Scholar]
- 12.Archuleta TL, Lemieux AM, Saengsirisuwan V, Teachey MK, Lindborg KA, et al. Oxidant stress-induced loss of IRS-1 and IRS-2 proteins in rat skeletal muscle: role of p38 MAPK. Free Radic Biol Med. 2009;47:1486–1493. doi: 10.1016/j.freeradbiomed.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 14.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH2-terminal Kinase Promotes Insulin Resistance during Association with Insulin Receptor Substrate-1 and Phosphorylation of Ser307. Journal of Biological Chemistry. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 15.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, et al. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 16.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 17.Levine RL, Berlett BS, Moskovitz J, Mosoni L, Stadtman ER. Methionine residues may protect proteins from critical oxidative damage. Mechanisms of Ageing and Development. 1999;107:323–332. doi: 10.1016/s0047-6374(98)00152-3. [DOI] [PubMed] [Google Scholar]
- 18.Pierce A, Mirzaei H, Muller F, De Waal E, Taylor AB, et al. GAPDH is conformationally and functionally altered in association with oxidative stress in mouse models of amyotrophic lateral sclerosis. J Mol Biol. 2008;382:1195–1210. doi: 10.1016/j.jmb.2008.07.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertelsen M, Anggard EE, Carrier MJ. Oxidative stress impairs insulin internalization in endothelial cells in vitro. Diabetologia. 2001;44:605–613. doi: 10.1007/s001250051667. [DOI] [PubMed] [Google Scholar]
- 20.Tirosh A, Potashnik R, Bashan N, Rudich A. Oxidative stress disrupts insulin-induced cellular redistribution of insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activation and GLUT4 translocation. J Biol Chem. 1999;274:10595–10602. doi: 10.1074/jbc.274.15.10595. [DOI] [PubMed] [Google Scholar]
- 21.Lee BC, Gladyshev VN. The biological significance of methionine sulfoxide stereochemistry. Free Radic Biol Med. 2011;50:221–227. doi: 10.1016/j.freeradbiomed.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadtman ER, Van Remmen H, Richardson A, Wehr NB, Levine RL. Methionine oxidation and aging. Biochim Biophys Acta. 2005;1703:135–140. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Hansel A, Kuschel L, Hehl S, Lemke C, Agricola H-J, et al. Mitochondrial targeting of the human peptide methionine sulfoxide reductase (MSRA), an enzyme involved in the repair of oxidized proteins. The FASEB Journal. 2002;16:911–913. doi: 10.1096/fj.01-0737fje. [DOI] [PubMed] [Google Scholar]
- 24.Vougier S, Mary J, Friguet B. Subcellular localization of methionine sulphoxide reductase A (MsrA): evidence for mitochondrial and cytosolic isoforms in rat liver cells. Biochem J. 2003;373:531–537. doi: 10.1042/BJ20030443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:15036–15040. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindgren CM, Heid IM, Randall JC, Lamina C, Steinthorsdottir V, et al. Genome-Wide Association Scan Meta-Analysis Identifies Three Loci Influencing Adiposity and Fat Distribution. PLoS Genet. 2009;5:e1000508. doi: 10.1371/journal.pgen.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bille DS, Banasik K, Justesen JM, Sandholt CH, Sandbæk A, et al. Implications of Central Obesity-Related Variants in LYPLAL1, NRXN3, MSRA, and TFAP2B on Quantitative Metabolic Traits in Adult Danes. PLoS One. 2011;6:e20640. doi: 10.1371/journal.pone.0020640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scherag A, Dina C, Hinney A, Vatin V, Scherag S, et al. Two New Loci for Body-Weight Regulation Identified in a Joint Analysis of Genome-Wide Association Studies for Early-Onset Extreme Obesity in French and German Study Groups. PLoS Genet. 2010;6:e1000916. doi: 10.1371/journal.pgen.1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salmon AB, Perez VI, Bokov A, Jernigan A, Kim G, et al. Lack of methionine sulfoxide reductase A in mice increases sensitivity to oxidative stress but does not diminish life span. FASEB J. 2009;23:3601–3608. doi: 10.1096/fj.08-127415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zierath JR, Krook A, Wallberg-Henriksson H. Insulin action and insulin resistance in human skeletal muscle. Diabetologia. 2000;43:821–835. doi: 10.1007/s001250051457. [DOI] [PubMed] [Google Scholar]
- 31.Ellis L, Clauser E, Morgan DO, Edery M, Roth RA, et al. Replacement of insulin receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase activity and uptake of 2-deoxyglucose. Cell. 1986;45:721–732. doi: 10.1016/0092-8674(86)90786-5. [DOI] [PubMed] [Google Scholar]
- 32.Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, et al. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes. 1998;47:1562–1569. doi: 10.2337/diabetes.47.10.1562. [DOI] [PubMed] [Google Scholar]
- 33.Pessler D, Rudich A, Bashan N. Oxidative stress impairs nuclear proteins binding to the insulin responsive element in the GLUT4 promoter. Diabetologia. 2001;44:2156–2164. doi: 10.1007/s001250100024. [DOI] [PubMed] [Google Scholar]
- 34.Youngren JF, Paik J, Barnard RJ. Impaired insulin-receptor autophosphorylation is an early defect in fat-fed, insulin-resistant rats. Journal of Applied Physiology. 2001;91:2240–2247. doi: 10.1152/jappl.2001.91.5.2240. [DOI] [PubMed] [Google Scholar]
- 35.Pujol A, Cousin B, Burnol AF, Loizeau M, Picon L, et al. Insulin receptor kinase activity in muscles and white adipose tissue during course of VMH obesity. American Journal of Physiology - Endocrinology And Metabolism. 1992;262:E161–E166. doi: 10.1152/ajpendo.1992.262.2.E161. [DOI] [PubMed] [Google Scholar]
- 36.Goodyear LJ, Giorgino F, Sherman LA, Carey J, Smith RJ, et al. Insulin receptor phosphorylation, insulin receptor substrate-1 phosphorylation, and phosphatidylinositol 3-kinase activity are decreased in intact skeletal muscle strips from obese subjects. J Clin Invest. 1995;95:2195–2204. doi: 10.1172/JCI117909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arner P, Pollare T, Lithell H, Livingston JN. Defective insulin receptor tyrosine kinase in human skeletal muscle in obesity and type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1987;30:437–440. doi: 10.1007/BF00292549. [DOI] [PubMed] [Google Scholar]
- 38.Hayes GR, Lockwood DH. Role of insulin receptor phosphorylation in the insulinomimetic effects of hydrogen peroxide. Proceedings of the National Academy of Sciences. 1987;84:8115–8119. doi: 10.1073/pnas.84.22.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ihara Y, Yasuoka C, Kageyama K, Wada Y, Kondo T. Tyrosine phosphorylation of clathrin heavy chain under oxidative stress. Biochemical and Biophysical Research Communications. 2002;297:353–360. doi: 10.1016/s0006-291x(02)02195-2. [DOI] [PubMed] [Google Scholar]
- 40.Zhou J, He X, Huang K. Bidirectional regulation of insulin receptor autophosphorylation and kinase activity by peroxynitrite. Archives of Biochemistry and Biophysics. 2009;488:1–8. doi: 10.1016/j.abb.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 41.Van den Dobbelsteen DJ, Toxopeus C, van Holsteijn CWM, Nooy HA, Droog CI, et al. Effect of alkylating and redox cycling quinones on insulin receptor autophosphorylation. Toxicology in Vitro. 1994;8:563–567. doi: 10.1016/0887-2333(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 42.Zhu M, de Cabo R, Anson RM, Ingram DK, Lane MA. Caloric restriction modulates insulin receptor signaling in liver and skeletal muscle of rat. Nutrition. 2005;21:378–388. doi: 10.1016/j.nut.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 43.Yu BP. Aging and oxidative stress: Modulation by dietary restriction. Free Radical Biology and Medicine. 1996;21:651–668. doi: 10.1016/0891-5849(96)00162-1. [DOI] [PubMed] [Google Scholar]
- 44.Leconte I, Clauser E. Two sequences flanking the major autophosphorylation site of the insulin receptor are essential for tyrosine kinase activation. Biochem J. 1995;306(Pt 2):465–472. doi: 10.1042/bj3060465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cama A, Sierra MDLL, Ottini L, Kadowski T, Gorden P, et al. A Mutation in the Tyrosine Kinase Domain of the Insulin Receptor Associated with Insulin Resistance in an Obese Woman. Journal of Clinical Endocrinology & Metabolism. 1991;73:894–901. doi: 10.1210/jcem-73-4-894. [DOI] [PubMed] [Google Scholar]
- 46.Dorajoo R, Blakemore AIF, Sim X, Ong RTH, Ng DPK, et al. Replication of 13 obesity loci among Singaporean Chinese, Malay and Asian-Indian populations. Int J Obes. 2011 doi: 10.1038/ijo.2011.86. [DOI] [PubMed] [Google Scholar]
- 47.Uthus EO, Picklo MJ. Obesity reduces methionine sulphoxide reductase activity in visceral adipose tissue. Free Radical Research. 2011;45:1052–1060. doi: 10.3109/10715762.2011.591793. [DOI] [PubMed] [Google Scholar]
- 48.Hoehn KL, Salmon AB, Hohnen-Behrens C, Turner N, Hoy AJ, et al. Insulin resistance is a cellular antioxidant defense mechanism. Proceedings of the National Academy of Sciences. 2009;106:17787–17792. doi: 10.1073/pnas.0902380106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaudhuri AR, de Waal EM, Pierce A, Van Remmen H, Ward WF, et al. Detection of protein carbonyls in aging liver tissue: A fluorescence-based proteomic approach. Mech Ageing Dev. 2006;127:849–61. doi: 10.1016/j.mad.2006.08.006. [DOI] [PubMed] [Google Scholar]