

Abstract
Bone marrow–mesenchymal stem cells (BM-MSC) ameliorate renal dysfunction and repair tubular damage of acute kidney injury by locally releasing growth factors, including the insulin-like growth factor-1 (IGF-1). The restricted homing of BM-MSC at the site of injury led us to investigate a possible gene-based communication mechanism between BM-MSC and tubular cells. Human BM-MSC (hBM-MSC) released microparticles and exosomes (Exo) enriched in mRNAs. A selected pattern of transcripts was detected in Exo versus parental cells. Exo expressed the IGF-1 receptor (IGF-1R), but not IGF-1 mRNA, while hBM-MSC contained both mRNAs. R- cells lacking IGF-1R exposed to hBM-MSC-derived Exo acquired the human IGF-1R transcript that was translated in the corresponding protein. Transfer of IGF-1R mRNA from Exo to cisplatin-damaged proximal tubular cells (proximal tubular epithelial cell [PTEC]) increased PTEC proliferation. Coincubation of damaged PTEC with Exo and soluble IGF-1 further enhanced cell proliferation. These findings suggest that horizontal transfer of the mRNA for IGF-1R to tubular cells through Exo potentiates tubular cell sensitivity to locally produced IGF-1 providing a new mechanism underlying the powerful renoprotection of few BM-MSC observed in vivo.
Introduction
Cells communicate and exchange information by different ways, including the secretion of soluble factors, the cell-to-cell adhesion contact, and the intercellular exchange of organelles through nanotubular structures [1]. Recent studies have proposed that cell-derived small circular membrane vesicles, called exosomes (Exo) and microvesicles (MV), represent an additional mechanism of cell-to-cell communication by transfer of membrane components as well as the cytoplasmic content [2,3]. Shuttling of genetic material via cell membrane vesicles might have driven the unicellular organism evolution in the oceans. High frequency of horizontal gene transfer could explain the genomic flexibility of marine microbial populations that facilitated their adaptation to changing environmental conditions [4]. Cells physiologically release vesicles whose quantity generally increases upon cell activation after hypoxia, oxidative injury, exposure to complement proteins, and shear stress [2,5,6]. Two distinct populations of vesicles with peculiar membrane structure, mechanism of production, pathophysiological relevance, and different size have been described. Membrane fragments of 30–90 nm diameter derived from the endosomal membrane compartment after fusion of secretory granules with the plasma membrane are defined as Exo. Once released, Exo bind the recipient cells through receptor–ligand interactions or fuse with the target cell membrane transferring membrane components, including cell receptors [2], and discharging the portion of cytosol segregated within their lumen into the cytoplasm of recipient cells [7]. The molecular cargo content of Exo derives from active packaging of certain nucleic acid species leading to the presence of mRNAs in Exo that are not found in donor cells [8]. Furthermore, relatively large MV (100 nm–1 μm diameter) are formed from the surface membrane of activated cells in a calcium and calpain-dependent manner following a disordered function of phospholipid transporters that results in the budding of altered membrane that exposes phosphatidylserine in the outer leaflet. MV and Exo contain biomolecules, including mRNA and microRNA, packaged in a random process and their release is considered an expression of a pathological process in place. Molecular transfer from MV/Exo contributes to changes in the maturation and differentiation of target cells as MV/Exo released by endothelial progenitor cells trigger neo-angiogenesis in endothelial cells [9]. Consistently, embryonic stem (ES) cells released-MV/Exo are able to reprogram hematopoietic progenitor cells [10] and bone marrow–mesenchymal stem cells (BM-MSC)-derived MV/Exo have regenerative capacity [11]. BM-MSC contribute to organ repair by virtue of their unique tropism and proregenerative capacity [12–15]. In previous studies, we found that infusion of BM-MSC protected mice with cisplatin-induced acute kidney injury (AKI) from tubular damage and renal function deterioration by stimulating the proliferation of resident renal cells [12, 16]. The insulin-like growth factor-1 (IGF-1), highly expressed in BM-MSC as mRNA and protein, was a major mediator for such an effect as gene silencing of the IGF-1 in BM-MSC, significantly decreased in vitro proliferation and increased apoptosis of cocultured proximal tubular cells (proximal tubular epithelial cell [PTEC]) [17]. Consistently, IGF-1-silenced MSC displayed limited protection from functional and structural damage in mice with cisplatin-induced AKI [17]. The restricted homing of BM-MSC at the site of injury in the face of a robust functional recovery of the injured tissue [12,18] has raised the interest to investigate additional mechanisms that act in concert with the release of growth factors to explain the MSC's strong renoprotective effect. Here we sought to investigate whether a communication mechanism through Exo could operate between BM-MSC and damaged proximal tubular cells by shuttling mRNA.
Materials and Methods
Cell cultures and incubations
Human (h) MSC were obtained from BM aspirates collected from adult subjects (30–40 years old) within the Hematology Division, Azienda Ospedaliera, Ospedali Riuniti di Bergamo [19]. Human BM-MSC (hBM-MSC) were plated at 2×105 cells per cm2 in the Dulbecco's modified Eagle's medium–low-glucose (DMEM-low glucose; Gibco) containing 0.1 mM gentamicin and 1,000 IU of heparin in the presence of 5% platelet lysate as previously described [19]. Nonadherent cells were removed after 3–4 days, and a fresh medium was added. At subconfluence, cells were recovered after trypsin-EDTA treatment, expanded, and subsequently characterized. By FACS analysis, human BM-MSC showed positivity for the CD29, CD90, CD73, and CD105 markers, and negativity for the hematopoietic markers CD45, CD34, CD14, and HLA-DR. Human BM-MSC had the capacity to differentiate toward adipocytes and osteoblasts [19,20]. All the experiments were performed with hBM-MSC derived from a pool of 3 donors between passages 5 and 8.
Mouse MSC derived from BM (mBM-MSC) of 2-month-old C57BL6/J mice [16] were obtained by their tendency to adhere tightly to plastic culture dishes [21]. Cells were plated at the density of 1×106/cm2 in the DMEM plus 10% fetal calf serum (FCS; Invitrogen) and antibiotics [22]; after 6 h, nonadherent cells were removed. At 2 weeks, subconfluent mBM-MSC were immunodepleted of CD45+ cells, characterized for their ability to differentiate to adipocytes, osteoblats, and chondroblasts [16] and used to isolate Exo.
Normal human dermal fibroblasts (NHDF) were obtained by Lonza. R- cells, a mouse fibroblast cell line knockout for the IGF-1R, were provided by Dr. Renato Baserga (Thomas Jefferson University, Philadelphia, PA) and were grown as previously described [23].
Human PTECs (HK2; American Type Culture Collection), were grown as previously described [24]. For evaluation of horizontal transfer experiments, 5×105 cells were incubated with mBM-MSC-derived Exo for 1 and 3h in the serum-deprived DMEM. After 3 washing steps with a 1×phosphate buffer solution (PBS), total RNA was extracted from HK2 and used for evaluation of mouse IGF-1R expression by real-time polymerase chain reaction (PCR).
The mouse PTEC line was provided by Dr. Eric G. Neilson (Vanderbilt University, Nashville, TN). PTECs were grown as described previously [25]. For in vitro studies, PTECs were seeded at 46×103 cells/cm2 and 24 h later incubated with the DMEM plus 2% FCS alone (test medium) or in the presence of 2.5 μM cis-platinum (II)-diamine dichloride (cisplatin; Sigma-Aldrich) for 6 h [17]. To study the effect of Exo on tubular cell proliferation in coculture, cisplatin-pretreated PTECs were incubated with a test medium or hBM-MSC-derived Exo (corresponding to a final concentration of 1 μg/mL of Exo proteins) or IGF-1 (5 ng/mL) [17] or both for 4 days. PTECs in the test medium served as controls. After 4 days, PTEC proliferation was estimated by cell count using trypan blue dye exclusion (Sigma-Aldrich).
Isolation of Exo
Enriched preparations of Exo were purified as previously described [8,9,11]. Briefly, hBM-MSC were cultured for 48 h in a medium containing platelet lysate previously ultracentrifugated overnight at 100.000 g at 4°C to remove exogenous MV and Exo. Mouse BM-MSC or NHDF were cultured for 48 h in a medium containing FCS ultracentrifugated as above. Exo were then obtained from conditioned supernatants of human or mouse BM-MSC and NHDF cultured for 18 h in the DMEM-low-glucose medium deprived of platelet lysate or FCS, respectively. After centrifugation at 2000 g for 20 min and filtration through 0.2-μm filters to remove cell debris and particles larger than 200 nm, cell-free supernatants were ultracentrifuged at 100.000 g for 1 h at 4°C, washed in PBS, and submitted to a second ultracentrifugation at the same condition. The Exo pellet was suspended in 100 μL PBS and the protein content quantified by the Bradford method (BioRad). For in vitro experiments, the Exo pellet was suspended in the culture medium. In selected experiments, Exo were treated with 10 μg/mL of RNase A (Ambion Inc.) for 1 h at 37°C, the reaction was stopped by the addition of a 10 U/mL RNase inhibitor (Ambion), and Exo were washed by ultracentrifugation. The effectiveness of RNase treatment was evaluated after RNA extraction using the Trizol reagent (Invitrogen) and submitted to reverse transcription (RT)-PCR analysis.
Electron microscopy analysis
The Exo pellet from hBM-MSC was fixed in 2% paraformaldehyde, and then loaded to copper grids (100 mesh) coated with Formvar. After washing, the grids were contrasted in 2% uranyl acetate, dried, and then examined by transmission electron microscopy (Morgagni 268D; Philips). The identity of the vesicles as Exo has been confirmed by the presence of the tetraspan surface proteins CD63 and CD9 by immunogold labeling of the grids over night at room temperature with primary antibodies (dilution 1:100 for CD63, BD Pharmingen; dilution 1:100 for CD9, Santa Cruz Biotechnology, Inc). In addition, the presence of the IGF-1R protein has been evaluated by immunogold labeling of the grids over night at room temperature with a primary antibody (dilution 1:50, Abcam, ab5497). As a positive control, immunogold labeling for IGF-1R was concomitantly performed on thin sections of human renal tissue obtained from a patient who had undergone nephrectomy for kidney adenocarcinoma. The grids were then exposed for 1 h to species-specific anti-IgG antibodies conjugated to colloidal particles: 6 nm for CD9 and 12 nm for CD63 and IGF-1R detection. Human BM-MSC cultured on filters were fixed by immersion in 2.5% glutaraldehyde in a 0.1 M cacodylate buffer (pH 7.4) for 1 h at 4°C, and then washed in the cacodylate buffer. Strips of filter of 2 mm×1 cm were cut with a razor blade and subsequently postfixed in 1% osmium tetroxide for 1 h, dehydrated through ascending grades of alcohol, and embedded in Epon resin. Semithin sections were stained with toluidine blue in borax and examined by light microscopy. Ultrathin sections (60 to 100 nm) were cut on an ultramicrotome (Leica Microsystems EM UC7), collected on copper grids, and stained with uranyl acetate and lead citrate, and then examined by transmission electron microscopy (Morgagni 268D; Philips).
Quantification of DNA and RNA in Exo
DNA and RNA from hBM-MSC-derived Exo were isolated using Trizol (Invitrogen) according to the manufacturer's protocol or using the RNeasy mini kit (Qiagen), suspended in 10 μL RNase-free water, and quantified by Nanodrop ND-1000.
Reverse transcription–polymerase chain reaction end point
RT-PCR analysis for IGF-1 and IGF-1R was performed on hBM-MSC or NHDF and their relative-derived Exo. GAPDH gene expression has been evaluated on hBM-MSC and their Exo, treated or not with RNase A. First-strand cDNA was produced from 0.5 μg total RNA using the SuperScript II First-Strand Synthesis Systems Kit (Invitrogen). No enzyme was added for reverse transcriptase-negative controls (RT-). The PCR protocol was 4 min at 94°C; 30 s at 94°C, 45 s at 56°C, 1 min at 72°C for 35 cycles; 72°C 5 min. The following primers were used: human IGF-1 (transcript of 349 bp), forward 5′-TGGATGCTCTTCAGTTCGTG-3′ and reverse 5′-ACTCGTGCAGAGCAAAGGAT-3′; human IGF-1R (transcript of 351 bp), forward 5′-AACCCCAAGACTGAGGTGTG-3′ and reverse 5′-ATCGATGCGGTACAATGTGA-3′; GAPDH (transcript of 450 bp), forward 5′-ACCACAGTCCATGCCATCAC-3′ and reverse 5′-TCCACCACCCTGTTGCTGTA-3′. Samples were then separated on a 1.5% agarose gel. To evaluate the integrity of the IGF-1R mRNA into the Exo, first-strand cDNA was produced from 0.2 μg total RNA using the SuperScript II First-Strand Synthesis Systems Kit (Invitrogen). cDNA was submitted to PCR using the following primers: forward 5′-AAAAGGAATGAAGTCTGGCT-3′ and reverse 5′-GGATCCAAGGATCAGCAGGT-3′ (transcript of 4122 bp). The PCR protocol was 4 min at 94°C; 45 s at 94°C, 45 s at 58°C, 5 min at 72°C for 40 cycles; 72°C 10 min.
Quantitative real-time PCR
Total RNA was extracted from cells and Exo using the Trizol reagent and contaminating genomic DNA in murine and human cells were removed by RNase-free DNase (Promega) for 1h at 37°C. The first-strand cDNA (1μg for R- and HK2 cells or 0.15 μg for hBM-MSC silenced for IGF-1R and their respective Exo) was produced using a mixture of oligo-dT (0.25 μg) and random examers oligonucleotides (50 ng) and 200 U of SuperScript II RT (Invitrogen) for 1 h at 42°C. No enzyme was added for reverse transcriptase-negative controls (RT-).
Amplification was performed on the 7300 Real-Time PCR System with the SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer's protocol. After an initial hold of 2 min at 50°C and 10 min at 95°C, the samples were cycled 40 times at 95°C for 15 s and 60°C for 60 s to reach the plateau. The comparative Ct method normalizes the number of target gene copies to the housekeeping gene as 18S (ΔCt). Gene expression was then evaluated by the quantification of cDNA corresponding with the target gene relative to a calibrator sample serving as a physiologic reference (ΔΔCt). On the basis of exponential amplification of the target gene as well as the calibrator, the amount of amplified molecules at the threshold cycle is given by: 2−ΔΔCt. The following primers were used: human IGF-1 receptor (300 nM): forward 5′-CCGGAATTGCATGGTAGCC-3′ reverse 5′-GCGTCATACCAAAATCTCCGA-3′; mouse IGF-1 receptor (300 nM): forward 5′-TAGGAACTGCATGGTAGCC-3′ reverse 5′-GTGTCATACCGAAATCTCCAA-3′; 18S (50 nM) forward 5′- ACGGCTACCACATCCAAGGA-3′, reverse 5′-CGGGAGTGGGGTAATTTGCG-3′; human BCL2-like1 (300 nM): forward 5′-TTCTCCTTCGGCGGGGCACT-3′; reverse 5′- AAGTATCCCAGCCGCCGTTCT-3′.
For real-time PCR array, total RNA was extracted from human and mouse BM-MSC and their respective Exo using the Qiagen RNeasy Mini kit, and cDNA (0.5–1 μg) was synthesized with the RT2 First-Strand Kit following the manufacturer's procedure. The human Insulin Signaling Pathway RT2 Profiler PCR Array (PAHS-030A; SABiosciences, Qiagen), the human PI3K/AKT Signaling Pathway RT2 Profiler (PAHS-058A), the mouse Insulin Signaling Pathway RT2 Profiler PCR Array (PAMM-030A), and the mouse PI3K/AKT Signaling Pathway RT2 Profiler (PAMM-058A) were used.
Immunofluorescence
After 24 h exposition to hBM-MSC-derived exosomes (hExo), R- cells were fixed in 2% paraformaldehyde plus 4% sucrose in PBS, pH 7.4, for 10 min at 37°C, and then permeabilized with 0.3% Triton for 10 min at room temperature. After 3 washings with PBS, nonspecific binding sites were saturated in a blocking solution (5% FCS, 2% bovine serum albumin, and 0.2% bovine gelatin in PBS) for 45 min at room temperature. R- cells were then incubated overnight at 4°C with a rabbit polyclonal anti-IGF-1Rβ antibody detecting the cytoplasmic domain of the receptor (Santa Cruz Biotechnology, sc-713, dilution 1:100). A FITC-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, dilution 1:50) was then added for 1 h at room temperature. DAPI staining was used to visualize nuclei. Representative images were acquired with an inverted confocal laser-scanning microscope. Human BM-MSC expressing IGF-1R were used as a positive control. A negative control was performed on R- not receiving the Exo and on R- omitting the primary antibody. The number of cells positive for IGF-1R was evaluated in at least 10 randomly selected high-power microscope fields (×40) and related to the total number of Dapi-positive cells in each field. Percentage is expressed as mean±SE of 3 independent experiments.
Silencing of IGF-1 receptor
Human BM-MSC were transfected with ON-TARGETplus SMARTpool siRNA (200pmoli) duplex specific for the target sequence NM_000875 (L-003012-00-00050, 5 nmol Dharmacon Inc.), or with control nontarget siRNA (Ambion, Silencer Select Negative Control #2 siRNA) using the Amaxa Human MSC Nucleofector Kit (VPE-1001 LONZA) according to the manufacturer's protocol. After transfection, cells were cultured for 24 h in the DMEM containing platelet lysate previously ultracentrifugated. Exo were then obtained from conditioned supernatants of hBM-MSC maintained for 18 h in the DMEM-low-glucose medium deprived of platelet lysate.
Statistical analysis
Results are expressed as mean±S.E. Analysis of variance (ANOVA) with Bonferroni or Tukey correction, or the Student's t test for unpaired data were used, as appropriate. Statistical significance is defined as P<0.05.
Results
Characterization of human BM-MSC-derived Exo
In our experimental conditions, hBM-MSC released Exo, which were identified in the conditioned medium by electron microscopy as rounded structures of approximately 40–100 nm (Fig. 1A, B). The expression of the CD63 and CD9 tetraspan surface proteins by immunogold labeling confirmed the presence of Exo in the preparations (Fig. 1C, D). Electron microscopy performed on the hBM-MSC monolayer cultured overnight in serum-free conditions showed also the presence of larger vesicles, possibly MV or multivesicular bodies, in the cell cytoplasm (Fig. 1E) that were released from the surface of hBM-MSC (Fig. 1F).
FIG. 1.

Electron microscopy analysis of exosomes. (A and B) Representative micrographs of transmission electron microscopy on human exosome preparations. Electron micrographs of CD63 (C) and CD9 (D) immunogold labeling of exosomes derived from human bone marrow–mesenchymal stem cells (hBM-MSC). (E and F) Representative micrographs of transmission electron microscopy on hBM-MSC monolayer releasing microvesicles and exosomes.
The concentration of Exo total proteins ranged from 12 to 25 ng/μL. Besides protein, Exo contain nucleic acids [8,9]. No DNA was found in Exo isolated from hBM-MSC either by nanodrop spectrophotometer quantification or 1% agarose gel electrophoresis (data not shown). By contrast, substantial amount of mRNA was detected whose concentration ranged from 30 to 150 ng/μL. These data are in line with a very recently published study [26]. To further confirm the presence of mRNA in Exo, purified preparations were treated with RNase A before RNA extraction. RT-PCR analysis for GAPDH did not show any band in the RNase A-treated Exo (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/scd).
Analysis of the mRNA content in Exo
Our previous observation demonstrated that IGF-1 released by mBM-MSC recruited at the site of injury was one of the major mediators in inducing kidney recovery after acute injury [17]. Starting from this point, we performed PCR arrays to determine the expression profile of genes involved in the insulin signaling and in the PI3K-AKT signaling pathways in hBM-MSC and in hExo. A representative scatter plot showing the correlation in the gene expression levels (2−ΔCt) of the insulin-signaling pathway within donor cells and Exo is shown in Fig. 2A. Relative expression levels for each gene from hBM-MSC are plotted against the corresponding expression in Exo in triplicate experiments. The points that fall between the 2 external lines represent mRNAs with similar levels of expression in hBM-MSC and hExo, while those falling above or below the lines represent genes that are more expressed in hBM-MSC or in hExo, respectively. The majority of genes showed comparable expression levels between samples in both arrays. In the IGF-1 signaling pathway, hBM-MSC expressed both IGF-1 and IGF1-receptor (IGF-1R) mRNAs, while Exo contained the IGF-1R, but not the IGF-1 transcript. These results were further confirmed by RT-PCR analysis (Fig. 2B). To assess the integrity of IGF-1R mRNA detected in hBM-MSC-derived Exo, RT-PCR analysis with primers that covered the entire length of the human IGF-1R mRNA was performed. As shown in Fig. 2D, a band of the predicted size was detected by gel electrophoresis in hExo.
FIG. 2.
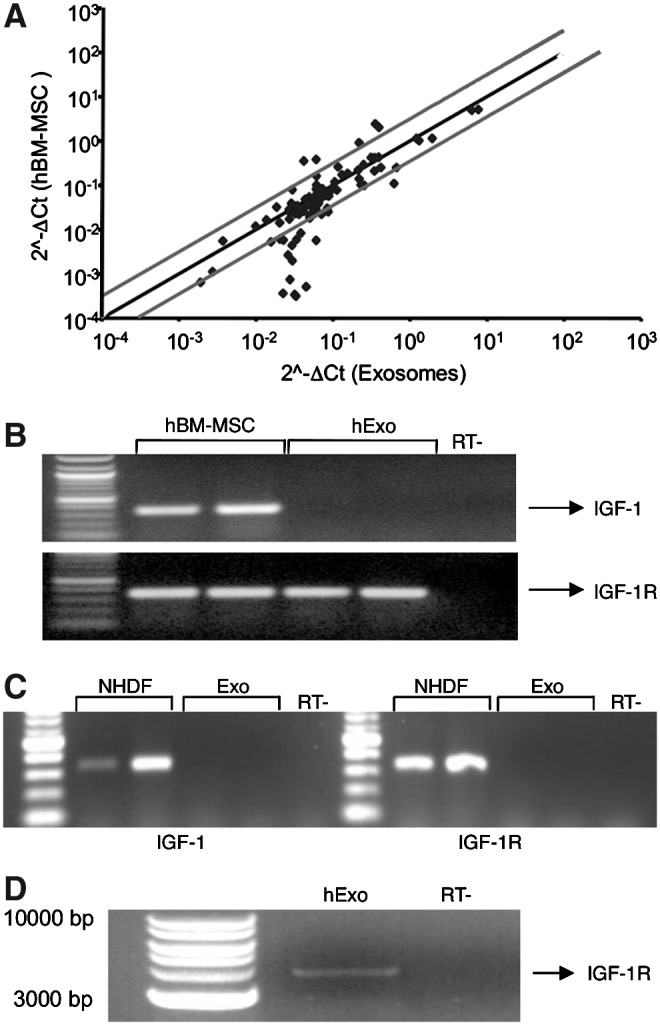
Gene expression profile in hBM-MSC and normal human dermal fibroblasts (NHDF) and their relative exosomes. (A) Representative scatter plot generated by the polymerase chain reaction (PCR) array to determine the expression profile of genes involved in the insulin-signaling pathway in hBM-MSC and their corresponding exosomes. (B) Reverse transcription (RT)-PCR analysis for insulin-like growth factor-1 (IGF-1) and IGF-1 receptor (IGF-1R) on hBM-MSC and their respective exosomes and (C) on NHDF and their respective exosomes. (D) RT-PCR analysis for human IGF-1R full length (transcript of 4122 bp) in human exosomes (hExo). RT- represents negative controls obtained by omitting reverse transcriptase enzyme.
To assess whether segregation of IGF-1/IGF-1R transcripts was confined to hExo, PCR arrays were also performed in Exo derived from mouse BM-MSC. Mouse Exo carried IGF-1R mRNA, but not the IGF-1 transcript, suggesting that segregation of mRNAs within the insulin-signaling pathway was shared between species. Transfer of selected transcripts was instead peculiar for BM-MSC since Exo derived from NHDF did not contain either IGF-1 or its receptor (Fig. 2C). The presence of IGF-1R into the hBM-MSC-derived Exo was not a random event since other receptors, such as the insulin receptor (INSR), or the peroxisome proliferator-activated receptor gamma (PPARG) were not detected in the Exo.
Exo-mediated horizontal transfer of mRNA as mechanism of intercellular communication
To evaluate whether IGF-1R mRNA could be shuttled from Exo to recipient cells, we elected to perform experiments in the R- mouse fibroblast cell line, obtained from mouse embryos homozygous for a targeted disruption of the Igf1r encoding the type 1 insulin-like growth factor receptor [23]. R- cells were incubated with hExo for 3h in a serum-free medium (Fig. 3A). To discard contaminant hExo not incorporated in the cells, 3 washing steps were performed at the end of incubation and total RNA was extracted from R- cells or Exo and analyzed for IGF-1R gene expression by RT-PCR analysis using primers specific for the human transcript. As shown in Fig. 3B, the IGF-1R transcript was detected in hExo and R- cells incubated with human Exo, but not in R- control cells.
FIG. 3.

Horizontal transfer of mRNA through human exosomes. (A) Experimental design of horizontal transfer. (B) RT-PCR for human IGF-1R on mRNA extracted from mouse R- cells exposed or not for 3 h to hBM-MSC-derived exosomes using primers specific for the human IGF-1R sequence. As a positive control, RT-PCR was performed on mRNA extracted from hExo. A transcript of 350 bp was clearly detected both in R- cells exposed to hExo and in exosome preparation, but not in untreated mouse R- cells. (C) Immunofluorescence staining for human IGF-1R in mouse R- cells exposed or not to hExo. The specificity of the staining was confirmed on hBM-MSC (a). Exosome-derived expression of hIGF-1R was detected in R- cells exposed to hExo (b). Negative control was performed on R- cells not receiving the hExo (c). Original magnification,×400. (D) Representative 4% agarose gel electrophoresis of the real-time PCR amplification products (expected size of 60 bp) and their relative quantification (bottom) for mouse IGF-1R on mRNA from HK2 untreated or exposed for 1 or 3 h to mouse BM-MSC-derived exosomes. RT- represents negative controls. Results are expressed as mean±S.E (n=3). Analysis of variance with Bonferroni correction was performed; *P<0.05 and **P<0.01 versus HK2.
To demonstrate that the transferred mRNA was intact and functional, we performed immunofluorescence experiments to detect the translated protein in the target cells. Due to the high homology between the human and mouse IGF-1R proteins, the commercially available antibodies for IGF-1R do not discriminate the 2 sequences, a problem that was overcome by using R- as recipient cells. R- cells were exposed to human Exo and immunofluorescence was performed 24 h later. As depicted in Fig. 3C, the staining of IGF-1R was clearly present in the R- cells exposed to human Exo (panel b), but not in untreated cells (panel c). Human BM-MSC, taken as a positive control, show intense staining of IGF-1R (panel a). No staining was observed in the negative control confirming the specificity of the signal. The percentage of R- cells that expressed the Exo-derived hIGF-1R protein was on average 24.2%±7.7%. To assess whether the IGF-1R protein could be present in Exo, contributing to a direct transfer of the protein, Exo were labeled for IGF-1R by immunogold, but no labeling was observed (data not shown).
Horizontal transfer of IGF-1R mRNA also occurred when mouse Exo were added to HK2 cells for 1 and 3 h. Using specific primers for mouse sequence, IGF-1R expression increased in HK2 exposed to mouse Exo. The increase was time-dependent suggesting at best that IGF-1R expression was due to the mRNA transfer instead of residual Exo RNA. As expected, no mouse transcript was detected in control HK2 (Fig. 3D).
Human BM-MSC-derived Exo induce regeneration of damaged proximal tubular epithelial cells
The ability of hBM-MSC-derived Exo to stimulate proliferation was investigated using an in vitro model of cisplatin-induced toxicity in mouse PTEC [17]. Mouse PTEC were exposed to cisplatin for 6 h. After drug withdrawal, PTEC were incubated with Exo from the hBM-MSC-conditioned medium, or with 5 ng/mL of IGF-1, or both. Proliferation of damaged PTEC, evaluated 4 days after the beginning of the incubation, was significantly increased following the addition of hExo or IGF-1 alone. Coincubation of damaged PTEC with hExo and soluble IGF-1 further enhanced cell proliferation in respect to single treatments (Fig. 4).
FIG. 4.
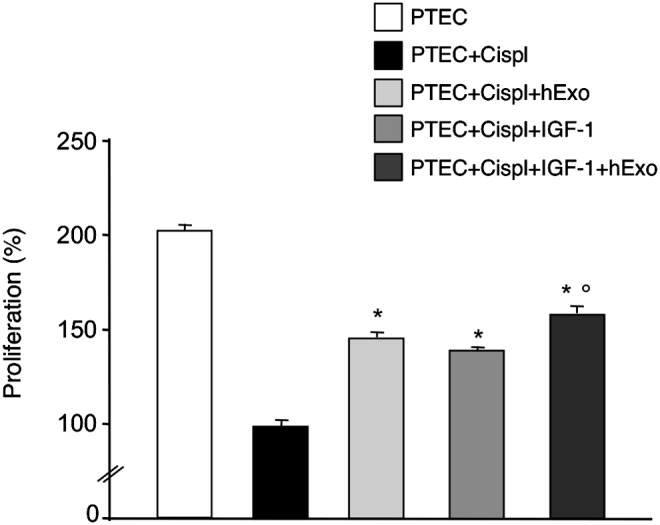
Proliferation of damaged mouse proximal tubular epithelial cells (PTEC). PTEC were exposed to cisplatin 2.5 μM for 6h, and then incubated with test medium or hBM-MSC-derived exosomes or IGF-1 (5 ng/mL) or both for 4 days. PTEC proliferation was estimated by cell count using trypan blue dye exclusion and PTEC+cispl was taken as 100%. Results are expressed as mean±S.E (n=3). Analysis of variance with Tukey correction was performed; *P<0.01 versus PTEC+cispl; °P<0.05 versus PTEC+cispl+hExo and PTEC+cispl+IGF-1.
Gene silencing of IGF-1R in human BM-MSC reduces the proliferative effect of Exo
To study the possible role of IGF-1R mRNA delivered to PTEC by Exo in potentiating the proliferative effect of IGF-1 on PTEC, we silenced the expression of IGF-1R in hBM-MSC before Exo purification. Preliminary experiments were performed to optimize the dose of siRNA to silence IGF-1R expression in hBM-MSC (Fig. 5A). Real-time RT-PCR experiments showed 80% inhibition of IGF-1R expression in hBM-MSC and no transcript was detected in Exo preparations (Fig. 5B). When damaged PTEC cells were exposed to Exo derived from IGF-1R-silenced hBM-MSC (hExo-siIGF-1R), IGF-1-induced proliferation was significantly reduced as compared to that of cells incubated with Exo from scramble siRNA-treated hBM-MSC (hExo-siScramble) (Fig. 6). To exclude the possibility that gene silencing could affect the Exo cargo content, we compared the expression of BCL2-like1 as the gene involved in the regenerative activity of BM-MSC, in IGF-1R-silenced or not Exo preparations. No significant difference in BCL2-like1 expression was observed in Exo preparations silenced or not for IGF-1R (data not shown).
FIG. 5.
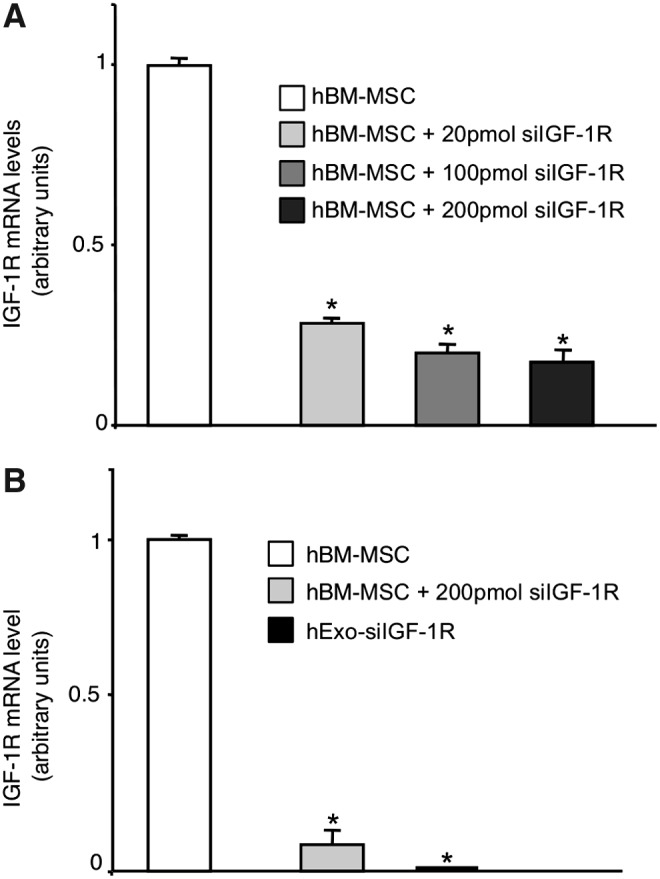
Gene silencing of IGF-1R in hBM-MSC. (A) Different amounts of small interfering RNA (siRNA) specific for human IGF-1R were tested to reach the highest efficiency of silencing. Results are expressed as mean±S.E (n=3). Analysis of variance with Bonferroni correction was performed; *P<0.01 versus hBM-MSC. (B) Evaluation of IGF-1R mRNA in hBM-MSC and their derived exosomes after gene silencing (hExo-siIGF-1R). Analysis of variance with Bonferroni correction was performed; *P<0.001 versus hBM-MSC.
FIG. 6.
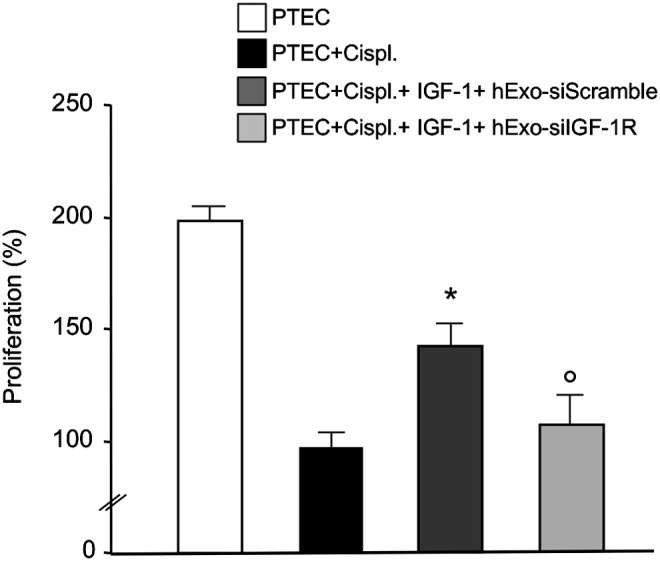
Effect of IGF-1R silencing on proliferation of damaged mouse PTEC. PTEC were exposed to IGF-1 and exosomes derived from hBM-MSC treated with scramble siRNA (hExo-siScramble) or siRNA specific for IGF-1R (hExo-siIGF-1R). PTEC were exposed to medium or cisplatin 2.5 μM for 6h, and then incubated with test medium or with IGF-1 and hExo preparations. PTEC proliferation was estimated after 4 days by cell count using trypan blue dye exclusion and PTEC+cispl was taken as 100%. Results are expressed as mean±S.E (n=4). Analysis of variance with Bonferroni correction was performed; *P<0.05 versus PTEC+cispl, °P<0.05 versus PTEC+cispl+IGF-1+hExo-siScramble.
Discussion
The present study demonstrates that hBM-MSC release into the culture medium heterogeneous populations of MV and Exo that do not contain DNA, but are engulfed with mRNA. Our previous data showed that BM-MSC release anti-inflammatory and prosurvival soluble factors, including TGF-β, IL-10, and IGF-1, the latter representing the best candidate for proximal tubular cell regeneration after AKI based on its proliferative and antiapoptotic properties [17]. Starting from this observation, we hypothesized that the effect of BM-MSC-released IGF-1 protein on proximal tubular cells could be amplified by increased expression of IGF-1 on tubular cells via Exo-mediated mRNA transfer. This, however, was not the case since analysis of the cargo content of hBM-MSC-derived Exo by PCR array did not show the presence of the IGF-1 transcript. We therefore reasoned that the paracrine effect of BM-MSC-derived IGF-1 on proximal tubular cells could be helped by the transfer of IGF-1R mRNA to the target cells. To lend further support to this hypothesis, PCR array data showed the presence of the IGF-1R mRNA in Exo from hBM-MSC that also was found to be intact by RT-PCR analysis.
We next evaluated whether the IGF-1R mRNA in Exo was horizontally transferred in recipient cells taking advantage from the availability of the IGF-1R null cells (R-). The R-fibroblast cell line, obtained from a mouse embryo lacking IGF-1R, acquired the capability to express the IGF-1R protein after incubation with Exo derived from hBM-MSC. These data would indicate that the shuttling of IGF-1R mRNA from Exo to R- cells occurred and was accompanied by a cell phenotype change due to the translation of the transcript into the corresponding protein.
Selective differences in the expression of IGF-1/IGF-1R mRNAs in Exo derived from BM-MSC and parental cells as well as the horizontal transfer of mRNAs between Exo and proximal tubular cells is not restricted to hBM-MSC, but is a shared phenomenon with mouse BM-MSC. These data point to a novel mechanism that may operate in BM-MSC of different species accounting for their regenerative effect. Selective mRNA transfer into Exo supported the notion that the mRNA contained in the Exo is not an exact copy of the mRNAs of the donor cells as previously observed in mast cells [8]. This observation was further supported by the finding that mRNAs for other receptors in the insulin-signaling pathway like the INSR or the PPARG were not found in hBM-MSC-derived Exo. Specific targeting of the IGF-1R mRNA sequence in the Exo is specific for BM-MSC since Exo derived from NHDF did not contain either IGF-1 or its receptor mRNAs. It is still unclear why a set of specific mRNA transcripts is differentially shuttled to Exo in various cell types. Understanding such a mechanism would help to understand the function of RNA delivery to secreted vesicles.
The use of an in vitro model of cisplatin-induced toxicity in proximal tubular cells allowed to demonstrate that the transfer of IGF-1R mRNA through Exo resulted into a functional effect as documented by increased proliferation of the damaged proximal tubular cells exposed to Exo. Silencing the expression of IGF-1R in hBM-MSC before Exo production strongly limited proliferation of tubular cells induced by Exo further reinforcing the concept of IGF-1R being involved in the functional effect. We cannot exclude that gene silencing could have altered the cargo content of Exo, modulating the expression of genes that exacerbate the inhibitory effect on tubular cell proliferation, although no significant changes in the expression of the antiapoptotic gene BCL2-like1 were observed in IGF-1R-silenced Exo in respect to unmodified Exo. Addition of exogenous IGF-1 induced an increase in the growth of cisplatin-treated tubular cells that was further enhanced by cell exposure to the IGF-1 protein and Exo in respect to treatments given alone. It is conceivable that sensitivity of proximal tubular cells to IGF-1 could be enhanced by the transfer of IGF-1R mRNA via Exo. This phenomenon would explain the ability of a low number of MSC engrafting the kidney to promote a prompt recovery from AKI [17].
The present data provide for the first time the evidence of a specific mechanism through which BM-MSC contribute to repair the injured tissue by virtue of a combined trophic effect induced by a soluble factor and the expression of the corresponding receptor in the target cells after the transfer of genetic material accomplished by Exo. Exo have the capacity to communicate messages to repair damaged cells as demonstrated for mast cells that exposed to oxidative stress-released Exo containing protective signals making the recipient cell more tolerant to oxidative processes and to cell death [27]. Besides functional mRNA, Exo contain microRNAs (miRNAs) [28,29], small noncoding RNAs that regulate gene expression at the post-transcriptional level [30]. MicroRNAs control cell fate and behavior by repressing the translation of selected mRNAs in stem cells as well as in differentiating daughter cells [31]. The presence of miRNAs into Exo from mast cells [8], ES cells [32], and BM-MSC [28,29] has been taken to suggest that they act as regulators of cell–cell communication by modulating the fate and behavior of target cells via epigenetic changes of their transcriptome. Whether the regenerative potential of BM-MSC can be attributed to a fine regulation of protein translation by miRNAs will be a matter of future investigation. In conclusion, BM-MSC recruited to the site of injury may reprogram the damaged cells stimulating their proliferation and favoring tissue regeneration through the contribution of growth factors and the transfer of genetic material by Exo both locally released by BM-MSC.
Supplementary Material
Acknowledgments
We thank Dr. Elena Gagliardini and Daniela Cavallotti for the excellent technical assistance. We are greatly indebted to Dr. Renato Baserga (Thomas Jefferson University, Philadelphia, PA) for providing the R- mouse cell line. C.R. is a recipient of a fellowship from Fondazione Aiuti per la Ricerca sulle Malattie Rare (ARMR), Bergamo, Italy. This work was partially supported by a grant from Genzyme Renal Innovations Program (GRIP) and from Ministero della Salute, Bando Cellule Staminali 2008, Codice Progetto B11J11001110002. Part of the research leading to these results has received funding from the European Community under the European Community's Seventh Framework Programme (FP7/2007–2013), grant number 223007, STAR-T REK project.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Rustom A. Saffrich R. Markovic I. Walther P. Gerdes HH. Nanotubular highways for intercellular organelle transport. Science. 2004;303:1007–1010. doi: 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- 2.Ratajczak J. Wysoczynski M. Hayek F. Janowska-Wieczorek A. Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia. 2006;20:1487–1495. doi: 10.1038/sj.leu.2404296. [DOI] [PubMed] [Google Scholar]
- 3.Camussi G. Deregibus MC. Bruno S. Cantaluppi V. Biancone L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010;78:838–848. doi: 10.1038/ki.2010.278. [DOI] [PubMed] [Google Scholar]
- 4.McDaniel LD. Young E. Delaney J. Ruhnau F. Ritchie KB. Paul JH. High frequency of horizontal gene transfer in the oceans. Science. 2010;330:50. doi: 10.1126/science.1192243. [DOI] [PubMed] [Google Scholar]
- 5.Denzer K. Kleijmeer MJ. Heijnen HF. Stoorvogel W. Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci. 2000;19(113 Pt):3365–3374. doi: 10.1242/jcs.113.19.3365. [DOI] [PubMed] [Google Scholar]
- 6.Fevrier B. Raposo G. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol. 2004;16:415–421. doi: 10.1016/j.ceb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Cocucci E. Racchetti G. Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Valadi H. Ekstrom K. Bossios A. Sjostrand M. Lee JJ. Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 9.Deregibus MC. Cantaluppi V. Calogero R. Lo Iacono M. Tetta C. Biancone L. Bruno S. Bussolati B. Camussi G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood. 2007;110:2440–2448. doi: 10.1182/blood-2007-03-078709. [DOI] [PubMed] [Google Scholar]
- 10.Ratajczak J. Miekus K. Kucia M. Zhang J. Reca R. Dvorak P. Ratajczak MZ. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006;20:847–856. doi: 10.1038/sj.leu.2404132. [DOI] [PubMed] [Google Scholar]
- 11.Bruno S. Grange C. Deregibus MC. Calogero RA. Saviozzi S. Collino F. Morando L. Busca A. Falda M, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20:1053–1067. doi: 10.1681/ASN.2008070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morigi M. Introna M. Imberti B. Corna D. Abbate M. Rota C. Rottoli D. Benigni A. Perico N, et al. Human bone marrow mesenchymal stem cells accelerate recovery of acute renal injury and prolong survival in mice. Stem Cells. 2008;26:2075–2082. doi: 10.1634/stemcells.2007-0795. [DOI] [PubMed] [Google Scholar]
- 13.Chhabra P. Brayman KL. The use of stem cells in kidney disease. Curr Opin Organ Transplant. 2009;14:72–78. doi: 10.1097/MOT.0b013e328320d2f5. [DOI] [PubMed] [Google Scholar]
- 14.Aejaz HM. Aleem AK. Parveen N. Khaja MN. Narusu ML. Habibullah CM. Stem cell therapy-present status. Transplant Proc. 2007;39:694–699. doi: 10.1016/j.transproceed.2007.01.069. [DOI] [PubMed] [Google Scholar]
- 15.Salem HK. Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells. 2010;28:585–596. doi: 10.1002/stem.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morigi M. Imberti B. Zoja C. Corna D. Tomasoni S. Abbate M. Rottoli D. Angioletti S. Benigni A, et al. Mesenchymal stem cells are renotropic, helping to repair the kidney and improve function in acute renal failure. J Am Soc Nephrol. 2004;15:1794–1804. doi: 10.1097/01.asn.0000128974.07460.34. [DOI] [PubMed] [Google Scholar]
- 17.Imberti B. Morigi M. Tomasoni S. Rota C. Corna D. Longaretti L. Rottoli D. Valsecchi F. Benigni A, et al. Insulin-like growth factor-1 sustains stem cell mediated renal repair. J Am Soc Nephrol. 2007;18:2921–2928. doi: 10.1681/ASN.2006121318. [DOI] [PubMed] [Google Scholar]
- 18.Togel F. Hu Z. Weiss K. Isaac J. Lange C. Westenfelder C. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol. 2005;289:F31–F42. doi: 10.1152/ajprenal.00007.2005. [DOI] [PubMed] [Google Scholar]
- 19.Capelli C. Domenghini M. Borleri G. Bellavita P. Poma R. Carobbio A. Mico C. Rambaldi A. Golay J. Introna M. Human platelet lysate allows expansion and clinical grade production of mesenchymal stromal cells from small samples of bone marrow aspirates or marrow filter washouts. Bone Marrow Transplant. 2007;40:785–791. doi: 10.1038/sj.bmt.1705798. [DOI] [PubMed] [Google Scholar]
- 20.Capelli C. Gotti E. Morigi M. Rota C. Weng L. Dazzi F. Spinelli O. Cazzaniga G. Trezzi R, et al. Minimally manipulated whole human umbilical cord is a rich source of clinical-grade human mesenchymal stromal cells expanded in human platelet lysate. Cytotherapy. 2011;13:786–801. doi: 10.3109/14653249.2011.563294. [DOI] [PubMed] [Google Scholar]
- 21.Pereira RF. Halford KW. O'Hara MD. Leeper DB. Sokolov BP. Pollard MD. Bagasra O. Prockop DJ. Cultured adherent cells from marrow can serve as long-lasting precursor cells for bone, cartilage, and lung in irradiated mice. Proc Natl Acad Sci U S A. 1995;92:4857–4861. doi: 10.1073/pnas.92.11.4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phinney DG. Kopen G. Isaacson RL. Prockop DJ. Plastic adherent stromal cells from the bone marrow of commonly used strains of inbred mice: variations in yield, growth, and differentiation. J Cell Biochem. 1999;72:570–585. [PubMed] [Google Scholar]
- 23.Sell C. Dumenil G. Deveaud C. Miura M. Coppola D. DeAngelis T. Rubin R. Efstratiadis A. Baserga R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol. 1994;14:3604–3612. doi: 10.1128/mcb.14.6.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morigi M. Macconi D. Zoja C. Donadelli R. Buelli S. Zanchi C. Ghilardi M. Remuzzi G. Protein overload-induced NF-kappaB activation in proximal tubular cells requires H(2)O(2) through a PKC-dependent pathway. J Am Soc Nephrol. 2002;13:1179–1189. [PubMed] [Google Scholar]
- 25.Haverty TP. Kelly CJ. Hines WH. Amenta PS. Watanabe M. Harper RA. Kefalides NA. Neilson EG. Characterization of a renal tubular epithelial cell line which secretes the autologous target antigen of autoimmune experimental interstitial nephritis. J Cell Biol. 1988;107:1359–1368. doi: 10.1083/jcb.107.4.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eldh M. Lotvall J. Malmhall C. Ekstrom K. Importance of RNA isolation methods for analysis of exosomal RNA: evaluation of different methods. Mol Immunol. 2012;50:278–286. doi: 10.1016/j.molimm.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Eldh M. Ekstrom K. Valadi H. Sjostrand M. Olsson B. Jernas M. Lotvall J. Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS One. 2010;5:e15353. doi: 10.1371/journal.pone.0015353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen TS. Lai RC. Lee MM. Choo AB. Lee CN. Lim SK. Mesenchymal stem cell secretes microparticles enriched in pre-microRNAs. Nucleic Acids Res. 2010;38:215–224. doi: 10.1093/nar/gkp857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collino F. Deregibus MC. Bruno S. Sterpone L. Aghemo G. Viltono L. Tetta C. Camussi G. Microvesicles derived from adult human bone marrow and tissue specific mesenchymal stem cells shuttle selected pattern of miRNAs. PLoS One. 2010;5:e11803. doi: 10.1371/journal.pone.0011803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filipowicz W. Bhattacharyya SN. Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 31.Gangaraju VK. Lin H. MicroRNAs: key regulators of stem cells. Nat Rev Mol Cell Biol. 2009;10:116–125. doi: 10.1038/nrm2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan A. Farber EL. Rapoport AL. Tejada D. Deniskin R. Akhmedov NB. Farber DB. Transfer of microRNAs by embryonic stem cell microvesicles. PLoS One. 2009;4:e4722. doi: 10.1371/journal.pone.0004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.