

Abstract
The study of nematode genomes over the last three decades has relied heavily on the model organism Caenorhabditis elegans, which remains the best-assembled and annotated metazoan genome. This is now changing as a rapidly expanding number of nematodes of medical and economic importance have been sequenced in recent years. The advent of sequencing technologies to achieve the equivalent of the $1000 human genome promises that every nematode genome of interest will eventually be sequenced at a reasonable cost. As the sequencing of species spanning the nematode phylum becomes a routine part of characterizing nematodes, the comparative approach and the increasing use of ecological context will help us to further understand the evolution and functional specializations of any given species by comparing its genome to that of other closely and more distantly related nematodes. We review the current state of nematode genomics and discuss some of the highlights that these genomes have revealed and the trend and benefits of ecological genomics, emphasizing the potential for new genomes and the exciting opportunities this provides for nematological studies.
Keywords: ecological genomics, evolution, genomics, nematodes, phylogenetics, proteomics, sequencing
Nematoda is one of the most expansive phyla documented with free-living and parasitic species found in nearly every ecological niche(Yeates, 2004). Traditionally, nematode phylogeny was based on classical and often incomplete understanding of morphological traits, but traditional systems have been revised and supplemented by a growing body of insight from molecular phylogenetics that is primarily based on ribosomal DNA for higher level taxonomic studies (De Ley and Blaxter, 2002; De Ley and Blaxter, 2004; Holterman et al., 2006). The study of the evolutionary relationships between species in vertebrates and in arthropods is transitioning to the comparative analysis of entire genomes due to the exponentially decreasing cost of sequencing and the study of nematodes is now following the same path (Lin et al., 2007; Blaxter, 2011; Burgess, 2011). While the model organism Caenorhabditis elegans was the first metazoan sequenced (Consortium, 1998), there have been only a few additional nematodes sequenced until recently and many representative clades and ecological niches remain unexplored. There are several advantages to whole genome sequencing for nematology. The simplest and most obvious is that the complete genome harbors the full repertoire of genes that are the inherited common core of any given species. Furthermore, the genome contains the structural and regulatory elements that lie in and between genes, even if we cannot yet identify them all. The genome also provides the foundation for future experimentation such as transformation and RNA interference (RNAi). The genome is the natural framework for indexing and organizing the massive genetic content of species within a phylum. The genetic ‘blueprint’ represented by a genome may prove to be the most valuable and enduring piece of knowledge we can currently obtain for any particular life form (Consortium, 1998).
As in many other fields of biology, the nematode C. elegans has proven invaluable as a model for genomic analysis, and thousands of investigators have contributed to our understanding of its 20,431 protein-coding genes (Consortium, 1998; Harris et al., 2010; WormBase release WS225). This is likely for the same reasons that make this hermaphrodite so powerful and useful in genetics: 1) its ease of culture, 2) its simple, rapid, invariant development, 3) many biological principles are universal, even if specific details are not, and 4) the more detailed our understanding of any biological phenomenon, the more interesting it tends to become (Horvitz and Sternberg, 1982). While sequencing efforts have expanded exponentially as technology improves and the cost continues to diminish, the finished C. elegans genome remains unrivaled in completeness compared to other metazoans. This is not likely to change, due partly to differences in technology but primarily because closing the remaining gaps in genomic sequence is a prolonged and expensive process with diminishing biological return (Consortium, 1998). The top-down approach of completing genome sequences by breaking the genome down into large, known fragments, which provide a physical map, and the subsequent sequencing of those fragments in their entirety, will probably not be common until new technologies sharply reduce the costs of finishing genomes.
Over the last two decades, sequencing technology has advanced from relying on the hierarchical sequencing and assembly of cloned fragments of DNA (i.e. automated Sanger sequencing as used in the C. elegans project), to the shotgun, high-throughput ∼500 bp reads produced by 454 Roche sequencing and the even cheaper ≤ 150 bp reads produced by Illumina sequencing (Mardis, 2008; Jex et al., 2011; Werner et al., 2011). Due to the rapid pace of sequencing technology development and turnover, we will refer to the newer technologies as ‘next-generation’ (next-gen) technologies throughout rather than focus on any specific platform. These next-gen technologies are driven with the eventual goal to achieve a ≤ $1000 human genome to enable health applications. Given that the typical nematode genome is less than 1/15 of the size of the 3.2 Gb human genome (see Table 1 for nematode genome sizes), sequencing nematode genomes is already affordable and, as technology improves, could become monetarily negligible. Current next-gen technologies use DNA fragments of various size to generate sequence, which range from less than 500 bp up to ≤20 kb, and can produce either single or paired end reads (either one or both ends of prepared fragments can be sequenced (Fig. 1)). Next-gen sequencing technologies generate many more sequencing reads that have a higher error rate than traditional Sanger sequencing, but this is balanced by higher overall coverage (whereas 2 Gb of generated sequence would provide 20-fold coverage of a 100 Mb genome, 10 Gb of generated sequence would provide 100-fold coverage). When considering these sequencing technologies it is important to distinguish fragment size and read length as distinct variables that will affect the resulting assembly, because it is easy to sometimes conflate or combine these separate aspects. Fragment size refers to the length of the DNA insert, from which sequence will be generated either from one or both sides, while read length refers to how many base pairs are actually being sequenced from one or both sides of the fragment (Fig. 1).
Table 1.
Genome statistics for published and selected forthcoming whole nematode genomes. Additional genomes planned and in progress can be viewed at www.nematodes.org/nematodegenomes/index.php/959_Nematode_Genomes.
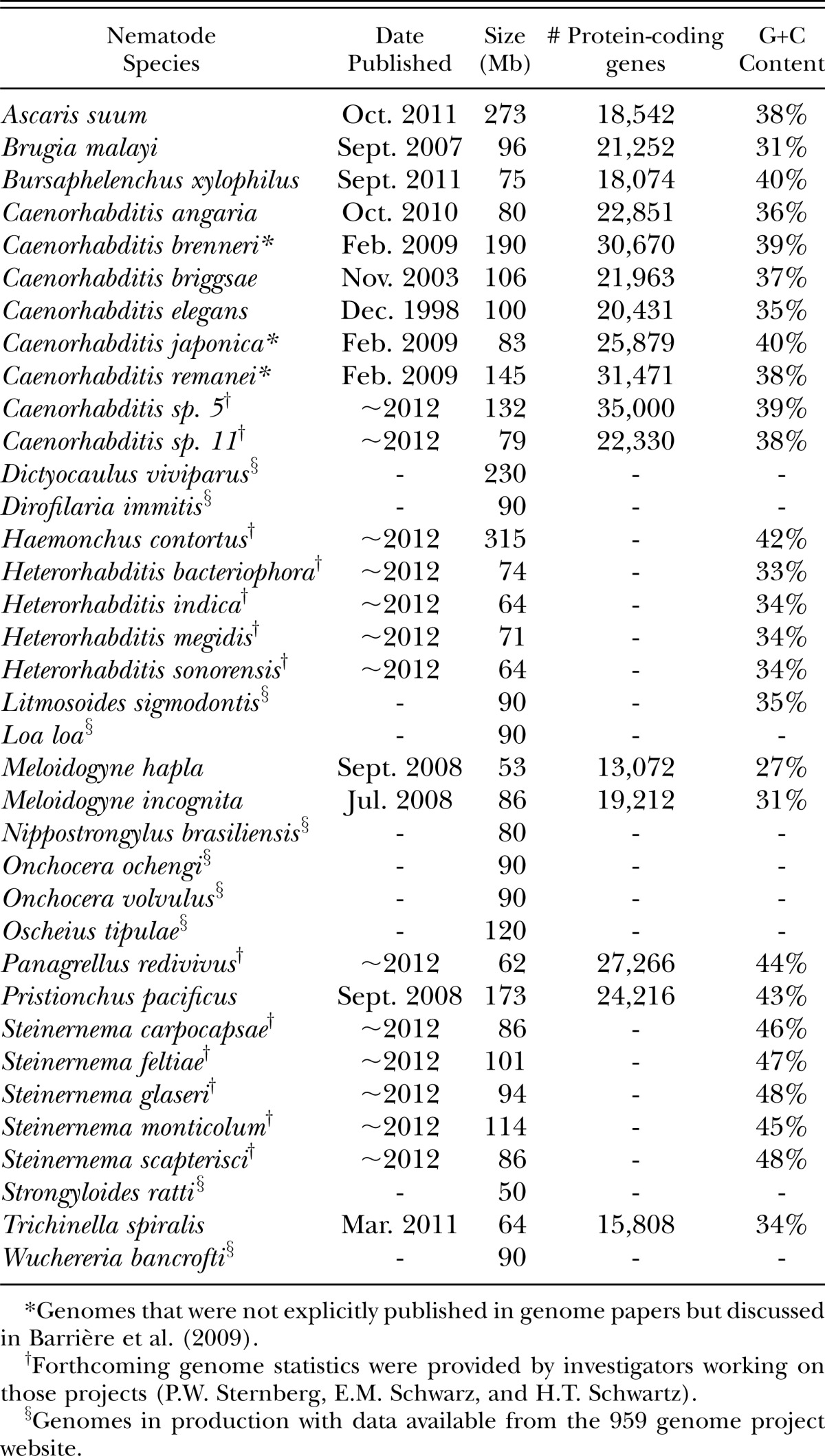
Fig. 1.

Hierarchical and shotgun sequencing. A) A shortened diagram of the hierarchical (top-down) sequencing technique used for the C. elegans genome. The genomic DNA was broken into large fragments (40-100 kb) that formed a physical map. The order of the fragments was known before they were actually sequenced. The fragments were then fully sequenced and assembled, resulting in six chromosomal contigs. B) A diagram of the shotgun-sequencing techniques used to prepare genomic DNA for sequencing using 454 Roche or Illumina short-read technology. The genomic DNA is sheared into approximately sized fragments of 0.5 to 1 kb. These fragments then have primers attached to one or both ends, depending on whether they will be run on a paired-end sequencer. The fragments are not sequenced in their entirety, but 50-500 bp of one or both ends of each fragment are sequenced. The resulting short reads are then assembled, with some gaps remaining as shown (gaps are represented by the dotted lines). These reads are then assembled into hundreds up to thousands of larger contigs (contiguous sequence). C) Jumping libraries are used in short-read sequencing to improve assembly quality. During the size selection, larger fragment sizes are selected and sequenced. Only one size is selected per library, but the sizes range from 2 kb to 20 kb. Read assembly is facilitated by knowing the approximate distance between the paired end reads, helping to overcome issues of repeats and homopolymeric regions, jumping large regions (as large as the insert length). Assembly quality is improved as multiple previously unconnected contigs are now known to connect, just as contig 1 and contig 2 are joined to form a larger contig called ‘scaffold 1’ in the figure.
The hierarchical application of Sanger sequencing to the assembly of the C. elegans genome helped to facilitate completeness and circumvented the potential problems of long repeats, homopolymeric regions, and low G+C content, along with the community effort of researchers, which was crucial and is ongoing (Consortium, 1998; Consortium, 1999). Next-gen technologies are more affordable and allow for much higher fold coverage of genomes, leading to hundreds of millions of genomic reads. In contrast to the hierarchical approach previously used, the shotgun strategies in favor today are based on breaking the entire genome into many more small fragments. These require more computational effort to assemble into multigenic sized contigs, let alone chromosomes (Fig. 1) (Mardis, 2008). The contiguity of the resulting draft genomes can be dramatically improved by library construction with inserts of larger but approximately known sizes as well as ‘jumping libraries’ (Fig. 1C) (Collins et al., 1987; Richards et al., 1988).
Since the first nematode genome was first published in 1998, twelve more whole nematode genomes have been sequenced and made publicly available (Consortium, 1998; Stein et al., 2003; Ghedin et al., 2007; Abad et al., 2008; Dieterich et al., 2008; Opperman et al., 2008; Barrière et al., 2009; Mortazavi et al., 2010 Jex et al., 2011; Kikuchi et al., 2011; Mitreva et al., 2011). There are at least 13 more nematode genomes scheduled for release in 2012, and several others in preparation (Table 1) (Kumar et al., 2012). Because Nematoda is so ecologically diverse and species-rich (1 to 10 million species (Lambshead, 1993; Lambshead, 2004)), phylogenetic relationships along with human health and agricultural considerations should inform sequencing efforts.
The current view of nematode genealogical relationships divides the phylum into 3 major clades: Enoplia, Dorylaimia, and Chromadoria (De Ley and Blaxter, 2002; De Ley and Blaxter, 2004; Hoda, 2011). Chromadoria is further broken down into 10 clades, which together with Enoplia and Dorylaimia form a total of 12 major monophyletic branches within the phylum (Fig. 2) (Holterman et al., 2006; van Megen et al., 2009; Blaxter, 2011). Sequencing efforts so far have focused on nematodes in the crown clades of Chromadoria, which include C. elegans as well as most medically and agriculturally relevant species (Fig. 2). A systematic genomic survey of the phylum would facilitate a better understanding of the evolution of Nematoda, enhance comparative studies, and could illuminate striking differences across the phylum such as differences in parasitic lifestyle (e.g. endoparasitic vs. ectoparasitic) or mode of reproduction (e.g. amphimictic vs. parthenogenetic) as well as developmental differences (e.g. asymmetric vs. symmetric cleavages; presence vs. absence of a prominent coeloblastula (Schierenberg, 2005)), among others.
Fig. 2.

A phylogenetic representation of sequenced and selected forthcoming nematode genomes. This diagram depicts twelve monophyletic clades representing the phylum Nematoda. Sequencing efforts have focused on a few select crown clades of Chromadoria. The clade designations are after Holterman et al. (2006). Taxa with published genomes have bolded names while taxa for which genomes are underway and scheduled for release in 2012 are regular typeface. Full genus names can be found in Table 1. Forthcoming genomes were selected from genomes with data available from the 959 genome project (Kumar et al., 2012) and from genome projects of which the authors had knowledge.
What are the benefits of genomics for nematologists? Herein we briefly review the basic information provided by most nematode genome analyses. We discuss the highlights of the 13 available nematode genomes, how their utility increases as the number of possible comparisons increases, and how the focus of nematode genomics is changing to emphasize the specific biology and ecology of each species. We finish by illustrating the potential benefit of sequencing additional nematode genomes, using as an example the prospects of entomopathogenic nematode genomes and discussing how they can contribute to our understanding of parasitism, mutualism, and nematode biology in general.
The Steps in Sequencing a Genome
With such a diversity of nematodes to choose from, which nematodes should be sequenced first? In addition to the above-mentioned biological motivations of phylogenetic position and human health and agricultural concerns, there are practical considerations such as the availability and homogeneity of material. Culturability is also a consideration, especially if investigators are interested in the transcriptome and subsequent experimentation. Adding transcriptional data can dramatically improve gene predictions and assembly quality (Mortazavi et al., 2008; Mortazavi et al., 2010). Whole-genome amplification techniques may make it possible to analyze interesting-but-unculturable nematodes in a cost-effective way. However, such amplification techniques may introduce additional problems such as polymorphisms and amplification errors, while culturable worms escape these difficulties since they can provide large quantities of DNA (typically 5 micrograms are needed to robustly construct a representative DNA library, which corresponds to ∼50,000 worms for C. elegans) and can be inbred to decrease heterozygosity. While the study of sequence variation within a species is of great importance, the same variation can make it difficult to assemble a genome de novo without producing assembly errors. Therefore every effort should be undertaken, if possible, to inbreed the strains used, to minimize polymorphisms. The genomic value of a culturable worm increases with complementary transcriptome data and the possibility of further experimentation. In fact, the implementation of some experimental techniques such as RNAi may depend on optimized culturing techniques that do not stress the nematodes being cultured (Dalzell et al., 2011). We believe that there are plenty of interesting culturable nematodes that can shed light on the evolution of the phylum and thus should be prioritized to fill sequencing pipelines. While the bulk of our discussion below focuses on genomic libraries, RNA-seq libraries for transcriptome sequencing can be built from as little as 100 ng of total RNA thus lowering the numbers of worms needed to collect data. As next-gen technologies mature, we can expect that the starting amounts of material necessary will decrease.
Once a suitable nematode is identified, the simplified, general pipeline for genomic sequencing is as follows: 1) extraction and purification of genomic DNA, 2) selection of a sequencing platform, 3) library construction, 4) sequencing, 5) assembly of the sequence into as long and as few contigs as possible, 6) gene predictions and subsequent annotation.
DNA extraction and purification. There are numerous DNA extraction and purification methods and proprietary kits that have been tested and are known to work well both for populations and individual nematodes (Williams et al., 1992; Jones et al., 2006; Adams et al., 2007).
Selection of a sequencing platform. Careful consideration should be given to selecting the appropriate sequencing technology and accompanying parameters, such as read length and fragment size. A common priority is to select the most cost-effective source of high-quality sequence while simultaneously collecting as many reads as possible to ensure good coverage. Good assemblies with short-read technologies typically require 100x average coverage to compensate for high error rates. Coverage takes into account the size of the genome and the length of sequenced reads; for a 100 Mb genome, 100 million 100 bp reads are needed to achieve 100x coverage. Matters are further complicated by the effect of GC-content (GC content of the genome is the percentage of guanines and cytosines) on the coverage in some next-gen technologies, which necessitate greater overall sequencing depth (i.e. more sequencing reads) to cover GC-poor regions well (Mortazavi et al., 2010). Certain sequencing platforms may be advisable for particularly GC-poor genomes (e.g. <35%), such as 454.
Library construction. Good library construction is often a critical step, depending on the sequencing technology used (Zheng et al., 2011). A genomic library is essentially genomic DNA that has been sheared into fragments, which are then size selected for an approximate distribution. These fragments then have sequencing primers ligated to one or both ends (Fig. 1). Because of the massive number of reads, and increasingly longer read lengths, the construction of good libraries with a normally distributed fragment size can make the difference between good and poor quality assemblies. Libraries with average fragment sizes of 500 bp are sufficient to assemble most nematode-size gene loci onto a single contig(Mortazavi et al., 2008). Genomes that are rich in longer repeat sequences or gene clusters that are larger than the fragment lengths will benefit from additional jumping libraries, which are paired-end libraries that are typically 3-20 kb apart (Fig. 1C) (Jex et al., 2011). In addition to traditional genomic jumping libraries, transcriptome data can be used to scaffold expressed genes that are broken across multiple contigs (Mortazavi et al., 2010).
Sequencing. After a library is constructed, it is then sequenced, which is typically handled by dedicated facilities. The sequencing run may take 1 to 10 days, but this may be prolonged depending on facility scheduling considerations. The resulting raw reads each consist of a DNA sequence and a corresponding quality score; these can be used to filter all but the highest-quality reads, which will improve the overall assembly.
Genome assembly. Reads are assembled into contigs using one of several available programs such as Velvet and SOAPdenovo (Zerbino and Birney, 2008; Li et al., 2009). Genome assembly is a resource-intensive step that can require substantial memory, but the relatively small size of nematode genomes makes assembly practical on servers with 128 to 256 gigabytes of RAM. Assembly programs work by finding overlap between reads and connecting them into contigs, and by connecting contigs using the paired information from paired-end (or jumping libraries) into scaffolds (connected contigs). In an ideal situation, one contig or even one scaffold per chromosome would be recovered, but this has only been achieved for C. elegans and C. briggsae (Fig. 1A) (Consortium, 1998; Stein et al., 2003). Assembly programs are often run multiple times with different parameters to maximize several of the assembly metrics described in the basic genome statistics section below.
Gene prediction and genome annotation. Once reads have been assembled, gene-finding programs that identify protein coding or non-protein coding genes such as Augustus and trnaScan are used to annotate the genome (Fig. 3) (Lowe and Eddy, 1997; Stanke et al., 2004). Perhaps the most helpful additional dataset for this step is transcriptome data that is generated by high-throughput sequencing of mRNA (RNA-seq). This provides expression data and identifies bona fide transcripts (either full length or fragments) directly. These data can also be used to train prediction software, thus facilitating more reliable gene predictions (Mortazavi et al., 2008; Mortazavi et al., 2010). The transcriptome provides interesting biological data about global gene expression and can be applied to nematodes at specific stages such as infective juveniles or embryos. RNA-seq data for any biological sample, whether strain (e.g. drug-resistant mutant compared to the wild-type) or stage-specific, can be used to identify genes with expression patterns of interest.
Fig. 3.

Gene annotation. A) An overview of gene architecture, showing a stretch of DNA demarcated into protein-coding exons and non-coding introns. B) An example of gene annotation, proceeding from identification of genes from the genome either predicted de novo from gene-like patterns in the sequence or by comparison to known genes. In this case, the daf-16 gene is shown with its introns and exons. Below that is a transcript from the daf-16 gene, with the open reading frame (ORF) along with the upstream and downstream untranslated regions (UTR). The genome is also scanned for known protein domains; in this case, daf-16 has a single protein domain, although other proteins may contain several domains.
Basic Genomic Metrics
The quality of a genome assembly can be assessed by metrics such as the total size of the genome relative to the fold coverage. This is estimated by dividing the total number of assembled nucleotides by the genome size, which varies from 50-315 Mb for published and forthcoming nematode genomes (Table 1). For example, the Ascaris suum genome was sequenced with ∼80 fold coverage, meaning that the 309 megabase genome was assembled from about 25 gigabases of sequence (Jex et al., 2011). The GC content of the genome is usually reported, and varies between 27-48% among published and forthcoming nematode genomes (Table 1). Other commonly reported quality metrics of genomic assemblies address contiguity and completeness. One commonly used metric is the ‘N50’ value, which indicates that half of the genome is in contigs at least as large as that value. For instance, the N50 of the A. suum genome is 408 kb, meaning that half of the assembly is in contigs at least 408 kb in length (Jex et al., 2011). Also important is the number of predicted protein coding genes, ranging from 13,000-45,000 among published and forthcoming genomes (Table 1). There are several other genomic statistics that have become potentially useful in comparisons such as gene density, number of transfer RNAs, and the percentage of high copy repeated sequences in the genome (Consortium, 1998; Stein et al., 2003; Ghedin et al., 2007).
Quality assessments of genomic assembly provide confidence and a framework for interpreting subsequent analyses while other genomic metrics provide more information about the biological content of the genome. For instance, all known metazoan genomes require a certain number of tRNAs for codon recognition and for shuttling specific amino acids during translation, such that the number of tRNAs, tRNA pseudo-genes, and tRNA-derived repeats found in a genome assembly can serve as a rough estimate of completeness(Bermudez-Santana et al., 2010).
How Protein Sequences are Analyzed and What They Reveal About Your Nematode of Choice
Annotation of nematode whole genomic sequence is complicated by several factors, including the structural complexity of introns, alternative RNA splicing, variable gene density, transplicing, and the presence of operons. Fortunately, annotation efforts on novel nematode species can leverage the excellent annotation of the C. elegans genome. These annotations are carefully curated and maintained in WormBase (www.wormbase.org), an expandable model for genome curation and annotation that already includes many available nematode genomes including Ascaris suum, Brugia malayi, Bursaphelenchus xylophilus, Meloidogyne hapla, Meloidogyne incognita, and many others. WormBase, with its established infrastructure and fulltime maintenance could serve as a repository for all nematode genomes and subsequent annotation (Harris et al., 2010). As more genomes are sequenced and annotated, it has become clear that the availability of transcriptome data (e.g. RNA-seq; see above) is paramount for more accurate and comprehensive gene predictions, as well as elucidating biological function. While RNA requires more careful handling to avoid degradation, the reverse transcribed cDNA can be sequenced in the exact same manner as genomic DNA and for a similar cost.
While the specific details of annotation for each nematode genome differ, a general approach to protein analysis involves the following: identification of the protein-coding gene set, characterization by protein domain analysis and comparison to other protein databases, and comparative analysis with other nematodes and beyond. The identification of protein-coding genes is done using one or multiple gene prediction software packages, which generate ab initio predictions using machine-learning methods such as Hidden Markov Models to identify open reading frames indicative of protein coding genes. The accuracy of these predictions can be improved by training the prediction software on experimental datasets such as ESTs, cDNA, protein similarity matches, and RNA-seq datasets. In particular, RNA-seq data can be used to partially or fully confirm gene-finder predictions (Stein et al., 2003; Pevsner, 2009b; Mortazavi et al., 2010; Jex et al., 2011). While computationally intensive, gene finding requires fewer resources than assembly.
As part of the annotation process, genes and proteins of the newly sequenced genome are evaluated by comparison to previously annotated genes and proteins from databases and genomes. Such evaluations identify putative homologous genes and proteins by sequence similarity. Homologous genes can be subdivided into orthologs and paralogs, depending on their history (Fitch, 1970). Orthologs are homologous sequences in different species that descended from a common ancestral gene during speciation, such that the ortholog of a gene in one species is the gene in the second species that shares decent from a common ancestral gene and is uniquely closely related to the gene in the first species. For example, the last common ancestor of Pristionchus pacificus and C. elegans may have possessed only one copy of the daf-16 gene, which encodes a transcription factor in the insulin/IGF-1-mediated signaling pathway, and each of these extant species has one copy of daf-16, making these genes daf-16 orthologs (Riddle et al., 1981; Sonnhammer and Durbin, 1997; Consortium, 1998; Dieterich et al., 2008; Pevsner, 2009a) (Fig. 4A). We make this inference about C. elegans and P. pacificus knowing that both of these species as well as an outgroup taxon (in this case A. suum) all only have one copy of daf-16.
Fig. 4.

Distinction between orthologous and paralogous genes. A) Orthologs are homologous sequences in different species that descend from a common ancestral gene during speciation. The daf-16 gene in an ancestral nematode was conserved in both extant lineages resulting from a speciation event that lead to P. pacificus and C. elegans. Ppa-daf-16 is the conserved daf-16 gene in P. pacificus and Cel-daf-16 is the conserved gene in C. elegans. B) Paralogs are homologous sequences within a species, having arisen by gene duplication or similar event. While Ppa-dsh-1 and Cel-dsh-1 are orthologs in this example, having both been conserved from the same parental dsh-1 copy, Cel-dsh-1 and Cel-dsh-2 are paralogs, having been duplicated within C. elegans. C) Neighbor-joining tree generated from gene comparisons of dsh-1 homologs between A. suum, P. pacificus, and C. elegans, providing evidence that Cel-dsh-1 is the ortholog of Ppa-dsh-1 while Cel-dsh-1 and Cel-dsh-2 are paralogs. The small bar at the bottom center of the tree shows the approximate distance equal to 0.08 nucleotide changes. A. suum was used as the outgroup taxon (see Figure 2). The tree was made from an alignment of the full proteins in MUSCLE and subsequently analyzed using default parameters of the ‘Dnadist’ and ‘Neighbor’ programs from PHYLIP 3.68 software package (Edgar, 2004; Felsenstein, 1989). D) A 34 amino acid window of the protein alignment from which the tree in part C was generated. It shows the sequence conservation of the dsh-1 orthologs and the subsequent divergence of Cel-dsh-2 from the other sequences. Areas of sequence divergence are highlighted in grey while asterisks (*) indicate conserved amino acid identity across all four genes. Hyphens (-) indicate gaps in the alignment, likely the result of insertion/deletion events. A. suum serves as the outgroup taxon.
Paralogs are homologous sequences within a species, having arisen by gene duplication. Paralogs are thought a priori to share similar function, but this may not always be the case, as gene duplication and subsequent modification is thought to be the major way organisms evolve genes with novel functions (Graur and Li, 2000a). For example P. pacificus contains a single copy of the gene dsh-1, which encodes a signaling protein involved in embryogenesis, while C. elegans has two paralagous copies of the dishevelled gene, dsh-1 and dsh-2. Relative to the outgroup A. suum, there appears to have been a duplication event in the C. elegans lineage since it diverged from P. pacificus; the last common ancestor of P. pacificus and C. elegans likely also possessed a single copy of this gene (Fig. 4B) (Eisenmann, 2005; Pevsner, 2009a). Based on higher sequence conservation with the sole P. pacificus protein, only Cel-dsh-1 is considered to be a genuine ortholog of Ppa-dsh-1, though experimental confirmation of conserved function would validate this inference (Fig. 4C and 4D).
Once a gene set has been identified, putative functions are ascribed by database searching and similarity comparisons of the proteins from the new genome to those with known function. Commonly used databases include the NCBI BLAST database, the EMBL-EBI InterProScan, Pfam, and Gene Ontology databases (Zbodnov and Apweiler, 2001; Bateman et al., 2004; Ye et al., 2006; Thomas et al., 2007; Pevsner, 2009b). This initial assignment of protein function is based on the assumption of homology by sequence or domain similarity. In essence, the proteome (the full complement of protein coding genes) that results from whole genome sequencing and annotation has functions ascribed to its individual protein-coding sequences by comparing them to a number of different databases in search of sequence or domain similarity (Dieterich et al., 2008). When a protein sequence from the genomic dataset has the highest degree of similarity to one sequence in another genome, it is a priori assumed to be homologous or to be derived from shared ancestry. The protein is further inferred to have similar function. In molecular phylogeny, homology infers shared ancestry. One important caveat of identifying homologs by sequence similarity is that it is not uncommon for two proteins to share functional similarity without shared ancestry, as a result of convergent evolution (Graur and Li, 2000a; Graur and Li, 2000b; Pevsner, 2009a). For example, Heterorhabditis and Steinernema nematodes utilize a specific type of insect parasitism and are known as entomopathogenic nematodes (EPNs), a characteristic they share not through ancestry but convergent evolution (Adams et al., 2007). A notable molecular example is the convergent evolution of nearly identical antifreeze proteins in both Antarctic notothenioid fishes and Arctic cod, which show remarkable sequence and functional similarity that is due to evolutionary convergence rather than shared ancestry (Chen et al., 1997). Another nematode example of convergence is the hermaphroditism of C. elegans and C. briggsae, which though outwardly similar as self-fertile hermaphrodites, have different molecular mechanisms for achieving this mode of reproduction (Hill et al., 2006). The opposite caveat is also true; proteins of shared ancestry do not necessarily share similar function (Sangar et al., 2007).
Orthologous gene associations across multiple genomes can provide powerful evolutionary insights into biological functions of individual genes as well as the evolution of species. They can be used to identify conserved genes, as in the case of pan-nematode genes or clade-specific genes. The identification of widely conserved or more specific genes serves as the basis for designing molecular diagnostic tools and elucidating the relationships between species. Multigene analyses from EST datasets have previously been successfully used to inform nematode phylogeny, and additional whole genome sequencing could identify new diagnostic markers to overcome sequencing identification difficulties and lack of phylogenetic resolution in some vexing taxa such as the tylenchids (Scholl and Bird, 2005; Adams et al., 2009). Furthermore, such comparisons can be used in pursuit of non-conserved taxon-specific genes, which may reveal something about the particular biology and adaptations of individual species. For example, Kikuchi et al. (2011), in conjunction with publishing the Bursaphelencus xylophilus genome included an orthology analysis across 10 nematode genomes. Although the genes shared across the 10 species did not fit an obvious phylogenetic pattern, the comparison revealed several gene families that are broadly conserved as well as small groups of genes shared between pairs or groups of nematodes that may be involved in the ecologies of those species. For example, 144 genes are shared exclusively between P. pacificus and B. xylophilus (Kikuchi et al., 2011). These nematodes occupy different ecological niches (one is necromenic and the other is a migratory endoparasite of plants), but they both share a close association with insects during their lifecycle. Kikuchi et al. (2011) suggest that these genes are candidates for being involved in that association. The case for such a conclusion would be stronger if genome comparisons could show that the last common ancestor of both species also shared an association with insects.
Orthology analyses can also be used to explore the conservation of important biological pathways, such as sex determination, dauer formation, or the RNAi pathway. Because of the extent of detailed genetic exploration in C. elegans, a common starting place is to identify pathways of interest in C. elegans and search for their orthologs in another nematode of interest, though these results should be interpreted conservatively. For example, the RNAi pathway in C. elegans has been well-studied and found to be quite complex, with at least 77 genes known to be involved in core aspects of the process (Dalzell et al., 2011). As a powerful reverse genetics technique, RNAi is a commonly examined pathway in newly sequenced genomes and has been developed as an experimental tool in both plant- and animal-parasitic nematodes including Globodera pallida, Heterordera glycines, M. incognita, and B. malayi (Urwin et al., 2002; Aboobaker and Blaxter, 2003; Dalzell et al., 2010). It may even have practical utility in agriculture in controlling plant-parasitic nematodes or at least increasing plant resistance (Huang et al., 2006; Yadav et al., 2006). How many of the 77 known RNAi effector genes are absolutely necessary for RNAi in general and how many are part of the specific mechanism of RNAi in C. elegans? For instance, sid-1 is necessary for systemic RNAi in C. elegans, but systemic RNAi has been reported in several other species that do not seem to contain an identifiable homolog of sid-1, including B. malayi, Globodera and Meloidogyne spp., Pristionchus pacificus, and Panagrolaimus superbus (Aboobaker and Blaxter, 2003; Kimber et al., 2007; Shannon et al., 2008; Rosso et al., 2009; Dalzell et al., 2010; Cinkornpumin and Hong, 2011). The successful application of experimental RNAi in species that are apparently missing some genes required for systemic RNAi in C. elegans implies that either these genes are rapidly evolving or have only become necessary in C. elegans, or that an alternate pathway exists (Aboobaker and Blaxter, 2003; Ghedin et al., 2007; Dalzell et al., 2011). Although RNAi has been shown to work in a number of both plant- and animal- parasitic nematodes, it is thought that culturability and the feasibility of maintaining non-stressful culturing conditions may better explain RNAi competencies than the disparity of RNAi effector genes across taxa(Dalzell et al., 2011). As more species are added to these types of genomic analyses and genetic experimentation in non-model systems continues to grow, our understanding of these processes and which parts are conserved, derived, or rapidly evolving will become more clear.
Operons
One striking feature of nematode genomes studied thus far is the presence of operons. Though originally thought to be a genomic feature unique to prokayrotes, operons have been found in nematodes as well as some ascidians and fruit flies (Blumenthal, 2004). Bacterial operons comprise 2 or more genes that are transcribed to form a single mRNA transcript (Fig. 5). In nematodes, multiple genes are transcribed into a single primary transcript, which is then processed into separate mRNAs; through RNA-splicing events, a spliced leader is added to the 5’ end of each downstream transcript in an operon (Fig. 5). In C. elegans, about 70% of mRNAs include a spliced leader, the majority of which (∼55%) are of the SL1 type. These SL1 spliced leaders are typically either from non-operonic transcripts or are from the first gene in an operon (Fig. 5) (Blumenthal, 2005). Downstream transcripts from within an operon each have an SL2 leader (Blumenthal, 2005). Operons can be inferred from the genome by the presence of very closely spaced genes in the same orientation in the genome and from the presence of SL2 spliced leaders. Apparent operons have been identified in all published nematode genomes with the exception of Trichinella spiralis, a highly unusual nematode, quite distantly related to all other sequenced nematodes and one of the world’s largest intracellular parasites (Fig. 2) (Despommier, 1990; Mitreva et al., 2011). Although T. spiralis is missing both canonical nematode trans-spliced leaders, SL1 and SL2, the presence of a number of other distinct spliced leader sequences leaves open the possibility that this species does contain operons. Additional nematode genomic data, especially from taxa in Enoplia, Doryliamia, and basal clades of Chromadoria, may reveal the untold story of operon evolution among nematodes (Fig. 2). Operons are thought to have evolved in nematodes to facilitate transitions from arrested development to rapid growth (Zaslaver et al., 2011).
Fig. 5.

Prokaryotic and eukaryotic operons. A) The prokaryotic operon model is that polycistronic mRNA is produced from a single promoter, containing several genes in the same transcript. Each protein is translated from a different location of a single mRNA. B) Eukaryotic operons also produce a single preRNA transcript with multiple genes, but are then processed to form mRNAs for each individual protein. As part of the processing each mRNA has a splice leader added to the 5’ end of the transcript. These mRNAs are then translated. Usually the first gene from the preRNA in a nematode operon transcript is spliced with the SL1 splice leader attached while all other downstream genes have the SL2 splice leader attached.
Genomes and Ecology
The first report of a nematode genome focused on the sequencing methodology, the development of physical and genetic maps, assembly, and annotation, as well as a comparison of the genome to prokaryotes and yeast (Consortium, 1998; Consortium, 1999). This comparison revealed that C. elegans has an unusually high number of nuclear hormone receptor proteins (NHRs), prompting researchers to propose that NHRs were perhaps important in the evolution of multicellularity (Consortium, 1998). Though originally thought to be normal among nematodes, it is now known that even among close relatives, C. elegans is an outlier in terms of its number of NHRs and G protein-coupled receptors (GPCRs) and in these respects is not an archetypical nematode(Blaxter, 1998; Blaxter, 2011). The anomalously high number of NHRs and GPCRs in the C. elegans genome was found by examining the top 20 most prevalent protein domains in the genome. Such comparisons of gene and domain prevalence among species may reveal important differences in the genome that ultimately underlie differences in the evolution, ecology, and lifestyles of nematodes. In this way, comparative genome analyses will serve as a tool for testing hypotheses about the ecology and evolution of related species; and the resolving power of such comparisons will increase with the addition of more sequenced taxa.
The sequencing of C. briggsae greatly enhanced our understanding of the C. elegans genome by providing strong evidence for 1,300 previously unidentified genes, thus demonstrating how sequencing closely-related species can enhance the annotation of genomes (Stein et al., 2003). Analysis of repeat regions revealed that C. elegans and C. briggsae have undergone rapid evolutionary turnover at the sequence level, providing evidence for a more recent divergence of these two nematodes compared to the evolutionary split between human and mouse lineages (∼40 million years ago for the nematodes and ∼75 million years ago for mouse/human). Similarly, the amino acid identity revealed between putative orthologs (∼80% for C. briggsae/C. elegans and ∼78.5% for mouse/human) supports this conclusion (Stein et al., 2003; Cutter, 2008).
As sequencing technology has advanced and costs have dropped, additional nematode genomes have been sequenced, including close relatives of C. elegans (C. angaria, C. brenneri, C. japonica, and C. remanei) and a handful of economically important parasites such as Bursaphelenchus xylophilus, Meloidogyne incognita, and M. hapla (Ghedin et al., 2007; Abad et al., 2008; Opperman et al., 2008; Barrière et al., 2009; Mortazavi et al., 2010; Jex et al., 2011; Kikuchi et al., 2011; Mitreva et al., 2011). One of the rationales for vertebrate-parasitic nematode sequencing projects (B. malayi, A. suum, and T. spiralis) was the identification of candidate genes to target pharmacologically (Ghedin et al., 2007; Jex et al., 2011; Mitreva et al., 2011). This is a particularly important avenue of research given the large number of humans affected by nematode diseases and our current reliance on a small pool of drugs, whose effectiveness is at risk due to increasing resistance (Keiser and Utzinger, 2010). In addition to identifying new drug targets, these genomic analyses identified genes likely to be involved in the vertebrate-parasitic lifestyle, or perhaps parasitism in general. The abundance and diversity of secreted proteases and protease inhibitors in these genomes was an interesting result and has produced a long list of genes that are candidates to be involved in invasion of host tissues and degradation or evasion of host immune responses. The B. malayi genome’s lack of key metabolic enzymes provided evidence for this nematode’s reliance on host- or Wolbachia-supplied molecules for purine, riboflavin, and heme biosynthesis (Ghedin et al., 2007). Due to the basal position of T. spiralis in Dorylaimia (Fig. 2), its genome was compared to all other available nematode genomes to identify pan-Nematoda-specific conservation. The resulting list of genes and proteins may have fundamental importance in all nematodes and points to potential targets for control of parasitic nematodes throughout the phylum(Mitreva et al., 2011). Because of the highly specific and derived lifestyle of T. spiralis, which is an intracellular parasite, it is likely that examination of additional basal taxa will improve and solidify a pan-Nematoda candidate gene list, which, in addition to providing potential pharmacological targets could be used to inform deeper level phylogenetic studies.
Root-knot nematodes are among the most agriculturally devastating plant pathogens known in any phylum (Trudgill and Blok, 2001; Abad et al., 2008). This motivated the sequencing of Meloidogyne incognita, closely followed by Meloidogyne hapla (Abad et al., 2008; Opperman et al., 2008). These genomes have provided intriguing insights into the adaptive strategies used by metazoans to circumvent immunity and successfully parasitize plants (Abad et al., 2008; Opperman et al., 2008). They also provided evidence to support the long-suspected role of horizontal gene transfer (HGT) in the evolution of plant parasitism (Smant et al., 1998; Popeijus et al., 2000). Both of these parasites seem to have benefitted from the acquisition of plant cell wall-degrading enzymes that appear bacterial in origin. The idea that nematodes can acquire and utilize such enzymes in a cross-kingdom way was further bolstered by similar findings from genomic analyses of the mycophagous plant parasite B. xylophilus and the necromenic species P. pacificus (Dieterich et al., 2008; Kikuchi et al., 2011). Recent follow-up work on HGT in multiple Pristionchus and related species utilized genome, transcriptome, and EST data sets, and revealed functional laterally acquired cellulase genes in several diplogastrid species, notable turnover of cellulase genes inferred from elevated gene birth and death rates, and showed evidence for selective forces working on individual cellulase genes with a high degree of specificity (Mayer et al., 2011). Moreover, some cellulases found in B. xylophilus have not been found in any other nematode and appear fungal in origin, providing evidence that, if these genes are the result of HGT and not the independently arising result of convergent evolution, nematodes may not be limited to bacteria as sources of adaptational armament (Kikuchi et al., 2011). The evidence for HGT in multiple distantly related nematodes (Bursaphelenchus, Koerneria, Meloidogyne, and Pristionchus) suggests that this mode of gene acquisition may play a broadly significant role in nematode adaptation and evolution (Fig. 2).
One clear theme that has emerged from genomic comparisons is that there may not be an archetypal nematode (Blaxter, 1998; Blaxter, 2011). For example, the massive expansions in GPCRs and NHRs reported in C. elegans are thus far not replicated in the genomes of any other sequenced nematodes, and likely play a significant role in C. elegans’ natural ecology, which has only recently been explored through modern investigation (Kiontke and Sudhaus, 2006; Troemel et al., 2008; Félix and Braendle, 2010). As more nematode species are fully sequenced, it is becoming clear that the ecology and specific biology of each species will become increasingly valuable in the interpretation and use of these genomes. While earlier reports of nematode genomes focused heavily on sequencing methodologies and the technical details of gene prediction and annotation, more recent studies have highlighted genomes in the context of nematode ecology and evolution; this trend is likely to continue. For instance, P. pacificus is an omnivorous feeder, necromenic but not parasitic. It associates with arthropods and waits for them to die, feasting on the microbial and fungal bloom resulting from the arthropod host’s death (Herrmann et al., 2006; Kiontke and Sudhaus, 2006). A broad view of the P. pacificus genome reveals expansions in protein families playing key roles in stress tolerance and the metabolism of xenobiotics (foreign chemical compounds; e.g. host defense molecules) (Dieterich et al., 2008). Tolerance to low oxygen concentrations and toxic host enzymes as well as complex metabolic pathways and other morphological adaptations were predicted to assist this nematode in its lifestyle, but prior to its genome being sequenced the molecular architecture of these adaptations could only be speculative (Dieterich et al., 2008). The genetic underpinnings of necromeny in P. pacificus and its adaptation to this particular niche have been revealed through its genome. These findings lead to additional genomically-generated hypotheses and sow fertile ground for future experimentation.
Ecological genomics is a burgeoning field aimed at understanding the genetic mechanisms that underlie organismal responses and adaptations to their natural environments (Ungerer et al., 2008). Model organisms, often chosen for ease of culture and a host of other traits that favor laboratory growth and experimentation, usually lack the extensive ecological context and framework that has been painstakingly built for many non-model systems. In contrast, many organisms used in ecological studies do not have the extensive experimental tool development (e.g. transformation and RNAi) or genetic pathways and interactions mapped out as in model systems. The time is ripe for dramatic expansion of ecological studies using model systems and genomic/transcriptomic sequencing and accompanying tool development to be done in favored ecological systems (Elmer and Meyer, 2011). Nematodes are in a superb position to see progress in both areas, with several well-developed model systems being explored from an ecological context (e.g. Kiontke and Sudhaus, 2006; Rae et al., 2008; Troemel et al., 2008; Félix and Braendle, 2010; Mayer and Sommer, 2011) and for nematode species for which archives of ecological data have been accumulated to be scrutinized from a genomic context (e.g. Ciche and Sternberg, 2007; Hallem et al., 2011).
Entomopathogenic Nematodes as an Example of Question-Driven Genomics
Nematode genomics, now highlighting specific aspects of organismal biology, life history traits, and ecology and evolution, provides opportunity for researchers to utilize the powerful broad view of sequencing to learn more about their nematode of choice. As an illustrative example of ecological genomics and what could be accomplished for every niche occupied by nematodes, we conclude by discussing some of the interesting genomic insights that can be gleaned from examining the forthcoming entomopathogenic nematode genomes.
EPNs occupy an interesting niche somewhere between parasitoids and pathogens, utilizing insect-pathogenic bacteria to facilitate their form of parasitism, acting as a vector for the bacteria and, working together as a complex, the nematode and bacteria rapidly kill their host (Kaya and Gaugler, 1993; Dillman et al., 2012). This very specific form of parasitism seems to have arisen at least twice among nematodes, in Heterorhabditidae and Steinernematidae, which are not closely related. The genomic sequencing of heterorhabditid and steinernematid nematodes will provide the framework for a genetic comparison of the evolution of entomopathogeny in these lineages (Elmer and Meyer, 2011). In contrast to the vertebrate- and plant-parasitic nematode genome studies, which compare organisms that obtain resources by different means, the intra-guild comparisons of EPN genomes will focus on species that exploit the same kind of environmental resources in similar ways (Root, 1967; Fauth et al., 1996). A genomic comparison of EPNs from multiple genera has the advantage of decades of ecological research and will increase our understanding of adaptation and convergent evolution in addition to revealing just how similar or different this niche exploitation is at the genetic level.
EPNs have rapidly become models for studying parasitism and mutualism. The genetic components of their association with symbiotic bacteria have been heavily studied from the bacterial side, but largely neglected in terms of the nematode’s contribution (Ciche and Sternberg, 2007; Chaston and Goodrich-Blair, 2010). Genome-wide expression analysis against the backdrop of the genomic sequence could shed light on what, if any, contribution is made by the nematodes to symbiosis. Within Steinernema, there are more than 60 described species (Nguyen et al., 2007a; Nguyen et al., 2007b; Nguyen et al., 2008; Tarasco et al., 2008; Edgington et al., 2009; Spiridonov et al., 2010; Khatri-Chhetri et al., 2011; Nguyen and Buss, 2011; Stokwe et al., 2011). Though only a handful of these have been tested, the host-range and specificity of insects they can infect is diverse and varied. A striking example is S. carpocapsae, which is the most heavily studied steinernematid. With an extremely broad host range, S. carpocapsae is capable of infecting more than 250 species of insects across 10 orders, although some infections were only demonstrated under laboratory conditions (Poinar, 1979). Closely related to S. carpocapsae is S. scapterisci, which is known to have a much narrower host range and seems to be a cricket specialist (Nguyen and Smart, 1991; Frank, 2009). The wide view afforded by protein family abundances revealed by genomes will provide testable hypotheses about the breadth of EPN host-range and the specificity of some EPNs for certain insect hosts, beyond what is currently known.
EPN research has also seen recent developments in the neuronal basis of behavior and the molecular mechanisms underlying host tissue invasion and death (Toubarro et al., 2010; Hallem et al., 2011). Understanding protein domain abundance against this backdrop will likely hone existing hypotheses and direct future experimentation, leading to a deepening of our knowledge in both of these areas of research. Along with the broad overview on the architecture of parasitism, it is anticipated that EPN genomes will provide insights to the above mentioned and other aspects of EPN ecology. A hopeful expectation of most new nematode genomes is that they will pave the way for techniques such as transformation and RNAi to be used in experimentally testing the genomically generated hypotheses, as exemplified with P. pacificus (Dieterich et al., 2008; Cinkornpumin and Hong, 2011).
Conclusion
Many new nematode genomic sequencing projects are underway, and improving technologies means still more will become feasible and affordable. These widening horizons are generating a need for more nematodes to be cultured and have their DNA harvested. More importantly, it opens the door for collaborations between genomicists and nematologists. We expect that fruitful collaborations will entail far more than merely providing material and could include various aspects such as (a) knowledge of the ecological background and candidate pathways or biological phenomena to explore within the sequence, (b) phylogenetic knowledge of sister taxa or associated nematodes for comparison or particularly informative developmental stages for transcript analysis, and (c) interesting morphological features that remain to be genetically explored. We urge the members of the Society of Nematologists to utilize their expertise and the wealth of their collective ecological knowledge to contribute to sequencing efforts and to adopt genomics into the toolkit of nematology. As nematology stands at the precipice of genomic grandeur, with 959 nematode genomes planned (a number chosen to reference the 959 somatic cells of C. elegans (Kumar et al., 2012)), we will soon be suffused with genomic data, offering the potential to discover long-sought answers to the biology, ecology, and evolution of genomes, and promising in turn to raise many more new questions.
Acknowledgments
We would like to thank all the researchers who have contributed to the wealth of literature from which we have drawn and from which we have been stimulated, enlightened, and encouraged. We also wish to thank Ganpati Jagdale and Parwinder Grewal for organizing the “EPNs as model systems in stress physiology and evolutionary biology” symposium at the 2011 Society of Nematologists annual meeting, and for inviting the authors to contribute. We express gratitude to Hillel Schwartz, Jagan Srinivasan, James Baldwin, Mihoko Kato, Margaret Ho, and two reviewers for helpful comments and discussion on the manuscript. ARD was supported by a United States Public Health Service Training Grant (T32GM07616). PWS is an investigator with the Howard Hughes Medical Institute.
Abbreviations and Definitions
bp - base pairs.
ng - nanograms.
Mb - megabase pairs, which corresponds to million base pairs.
Gb - gigabase pairs, which corresponds to billion base pairs.
Next-gen - next generation sequencing technologies
Homopolymeric regions - long stretches of a single nucleotide
Contigs - contiguous stretches of genomic sequence.
Operons - single transcription units that give rise to multiple independent proteins.
Guild - a group of species, regardless of phylogenetic relationship or geographic distribution that use the same kind of environmental resources in a similar way.
GC content - the percentage of guanines and cytosines.
RNA-seq - a technique that involves the conversion of isolated RNA transcripts into cDNA copies and the subsequent sequencing of these cDNAs using high-throughput sequencing technology. This is also known as whole transcriptome shotgun sequencing and can provide a glimpse of the overall transcriptome of an organism or it can be done on specific stages or under certain physiological or environmental conditions to reveal changes in transcription during biologically interesting phenomena.
NHRs - nuclear hormone receptors, which are transcription factors that are involved in regulating gene expression and thought to be important in the evolution of multicellularity.
GPCRs - G-protein-coupled receptors. These 7-transmembrane receptors mediate chemoreception in nematodes.
Literature Cited
- Abad P, Gouzy J, Aury JM, Castagnone-Sereno P, Danchin EGJ, Deleury E, Perfus-Barbeoch L, Anthouard V, Artiguenave F, Blok VC. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nature Biotechnology. 2008;26:909–915. doi: 10.1038/nbt.1482. [DOI] [PubMed] [Google Scholar]
- Aboobaker AA, Blaxter M. Use of RNA interference to investigate gene function in the human filarial nematode parasite Brugia malayi. Molecular Biochemical Parasitology. 2003;129:41–51. doi: 10.1016/s0166-6851(03)00092-6. [DOI] [PubMed] [Google Scholar]
- Adams BJ, Dillman AR, Finlinson C. 2009. Molecular taxonomy and phylogeny. Pp. 119–138 in R. N. Perry M. Moens and J. L. Starr, eds. Root-knot Nematodes, CABI. [Google Scholar]
- Adams BJ, Peat SM, Dillman AR. 2007. Phylogeny and evolution. Pp. 693–733 in K. B. Nguyen and D. J. Hunt, eds. Entomopathogenic nematodes: Systematics, phylogeny, and bacterial symbionts., vol. 5. Leiden-Boston: Brill. [Google Scholar]
- Barrière A, Yang S-P, Pekarek E, Thomas CG, Haag ES, Ruvinsky I. Detecting heterozygosity in shotgun genome assemblies: Lessons from obligately outcrossing nematodes. Genome Research. 2009;19:470–480. doi: 10.1101/gr.081851.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Research. 2004;1:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez-Santana C, Attolini CS, Kirsten T, Engelhardt J, Prohaska SJ, Steigele S, Stadler PF. Genomic organization of eukaryotic tRNAs. BMC Genomics. 2010;11:270. doi: 10.1186/1471-2164-11-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaxter M. Caenorhabditis elegans is a nematode. Science. 1998;282:2041–2046. doi: 10.1126/science.282.5396.2041. [DOI] [PubMed] [Google Scholar]
- Blaxter M. Nematodes: The worm and its relatives. PLoS Biology. 2011;9:1–9. doi: 10.1371/journal.pbio.1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal T. Operons in eukaryotes. Briefings in Functional Genomics and Proteomics. 2004;3:199–211. doi: 10.1093/bfgp/3.3.199. [DOI] [PubMed] [Google Scholar]
- Blumenthal T. 2005. Trans-splicing and operons. WormBook, ed. The C. elegans Reasearch Community, WormBook, doi/10.1895/wormbook.1.5.1, http://www.wormbook.org.
- Burgess DJ. Comparative genomics: Mammalian alignments reveal human functional elements. Nature Reviews Genetics. 2011;12:806–807. doi: 10.1038/nrg3112. [DOI] [PubMed] [Google Scholar]
- Chaston J, Goodrich-Blair H. Common trends in mutualism revealed by model associations between invertebrates and bacteria. FEMS Microbiology Reviews. 2010;34:41–58. doi: 10.1111/j.1574-6976.2009.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, DeVries AL, Cheng C-HC. Convergent evolution of antifreeze glycoproteins in Antarctic notothenioid fishes and Arctic cod. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:3817–3822. doi: 10.1073/pnas.94.8.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciche TA, Sternberg PW. Postembryonic RNAi in Heterorhabditis bacteriophora: a nematode insect parasite and host for insect pathogenic symbionts. BMC Developmental Biology. 2007;7:101. doi: 10.1186/1471-213X-7-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinkornpumin JK, Hong RL. 2011 doi: 10.3791/3270. RNAi mediated gene knockdown and transgenesis by microinjection in the necromenic nematode Pristionchus pacificus. Journal of Visualized Experiments 56: e3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Drumm ML, Cole JL, Lockwood WK, Van de Woude GF, Iannuzzi MC. Contruction of a general human chromosome jumping library, with application to cystic fibrosis. Science. 1987;235:1046–1049. doi: 10.1126/science.2950591. [DOI] [PubMed] [Google Scholar]
- Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Consortium. How the worm was won. Trends in Genetics. 1999;15:51–58. doi: 10.1016/s0168-9525(98)01666-7. [DOI] [PubMed] [Google Scholar]
- Cutter AD. Divergence times in Caenorhabditis and Drosophila inferred from direct estimates of the neutral mutation rate. Molecular Biology and Evolution. 2008;25:778–786. doi: 10.1093/molbev/msn024. [DOI] [PubMed] [Google Scholar]
- Dalzell JJ, McMaster S, Fleming CC, Maule AG. Short interfering RNA-mediated gene silencing in Globodera pallida and Meloidogyne incognita infective stage juveniles. International Journal of Parasitolology. 2010;40:91–100. doi: 10.1016/j.ijpara.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Dalzell JJ, McVeigh P, Warnock ND, Mitreva M, Bird DM, Abad P, Fleming CC, Day TA, Mousley A, Marks NJ. 2011 doi: 10.1371/journal.pntd.0001176. RNAi effector diversity in nematodes. PLoS Neglected Tropical Diseases 5(6): e1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ley P, Blaxter M. 2002. Systematic position and phylogeny. In: The Biology of Nematodes, ed. D. Lee, pp. 1–30. London: Taylor & Francis. [Google Scholar]
- De Ley P, Blaxter M. 2004. A new system for Nematoda: combining morphological characters with molecular trees, and translating clades into ranks and taxa. Pp. 633–653 in R. Cook and D. J. Hunt, eds. Nematology Monographs and Perspectives, vol. 2. Leiden: E.J. Brill. [Google Scholar]
- Despommier DD. Trichinella spiralis: The worm that would be virus. Parasitology Today. 1990;6:193–196. doi: 10.1016/0169-4758(90)90355-8. [DOI] [PubMed] [Google Scholar]
- Dieterich C, Clifton SW, Schuster LN, Chinwalla A, Delehaunty K, Dinkelacker I, Fulton L, Fulton R, Godfrey J, Minx P. The Pristionchus pacificus genome provides a unique perspective on nematode lifestyle and parasitism. Nature Genetics. 2008;40:1193–1198. doi: 10.1038/ng.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillman AR, Chaston JM, Adams BJ, Ciche TA, Goodrich-Blair H, Stock SP, Sternberg PW. 2012 doi: 10.1371/journal.ppat.1002527. An entomopathogenic nematode by any other name. PLoS Pathogens 8(3): e1002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington S, Buddie AG, Tymo LM, Hunt DJ, Nguyen KB, France AI, Merino LM, Moore D. Steinernema australe n. sp. (Panagrolaimorpha: Steinernematidae) a new entomopathogenic nematode from Isla Magdalena, Chile. Nematology. 2009;11:699–717. [Google Scholar]
- Eisenmann D. 2005. Wnt signaling. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.7.1, http://www.wormbook.org.
- Elmer KR, Meyer A. Adaptation in the age of ecological genomics: insights from parallelism and convergence. Trends in Ecology and Evolution. 2011;26:298–306. doi: 10.1016/j.tree.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Fauth JE, Bernardo J, Camara M, Resetarits WJ, Van Buskirk J, McCollum SA. Simplifying the jargon of community ecology: A conceptual approach. The American Naturalist. 1996;147:282–286. [Google Scholar]
- Félix MA, Braendle C. The natural history of Caenorhabditis elegans. Current Biology. 2010;20:965–969. doi: 10.1016/j.cub.2010.09.050. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Fitch WM. Distinguishing homologous from analogous proteins. Systematic Zoology. 1970;19:99–113. [PubMed] [Google Scholar]
- Frank JH. 2009. Steinernema scapterisci as a biological control agent of Scapteriscus mole crickets. Pp. 115–131 in A. E. Hajek, T. R. Glare, and M. O’Callaghan, eds. Progress in biological control: Use of microbes for control and eradication of invasive arthropods, vol 6. Springer. [Google Scholar]
- Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, Allen JE, Delcher AL, Guiliano DB, Miranda-Saavedra M. Draft genome of the filarial nematode parasite Brugia malayi. Science. 2007;317:1756–1760. doi: 10.1126/science.1145406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graur D, Li W-H. 2000a. Gene duplication, exon shuffling, and concerted evolution. Pp 249–322 in D. Grauer and W.-H. Li, eds. Fundamentals of molecular evolution, 2nd Edition. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Graur D, Li W-H. 2000b. Genome evolution. Pp 367–427 in D. Grauer and W.-H. Li, eds. Fundamentals of molecular evolution, 2nd Edition. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Hallem EA, Dillman AR, Hong AV, Zhang Y, Yano JM, DeMarco SF, Sternberg PW. A sensory code for host seeking in parasitic nematodes. Current Biology. 2011;21:377–383. doi: 10.1016/j.cub.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, Antoshechkin I, Bieri T, Blasiar D, Chan J, Chen WJ, De La Cruz N, Davis P, Duesbury M, Fang RH. WormBase: a comprehensive resource for nematode research. Nucleic Acids Research. 2010;38:D463–D467. doi: 10.1093/nar/gkp952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann M, Mayer WE, Sommer RJ. Nematodes of the genus Pristionchus are closely associated with scarab beetles and the Colorado potato beetle in Western Europe. Zoology. 2006;109:96–108. doi: 10.1016/j.zool.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Hill RC, de Carvalho CE, Salogiannis J, Schlager B, Pilgrim D, Haag ES. Genetic flexibility in the convergent evolution of hermaphroditism in Caenorhabditis nematodes. Developmental Cell. 2006;10:531–8. doi: 10.1016/j.devcel.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Hoda M. Phylum Nematoda Cobb 1932. Zootaxa. 2011;3148:63–95. [Google Scholar]
- Holterman M, van der Wurff A, van den Elsen S, van Megen H, Bongers T, Holovachov O, Bakker J, Helder J. Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown clades. Molecular Biology and Evolution. 2006;23:1792–1800. doi: 10.1093/molbev/msl044. [DOI] [PubMed] [Google Scholar]
- Horvitz HR, Sternberg PW. Nematode postembryonic cell lineages. Journal of Nematology. 1982;14:240–248. [PMC free article] [PubMed] [Google Scholar]
- Huang G, Allen R, Davis EL, Baum TJ, Hussey RS. Engineering broad root-knot resistance in transgenic plants by RNAi silencing of a conserved and essential root-knot nematode parasitism gene. Proceedings of the National Academy of Science USA. 2006;103:14302–14306. doi: 10.1073/pnas.0604698103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jex AR, Liu S, Li B, Young ND, Ross SH, Li Y, Yang L, Zeng N, Xu X, Xiong Z. Ascaris suum draft genome. Nature. 2011;479:529–533. doi: 10.1038/nature10553. [DOI] [PubMed] [Google Scholar]
- Jones KL, Todd TC, Herman MA. Development of taxon-specific markers for high-throughput screening of microbial-feeding nematodes. Molecular Ecology Notes. 2006;6:712–714. [Google Scholar]
- Kaya HK, Gaugler R. Entomopathogenic nematodes. Annual Review of Entomology. 1993;38:181–206. [Google Scholar]
- Keiser J, Utzinger J. The Drugs We Have and the Drugs We Need Against Major Helminth Infections. Advances in Parasitology. 2010;73:197–230. doi: 10.1016/S0065-308X(10)73008-6. [DOI] [PubMed] [Google Scholar]
- Khatri-Chhetri HB, Waeyenberge L, Spiridonov SE, Manandhar HK, Moens M. Steinernema everestense n. sp. (Rhabditida: Steinernematidae), a new species of entomopathogenic nematode from Pakhribas, Dhunkuta, Nepal. Nematology. 2011;13:443–462. [Google Scholar]
- Kikuchi T, Cotton JA, Dalzell JJ, Hasegawa K, Kanzaki N, McVeigh P, Takanashi T, Tsai IJ, Assefa SA, Cock PJA. 2011 doi: 10.1371/journal.ppat.1002219. Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus. PLoS Pathogens 7: e1002219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimber MJ, McKinney S, McMaster S, Day TA, Fleming CC, Maule AG. flp gene disruption in a parasitic nematode reveals motor dysfunction and unusual neuronal sensitivity to RNA interference. The Federation of American Societies for Experimental Biology Journal. 2007;21:1233–43. doi: 10.1096/fj.06-7343com. [DOI] [PubMed] [Google Scholar]
- Kiontke K, Sudhaus W. 2006. Ecology of Caenorhabditis species. In WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.37.1, http://www.wormbook.org. [DOI] [PMC free article] [PubMed]
- Kumar S, Schiffer PH, Blaxter M. 959 nematode genomes: a semantic wiki for coordinating seqeuncing projects. Nucleic Acids Research. 2012;40:D1295–D1300. doi: 10.1093/nar/gkr826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambshead PJ. Recent developments in marine benthic biodiversity research. Oceanis. 1993;19:5–24. [Google Scholar]
- Lambshead PJ. 2004. Marine nematode biodiversity. Pp. 4554–4558 in Z. X. Chen Y. Chen S. Y. Chen and D. W. Dickson, eds. Nematode morphology, physiology, and ecology. CABI. [Google Scholar]
- Li R, Yu C, Li L, Lam T-W, Yiu S-M, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Lin MF, Carlson JW, Crosby MA, Matthews BB, Yu C, Park S, Wan KH, Schroeder AJ, Gramates LS, St. Pierre SE. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Research. 2007;17:1823–1836. doi: 10.1101/gr.6679507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. Next-generation DNA sequencing methods. Annual Review of Genomics and Human Genetics. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- Mayer MG, Sommer RJ. Natural variation in Pristionchus pacificus dauer formation reveals cross-preference rather than self-preference of nematode dauer pheromones. Proceedings of the Royal Society B: Biological Sciences. 2011;278:2784–90. doi: 10.1098/rspb.2010.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer WE, Schuster LN, Bartelmes G, Dieterich C, Sommer RJ. Horizontal gene transfer of microbial cellulases into nematode genomes is associated with functional assimilation and gene turnover. BMC Evolutionary Biology. 2011;11:13. doi: 10.1186/1471-2148-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitreva M, Jasmer DP, Zarlenga DS, Wang Z, Abubucker S, Martin J, Taylor CM, Yin Y, Fulton L, Minx P. The draft genome of the parasitic nematode Trichinella spiralis. Nature Genetics. 2011;43:228–236. doi: 10.1038/ng.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Schwarz EM, Williams BA, Schaeffer L, Antoshechkin I, Wold B, Sternberg PW. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Research. 2010;20:1740–1747. doi: 10.1101/gr.111021.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nguyen KB, Buss EA. Steinernema phyllophagae n. sp. (Rhabditida: Steinernematidae), a new entomopathogenic nematode from Florida, USA. Nematology. 2011;13:425–442. [Google Scholar]
- Nguyen KB, Hunt DJ, Mracek Z. 2007a. Steinernematidae: species and descriptions. Pp. 121–609 in K. B. Nguyen and D. J. Hunt, eds. Entomopathogenic Nematodes: Systematics, phylogeny and bacterial symbionts.Boston: Brill. [Google Scholar]
- Nguyen KB, Puza V, Mracek Z. Steinernema cholashanense n. sp. (Rhabditida, Steinernematidae) a new species of entomopathogenic nematode form the province of Sichuan, Chola Shan Mountains, China. Journal of Invertebrate Pathology. 2008;97:251–264. doi: 10.1016/j.jip.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Nguyen KB, Smart GC. Pathogenicity of Steinernema scapterisci to selected invertebrates. Journal of Nematology. 1991;23:7–11. [PMC free article] [PubMed] [Google Scholar]
- Nguyen KB, Stuart RJ, Andalo V, Gozel U, Rogers ME. Steinernema texanum n. sp. (Rhabditida: Steinernematidae), a new entomopathogenic nematode from Texas, USA. Nematology. 2007b;9:379–396. [Google Scholar]
- Opperman CH, Bird DM, Williamson VM, Rokhsar DS, Burke M, Cohn J, Cromer J, Diener S, Gajan J, Graham S. Sequence and genetic map of Meloidogyne hapla: A compact nematode genome for plant parasitism. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14802–14807. doi: 10.1073/pnas.0805946105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevsner J. 2009a. Pairwise sequence alignment. Pp. 47–97 in Bioinformatics and functional genomics, 2nd edition. Hoboken: John Wiley & Sons. [Google Scholar]
- Pevsner J. 2009b. Protein analysis and proteomics. Pp. 379–416 in Bioinformatics and functional genomics, 2nd edition. Hoboken: John Wiley & Sons. [Google Scholar]
- Poinar GO., Jr . Nematodes for biological control of insects. Boca Raton: CRC Press; 1979. [Google Scholar]
- Popeijus H, Overmars H, Jones J, Blok VC, Goverse A, Helder J, Schots A, Bakker J, Smant G. Degradation of plant cell walls by a nematode. Nature. 2000;406:36–37. doi: 10.1038/35017641. [DOI] [PubMed] [Google Scholar]
- Rae R, Riebesell M, Dinkelacker I, Wang Q, Herrmann M, Weller AM, Dieterich C, Sommer RJ. Isolation of naturally associated bacteria of necromenic Pristionchus nematodes and fitness consequences. Journal of Experimental Biology. 2008;211:1927–1936. doi: 10.1242/jeb.014944. [DOI] [PubMed] [Google Scholar]
- Richards JE, Gilliam TC, Cole JL, Drumm ML, Wasmuth JJ, Gusella JF, Collins FS. Chromosome jumping from D4S10 (G8) toward the Huntington disease gene. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:6437–6441. doi: 10.1073/pnas.85.17.6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle DL, Swanson MM, Albert PS. Interacting genes in nematode dauer larva formation. Nature. 1981;290:668–671. doi: 10.1038/290668a0. [DOI] [PubMed] [Google Scholar]
- Root RB. The niche exploitation pattern of the blue-gray gnatcatcher. Ecological Monographs. 1967;37:317–350. [Google Scholar]
- Rosso MN, Jones JT, Abad P. RNAi and functional genomics in plant parasitic nematodes. Annual Review of Phytopathology. 2009;47:207–232. doi: 10.1146/annurev.phyto.112408.132605. [DOI] [PubMed] [Google Scholar]
- Sangar V, Blankenberg DJ, Altman N, Lesk AM. Quantitative sequence-function relationships in proteins based on gene ontology. BMC Bioinformatics. 2007;8:294. doi: 10.1186/1471-2105-8-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierenberg E. Unusual cleavage and gastrulation in a freshwater nematode: developmental and phylogenetic implications. Development Genes and Evolution. 2005;215:103–108. doi: 10.1007/s00427-004-0454-9. [DOI] [PubMed] [Google Scholar]
- Scholl EH, Bird DM. Resolving tylenchid evolutionary relationships through multiple gene analysis derived from EST data. Molecular Phylogenetics and Evolution. 2005;36:536–545. doi: 10.1016/j.ympev.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Shannon AJ, Tyson T, Dix I, Boyd J, Burnell AM. Systemic RNAi mediated gene silencing in the anhydrobiotic nematode Panagrolaimus superbus. BMC Molecular Biology. 2008;9:58. doi: 10.1186/1471-2199-9-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smant G, Stokkermans JPWG, Yan Y, De Boer JM, Baum TJ, Wang X, Hussey RS, Gommers FJ, Henrissat B, Davis EL. Endogenous cellulases in animal: Isolation of β-1,4-endoglucanase genes from two species of plant-parasitic cyst nematodes. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4906–4911. doi: 10.1073/pnas.95.9.4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Durbin R. Analysis of protein domain families in Caenorhabditis elegans. Genomics. 1997;46:200–216. doi: 10.1006/geno.1997.4989. [DOI] [PubMed] [Google Scholar]
- Spiridonov SE, Waeyenberge L, Moens M. Steinernema schliemanni sp. n. (Steinernematidae; Rhabditida): a new species of steinernematids of the 'monticolum' group from Europe. Russian Journal of Nematology. 2010;18:175–190. [Google Scholar]
- Stanke M, Steinkamp R, Waack S, Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Research. 2004;32:W309–W312. doi: 10.1093/nar/gkh379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, Bao ZR, Blasiar D, Blumenthal T, Brent MR, Chen NS, Chinwalla A, Clarke L, Clee C, Coghlan A. 2003 The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. PLoS Biology 1(2): e45. [Google Scholar]
- Stokwe NF, Malan AP, Nguyen KB, Knoetze R, Tiedt L. Steinernema citrae n. sp. (Rhabditida: Steinernematidae), a new entomopathogenic nematode from South Africa. Nematology. 2011;13:569–587. [Google Scholar]
- Tarasco E, Mracek Z, Nguyen KB, Triggiani O. Steinernema ichnusae sp. n. (Nematoda: Steinernematidae) a new entomopathogenic nematode from Sardinia Island (Italy) Journal of Invertebrate Pathology. 2008;99:173–185. doi: 10.1016/j.jip.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Thomas PD, Mi H, Lewis S. Ontology annotation: Mapping genomic regions to biological function. Current Opinion in Chemical Biology. 2007;11:4–11. doi: 10.1016/j.cbpa.2006.11.039. [DOI] [PubMed] [Google Scholar]
- Toubarro D, Lucena-Robles M, Nascimento G, Santos R, Montiel R, Verissimo P, Pires E, Faro C, Coelho AV, Simoes N. Serine Protease-mediated Host Invasion by the Parasitic Nematode Steinernema carpocapsae. Journal of Biological Chemistry. 2010;285:30666–30675. doi: 10.1074/jbc.M110.129346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER, Félix MA, Whiteman NK, Barriere A, Ausubel FM. 2008 doi: 10.1371/journal.pbio.0060309. Microsporidia are natural intracellular parasites of the nematode C. elegans. PLoS Biol 6: e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudgill DL, Blok VC. Apomictic, polyphagous rook-knot nemtaodes: exceptionally successful and damaging biotrophic root pathogens. Annual Review of Phytopathology. 2001;39:53–77. doi: 10.1146/annurev.phyto.39.1.53. [DOI] [PubMed] [Google Scholar]
- Ungerer MC, Johnson LC, Herman MA. Ecological genomics: understanding gene and genome function in the natural environment. Heredity. 2008;100(2):178–83. doi: 10.1038/sj.hdy.6800992. [DOI] [PubMed] [Google Scholar]
- Urwin PE, Lilley CJ, Atkinson HJ. Ingestion of double-stranded RNA by preparasitic juvenile cyst nematodes leads to RNA interference. Molecular Plant Microbe Interactions. 2002;15:747–52. doi: 10.1094/MPMI.2002.15.8.747. [DOI] [PubMed] [Google Scholar]
- van Megen H, van Den Elsen S, Holterman M, Karssen G, Mooyman P, Bongers T, Holovachov O, Bakker J, Helder J. A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology. 2009;11:927–950. [Google Scholar]
- Werner JJ, Zhou D, Caporaso JG, Knight R, Angenent LT. 2011. Comparison of Illumina paired-end and single-direction sequencing for microbial 16S rRNA gene amplicon surveys. The International Society for Microbial Ecology Journal, doi: 10.1038/ismej.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BD, Schrank B, Huynh C, Shownkeen R, Waterston RH. A genetic mapping system in Caenorhabditis elegans based on polymorphic sequence-tagged sites. Genetics. 1992;131:609–24. doi: 10.1093/genetics/131.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav BC, Veluthambi K, Subramaniam K. Host-generated double stranded RNA induces RNAi in plant-parasitic nematodes and protects the host from infection. Molecular and Biochemical Parasitology. 2006;148:219–22. doi: 10.1016/j.molbiopara.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Ye J, McGinnis S, Madden TL. BLAST: improvements for better seqeunce analysis. Nucleic Acids Research. 2006;34:W6–W9. doi: 10.1093/nar/gkl164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeates GW. 2004. Ecological and behavioral adaptations. Pp. 1–24 in R. Gaugler and A. L. Bilgrami, eds. Nematode Behavior. Cambridge: CABI. [Google Scholar]
- Zaslaver A, Baugh LR, Sternberg PW. Metazoan operons accelerate recovery from growth-arrested states. Cell. 2011;145:981–992. doi: 10.1016/j.cell.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbodnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Advani A, Melefors O, Glavas S, Nordstrom H, Ye W, Engstrand L, Andersson AF. Titration-free 454 sequencing using Y adapters. Nature Protocols. 2011;6:1367–76. doi: 10.1038/nprot.2011.369. [DOI] [PubMed] [Google Scholar]