

Abstract
Background
Aberrant activation of beta-catenin/TCF4 and STAT3 signaling in glioblastoma multiforme (GBM) has been reported. However, the molecular mechanisms related to this process are still poorly understood.
Methods
Genome-wide screening of the binding characteristics of the transcription factors TCF4 and STAT3 in GBM cells was performed by chromatin immunoprecipitation sequencing (ChIP-seq) assay. Hierarchical clustering was used to analyze the association of TCF4 and STAT3 coregulated genes with The Cancer Genome Atlas (TCGA) GBM subtypes (classical, mesenchymal, neural, and proneural). New molecular classification of GBM was proposed and validated in Western and Asian populations.
Results
We identified 1250 overlapping putative target genes that were coregulated by TCF4 and STAT3. Further, the coregulated genes had the potential to guide TCGA GBM subtypes. Finally, we proposed a new molecular classification of GBM into 2 subtypes (proneural-like and mesenchymal-like) and showed that the new classification could be applied to both Western and Asian populations. In addition, the GBM response to temozolomide therapy differed depending on its subtype; mesenchymal-like GBM benefited, while there was no benefit for proneural-like GBM.
Conclusions
This is the first comprehensive study to combine a ChIP-seq assay of TCF4 and STAT3 and data mining of patient cohorts to derive molecular subtypes of GBM.
Keywords: ChIP-seq, glioblastoma, molecular subtype, STAT3, TCF4
Glioblastoma multiforme (GBM) is the most aggressive malignant brain tumor in humans, with a median survival time of ∼14 months despite multimodal treatment.1 Deregulation of beta-catenin/TCF4 and STAT3 signaling pathways were reported to contribute significantly to GBM development.2–5 However, the mechanisms of beta-catenin/TCF4 and STAT3 signaling pathways involved in GBM tumorigenesis remain unclear.
Recent reports have shown that crosstalk between the beta-catenin/TCF4 and STAT3 signaling pathways is involved in the development of multiple tumors.6–8 In human esophageal squamous cell carcinoma, beta-catenin increased STAT3 mRNA and protein expression by direct interaction with the STAT3 promoter, which specifically bound to TCF4.6 In breast cancer, STAT3 was reported to upregulate the protein expression and transcriptional activity of beta-catenin by binding to the promoter of beta-catenin.7 Our previous research also demonstrated that in GBM, downregulation of beta-catenin induced a reduction of STAT3 mRNA and protein expression levels, whereas inhibition of STAT3 repressed beta-catenin expression.9,10 Moreover, the beta-catenin/TCF4 and STAT3 signaling pathways were found to synergistically modulate the AKT pathway.10–13 In GBM cells, beta-catenin/TCF4 signaling interacted with the AKT pathway by binding to the promoters of AKT1 and AKT2,11,12 while the inhibition of STAT3 sensitized temozolomide (TMZ)-induced cell death, at least in part, by blocking the AKT pathway.10 However, at the genome-wide level, the mechanisms by which the beta-catenin/TCF4 and STAT3 transcription factors coregulate genes toward GBM development have never been reported.
To investigate this problem, we applied chromatin immunoprecipitation sequencing (ChIP-seq) technology to study TCF4 and STAT3 regulatory mechanisms in GBM cells. We discovered that genes coregulated by TCF4 and STAT3 and developmental genes in the nervous system could clearly guide The Cancer Genome Atlas (TCGA) classification of GBM into 4 subtypes (classical, mesenchymal, neural, and proneural).14 Further, we describe a novel classification of GBM into 2 major types, mesenchymal-like and proneural-like, which was validated in 3 independent large GBM gene expression cohorts from Western or Asian populations.
Materials and Methods
Cell Culture
Human glioblastoma cells (U87) were obtained from the Chinese Academia Sinica Cell Repository. Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and incubated at 37°C with 5% CO2.
ChIP-seq and Identification of Binding Sites
Genomic DNA was sonicated in 1.5-mL tubes into 100- to 500-bp fragments. Immunoprecipitations were performed with anti-TCF4 (Upstate) and anti-STAT3 (Santa Cruz), with immunoglobulin G as negative control. DNA libraries were generated following the Illumina protocol for preparing samples for ChIP-seq of DNA. The entire amount of eluted DNA was used to construct libraries. During this procedure, quantitative reverse transcriptase PCR was carried out to check the internal standards after enrichment and library preparation. Then DNA fragments 150–400 bp long were gel purified following the adaptor ligation step. The PCR-amplified DNA libraries were quantified on an Agilent 2100 Bioanalyzer and diluted for cluster generation.
An Illumina GA II was employed to sequence the libraries. The TCF4 and STAT3 sequences were aligned to the reference human genome (University of California Santa Cruz [UCSC] hg18; National Center for Biotechnology Information [NCBI] Build 36) using the Burrows-Wheeler Aligner software program15; only uniquely mapped nonduplicate reads were retained. Binding sites were identified using the Model-based Analysis for ChIP Sequencing, a Poisson-based method, with a P-value cutoff of .001 and Mfold values adjusted to ensure >1000 peaks to build the model.16 Association of binding sites with genomic features was performed by overlapping defined sets of binding sites with known genomic features obtained from the UCSC tables for assembly hg18, that is, full-length RefSeq gene, exon, intron, promoter region defined as ±2 kilobases (kb) from the transcriptional start site (TSS), and long-distance regulatory region from 50 kb to 2 kb upstream of the TSS and from the transcription end site (TES) to 50 kb downstream of the TES.
ChIP-PCR
Briefly, primers were constructed to cover regions that were sequenced in the ChIP-seq experiment. Immunoprecipitated DNA was purified and eluted in 50 μL water. For quantitative PCR analysis, 1 μL of ChIP DNA or 2 ng input DNA was used. Each target site was calculated as 2 to the power of the cycle threshold difference between input DNA and ChIP samples. Enrichments at target sites were compared with negative/unbound control region GAPDH. For a complete list of the primers used to amplify those regions, see Supplementary material, Table S1.
Patients and Samples
A total of 3 large gene expression profiling cohorts of gliomas were used in this study. Gene expression data of TCGA (173 core GBM samples) and the validation data set (260 GBM samples) were downloaded from the TCGA database (http://tcga-data.nci.nih.gov/docs/publications/gbm_exp/). Additionally, we performed gene expression profiling upon 220 glioma samples collected from the Chinese Glioma Genome Atlas (CGGA, http://www.cgcg.org.cn/), including 58 astrocytomas, 17 oligodendrogliomas, 22 oligoastrocytomas, 8 anaplastic astrocytomas, 11 anaplastic oligodendrogliomas, 15 anaplastic oligoastrocytomas, 4 secondary GBMs, and 85 primary GBMs. All 220 Chinese glioma patients underwent surgical resection between January 2005 and December 2009 and subsequently received radiation therapy and/or alkylating agent–based chemotherapy. This study was approved by the institutional review boards of all hospitals involved in the study, and written informed consent was obtained from all patients selected.
RNA Extraction and Whole Genome Gene Profiling of 220 Chinese Gliomas
After surgery, all tissue samples were immediately snap-frozen in liquid nitrogen. A frozen section stained with hematoxylin and eosin was prepared from each sample to assess the percentage of tumor cells before RNA extraction. Total RNA was extracted from the frozen tumor samples, and the RNA concentration and quality were measured using a NanoDrop ND-1000 spectrophotometer.
Microarray analysis was performed on all 220 samples using the Whole Human Genome Array (Agilent) according to the manufacturer's instructions. The integrity of the total RNA was checked using an Agilent 2100 Bioanalyzer. Complementary DNA and biotinylated cRNA were synthesized and hybridized to the array. Data were acquired using the Agilent G2565BA Microarray Scanner System and Agilent Feature Extraction Software v9.1. Probe intensities were normalized using GeneSpring GX v.11.0 (Agilent).
Pyrosequencing for IDH1 Mutation and MGMT Promoter Methylation
For IDH1 mutation analysis, genomic DNA was isolated from frozen tumor tissues using the QIAamp DNA Mini Kit (Qiagen). The primers used were forward 5′-GCTTGTGAGTGGATGGGTAAAAC-3′ and reverse 5′-biotin-TTGCCAACATGACTTACTTGATC-3′. For MGMT promoter methylation analysis, bisulite modification of the DNA was performed using the EpiTect Kit (Qiagen). The primers used were forward 5′-GTTTYGGATATGTTGGGATA-3′ and reverse 5′-biotin-ACCCAAACACTCACCAAATC-3′. Pyrosequencing analysis of IDH1 mutation and MGMT promoter methylation was performed by Gene Tech.
Denaturing High Performance Liquid Chromatography (DHPLC) Analysis for 1p/19q Status
The microsatellite markers D1S548 (1p36.23), D1S1608 (1p36.32) and D1S1592 (1p36.13) were used to identify LOH 1p. To determine LOH 19q, the markers D19S431 (19q12), D19S433 (19q12) and D19S601 (19q13.41) were used. The genetic location, primer sequences, and size of the product of each marker were obtained from the Genome Database (http://www.gdb.org). Primers were synthesized commercially (Invitrogen Inc., Carlsbad, CA, USA). Five microliters of crude PCR product was used for DHPLC analysis on the automated WAVE DNA Fragment Analysis System (Transgenomic Inc.).
Immunohistochemistry
Briefly, surgical specimens were fixed in formalin, routinely processed, and paraffin embedded. Five-micron-thick sections were prepared, and immunohistochemical staining with streptavidin-biotin immunoperoxidase assay was performed using antibodies against MGMT, EGFR, and Ki67 (1:100, Santa Cruz Biotechnology). The staining intensity was scored by 2 experienced pathologists without knowledge of clinical information on a scale of 0 = negative, 1 = slight positive, 2 = moderate positive, and 3 = intense positive. A score of 0 and 1 or 2 and 3 indicated low or high expression, respectively. Controls without primary antibody and positive control tissues were included in all experiments to ensure the quality of staining. In case of a discrepancy, the 2 observers simultaneously reviewed the slides to achieve a consensus.
Hierarchical Clustering, Gene Ontology, and Statistical Analysis
Each gene expression profiling cohort was median centered. Gene sets were used in an average-linkage hierarchical cluster analysis, and visualized results were generated by TreeView. Gene ontology (GO) analysis was performed using tools from the Database for Annotation, Visualization and Integrated Discovery (DAVID) for functional annotation (http://david.abcc.ncifcrf.gov/). Significance analysis of microarrays (SAM) software was used (false discovery rate <0.05, fold >1.5) to identify significant genes.17 Student's t-test was used to determine significant differences. The kappa coefficient was used to measure agreement between observations corrected for what might be expected to occur by chance.18 Kappa = .20 was interpreted as slight agreement, κ = .21–.40 as fair agreement, κ = .41–.60 as moderate agreement, κ = .61–.80 as substantial agreement, and κ = .81–1.00 as almost perfect agreement. Kaplan–Meier survival analysis was used to estimate survival distributions, and a log-rank test was used to assess the statistical significance between stratified survival groups, using GraphPad Prism 5.0 statistical software.
Results
Identification of Putative TCF4 and STAT3 Binding Sites
To identify genes regulated by TCF4 and STAT3 at the genome-wide level, we performed ChIP-seq on human GBM U87 cells using TCF4- and STAT3-specific antibodies. A total of 14.4 million 49-bp short reads were generated per sample. We mapped the short reads to the human reference genome (UCSC hg18) using the Burrows-Wheeler Aligner program.15 After removing duplication caused by PCR amplification, we obtained a total of nearly 10 million uniquely mapped nonduplicate reads (10.5 million for TCF4, 9.1 million for STAT3). To determine the binding sites, we used Model-based Analysis for ChIP Sequencing16 to analyze the uniquely mapped nonduplicate reads. We obtained 8307 and 6908 candidate binding sites for TCF4 and STAT3, respectively (Supplementary material, Tables S2 and S3). We validated 7 randomly selected binding sites each for TCF4 and STAT3, with the use of ChIP-PCR. Our ChIP-PCR validation rate was high: 6/7 for TCF4, and 7/7 for STAT3 (Fig. 1A).
Fig. 1.

Identification of TCF4 and STAT3 binding loci on the human reference genome (UCSC hg18). (A) ChIP validation of TCF4 and STAT3 binding sites using qPCR. Relative enrichment was calculated over input DNA. Each data point represents the average of triplicate ChIP experiments. Immunoglobulin (Ig)G was used as a nonspecific control ChIP. GAPDH promoter regions were used as negative control regions for TCF4 and STAT3 binding. (B) Distribution of the location of TCF4 and STAT3 binding loci based on the closest transcription start site of a known RefSeq gene. (C) Genomic annotation of TCF4 and STAT3 binding sites based on known RefSeq genes.
Preferential Binding in the Vicinity of the Transcriptional Start Site
The identification of the potential binding sites allowed us to determine the regions in the genome where TCF4 and STAT3 were likely to bind. We examined the frequency distribution of TCF4 and STAT3 binding sites around the TSSs that were closest to them using a 1-kb window. We discovered that the binding sites of both TCF4 and STAT3 clustered close to TSSs with a low frequency of binding that extended to distances that were quite far removed from them (Fig. 1B). Approximately 36.6% (TCF4) and 34.1% (STAT3) of the potential binding sites were located within 15 kb of a TSS; indeed, 15.9% (TCF4) and 14.2% (STAT3) of the binding sites were within 5 kb of a TSS, indicating a tendency for the TCF4 and STAT3 transcription factors to preferentially bind genomic regions around the TSS.
Genomic Distribution of Binding Sites
To comprehensively understand the genomic distribution of the binding sites, we associated a binding site with a nearest known RefSeq gene, requiring that the binding site be located within 50 kb upstream and 50 kb downstream of the nearest gene coding region. We defined the promoter regions and long-distance regulatory regions of a gene as described in Materials and Methods. We discovered that the genomic distributions of the potential TCF4 and STAT3 binding sites were similar. Approximately 63% of the binding sites were located within ±50 kb of the coding regions, that is, 3% in promoter, 2% in exon, 36% in intron, and 22% in long-distance regulatory regions for TCF4; and 3% in promoter, 2% in exon, 37% in intron, and 21% in long-distance regulatory regions for STAT3 (Fig. 1C). Notably, a substantial proportion of the binding sites were identified in the long-distance regulatory regions, suggesting that TCF4 and STAT3 also regulate genes from sites that are distant from the proximal promoter regions.
Genes Potentially Targeted by Both TCF4 and STAT3 Were Enriched in Nervous System Development
By associating binding sites with their nearest genes, we identified a total of 3812 and 3165 putative target genes for TCF4 and STAT3, respectively. The target genes included EGFR,19 VEGF,20,21 and IL6, 22 which had been reported previously to be transcribed by TCF4 and STAT3. We found that 1250 of the target genes were targeted by both TCF4 and STAT3 (Supplementary material, Table S4).
To investigate the biological processes that may be coregulated by TCF4 and STAT3, we performed GO enrichment analysis upon the cotargeted genes using DAVID with P = .05.23 We found that the genes targeted by both TCF4 and STAT3 were enriched in developmental processes of the nervous system, including neuron differentiation, neuron migration, neuron projection morphogenesis, cell morphogenesis involved in neuron differentiation, and neuron development (Supplementary material, Table S5). Interestingly, other enriched terms were related to adhesion processes, metabolic processes, signal transduction processes, and transcription processes.
GBMs Were Classified Into 4 Subtypes Associated With Nervous System Development Based on Genes Targeted by Both TCF4 and STAT3
TCGA researchers examined 840 genes to classify 202 GBM samples in the TCGA cohort into 4 subtypes (classical, mesenchymal, neural, and proneural), each having distinct gene expression patterns and clinical characteristics.14 To explore whether the TCF4 and STAT3 cotargeted genes could separate GBM into different subtypes, we performed hierarchical clustering on 173 core GBM samples in the TCGA cohort using the 1250 genes that were cotargeted by TCF4 and STAT3. We found that 801 of the cotargeted genes were covered in the TCGA cohort (Supplementary material, Table S4). By performing an agreement analysis, we discovered that these cotargeted genes could classify the 173 GBM samples from the TCGA cohort into 4 subtypes (Fig. 2A) that were highly consistent with the TCGA GBM subtypes (percentage agreement, 90.8%; κ = .874).
Fig. 2.

Identification of TCGA GBM subtypes using TCF4 and STAT3 cotargeted genes. (A) Hierarchical clustering of 173 TCGA core GBM samples using 801 cotargeted genes. (B) Hierarchical clustering of 173 TCGA core GBM samples using 132 differentially expressed cotargeted genes. Kappa agreement analysis was performed to compare the 4 GBM subtypes with TCGA subtypes.
Next, from 801 of the cotargeted genes, we selected 132 genes that were differentially expressed among 4 TCGA GBM subtypes (false discovery rate <0.05; fold change >1.5) (Supplementary material, Table S4). The hierarchical clustering of the previously selected 173 core GBM samples was repeated with the 132 cotargeted genes. Again, 4 tumor subtypes, consistent with the TCGA GBM subtypes, were found (percentage agreement, 93.1%; κ = 0.905) (Fig. 2B). Functional annotation by DAVID of the 132 cotargeted genes demonstrated that they were highly enriched in developmental processes of the nervous system (Supplementary material, Table S6).
To test whether the genes that were associated with nervous system development could classify GBM into the different subtypes, we selected the top 100 genes that were statistically enriched in developing astrocytes and the top 100 genes that were statistically enriched in oligodendrocyte progenitor cells from the Human Brain Transcriptome database24 and used them to cluster the 173 GBM samples from the TCGA cohort. We found that the GBM in the TCGA cohort clustered into 4 subtypes (Fig. 3) that, again, were largely consistent with the TCGA GBM subtypes (percentage agreement, 80.9%; κ = 0.742).
Fig. 3.
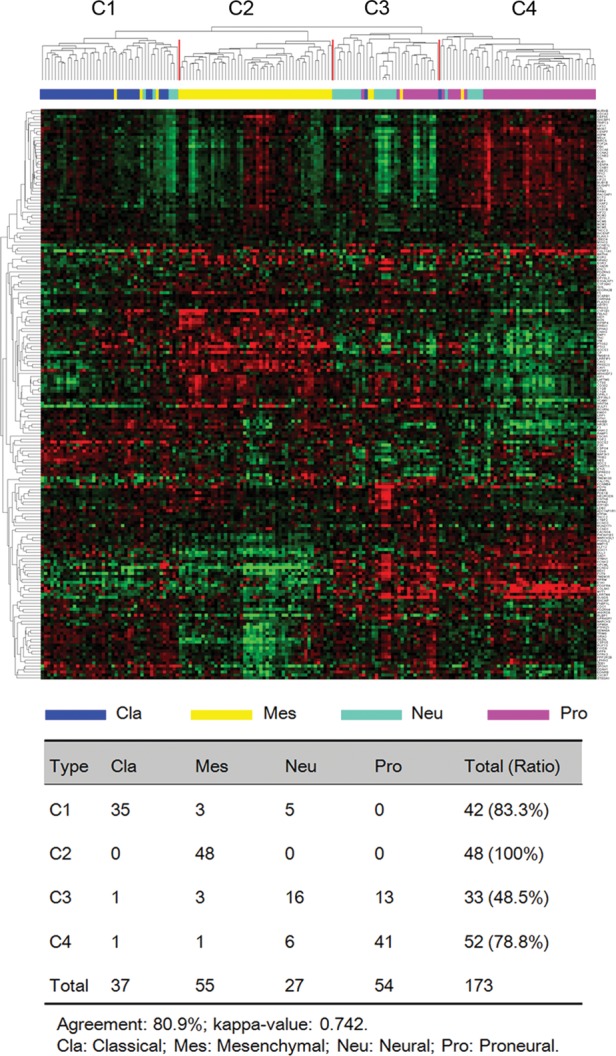
Identification of TCGA GBM subtypes using cotargeted genes associated with nervous system development. Hierarchical clustering of 173 TCGA core GBM samples using 200 genes associated with nervous system development. Kappa agreement analysis was performed to compare the 4 GBM subtypes with TCGA subtypes.
New Molecular Classification of GBM as Mesenchymal-like and Proneural-like Subtypes
We observed that the classical and mesenchymal TCGA GBM subtypes clustered together, each having had its mortality reduced by aggressive treatment (P = .02 for classical, P = .02 for mesenchymal), while neural and proneural subtypes clustered together, each having no statistically significant alteration of survival by aggressive treatment (P = .1 for neural, P = .4 for proneural).14 Additionally, for the 4 GBM subtypes in the TCGA cohort that were defined by the TCF4 and STAT3 cotargeted genes, we discovered that the GBM subtypes that had high agreement with the classical and mesenchymal subtypes clustered together, and those that were highly consistent with the neural and proneural subtypes clustered together (Fig. 2). A similar result was observed for the GBM subtypes defined by developmental genes in the nervous system (Fig. 3). Based on the aggressive treatment efficacy and clustering signature, we divided the 173 GBM specimens from the TCGA cohort into 2 major types: mesenchymal-like (containing the classical and mesenchymal subtypes) and proneural-like (containing the neural and proneural subtypes). We found that the age at diagnosis for GBM in the proneural-like subtype was younger than for GBM in the mesenchymal-like subtype (P = .044) (Supplementary material, Table S7). Mutations in the IDH1, TP53, PIK3R1, and NF1 genes and a majority of the copy number alterations were different in the 2 newly defined subtypes (Supplementary material, Tables S8 and S9).
Using SAM upon 173 core GBM expression profilings in the TCGA cohort, we identified a total of 142 genes from 801 cotargeted genes that were differentially expressed between mesenchymal-like and proneural-like subtypes (false discovery rate <.05; fold change >1.5). Hierarchical clustering with these 142 genes classified the 173 GBM samples in the TCGA cohort into the 2 subtypes that had high agreement with the mesenchymal-like and proneural-like subtypes defined by the 840 genes from TCGA (percentage agreement, 95.4%; κ = .907) (Fig. 4A). These findings confirmed the classification of the 142 genes as mesenchymal-like and proneural-like subtypes. Because 109 out of the 173 GBM samples had clinical information available, we performed a survival analysis of the 109 GBM samples, which revealed that there was no statistically significant difference between mesenchymal-like and proneural-like subtypes (P = .8013) (Fig. 4B). However, in the mesenchymal-like subtype, the GBM patients who had received TMZ therapy had statistically significant better overall survival (P = .037) (Fig. 4B). In the proneural-like subtype, TMZ therapy did not improve the overall survival of GBM patients (P = .8073) (Fig. 4B), suggesting that the 142 cotargeted genes could be used to help in selecting a therapeutic regimen.
Fig. 4.

Molecular classification of GBM samples into mesenchymal-like and proneural-like GBM subtypes. (A) Hierarchical clustering of 173 TCGA core GBM samples using 142 differentially expressed cotargeted genes. (B) Evaluation of the survival of 109 GBM patients by tumor subtype and TMZ treatment type. Kaplan–Meier analysis was used. (C) Gene order of 142 genes from the TCGA samples was maintained in the validation data set (n = 260). Kappa agreement analysis was performed to compare these subtypes with TCGA subtypes.
In the TCGA database, there were validation data: another independent GBM cohort that consisted of 260 GBM samples collected from the public domain.25–28 When hierarchical clustering was performed with the 260 GBM samples using the 142 cotargeted genes, the results identified 2 subtypes that were similar to the mesenchymal-like or proneural-like subtypes defined by the 840 genes from TCGA (percentage agreement, 86.2%; κ = .727) (Fig. 4C).
Mesenchymal-like and Proneural-like GBM Subtype Signatures in Chinese GBM Samples
The TCGA data and the validation data were from Western populations; therefore, the existence of mesenchymal-like and proneural-like subtypes in Asian populations was still unclear. Therefore, we used 220 glioma samples from Chinese patients to validate the molecular subtyping system proposed based on the results obtained using the 142 TCF4 and STAT3 cotargeted genes. We found 141 of the 142 cotargeted genes in the Chinese cohort. Hierarchical clustering of the 220 gliomas resulted in 2 subtypes with gene expression patterns similar to the mesenchymal-like and proneural-like subtypes (Fig. 5A). We discovered that 72 of 89 GBM samples clustered in the mesenchymal-like subtype and 17 of 18 oligodendrogliomas and 16 of 21 oligoastrocytomas clustered in the proneural-like subtypes, revealing that the mesenchymal-like subtype was enriched in high-grade glioma, while the proneural-like subtype was enriched in low-grade glioma. Kaplan–Meier analysis demonstrated that the gliomas that clustered in the proneural-like subtype had a significantly better overall survival rate (P = .0164) (Fig. 5B).
Fig. 5.

Mesenchymal-like and proneural-like GBM subtypes in Chinese gliomas. (A) Identification of mesenchymal-like and proneural-like GBM subtypes by hierarchical clustering of 220 Chinese glioma samples. Gene order of 142 genes from the TCGA samples was maintained in the Chinese data set. (B) Evaluation of the survival of glioma patients by treatment type and tumor subtype. Kaplan–Meier analysis was used. AA, anaplastic astrocytoma; AO, anaplastic oligodendroglioma; AOA, anaplastic oligoastrocytoma; A, astrocytoma; O, oligodendroglioma; OA, oligoastrocytoma.
Next, we explored the clinical characteristics of the 89 GBM samples in both the mesenchymal-like and proneural-like subtypes. We found that there was no significant difference in the overall survival rate between GBM in the mesenchymal-like and proneural-like subtypes (P = .3566); however, the mesenchymal-like GBM patients who had received TMZ therapy had a statistically significant better overall survival (P = .0013), while TMZ treatment did not significantly improve the overall survival in patients with the proneural-like GBM subtype (P = .3258). These results suggest that the mesenchymal-like and proneural-like subtypes defined based on the 142 cotargeted genes could also help physicians select the most appropriate therapeutic regimen for the different GBM subtypes in patients from Chinese populations. In addition, we compared the clinical and pathological information for GBM in the mesenchymal-like and proneural-like subtypes. Gender, age, Karnofsky performance score, extent of resection, and overall survival were not statistically significantly different between the 2 subtypes (Table 1 and Supplementary material, Table S10). The status of MGMT promoter methylation, 1p and 19q loss, and MGMT, Ki67, and EGFR expression showed no differences between the 2 subtypes. Molecular pathology analysis demonstrated that 6 of 8 GBM tumors in the proneural-like subtype had IDH1 mutations (P = .010) (Table 1).
Table 1.
Clinical and pathology features of mesenchymal-like and proneural-like GBM
| Mesenchymal-like | Proneural-like | P | |
|---|---|---|---|
| n cases | 72 (16.1%) | 17 (83.9%) | |
| Gender, female/male | 28/44 | 9/8 | .290 |
| Age at diagnosis, y | 45.4 ± 12.3 | 46.3 ± 15.0 | .804 |
| KPS score, ≥80/<80 | 50/22 | 9/8 | .195 |
| Resection, total/subtotal | 41/31 | 7/10 | .241 |
| Overall survival, days | 458.3 ± 214.6 | 403.1 ± 237.9 | .354 |
| MGMT promoter methylation, unmethylated/methylated | 31/14 | 8/3 | .802 |
| IDH1 mutation, yes/no | 7/54 | 6/8 | .010 |
| 1p loss, yes/no | 6/30 | 1/6 | .874 |
| 19q loss, yes/no | 8/28 | 2/5 | .721 |
| MGMT, low/high | 19/50 | 7/10 | .273 |
| EGFR, low/high | 27/43 | 9/8 | .281 |
| Ki67, low/high | 18/52 | 7/10 | .210 |
Discussion
The critical role of beta-catenin/TCF4 and STAT3 as regulatory elements contributing to tumorigenesis has been reported in multiple cancer types. Here, we found the first evidence that TCF4 and STAT3 could cooperatively modulate target genes at the genome-wide level to promote the development of GBM. Our ChIP-seq studies on GBM U87 cells revealed that there was a statistically significant overlap (1250 genes) between genes targeted by TCF4 and genes targeted by STAT3. GO analysis also demonstrated striking similarities between biological processes for the 3812 TCF4 and 3165 STAT3 target genes (data not shown). The 1250 overlapping genes were found to be enriched in developmental processes of the nervous system, suggesting their potentially important functions in transforming cells in the nervous system toward malignancy.
Similar to previous findings for other transcription factors, including the estrogen receptor,29 p160 protein family,30 and SMAD4,31 we observed that a majority of the TCF4 and STAT3 binding loci on the reference genome were located more than 2 kb upstream of the 5′ TSS of a known RefSeq gene, indicating that they may be able to regulate many genes through long-distance regulatory regions in GBM cells. Our genome-wide mapping analysis also revealed the importance of the whole-genome-wide sequencing technologies because the promoter array technology (ChIP-chip) may miss target binding loci that are far from the 5′ TSS of a known gene.
Based on gene expression profiling, it has previously been suggested that GBM cells could be classified into different subtypes, each having its own unique clinical or molecular characteristics. Phillips et al28 reported 3 high-grade astrocytoma subsets and named them proneural, proliferative, and mesenchymal in recognition of the main features of the molecular signatures associated with outcome. In 2010, TCGA researchers described a robust gene expression–based molecular classification of GBM into proneural, neural, classical, and mesenchymal subtypes and integrated multidimensional genomic data to establish patterns of somatic mutations and DNA copy number.14 However, there is as yet no consensus on the number and signature of clinical transcriptional GBM subtypes. In our study, we chose TCF4 and STAT3 cotargeted genes to mine the TCGA GBM data and classified, by unsupervised clustering, GBM into 4 different subtypes that were in almost perfect agreement with the TCGA subtypes. Thus, this is the first study to use a subset of TCF4 and STAT3 cotargeted genes to classify GBM from the TCGA database.
This study is the first to use developmental genes in the nervous system for GBM molecular classification. In the 1980s, Pierce32 defined the cancer cell as being “controlled by the embryo,” thus emphasizing the association between carcinogenesis and embryo development. Since then, extensive studies have reported the existence of cancer stem cells, and much progress has been made toward elucidating the cellular origin of these tumors. In 2002–2003, Ignatova,33 Singh,34 and Hemmati35 and their colleagues first described stemlike cells that existed in brain tumors and termed them glioma stem cells. Glioma stem cells have been recognized as apex cells that share defining features with somatic stem cells in the hierarchical organization of glioma. It is still unknown where glioma stem cells are generated, but a number of possible sources have been proposed: neural stem cells, glial progenitor cells, or differentiated glioma cells. Thus, the particular cell type and processes that lead to oncogenic transformation have yet to be discovered. Here, we found that developmental genes in the nervous system could be applied to classify GBM into 4 subtypes, each of which has high agreement with the GBM subtypes defined by TCGA, suggesting that developmental genes in the nervous system drive GBM pathogenesis and molecular classification into subtypes.
By analyzing the GBM subtypes defined by the TCF4 and STAT3 cotargeted genes and developmental genes in the nervous system, we discovered that the proneural and mesenchymal subtypes had better agreement with TCGA subtypes. A comparison of the GBM subtypes defined by Phillips et al and by TCGA also suggested that there was a robust distinction between the proneural and mesenchymal GBM subtypes.36 In TCGA data, the response of the different GBM subtypes to aggressive therapy differed; the classical and mesenchymal subtypes benefited, while the proneural and neural subtypes did not. Thus, based on the clustering signature and on the treatment efficacy in the different subtypes, we proposed a new GBM classification with 2 major subtypes: mesenchymal-like (containing the classical and mesenchymal subtypes) and proneural-like (containing the neural and proneural subtypes). In further support of our GBM classification based on the results for 142 cotargeted genes, we used TCGA validation data and Chinese glioma data to classify GBM. The molecular classification clearly recapitulated the gene sample groups and the treatment response. The new GBM classification system awaits validation on larger GBM data sets in future studies.
In conclusion, the present study provides the first comprehensive genome-wide map of TCF4 and STAT3 targets in human GBM cells, which could be used to study the functions of the TCF4 and STAT3 transcription factors in tumorigenesis. To our knowledge, this is the first study to link TCF4 and STAT3 coregulated genes and developmental genes in the nervous system with a molecular classification of GBM, leading to new insights into GBM tumorigenesis and nervous system development. We have proposed a novel classification of GBM into 2 major subtypes with different treatment responses: proneural-like and mesenchymal-like, in Western and Asian populations. Thus, combining ChIP-seq to identify binding loci with molecular profiling of patient cohorts may become a powerful approach for identifying potential gene signatures with important biological and clinical roles.
Funding
This work was supported financially by grants from the National High Technology Research and Development Program 863 (no. 2012AA02A508), the International Science and Technology Cooperation Program of China (no. 2012DFA30470), the National Natural Science Foundation of China (nos. 81001128, 81172406), the Jiangsu Province Key Discipline of Medicine (no. XK201117), the Clinical Foundation of Jiangsu Province Science and Technology Commission (no. BL2012028), and the Specialized Research Fund for the Doctoral Program of Higher Education (no. 20111202110004).
Supplementary Material
Acknowledgments
We are grateful to Xiao-Long Fan (The Rausing Laboratory, Division of Neurosurgery, Lund University Hospital) for his assistance with the in vivo experiments.
Conflict of interest statement. None declared.
References
- 1.Van Meir EG, Hadjipanayis CG, Norden AD, et al. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–193. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang W, Xia Y, Ji H, et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang N, Wei P, Gong A, et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guryanova OA, Wu Q, Cheng L, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011;19:498–511. doi: 10.1016/j.ccr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan S, Zhou C, Zhang W, et al. Beta-Catenin/TCF pathway upregulates STAT3 expression in human esophageal squamous cell carcinoma. Cancer Lett. 2008;271:85–97. doi: 10.1016/j.canlet.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 7.Armanious H, Gelebart P, Mackey J, et al. STAT3 upregulates the protein expression and transcriptional activity of beta-catenin in breast cancer. Int J Clin Exp Pathol. 2010;3:654–664. [PMC free article] [PubMed] [Google Scholar]
- 8.Anand M, Lai R, Gelebart P. Beta-catenin is constitutively active and increases STAT3 expression/activation in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica. 2011;96:253–261. doi: 10.3324/haematol.2010.027086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yue X, Lan F, Yang W, et al. Interruption of beta-catenin suppresses the EGFR pathway by blocking multiple oncogenic targets in human glioma cells. Brain Res. 2010;1366:27–37. doi: 10.1016/j.brainres.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Chen L, Bao Z, et al. Inhibition of STAT3 reverses alkylator resistance through modulation of the AKT and beta-catenin signaling pathways. Oncol Rep. 2011;26:1173–1180. doi: 10.3892/or.2011.1396. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Huang K, Shi Z, et al. High beta-catenin/Tcf-4 activity confers glioma progression via direct regulation of AKT2 gene expression. Neuro Oncol. 2011;13:600–609. doi: 10.1093/neuonc/nor034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Huang K, Han L, et al. Beta-catenin/Tcf-4 complex transcriptionally regulates AKT1 in glioma. Int J Oncol. 2011;39:883–890. doi: 10.3892/ijo.2011.1104. [DOI] [PubMed] [Google Scholar]
- 13.Wang XH, Meng XW, Xing H, et al. STAT3 and beta-catenin signaling pathway may affect GSK-3beta expression in hepatocellular carcinoma. Hepatogastroenterology. 2011;58:487–491. [PubMed] [Google Scholar]
- 14.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33:159–174. [PubMed] [Google Scholar]
- 19.Tan X, Apte U, Micsenyi A, et al. Epidermal growth factor receptor: a novel target of the Wnt/beta-catenin pathway in liver. Gastroenterology. 2005;129:285–302. doi: 10.1053/j.gastro.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skurk C, Maatz H, Rocnik E, et al. Glycogen-synthase kinase3beta/beta-catenin axis promotes angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ Res. 2005;96:308–318. doi: 10.1161/01.RES.0000156273.30274.f7. [DOI] [PubMed] [Google Scholar]
- 21.Jung JE, Kim HS, Lee CS, et al. Caffeic acid and its synthetic derivative CADPE suppress tumor angiogenesis by blocking STAT3-mediated VEGF expression in human renal carcinoma cells. Carcinogenesis. 2007;28:1780–1787. doi: 10.1093/carcin/bgm130. [DOI] [PubMed] [Google Scholar]
- 22.Yoon S, Woo SU, Kang JH, et al. NF-kappaB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene. 2012;31:3467–3481. doi: 10.1038/onc.2011.517. [DOI] [PubMed] [Google Scholar]
- 23.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 24.Cahoy JD, Emery B, Kaushal A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murat A, Migliavacca E, Gorlia T, et al. Stem cell-related ‘self-renewal’ signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol. 2008;26:3015–3024. doi: 10.1200/JCO.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- 27.Sun L, Hui AM, Su Q, et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006;9:287–300. doi: 10.1016/j.ccr.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 29.Stender JD, Kim K, Charn TH, et al. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol. 2010;30:3943–3955. doi: 10.1128/MCB.00118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zwart W, Theodorou V, Kok M, et al. Oestrogen receptor-co-factor-chromatin specificity in the transcriptional regulation of breast cancer. EMBO J. 2011;30:4764–4776. doi: 10.1038/emboj.2011.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy BA, Deatherage DE, Gu F, et al. ChIP-seq defined genome-wide map of TGFbeta/SMAD4 targets: implications with clinical outcome of ovarian cancer. PLoS One. 2011;6:e22606. doi: 10.1371/journal.pone.0022606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pierce GB. The cancer cell and its control by the embryo. Rous-Whipple Award lecture. Am J Pathol. 1983;113:117–124. [PMC free article] [PubMed] [Google Scholar]
- 33.Ignatova TN, Kukekov VG, Laywell ED, et al. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 34.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 35.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huse JT, Phillips HS, Brennan CW. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia. 2011;59:1190–1199. doi: 10.1002/glia.21165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.