

Abstract
Background
Kallikreins have prognostic value in specific malignancies, but few studies have addressed their clinical significance to glioblastoma multiforme (GBM). Kallikrein 6 (KLK6) is of potential high relevance to GBM, since it is upregulated at sites of CNS pathology and linked to reactive astrogliosis. Here we examine the clinical value of KLK6 as a prognostic indicator of GBM patient survival and its activity in promoting resistance to cytotoxic agents.
Methods
The association between patient survival and levels of KLK6 immunoreactivity were investigated in 60 grade IV astrocytoma tumor specimens. Levels of KLK6 RNA were also evaluated in a separate set of GBM patient tumors (n = 23). Recombinant KLK6 or enforced KLK6 overexpression in GBM cell lines was used to evaluate effects on astrocytoma cell survival.
Results
A range of KLK6 expression was observed across grade IV tumors, with higher levels a poor prognostic indicator of patient survival (P = .02) even after adjusting for gender and Eastern Cooperative Oncology Group performance scores (P = .01). KLK6 reduced the sensitivity of GBM cell lines to cytotoxic agents, including staurosporine and cisplatin, and to the current standard of patient care: radiotherapy or temozolomide alone or in combination. The ability of KLK6 to promote resistance to apoptosis was dependent on activation of the thrombin receptor, protease activated receptor 1.
Conclusions
Taken together, these results indicate that elevated levels of KLK6 in GBM are likely to promote the resistance of tumor cells to cytotoxic agents and are an indicator of reduced patient postsurgical survival times.
Keywords: apoptosis, glioma, grade IV, malignancy, temozolomide
Glioblastoma multiforme (GBM) is classified as World Health Organization (WHO) grade IV astrocytoma and is the most common and malignant form of primary intracranial tumor in adults. GBM patients have an extremely poor prognosis, with a median survival of only 12 to 15 months.1 With many of the cellular and molecular signatures of GBM being discovered, it is clear that considerable heterogeneity exists within this tumor type. Heterogeneity is one factor contributing to the poor response of GBM to cell death–inducing agents and its tendency in some cases to be highly proliferative and invasive.2–4 Continued investigation regarding the molecular heterogeneity of GBM and the significance of this to tumor survival, dispersion, and therapy resistance will be essential to the development of new targeted therapeutic approaches to improve patient survival.
Kallikreins (KLKs) are a family of 15 secreted serine proteases whose genes are tandemly arranged on chromosome locus 19q13.4 and that compose the largest contiguous cluster of protease genes of any catalytic class in the human genome.5 KLKs are hormonally regulated, detected in a wide range of human tissues, and implicated in a broad spectrum of physiological activities, including blood pressure regulation, seminal clot liquefaction, tissue remodeling, peptide hormone processing, and inflammation.6 The association of several KLK gene family members with malignancy is also clearly recognized, most prominently in the case of prostate, ovarian, and breast cancers.7 For example, KLK3 (prostate-specific antigen) serves as an important prostate cancer serum marker.8 Elevations of KLK6 in ovarian cancer are associated with higher grade, later stage, and serous histotype, as well as unfavorable prognosis.9–12 KLK6 is also linked to poor survival in non-small-cell lung,13 gastric,14 and colon cancers.15,16 Despite the recognition of the prognostic significance of aberrant KLK levels in diverse cancers, the mechanism by which these secreted serine proteases contribute to tumor progression has been inadequately addressed.
While few studies have focused attention on the possible roles of KLKs in brain tumors, it is important to note that chromosome 19q has long been a region of interest to glioma scientists.17 Significantly, it is known that deletions in chromosome 19q are associated with prolonged survival in patients with oligodendroglioma.18 In GBM, deletion of the chromosome 19q KLK cluster is associated with longer patient survival; conversely, genetic gain is a poor prognostic indicator.19,20 While the role of KLKs in GBM has not been previously examined in detail, several studies have compared expression levels across WHO glioma grades I to IV. For example, among 73 intracranial tumors examined in one study, patients with tumors positive for KLK7 exhibited shortened survival relative to patients in which no KLK7 expression was detected. Across intracranial tumor types, both a significant downregulation21,22 and a significant upregulation of KLK6 have been reported.23
In this study, we have focused our efforts on determining the relationship between KLK6 and patient survival in 60 primary GBM tumor specimens and delineating the significance of elevated KLK6 to tumor cell survival. Results indicate that GBM patients with higher tumor KLK6 levels experience shorter postsurgical survival. Moreover, these studies demonstrate that KLK6 may drive poor GBM patient prognosis by promoting the survival of glioblastoma cells and their resistance to radiotherapy (RT) and temozolomide (TMZ). By further demonstrating that KLK6 can trigger prosurvival effects by activation of protease activated receptor (PAR)1, these studies point to both KLK6 and PAR1 as new potential therapeutic targets to sensitize GBM to cell death induced by conventional therapies, thereby improving patient survival.
Materials and Methods
KLK6 Immunoreactivity in Grades III and IV Astrocytoma
All glioma specimens used in these studies were examined for histopathological features by a neuropathologist and were determined to be WHO grade III or grade IV. Tissue microarrays were prepared from tumor core samples obtained from surgically resected, formalin-fixed paraffin-embedded primary glioma specimens (see Supplementary material, Table S1, for demographic data). All tumor samples were arrayed in triplicate and sectioned at 5 µM. Two arrays were used in these studies, the first containing 38 grade IV astrocytomas and the second 22 grade IV and 8 grade III astrocytomas. Demographic information and clinical follow-up data were obtained through review of medical records including age at surgery, gender, extent of resection, and Eastern Cooperative Oncology Group (ECOG) performance score (Supplementary material, Table S1). The use of human materials in this study was approved by the Mayo Clinic Institutional Review Board.
KLK6 immunoreactivity (IR) was detected using a KLK6-specific monoclonal antibody (MSP-3-3) as previously described in detail.24–27 Briefly, deparaffinized sections were treated with citrate solution (BioGenex), and purified primary antibody was applied to sections at a concentration of 10 µg/mL for 18 h at 4°C. After sequential application of biotinylated secondary antibody (Jackson Laboratories), followed by peroxidase-conjugated streptavidin, each for 1 h at 20°C (DAKO), IR was visualized using 3′,3′-diaminobenzadine tetrahydrochloride as the substrate. All immunostained sections were counterstained with Gills hematoxylin and imaged with a Bliss digital imaging system that pairs an Axioplan microscope (Zeiss) with a slide scanner (Bacus Laboratories). KLK6 IR in each tumor core was evaluated independently by 2 observers without knowledge of tumor type or demographic information. In each case, a score was assigned for staining intensity (range 1–3: low, medium, high) and for percent of the tumor core stained (range 1–4 in 25% increments). The final IR score for each tumor was assessed as the product of staining intensity and percent stained. Scores across multiple cores from the same tumor and across the 2 independent observers were averaged. Additional high-resolution images were prepared using 100X-oil immersion objective and an Olympus BX51 microscope fitted with an Olympus DP71 camera.
A 2-sample Student's t-test was used to compare differences in KLK6 IR between grade III and grade IV astrocytomas and to determine the significance of differences in KLK6 RNA levels between grade IV tumors and nondiseased brain samples. Cox proportional hazards regression was used to assess the association of KLK6 IR scores with survival across all 60 grade IV astrocytomas. Analysis of the martingale residuals from the Cox proportional hazards regression model as described by Therneau and Grambsh28 was used to assess the functional form of KLK6 IR scores. This analysis was used to dichotomize KLK6 IR scores into 2 discrete GBM patient populations: those with IR scores <10 and ≥10. Martingale residuals are similar to the difference between the observed and expected number of events. Both univariable and multivariable models including age, gender, and ECOG performance score as covariables were assessed. Grade IV patients with IR scores <10 or ≥10 were also used to determine cumulative survival probabilities using the Kaplan–Meier method. In all cases, P < .05 was considered statistically significant.
KLK6 RNA in Grade IV Astrocytoma
KLK6 RNA levels were determined in GBM tumor specimens that were snap frozen at the time of surgical resection (n = 23). To accurately identify the area of tumor tissue, a 5-μM frozen section from each specimen was stained with hematoxylin and eosin and the tumor area circled by a neuropathologist. Additional, 10-μM frozen sections were then macrodissected on dry ice and the tumor tissue only placed in Trizol Reagent (Invitrogen) prior to RNA extraction.29 Levels of KLK6 in RNA obtained from GBM were compared with those in 6 control nondiseased human total brain samples derived from 3 separate commercial sources (Ambion, Clontech, and Stratagene).
Real-time reverse transcription (RT)-PCR was used to determine KLK6 RNA expression levels in GBM patient and cell line samples. In each case, total RNA (0.5 μg) was amplified using Light Cycler-RNA Amplification Kit SYBR Green I (Roche Diagnostics). Levels of human KLK6 were normalized to levels of the constitutively expressed gene glyceraldehyde phosphate 3-dehydrogenase (GAPDH) to control for loading. Primer sequences used for amplification of KLK6 were forward 5′-TGCCAGGGTGATTCTGGG-3′ and reverse 5′-TGCAGACGTTGGTGTAGACT-3′; and for GAPDH forward 5′-ACCACCATGGAGAAGGC-3′ and reverse 5′-GGCATGGACTGTGGTCATGA-3′.30,31 Expression levels were quantified relative to standard curves created by amplification of vectors containing known dilutions of each gene of interest.
Astrocytoma Culture Conditions
U251 and SF767 astrocytoma cell lines were cultured in Dulbecco's modified Eagle’s medium (Invitrogen) supplemented with 10% fetal bovine serum (Atlanta Biologicals). The U251 cell line was provided by Dr R. Jenkins, and the SF767 cell line by Dr J. Sarkaria (Mayo Clinic). All cultures were maintained at 37°C in 95% air and 5% CO2. U251 astrocytoma cell transfection was accomplished using Lipofectamine 2000 (Invitrogen). To avoid the influence of potential contaminating proteases or protease inhibitors in serum-containing media, all experiments were completed under defined, serum-free conditions in neurobasal A media supplemented with 1% B-27, 0.5% N-2 (Invitrogen), 50 U/mL penicillin-streptomycin, 0.45% glucose, 2 mM Glutamax, 1 mM sodium pyruvate, and 50 μM β-mercaptoethanol.
Evaluation of the Effects of Elevated Levels of KLK6 on GBM Cell Survival
To determine the potential effects of increased KLK6 on GBM cell survival, we examined the ability of well-characterized apoptosis-inducing agents to elicit cell death in U251 astrocytoma cells treated with recombinant protease or stably transfected with a cytomegalovirus (CMV)-driven KLK6 expression construct. Recombinant KLK6 was heterologously expressed from insect cells, activated from its zymogen form, and purified as previously described.27,31–33 Astrocytoma cells were treated with KLK6 (1–10 μg/mL; 30–300 nM) for 2 h prior to application of cell death–inducing agents. Enforced expression of KLK6 was accomplished by transfection of U251 astrocytoma cells with a vector containing the full-length human KLK6 gene (NM_002774) fused to a green fluorescent protein (GFP) expression cassette under the control of a CMV promoter (pReceiver-MO3; GeneCopoeia).31 An empty pReceiver-MO3 GFP vector was used as a control. Cell lines stably expressing these vectors were selected using 500 μg/mL G418 (Mediatech). Overexpression was verified by examination of KLK6 RNA levels by real time RT-PCR and examination of GFP fluorescence in transfected cells.
To determine the possible role of PAR1 in mediating the cellular effects of KLK6 in GBM, knockdown of human PAR1 RNA was accomplished by transfection of U251 astrocytoma cells with PAR1 short hairpin (sh)RNA. Five knockdown vectors were screened and 2 identified, which produced significant knockdown of PAR1 RNA levels (NM_001992.x-2152s1c1 or NM_001992.x-1147s1c1). MISSION pLKO.1-puro nonmammalian shRNA control plasmid DNA (SHC002) was utilized as a transfection control (Sigma). Cells stably transfected with each vector were selected using 1 μg/mL puromycin. Knockdown of PAR1 was validated by determination of PAR1 RNA expression levels in RNA extracted from stably transfected cells using real-time RT-PCR with forward 5′-CTGTTGTCTGCCCGCAC-3′ and reverse 5′-ACTAATCTGTATTCAGTTAACCCACTT-3′ primers.
The effects of KLK6 on GBM cell survival were examined across multiple cell death paradigms using flow cytometric markers of cell death and apoptosis31 or colony-forming assays. The cell death paradigms examined included staurosporine and cisplatin, in addition to conventional therapies currently used in GBM patients, that is, RT, TMZ, or RT + TMZ.34 The dose chosen for each treatment was based on previously published reports and preliminary dose response assays using the cell growth conditions applied in this study.35–37 Staurosporine and cisplatin are widely used tools to dissect mechanisms of apoptosis, including those related to glioblastoma.38,39 Staurosporine is a pan–tyrosine kinase inhibitor and was applied to cells at 1–2 μM for 18 h prior to examination of cell death by flow cytometry. Cisplatin induces cell death by cross-linking DNA and was applied to cells at 2.5 μg/mL for 24 h in experiments examining cell death by flow cytometry, or at 0.1 μg/mL for 48 h in colony-forming assays (APP Pharmaceuticals). TMZ is an alkylating agent and was applied at 10 μM for 24 h (Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute). Radiation resulting in DNA damage was delivered using a cesium-137 source at a dose rate of 7.93 Gy/min for a total dose of either 2 or 5 Gy. Cell death in response to RT and TMZ was examined using clonogenicity assays. In each case, cells were treated in triplicate and experiments repeated independently at least 3 times with parallel results. Treatment groups were compared using 2-sample Student's t-tests with P < .05 considered significantly different.
To quantify cell death by flow cytometry, unfixed cells were labeled with 2 μg/mL propidium iodide (PI) or 6.5 μg/mL 7-aminoactinomycin D (7-AAD) at the end point of each experiment. In some cases, the extent of apoptosis was also evaluated by co-labeling cells with annexin V conjugated with fluorescein isothiocyanate (FITC; eBioscience), permitting identification of cells that were live (annexin V–FITC− and PI−), early apoptotic (annexin V–FITC+ and PI−), or dead (annexin V–FITC+ and PI+). Cells were analyzed using either a FACScan or FACSCalibur flow cytometer (BD Biosciences). FlowJo software was used to analyze raw data. All samples were run in triplicate and each experiment repeated independently at least 3 times.
Colony-forming assays were used to determine the effect of enforced KLK6 expression on U251 astrocytoma cell survival or sensitivity to cell death induced by cisplatin, RT, or TMZ alone, or RT + TMZ. Since the initial plating density could impact results, preliminary experiments were used to determine a single plating density that would yield a quantifiable number of nonoverlapping colonies across control and drug treatment conditions. Naive U251 cells, or those stably transfected with the KLK6 or control expression vectors, were plated in triplicate at cells per well of 1 × 103 (no treatment), 2 × 103 (single treatments), or 5 × 103 (combined treatments), in defined media on 24-well plates coated with poly-l-lysine (10 μg/mL). Two hours after plating, cells were treated with cisplatin (0.1 μg/mL), ionizing RT (2 or 5 Gy), and/or TMZ (10 µM) for 24 h. Media were replaced after treatment and surviving cells permitted to expand over a 14-day period before being fixed and stained with 10% crystal violet (Sigma) in 1% ethanol. Plates were air-dried and scanned (Epson Perfection v700 scanner) and the number of colonies counted by visual inspection without knowledge of the treatment groups. In each case, data are presented as the mean number of colonies formed as a ratio to the number of cells initially plated.
To further evaluate the effects of KLK6 on the apoptosis cascade, we examined its effects on cleavage of poly-ADP-ribose polymerase (PARP), one of the final substrates of caspase-3. U251 glioma cells were plated in serum-free media at 4 × 105 cells per well in 6-well plates and pretreated with recombinant KLK6 (10 µg/mL) for 1 h prior to application of 2 µM staurosporine for an additional 6 h. Protein lysates were then prepared using radioimmunoprecipitation assay buffer and proteins separated on sodium dodecyl sulfate–polyacrylamide gels prior to transfer to nitrocellulose membranes (Bio-Rad Laboratories). PARP was detected using a monoclonal antibody that was a gift from Dr S. Kauffman (Mayo Clinic).40 Equal loading was verified by reprobing blots for β-actin (Novus). In all cases, proteins of interest were detected on film using SuperSignal West Pico Chemiluminescent Substrate. For quantification, films were scanned and images quantified using Image Lab 2.0 software (Bio-Rad). Both the upper band corresponding to a 116-kDa intact form and the signature 89-kDa caspase-generated fragment were normalized to actin. All western blots were repeated 3 times using separate cell culture preparations, with similar results.41
Results
Kallikrein 6 Is Elevated in GBM and Is Associated With Reduced Patient Survival
The potential clinical significance of KLK6 in GBM was assessed by comparing the levels of KLK6 IR in primary grade IV (n = 60) and grade III (n = 8) tumor core samples contained on 2 tissue microarrays. KLK6 immunostaining was detectable in all tumor cores examined, with the level of IR (reflecting) markedly elevated in grade IV (7.7 ± 2.4 SEM) relative to grade III specimens (2.4 ± 1.5 SEM) (P < .001, 2-sample t-test) (Fig. 1A and B, Supplementary material, Table S2). In each case, KLK6 IR was largely cytoplasmic, with variable extracellular staining also observed, including staining of the interstitial stroma in some cases (Fig. 1C–L).
Fig. 1.

KLK6 is differentially expressed across grade III and grade IV astrocytoma and is associated with poor patient survival. Tissue microarrays containing grade III (A) and grade IV (B) surgically resected astrocytoma samples were evaluated for KLK6 IR (grade III 2.4 ± 1.5, grade IV 7.7 ± 2.4; mean ± SD), P < .001, 2-sample t-test (see Supplementary material, Tables S1–S3). Photomicrographs captured with the Bliss digital imaging system (20× objective, C–G) or using 100× oil (H–L) show examples of the range of KLK6 IR seen in grade III IR = 1.5 (C and H) or IR = 6 (D and I), and in grade IV IR = 2.5 (E and J), IR = 7 (F and K), or IR = 12 (G and L) astrocytoma specimens (scale bar, 100 µm). In the higher power images, the hematoxylin-stained nuclei can be readily distinguished from KLK6 IR seen in association with cellular cytoplasm or the surrounding matrix. (M) Kaplan–Meier curves for KLK6 IR scores of <10 or ≥10 in grade IV patients (P = .014, univariable analysis; see Table 1). (N) Histogram shows level of KLK6 RNA in nondiseased control human brain samples (mean 0.86 ± 0.3 SEM) and in grade IV tumor specimens (mean 21.1 ± 3.8 SEM), P < .05, 2-sample Student's t-test.
To determine the possible prognostic significance of elevated KLK6 in grade IV astrocytoma, we determined the relationship between KLK6 IR scores and patient survival. Univariable Cox proportional hazards regression analysis of KLK6 IR scores across all 60 GBM patient tumor specimens demonstrated that elevated levels were associated with significantly reduced patient survival (hazard ratio [HR] = 1.16, 95% confidence interval [CI] = 1.02–1.31, P = .02, Table 1). In multivariable Cox modeling, including gender and ECOG performance score, higher tumor KLK6 IR scores were a poor prognostic indicator of patient survival (HR = 1.15, 95% CI = 1.01–1.30, P = .03). When age was also included in the multivariable analysis, tumor KLK6 IR scores were no longer a significant predictor of patient survival (Table 1).
Table 1.
Significance of KLK6 IR scores to patient survival across all 60 grade IV specimens examined and for tumors with IR scores of <10 or ≥10
| N | Median Survival | Univariable Analysis |
Multivariable Analysis |
|||||
|---|---|---|---|---|---|---|---|---|
| Hazard Ratio | 95% CI | Pa | Hazard Ratio | 95% CI | Pa | |||
| Grade IV, IR score | 60 | 355 | 1.16 | 1.02–1.31 | .020 | 1.10 | 0.96–1.25 | .17b |
| n | 1.15 | 1.01–1.30 | .03c | |||||
| <10 | 48 | 408 | 1.72 | 0.84–3.54 | .14b | |||
| ≥10 | 12 | 276 | 2.36 | 1.19–4.68 | .014 | 2.40 | 1.20–4.78 | .01c |
aProportional hazards regression.
bAdjusted analysis includes age at surgery, ECOG performance score, and gender as covariables.
cAdjusted analysis includes performance score and gender as covariables.
The martingale residuals from the Cox proportional hazards regression model were used to dichotomize KLK6 IR scores into 2 discrete GBM patient populations (Fig. 1M, Table 1). Patients with KLK6 IR scores ≥10 (n = 12) had a median survival of 276 days, while patients with KLK6 IR scores <10 (n = 48) had a median survival of 408 days (HR = 2.36, 95% CI = 1.19–4.68, P = .014). In the multivariable model, including gender and performance scores, KLK6 IR scores ≥10 were associated with significantly reduced patient survival (HR = 2.40, 95% CI = 1.2–4.78, P = .01, Table 1). When age was included in the multivariable analysis, KLK6 IR scores ≥10 were no longer significantly associated with patient survival (HR = 1.72, 95% CI = 0.84–3.54).
To further investigate the elevations seen in KLK6 protein in GBM patient tumor samples, we examined KLK6 RNA levels in an independent set of pathologist-reviewed GBM tumor specimens (n = 23, Fig. 1N, Supplementary material, Table S3). GBM KLK6 RNA levels were compared with levels present in nondiseased whole human brain RNA samples (n = 6). Parallel to results at a protein level, GBM samples showed a broad range of KLK6 RNA expression (mean 21.08 ± 3.83 SEM, range 1.47–72.9). KLK6 RNA expression in GBM was significantly higher than that detected in healthy human brain by ∼20-fold (mean 0.86 ± 0.31 SEM; P < .05, range 0.21–2.21) (P < .05, 2-sample Student's t-test).
Recombinant KLK6 Promotes Survival of Astrocytoma Cells In Vitro
We recently demonstrated that KLK6 promotes the survival of Jurkat leukemia cells and primary lymphocytes in response to a wide range of apoptosis-inducing agents31 and therefore hypothesized that elevated levels of KLK6 in GBM enhance GBM cell survival, thereby reducing patient survival. To begin to address this hypothesis, we examined whether recombinant KLK6 alters the survival of U251 astrocytoma cells treated with staurosporine (Fig. 2). Treatment of U251 astrocytoma cells with either 5 or 10 µg/mL of recombinant KLK6 in conjunction with staurosporine resulted in a significant reduction in the number of PI+ dead cells and a corresponding increase in the number of PI− live cells, observed by flow cytometry (P = .02, 2-sample Student's t-test; Fig. 2A and B). KLK6 produced a parallel reduction in cell death in staurosporine-treated SF767 astrocytoma cells (P = .001, 2-sample Student's t-test; Fig. 2C). To determine the ability of KLK6 to alter apoptosis, U251 cells were treated with staurosporine in the presence of KLK6 and stained with annexin V in addition to PI prior to flow cytometry. As shown in Fig. 3A and B, KLK6 significantly increased the number of live cells (annexin V−, PI−) and decreased the number of early apoptotic (annexin V+, PI−) and dead cells (annexin V+, PI+) seen in response to staurosporine treatment (P ≤ .04, 2-sample Student's t-test). In the staurosporine model, KLK6 improved GBM cell survival and correspondingly decreased cell death by ∼20%. We note that PI or 7-AAD labeling in untreated cells that was run in parallel in each experiment did not exceed 2.1%.
Fig. 2.
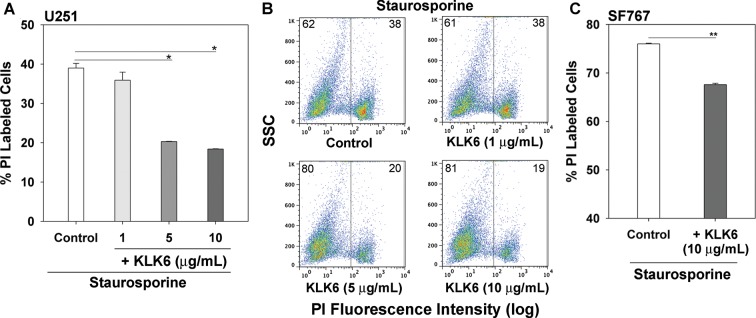
Recombinant KLK6 promotes the resistance of astrocytoma cell lines to staurosporine-induced cell death. (A) Treatment of U251 astrocytoma cells with 5 or 10 µg/mL of recombinant KLK6 significantly reduced the number of PI-labeled dead cells detected 18 h after the addition of staurosporine (1.0 µM). In the same experiment, 1.7% of untreated U251 cells were PI labeled. Data are mean ± SEM of triplicate cultures examined in parallel, *P = .02, 2-sample Student's t-test. Dot blots (B) show representative examples of flow cytometry data demonstrating the reduction in the percentage of PI-labeled dead cells seen in response to increasing concentrations of KLK6 (SSC, side scatter). (C) In parallel, treatment of SF767 astrocytoma cells with 10 µg/mL recombinant KLK6 significantly reduced the number of PI-labeled dead cells detected 18 h after addition of staurosporine (1.0 µM), **P = .001, 2-sample Student's t-test; 1.6% of untreated SF767 cells were PI labeled in the same experiment.
Fig. 3.
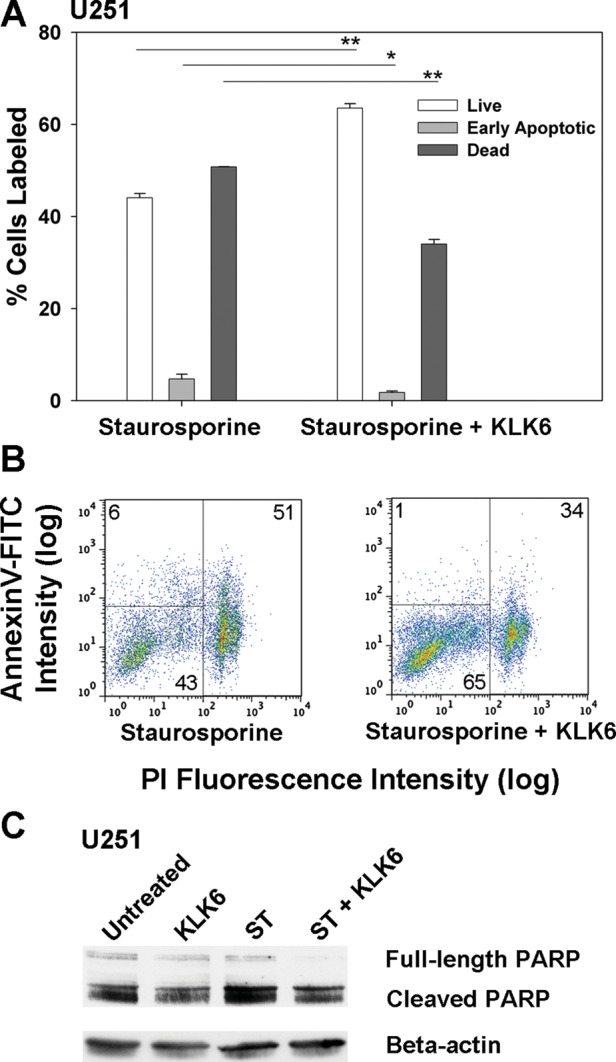
Recombinant KLK6 promotes resistance of U251 astrocytoma cells to apoptosis induced by staurosporine. Histogram (A) and corresponding dot plots (B) demonstrate that KLK6 significantly increases the number of live cells (annexin V–FITC− and PI−), while reducing the number of early apoptotic (annexin V–FITC+ and PI−) and dead cells (annexin V–FITC+ and PI+) seen in response to 1 µM staurosporine. Data shown are mean ± SEM of triplicate cultures examined in parallel, *P = .04, **P ≤ .001, 2-sample Student's t-test. In the same experiment, 82.8% of untreated U251 cells were annexin V–FITC− and PI−, 0.7% were annexin V–FITC+ and PI−, and 16% were annexin V–FITC+ and PI+. Western blot (C) shows increased levels of cleaved PARP (full-length 116 kDa, cleaved 89 kDa) after treatment with staurosporine (ST; 2 μM, 6 h). Pretreatment of cultures for 1 h with KLK6 (10 μg/mL) prior to application of ST reduced the amount of cleaved PARP observed (55% reduction; P = .01, Student's t-test). Actin was used as a control for loading, and results shown are representative of those from 3 separate experiments.
We applied western blot approaches to demonstrate that KLK6 significantly decreases PARP cleavage in U251 cells treated with staurosporine. We note that U251 cells showed a relatively high baseline level of PARP cleavage, as observed in previous studies.42,43 PARP is one of the final substrates cleaved by caspase-3 in the apoptosis cascade, and its cleavage was increased in U251 cells treated with staurosporine for 6 h. The amount of cleaved PARP was significantly reduced in cells cotreated with KLK6 (Fig. 3C); PARP cleavage was reduced to 45 ± 10.4% of control levels across 3 separate experiments (P = .01, Student's t-test).
KLK6-Mediated Survival of Astrocytoma Cells Depends in Part on Activation of PAR1
Emerging data indicate that at least some KLKs, including KLK6, are likely to mediate their cellular effects by activation of select PARs.27,31,44,45 To determine the role of PAR1 in mediating the prosurvival effects of KLK6 in U251 astrocytoma cells, PAR1 RNA levels were downregulated using shRNA targeting vectors (Fig. 4). Two PAR1 knockdown vectors were identified, which produced 48.5 ± 6.2% and 65.3 ± 8.0% knockdown of PAR1 RNA in the resultant antibiotic selected cell lines (shPAR1-3 and shPAR1-4), respectively. Recombinant KLK6 was able to promote survival of staurosporine-treated U251 cells transfected with a nontargeting control, but such prosurvival effects were reduced in each PAR1 knockdown, with reductions reaching the level of statistical significance in cells with the highest levels of enforced PAR1 knockdown (P < .03, 2-sample Student's t-test).
Fig. 4.

Knockdown of PAR1 blocks KLK6-mediated rescue of staurosporine-treated U251 astrocytoma cells. (A) U251 cells were stably transfected with a vector coding for an shRNA specific for human PAR1 (shPAR1-3 or shPAR1-4) or a no target control vector, and the ability of KLK6 (5 µg/mL) to reduce staurosporine (1 µM)-induced cell death was examined. Histogram shows that the ability of recombinant KLK6 to reduce cell death elicited by staurosporine was reduced by both PAR1-targeting vectors but only significantly blocked by the shPAR1-4 vector that produced the highest levels of knockdown (65%). In this experiment, untreated control cells contained 1.9%, 2.0%, and 1.9% PI+ cells in the case of no target control, shPAR1-3, and shPAR1-4 cells, respectively. (B) Knockdown of PAR1 RNA was determined by real-time RT-PCR (ratio of PAR1 copy number to GAPDH copy number calculated in each case and expressed as percent control). Data shown are mean ± SEM of triplicate samples examined in parallel, 2-sample Student's t-test: *P < .03, **P < .015.
Overexpression of KLK6 in Astrocytoma Cells Promotes Survival in Multiple In vitro Models of Cell Death
To validate the prosurvival effects generated by recombinant KLK6 in astrocytoma cells, stable U251 astrocytoma cell lines overexpressing human KLK6 were generated. Four unique clones were generated, each showing parallel effects across the experiments described (Figs 5–7). The KLK6 expression construct contains GFP C-terminal to the KLK6 insert. GFP was readily visualized in overexpressing cells in a perinuclear distribution, consistent with the localization of KLK6 to organelles involved in the secretory pathway46 (Fig. 5H). Significant levels of GFP were also visualized at the cell membrane in U251 KLK6-GFP transfected cells, demonstrating that KLK6 is being secreted and associates with the surface of glioma cells. By contrast, U251 cells transfected with a control GFP–expressing vector showed a uniform distribution of GFP within the cell cytoplasm without selective subcellular localization (Fig. 5D). Approximately 35% of cells in KLK6-CMV-transfected antibiotic-selected cultures and 40% of cells in GFP-CMV-transfected antibiotic-selected cultures were visually GFP+ at any given time. Significant elevations in KLK6 gene transcription were also readily detected by PCR in KLK6-GFP-transfected U251 astrocytoma cells. Parallel to results seen with recombinant KLK6, U251 cells with enforced KLK6 overexpression showed significantly improved survival when challenged with staurosporine (Fig. 5A and B; P = .02, 2-sample Student's t-test).
Fig. 5.

Overexpression of KLK6 in U251 astrocytoma cells promotes resistance to staurosporine-induced cell death. (A and B) U251 astrocytoma cells were stably transfected with a vector in which the human KLK6 gene was constitutively expressed N-terminal to GFP under the control of a CMV promoter (KLK6-CMV) or with an empty vector (Control-CMV) and examined for cell death in response to staurosporine (2.0 µM). Relative to controls, astrocytoma cells engineered to overexpress KLK6 showed significant reductions in staurosporine-induced cell death measured by uptake of 7-AAD using flow cytometry (SSC, side scatter). In this experiment, untreated Control-CMV and KLK6-CMV U251 cultures contained 2.1% 7-AAD+ cells. (C) Histogram shows ratio of the level KLK6 RNA to that of the constitutively expressed gene GAPDH in control and stably transfected U251 cells (A to C, KLK6-CMV-Clone 1). (D–K) Photomicrographs show the localization of GFP in U251 astrocytoma cells transfected with control (D–G) or the KLK6-CMV expression construct (H–K, KLK6-CMV-Clone 3). U251 cells were co-labeled with rhodamine-conjugated phalloidin to visualize actin (E and I) and counterstained with DAPI (4’,6-diamidino-2-phenylindole; F and J) to enumerate nuclei. Merged images are also shown (G and K). (K, scale bar = 100 µm for all images.) Data shown are mean ± SEM of triplicate samples examined in parallel, 2-sample Student's t-test: *P = .02 (A), **P = .001 (C). Results are representative of those seen in at least 3 independent cell culture experiments.
Fig. 6.

Overexpression of KLK6 in U251 astrocytoma cells promotes cell survival and resistance to cell death induced by radiotherapy or TMZ alone or in combination. Clonogenic assays were performed to determine the effect of KLK6 overexpression (KLK6-CMV) on the survival of U251 astrocytoma cells and their sensitivity to 2 or 5 Gy of radiation, to 10 µM TMZ, or to 2 Gy of radiation + 10 µM TMZ. The number of colonies formed in each case was directly compared with nontransfected control U251 cells and with U251 cells stably expressing an empty control vector (Control-CMV). In each case, plated cells (1 × 103 cells per well, A and B; 2 × 103 cells per well, C and D; 5 × 103 cell per well, E and F) were treated over a 24-h period and then allowed to expand for 2 wk prior to being fixed and stained with crystal violet. (A, C, and E) Histograms show the number of surviving colonies (± SEM) in triplicate; cultures treated in parallel expressed as a percent of cells plated initially. Mean colony counts ± SEM are listed for each cell line and treatment in Table 2. A significant increase in colony number was observed in KLK6-overexpressing cells under all treatment conditions examined (*P ≤ .02, **P ≤ .0005, 2-sample Student's t-test). (B, D, and F) Images of representative crystal violet stained colonies are shown for each treatment condition. Results shown were obtained using KLK6-CMV-Clone 1 (A and B), -Clone 2 (C and D), and -Clone 3 (E and F).
Fig. 7.
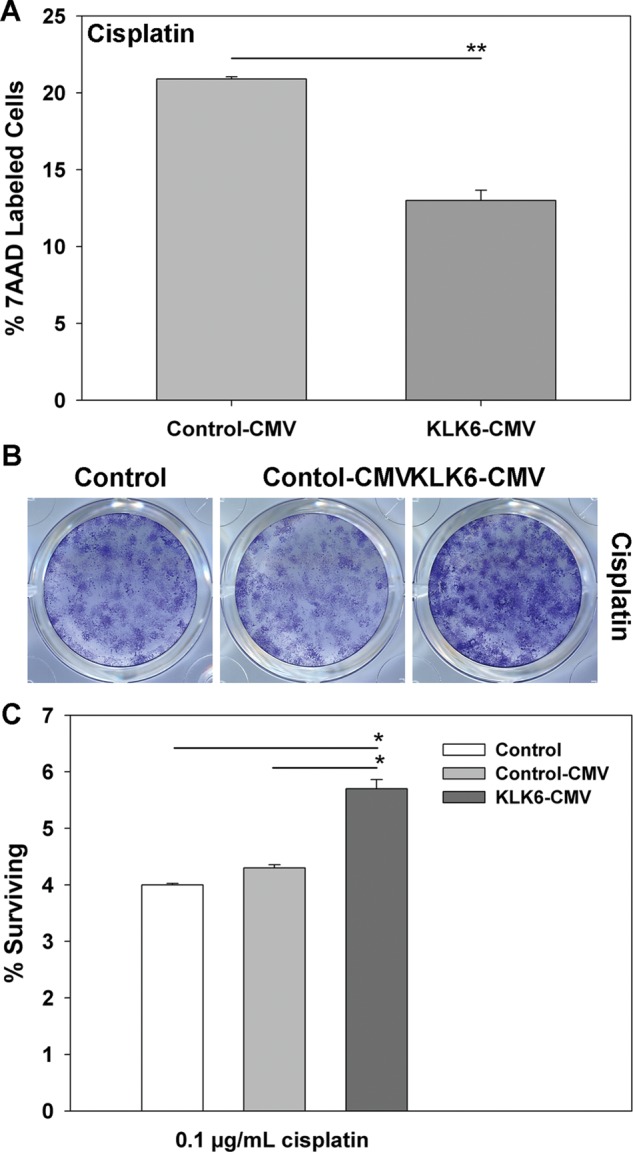
Overexpression of KLK6 promotes resistance of U251 astrocytoma cells to cell death induced by cisplatin. (A) Histogram shows the percent of cell death (7-AAD+ by flow cytometry) after exposure of U251 astrocytoma cells stably expressing an empty control CMV vector or a vector in which KLK6 is constitutively expressed under the control of CMV (KLK6-CMV-Clone 4) to cisplatin (2.5 μg/mL, 48 h). In the experiment shown in (A), untreated Control-CMV cultures contained 1.1% 7-AAD+ cells, while untreated KLK6-CMV cultures contained 1.9% 7-AAD+ cells. (B and C) U251 astrocytoma cells overexpressing KLK6 (KLK6-CMV-Clone 3) also showed significantly higher levels of survival when plated at a density of 2 × 103 cells per well, transiently treated with cisplatin (0.1 μg/mL, 24 h), and then allowed to expand for 2 wk in culture before being fixed and stained with crystal violet (controls were treated in parallel). Representative crystal violet stained colonies are shown in (B), and the mean percent surviving colonies relative to the number of cells plated (± SEM) of 3 triplicate cultures is shown in (C). Mean cell counts ± SEM are listed for each cell line and treatment in Table 2. (*P < .001, **P ≤ .005, 2-sample Student's t-test).
To determine whether elevations in KLK6 influence the sensitivity of GBM to conventional therapies, the effects of KLK6 overexpression on U251 astrocytoma cell survival were examined in response to treatment with radiation or TMZ alone or in combination (Fig. 6 and Table 2). Cell death in these models is much more protracted relative to that seen with staurosporine, and therefore clonogenicity assays were used to evaluate survival. Since KLK6-overexpressing cells showed enhanced colony formation under no treatment conditions (P ≤ .02, 2-sample Student's t-test), clonogenic assays across a given treatment were carried out with the same initial plating density (Fig. 6 and Table 2). KLK6-overexpressing U251 cells were more resistant to cell death when treated with either 2 Gy or 5 Gy of radiation (P ≤ .007, 2-sample Student's t-test). KLK6-overexpressing cells also showed significantly improved colony formation after treatment with 10 µM TMZ (P = .002, 2-sample Student's t-test). Finally, KLK6-overexpressing U251 cells were also more resistant to cell death in response to combined treatment with 10 µM TMZ plus 2 Gy radiation (P ≤ .002, 2-sample Student's t-test).
Table 2.
Overexpression of KLK6 in U251 astrocytoma cells promotes resistance to cell death
| Number of Cells Plated | Control | Control-CMV | KLK6-CMV | |
|---|---|---|---|---|
| No treatment | 1000 | 69.7 ± 4.1 | 69.0 ± 6.53 | 92.3 ± 2.33* |
| 2 Gy | 2000 | 80.0 ± 1.22 | 68.7 ± 5.34 | 106.0 ± 1.47** |
| 5 Gy | 2000 | 25.5 ± 1.19 | 16.0 ± 0.82 | 43.0 ± 0.82*** |
| 10 μM TMZ | 2000 | 35.8 ± 1.65 | 36.3 ± 1.38 | 65.5 ± 1.55*** |
| 2 Gy + 10 μM TMZ | 5000 | 85.7 ± 1.76 | 77.3 ± 0.67 | 139.8 ± 0.85** |
| 0.1 μg/mL cisplatin | 2000 | 80.5 ± 0.05 | 86.8 ± 1.11 | 113.3 ± 3.22** |
Clonogenenic assays were used to determine the effect of KLK6 overexpression (KLK6-CMV) on colony formation by U251 cells treated with radiation and/or TMZ, or cisplatin. The mean ± SEM number of colonies counted in each case is provided.
Cisplatin is another highly effective chemotherapeutic agent, although its lack of penetration of the blood–brain barrier currently limits its application in GBM. Supporting the robust and broad prosurvival effects elicited by KLK6, we show that KLK6-overexpressing U251 astrocytoma cells are more resistant to cisplatin-induced cell death when examined using markers of cell death in short-term flow cytometry studies (P = .005, 2-sample Student's t-test) or in long-term colony-forming assays (P ≤ .002, 2-sample Student's t-test; Fig. 7 and Table 2).
Discussion
Given the robust upregulation of KLK6 in reactive astrocytes27 and its potential involvement in malignancy,7 we have made a comprehensive examination of KLK6 expression and its functional significance in high-grade astrocytoma. Results demonstrate for the first time that KLK6 expression is highly elevated in a subset of GBM patient tumors and is associated with shortened postsurgery survival times. Moreover, our studies show that KLK6 promotes survival of glioblastoma cell lines in the face of several well-studied apoptosis-inducing agents, including RT and/or TMZ. Importantly, the ability of KLK6 to promote tumor cell survival was shown to be dependent on PAR1, such that both KLK6 and PAR1 represent potential new therapeutic targets for the development of approaches to sensitize GBM to cytotoxic agents, thereby improving response to therapy and potentially patient survival.
High-grade gliomas are molecularly heterogeneous, and the results presented here suggest that KLK6 is a marker of such heterogeneity, with elevated levels predictive of shortened postsurgery patient survival. Importantly, higher KLK6 IR scores were associated with reduced patient survival even after controlling for gender and ECOG performance score, the latter of which along with age is a robust predictor of patient survival.1 The risk of significantly reduced patient survival was more than 2-fold higher in GBM patients with high tumor KLK6 IR scores. However, when age was included in the multivariable analysis, KLK6 IR scores were no longer a significant predictor of survival. Age is known to be one of the most powerful prognostic indicators of GBM patient survival.1 While KLK6 levels may not be a stronger indicator of GBM patient survival than age, the significant association of elevated KLK6 with reduced patient survival highlights the likely participation of this unique serine protease in GBM malignancy.
KLK6 IR levels were ∼3-fold higher in grade IV compared with grade III glioma specimens. The likely association of KLK6 with tumor grade is further supported by a recent study of KLK6 RNA expression across intracranial grades I to IV tumors, which demonstrated that higher KLK6 levels are associated with higher tumor grade.23 In this study, the presence or absence of KLK6 expression across grades was found to be a significant predictor of disease-free patient survival. In the present study, we examined KLK6 RNA in an independent set of grade IV tumor specimens, and while we found a wide range of expression, reflecting KLK6 IR in the tissue microarrays, we were able to readily detect KLK6 in all GBM samples examined. Overall, KLK6 RNA levels were 20-fold higher in GBM relative to healthy brain samples. While 2 prior studies reported significant reductions in KLK6 in high-grade glioma,21,22 the present results, taken with the recent findings of Talieri et al,23 strongly support the association of elevated KLK6 with a more malignant glioma phenotype and with reduced patient survival. Corresponding to these findings in glioma, elevations in KLK6 are reported to be a poor prognostic indicator in ovarian,9–12 gastric,14 and colon cancers,15,16 in addition to non-small-cell lung cancer.13
We demonstrate that KLK6 promotes the survival of glioblastoma cell lines even in the presence of staurosporine and cisplatin as well as RT and/or TMZ. Since RT and TMZ are the current standards of care for GBM patients,47 additional studies will be needed to determine whether KLK6 may serve as a molecular target to improve the response of GBM to these and perhaps other cytotoxic therapies.
The hypothesis that KLK6 may contribute to reduced survival in GBM patients by promoting the resistance of glioblastoma cells to apoptosis was driven by our recent discovery that KLK6 promotes the survival of Jurkat leukemia cells.31 In these studies, we demonstrated that recombinant KLK6, or enforced KLK6 overexpression, promotes the survival of Jurkat T-cells, in addition to murine T- and B-cells, in response to a wide variety of apoptosis-inducing agents, including staurosporine, camptothecin, dexamethasone, and Fas ligand. Also, we demonstrated that KLK6-mediated prosurvival effects were associated with increases in Bcl-XL and suppression of the pro-apoptotic protein Bim. The likely anti-apoptotic effects of KLK6 are further supported by a recent study demonstrating that KLK6 shRNA results in an increase of active caspase-8 and -3 in colon cancer cell lines.15 The present study demonstrates that KLK6 reduces markers of apoptosis in glioblastoma cells treated with staurosporine, including annexin V staining and PARP cleavage. Together these studies support a novel model in which KLK6 drives malignancy by promoting resistance to apoptosis. Therefore, reduced survival in GBM patients with elevated levels of KLK6 may relate to a survival advantage that this protease confers at the level of glioblastoma cells.
The mechanism by which KLK6 promotes the resistance of tumor cells to apoptosis was shown to depend at least in part on activation of PAR1. As cell surface G-protein coupled receptors, PARs endow the cell with the ability to respond, or to overrespond, to the rapidly changing proteolytic microenvironment, such as that known to occur in GBM.48 Four PARs (PAR1–PAR4) have been identified, with PAR1 otherwise known as the thrombin receptor. Alterations in PAR signaling have already been implicated widely in disease, including roles in cancer, asthma, atherosclerosis, arthritis, and colitis.49 PAR1 is expressed in 76.9% of primary gliomas50 and is therefore positioned to be an important player in glioma biology, including as a mediator of the prosurvival effects of KLK6 that we demonstrate in this study.
A major advance in delineating the likely mechanism of action of KLKs in malignancy came with our recent discovery that KLK6 and other select KLKs cleave the extracellular domain of PAR1 and PAR2.27,31,44,45,51 Proteolytic cleavage of PAR reveals a tethered ligand that binds intramolecularly to elicit signaling.49 KLK6-mediated activation of PARs has already been shown to trigger signaling intermediates that profoundly affect cellular biology relevant to tumorigenesis,27,45 including signaling cascades that regulate sensitivity to apoptosis.31 In the present study, we used PAR1 shRNA to demonstrate that this receptor plays a key role in the mechanism by which KLK6 mediates its prosurvival effects in glioblastoma cells. Notably, prosurvival effects of PAR1 signaling through Bcl2 family members have also been observed in prostate cancer cells52 and through activation of Akt in breast cancer cell lines.53 In neu-oncogene–transformed murine astrocytes, KLK6 activates both PAR1 and PAR2 to trigger Ca2+ signaling and activation of the Akt and extracellular signal-regulated kinase signaling cascades.45 In primary murine immune cells, KLK6 increases Bcl-XL and suppresses Bim expression.31 Taken together, these data support the role of PAR1 in mediating the cellular effects of KLK6, including its prosurvival effects, for which PAR1 appears necessary. Additional studies will be needed to determine the extent to which PAR2, or other PARs, may also be involved in driving the malignant properties of glioma and the signaling intermediates involved.
While the present studies strongly support the concept that KLK6 drives glioma malignancy by promoting resistance to apoptosis, they do not rule out other possible mechanisms. KLK6 promotes production of interleukin-6 in primary murine astrocytes and their transformation from an epithelioid to a mesenchymal morphology.27 These changes are cardinal signs of astrogliosis54 and prominent features of GBM malignancy,2 and their regulation by KLK6 in glioblastoma warrants further investigation. Tumor invasivity is also a prominent feature of GBM and is well known to contribute to poor patient survival.1 As secreted serine proteases, KLK626,33,55 and other KLKs (see Sotiropoulou6 for review) are known to participate in turnover of extracellular matrix proteins and therefore are well positioned to mediate tumor invasion. A role for KLK6 in tumor invasion has already been demonstrated in other cancer types in vitro, including in colon,15,56 ovarian,57 and breast cancer cell lines.58 Interestingly, in melanoma cell lines the ability of KLK6 to promote invasion was shown to be PAR1 dependent.59 Importantly in this regard, PAR1 activation also promotes migration of renal carcinoma cells in vitro,60 invasion of breast carcinoma in vivo,61 and chemokinesis of human melanoma cells.62 Therefore, the possibility that KLK6 contributes to reduced survival in GBM patients not only by promoting tumor cell survival, but also by promoting tumor cell invasion, epithelial-mesenchymal transition, and interleukin-6 production27 makes it an attractive therapeutic target that warrants further investigation.
The results of this investigation strongly support the concept that elevations in GBM tumor KLK6 are a poor prognostic indicator of patient survival and correlate with glioma malignancy. In addition, these studies provide evidence to support a pathophysiological model in which KLK6 promotes tumor cell survival in an autocrine or paracrine fashion by activation of PAR1. Additional research will be needed to determine how best to incorporate other prominent features of glioma malignancy, including GBM dispersion, epithelial-mesenchymal transition, cytokine production, and immune modulation, into this emerging tumor malignancy model driven by KLK-PAR. Importantly, these studies suggest that KLK6 or PAR1 provides new and highly druggable targets for the development of adjunctive therapies to promote the sensitivity of GBM to the current standard of patient care, that is, RT and TMZ, and certainly in this regard additional studies are urgently needed.
Supplementary Material
Funding
This work was supported by a National Institutes of Health Brain Tumor SPORE grant career development award (grant no. P50CA108961; B. P. O, I. A. S.) and a National Institutes of Health Mayo Neuro-oncology Training Grant (nos. T32 NS07494, B. P. O, K. L. D; and R01NS052471, I. A. S.). A. R. P. was the recipient of a Mayo Clinic Summer Undergraduate Research Fellowship.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Jianmin Wu, Michael Panos, Sandy Passe, and Amanda Rynearson for technical assistance and Julie Hynes for abiding inspiration.
Conflict of interest statement. None declared.
References
- 1.Adamson C, Kanu OO, Mehta AI, et al. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs. 2009;18(8):1061–1083. doi: 10.1517/13543780903052764. [DOI] [PubMed] [Google Scholar]
- 2.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 3.Gravendeel LA, Kouwenhoven MC, Gevaert O, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69(23):9065–9072. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- 4.Colman H, Zhang L, Sulman EP, et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010;12(1):49–57. doi: 10.1093/neuonc/nop007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundwall A, Clauss A, Olsson AY. Evolution of kallikrein-related peptidases in mammals and identification of a genetic locus encoding potential regulatory inhibitors. Biol Chem. 2006;387(3):243–249. doi: 10.1515/BC.2006.032. [DOI] [PubMed] [Google Scholar]
- 6.Sotiropoulou G, Pampalakis G, Diamandis EP. Functional roles of human kallikrein-related peptidases. J Biol Chem. 2009;284(48):32989–32994. doi: 10.1074/jbc.R109.027946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emami N, Diamandis EP. Utility of kallikrein-related peptidases (KLKs) as cancer biomarkers. Clin Chem. 2008;54(10):1600–1607. doi: 10.1373/clinchem.2008.105189. [DOI] [PubMed] [Google Scholar]
- 8.Zhu X, Albertsen PC, Andriole GL, Roobol MJ, Schroder FH, Vickers AJ. Risk-based prostate cancer screening. Eur Urol. 2011;61(4):652–661. doi: 10.1016/j.eururo.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diamandis EP, Scorilas A, Fracchioli S, et al. Human kallikrein 6 (hK6): a new potential serum biomarker for diagnosis and prognosis of ovarian carcinoma. J Clin Oncol. 2003;21(6):1035–1043. doi: 10.1200/JCO.2003.02.022. [DOI] [PubMed] [Google Scholar]
- 10.Luo LY, Soosaipillai A, Grass L, Diamandis EP. Characterization of human kallikreins 6 and 10 in ascites fluid from ovarian cancer patients. Tumour Biol. 2006;27(5):227–234. doi: 10.1159/000094693. [DOI] [PubMed] [Google Scholar]
- 11.Shan SJ, Scorilas A, Katsaros D, Diamandis EP. Transcriptional upregulation of human tissue kallikrein 6 in ovarian cancer: clinical and mechanistic aspects. Br J Cancer. 2007;96(2):362–372. doi: 10.1038/sj.bjc.6603556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bayani J, Marrano P, Graham C, et al. Genomic instability and copy-number heterogeneity of chromosome 19q, including the kallikrein locus, in ovarian carcinomas. Mol Oncol. 2011;5(1):48–60. doi: 10.1016/j.molonc.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nathalie HV, Chris P, Serge G, et al. High kallikrein-related peptidase 6 in non-small cell lung cancer cells: an indicator of tumor proliferation and poor prognosis. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JJ, Kim JT, Yoon HR, et al. Upregulation and secretion of kallikrein-related peptidase 6 (KLK6) in gastric cancer. Tumour Biol. 2012 doi: 10.1007/s13277-011-0267-1. [DOI] [PubMed] [Google Scholar]
- 15.Kim JT, Song EY, Chung KS, et al. Up-regulation and clinical significance of serine protease kallikrein 6 in colon cancer. Cancer. 2011;117(12):2608–2619. doi: 10.1002/cncr.25841. [DOI] [PubMed] [Google Scholar]
- 16.Petraki C, Dubinski W, Scorilas A, et al. Evaluation and prognostic significance of human tissue kallikrein-related peptidase 6 (KLK6) in colorectal cancer. Pathol Res Pract. 2012;208(2):104–108. doi: 10.1016/j.prp.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 17.Cairncross G, Jenkins R. Gliomas with 1p/19q codeletion: a.k.a. oligodendroglioma. Cancer J. 2008;14(6):352–357. doi: 10.1097/PPO.0b013e31818d8178. [DOI] [PubMed] [Google Scholar]
- 18.Cairncross G, Berkey B, Shaw E, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24(18):2707–2714. doi: 10.1200/JCO.2005.04.3414. [DOI] [PubMed] [Google Scholar]
- 19.Burton EC, Lamborn KR, Feuerstein BG, et al. Genetic aberrations defined by comparative genomic hybridization distinguish long-term from typical survivors of glioblastoma. Cancer Res. 2002;62(21):6205–6210. [PubMed] [Google Scholar]
- 20.Vogazianou AP, Chan R, Backlund LM, et al. Distinct patterns of 1p and 19q alterations identify subtypes of human gliomas that have different prognoses. Neuro Oncol. 2010 doi: 10.1093/neuonc/nop075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yousef GM, Borgono CA, White NM, et al. In silico analysis of the human kallikrein gene 6. Tumour Biol. 2004;25(5–6):282–289. doi: 10.1159/000081393. [DOI] [PubMed] [Google Scholar]
- 22.Strojnik T, Kavalar R, Zajc I, Diamandis EP, Oikonomopoulou K, Lah TT. Prognostic impact of CD68 and kallikrein 6 in human glioma. Anticancer Res. 2009;29(8):3269–3279. [PubMed] [Google Scholar]
- 23.Talieri M, Zoma M, Devetzi M, Scorilas A, Ardavanis A. Kallikrein-related peptidase 6 (KLK6) gene expression in intracranial tumors. Tumour Biol. 2012 doi: 10.1007/s13277-012-0385-4. [DOI] [PubMed] [Google Scholar]
- 24.Scarisbrick IA, Asakura K, Blaber S, et al. Preferential expression of myelencephalon specific protease by oligodendrocytes of the adult rat spinal cord white matter. Glia. 2000;30:219–230. doi: 10.1002/(sici)1098-1136(200005)30:3<219::aid-glia2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 25.Scarisbrick IA, Isackson PJ, Ciric B, Windebank AJ, Rodriguez M. MSP, a trypsin-like serine protease, is abundantly expressed in the human nervous system. J Comp Neurol. 2001;431(3):347–361. [PubMed] [Google Scholar]
- 26.Scarisbrick IA, Blaber SI, Lucchinetti CF, Genain CP, Blaber M, Rodriguez M. Activity of a newly identified serine protease in CNS demyelination. Brain. 2002;125(Pt 6):1283–1296. doi: 10.1093/brain/awf142. [DOI] [PubMed] [Google Scholar]
- 27.Scarisbrick IA, Radulovic M, Larson N, et al. Kallikrein 6 is a novel molecular trigger of reactive astrogliosis. Biol Chem. 2012;393:355–367. doi: 10.1515/hsz-2011-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. New York: Springer; 2000. [Google Scholar]
- 29.Drucker KL, Kitange GJ, Kollmeyer TM, et al. Characterization and gene expression profiling in glioma cell lines with deletion of chromosome 19 before and after microcell-mediated restoration of normal human chromosome 19. Genes Chromosomes Cancer. 2009;48(10):854–864. doi: 10.1002/gcc.20688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scarisbrick IA, Blaber SI, Tingling JT, Rodriguez M, Blaber M, Christophi GP. Potential scope of action of tissue kallikreins in CNS immune-mediated disease. J Neuroimmunol. 2006;178(1–2):167–176. doi: 10.1016/j.jneuroim.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 31.Scarisbrick IA, Epstein B, Cloud BA, et al. Functional role of kallikrein 6 in regulating immune cell survival. PLoS One. 2011;6(3):e18376. doi: 10.1371/journal.pone.0018376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernett MJ, Blaber SI, Scarisbrick IA, Dhanarajan P, Thompson SM, Blaber M. Crystal structure and biochemical characterization of human kallikrein 6 reveals that a trypsin-like kallikrein is expressed in the central nervous system. J Biol Chem. 2002;277(27):24562–24570. doi: 10.1074/jbc.M202392200. [DOI] [PubMed] [Google Scholar]
- 33.Blaber SI, Scarisbrick IA, Bernett MJ, et al. Enzymatic properties of rat myelencephalon-specific protease. Biochemistry. 2002;41(4):1165–1173. doi: 10.1021/bi015781a. [DOI] [PubMed] [Google Scholar]
- 34.Stupp R, Dietrich PY, Ostermann Kraljevic S, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20(5):1375–1382. doi: 10.1200/JCO.2002.20.5.1375. [DOI] [PubMed] [Google Scholar]
- 35.Beier D, Rohrl S, Pillai DR, et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008;68(14):5706–5715. doi: 10.1158/0008-5472.CAN-07-6878. [DOI] [PubMed] [Google Scholar]
- 36.Gill JS, Windebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest. 1998;101(12):2842–2850. doi: 10.1172/JCI1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu C, Sarkaria JN, Petell CA, et al. Combination of measles virus virotherapy and radiation therapy has synergistic activity in the treatment of glioblastoma multiforme. Clin Cancer Res. 2007;13(23):7155–7165. doi: 10.1158/1078-0432.CCR-07-1306. [DOI] [PubMed] [Google Scholar]
- 38.Combs SE, Zipp L, Rieken S, et al. In vitro evaluation of photon and carbon ion radiotherapy in combination with chemotherapy in glioblastoma cells. Radiat Oncol. 2012;7:9. doi: 10.1186/1748-717X-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao X, Zhu H, Ali-Osman F, Lo HW. EGFR and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced mitochondrial translocalization: a potential mechanism of EGFR-driven antagonism of apoptosis. Mol Cancer. 2011;10:26. doi: 10.1186/1476-4598-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eischen CM, Kottke TJ, Martins LM, et al. Comparison of apoptosis in wild-type and Fas-resistant cells: chemotherapy-induced apoptosis is not dependent on Fas/Fas ligand interactions. Blood. 1997;90(3):935–943. [PubMed] [Google Scholar]
- 41.Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53(17):3976–3985. [PubMed] [Google Scholar]
- 42.Zhao W, Kridel S, Thorburn A, et al. Fatty acid synthase: a novel target for antiglioma therapy. Br J Cancer. 2006;95(7):869–878. doi: 10.1038/sj.bjc.6603350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuh B, Sobo M, Cen L, et al. LLL-3 inhibits STAT3 activity, suppresses glioblastoma cell growth and prolongs survival in a mouse glioblastoma model. Br J Cancer. 2009;100(1):106–112. doi: 10.1038/sj.bjc.6604793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oikonomopoulou K, Hansen KK, Saifeddine M, et al. Proteinase-activated receptors, targets for kallikrein signaling. J Biol Chem. 2006;281(43):32095–32112. doi: 10.1074/jbc.M513138200. [DOI] [PubMed] [Google Scholar]
- 45.Vandell AG, Larson N, Laxmikanthan G, et al. Protease-activated receptor dependent and independent signaling by kallikreins 1 and 6 in CNS neuron and astroglial cell lines. J Neurochem. 2008;107(3):855–870. doi: 10.1111/j.1471-4159.2008.05658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatebe H, Watanabe Y, Kasai T, et al. Extracellular neurosin degrades alpha-synuclein in cultured cells. Neurosci Res. 2010;67(4):341–346. doi: 10.1016/j.neures.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 48.Nakada M, Okada Y, Yamashita J. The role of matrix metalloproteinases in glioma invasion. Front Biosci. 2003;8:e261–e269. doi: 10.2741/1016. [DOI] [PubMed] [Google Scholar]
- 49.Adams MN, Ramachandran R, Yau MK, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130:248–282. doi: 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, Zhan H, Xu W, et al. Upregulation of matrix metalloproteinase-1 and proteinase-activated receptor-1 promotes the progression of human gliomas. Pathol Res Pract. 2011;207(1):24–29. doi: 10.1016/j.prp.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 51.Angelo PF, Lima AR, Alves FM, et al. Substrate specificity of human kallikrein 6: salt and glycosaminoglycan activation effects. J Biol Chem. 2006;281(6):3116–3126. doi: 10.1074/jbc.M510096200. [DOI] [PubMed] [Google Scholar]
- 52.Tantivejkul K, Loberg RD, Mawocha SC, et al. PAR1-mediated NFkappaB activation promotes survival of prostate cancer cells through a Bcl-xL-dependent mechanism. J Cell Biochem. 2005;96(3):641–652. doi: 10.1002/jcb.20533. [DOI] [PubMed] [Google Scholar]
- 53.Yang E, Boire A, Agarwal A, et al. Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Res. 2009;69(15):6223–6231. doi: 10.1158/0008-5472.CAN-09-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32(12):638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghosh MC, Grass L, Soosaipillai A, Sotiropoulou G, Diamandis EP. Human kallikrein 6 degrades extracellular matrix proteins and may enhance the metastatic potential of tumour cells. Tumour Biol. 2004;25(4):193–199. doi: 10.1159/000081102. [DOI] [PubMed] [Google Scholar]
- 56.Henkhaus RS, Gerner EW, Ignatenko NA. Kallikrein 6 is a mediator of K-RAS-dependent migration of colon carcinoma cells. Biol Chem. 2008;389(6):757–764. doi: 10.1515/BC.2008.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prezas P, Arlt MJ, Viktorov P, et al. Overexpression of the human tissue kallikrein genes KLK4, 5, 6, and 7 increases the malignant phenotype of ovarian cancer cells. Biol Chem. 2006;387(6):807–811. doi: 10.1515/BC.2006.102. [DOI] [PubMed] [Google Scholar]
- 58.Pampalakis G, Prosnikli E, Agalioti T, Vlahou A, Zoumpourlis V, Sotiropoulou G. A tumor-protective role for human kallikrein-related peptidase 6 in breast cancer mediated by inhibition of epithelial-to-mesenchymal transition. Cancer Res. 2009;69(9):3779–3787. doi: 10.1158/0008-5472.CAN-08-1976. [DOI] [PubMed] [Google Scholar]
- 59.Krenzer S, Peterziel H, Mauch C, et al. Expression and function of the kallikrein-related peptidase 6 in the human melanoma microenvironment. J Invest Dermatol. 2011;131(11):2281–2288. doi: 10.1038/jid.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bergmann S, Junker K, Henklein P, Hollenberg MD, Settmacher U, Kaufmann R. PAR-type thrombin receptors in renal carcinoma cells: PAR1-mediated EGFR activation promotes cell migration. Oncol Rep. 2006;15(4):889–893. [PubMed] [Google Scholar]
- 61.Arora P, Cuevas BD, Russo A, Johnson GL, Trejo J. Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene. 2008;27(32):4434–4445. doi: 10.1038/onc.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi X, Gangadharan B, Brass LF, Ruf W, Mueller BM. Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol Cancer Res. 2004;2(7):395–402. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.