

Abstract
A new rhodium carbenoid approach to access alkynoates has been developed. This transformation combines the addition of enol ethers at the vinylogous position of β-siloxy-substituted vinyldiazo derivatives and an unprecedented siloxy group migration to yield the products as single diastereomers.
Keywords: carbenoids, 2-alkynoate, siloxy-vinyldiazo, rhodium
2-Alkynoates represent a versatile class of synthetic intermediates in the field of organic synthesis1 and are useful precursors to numerous biologically active compounds.2 Despite significant interest from the synthetic community, facile incorporation of an alkynecarboxylate moiety is still an ongoing challenge. The difficulty associated with the direct alkylation of 2-alkynoates is due to their tendency to isomerize into the corresponding allenes under basic conditions, which are then prone to undergo conjugate addition.3 In practice, commonly reported procedures require a multi-step reaction sequence.4 A reasonably direct alternative approach to introduce the alkynecarboxylate group is the Nicholas reaction,5 which allows the functionalization of propargylic sites with a variety of nucleophiles.6 This approach requires the use of stoichiometric dicobalt reagents as well as the need to perform an oxidative decomplexation to regenerate the alkyne moiety following substitution. In this communication, we describe a new stereoselective approach that allows a straightforward access to highly functionalized alkyl 2-alkynoates by means of a rhodium-catalyzed transformation between silyl enol ethers and 3-siloxy-2-diazobutenoates [Eq. (1)].
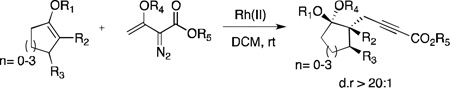 |
(1) |
The successful development of the aforementioned alkylation protocol is based on the discovery of an unusual transformation of rhodium-stabilized vinylcarbenoids. Transient vinylcarbenoids undergo a wide variety of synthetically useful reactions,7 and their chemistry is particularly rich because they display electrophilic character at both the carbenoid site and the vinylogous position.8 Especially versatile vinylcarbenoids are those derived from 3-siloxy-2-diazobutenoates. Reactions initiated at its carbenoid site have been used for stereoselective cyclopropanation10 and the tandem cyclopropanation/Cope rearrangement,11 leading to the synthesis of three-, five- or seven-membered carbocycles. The combination of siloxy α-diazoacetates with cinnamaldehydes offers access to complementary motifs.12 More recently, the addition of nitrones to the vinylogous position of this carbenoid as well as a number of unusual transformations have been reported.13 The key transformation behind the 2-alkynoate alkylation protocol also involves reaction at the vinylogous position of a vinylcarbenoid, but this is then followed by an unprecedented siloxy group migration.
We have had an extensive program to broaden the scope of substrates that undergo selective vinylogous addition over carbenoid-type transformations.8 During these studies we discovered that the Rh2(esp)29-catalyzed reaction of 1-(trimethylsiloxy)cyclohexene 1 with 3-siloxy-2-diazobutenoate 2 unexpectedly led to the formation of alkyne 3 as a single diastereomer in 53% yield (Scheme 1). The structure of compound 3 was unambiguously confirmed by single crystal X-ray diffraction.13
Scheme 1.

X-ray structure of product 3 formed from 2 and 1-(trimethylsiloxy)cyclohexene 1
This reaction was intriguing not only because the formation of 3 occurred via vinylogous addition, but also because the disiloxyketal functional group arose from the migration of the OTBS group from the vinylcarbenoid to the cyclohexyl moiety. To the best of our knowledge, such migration is unprecedented in the carbenoid literature. In order to evaluate the stereospecificity of the transformation, the reaction between the siloxycyclohexene and β-siloxy-vinyldiazoacetate was repeated with the TBS and TMS substituents interconverted (Table 1, entry 1). The diasteromer to 3 was isolated in a highly stereoselective manner.
Table 1.
Substrate scope.
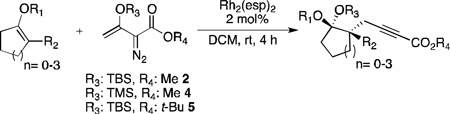 | ||||
|---|---|---|---|---|
| Entry | Substrate | Diazo | Product | Yield[a] [%] |
| 1 | 4 |  |
49[b] | |
| 2 | 2 |  |
79[b] | |
| 3 |  |
2 | 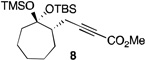 |
61[b] |
| 4 | 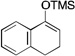 |
2 | 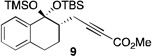 |
69[b] |
| 5 | 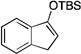 |
2 | 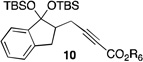 |
33[c] |
| 6 | 5 | 84[d] | ||
| 7 | 2 | 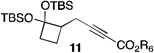 |
51[c,e] | |
| 8 | 5 | 98[d] | ||
| 9[f] | 2 | 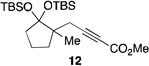 |
56 | |
| 10[f] | 2 | 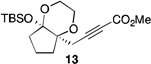 |
53[b] | |
Isolated yield.
d.r. > 20:1.
R4=Me.
R4=t-Bu.
Yield refers to the isolated alkynoate product which was separated from the cyclopropanated byproduct by column chromatography.
Reaction was run at reflux.
The scope of this transformation was then explored with a range of cyclic enol ethers. The reaction proceeded in moderate to excellent yields, and in all cases only a single diastereomer of the product was generated. With certain substrates, such as siloxyindene and siloxycyclobutene, the potentially competing products derived from carbenoid reactivity became apparent and resulted in lower isolated yields of the alkynoate products (Table 1, entries 5 and 7). We have demonstrated that increasing the size of the ester group in the vinylcarbenoid enhances the vinylogous reactivity.8c,e Indeed, when the reactions on these substrates were repeated using t-butyl 3-siloxy-2-diazobutenoates 5, alkynoates 10 and 11 were isolated as the sole products in 84 and 98% yield, respectively (entries 6 and 8). The vinylogous reactions to the carbenoid derived from 3-butenediazoacetate 2 are also applicable to tetrasubstituted vinyl ethers, leading to the formation of alkynoates containing two adjacent quaternary centers (entries 9 and 10). The relative configuration of products 6–13 was assigned by analogy to alkynoate 3 since an equivalent migration is presumed to be involved in the formation of those compounds.
It is well established that donor/acceptor carbenoids are more selective than their acceptor-only homologs.15 We have previously demonstrated that the reaction between donor/acceptor carbenoids and silyl enol ethers favors C—H insertion16 or the combined C—H functionalization/Cope rearrangement17 over cyclopropanation. In this particular alkynoate transformation we rationalize that the open/unsubstituted vinylogous position of the vinylcarbenoid is responsible for the vinylogous addition by the silyl enol ether. In addition, the use of sterically demanding nucleophiles, essentially tri- and tetra-substituted olefins, is certainly accountable for the enhancement of vinylogous reactivity.8e,g A plausible mechanism for the alkynoate formation is detailed in Scheme 2. Addition of the silyl enol ether to the unsubstituted γ-position of the activated β-siloxy-vinylcarbenoid would result in the formation of the zwitterionic intermediate 14. Subsequent direct [1,4]-siloxy group transfer or stepwise addition to the oxocarbenium ion via intermediate 15 followed by β-elimination would lead to the alkynoate product. Previous studies have shown that rhodium-stabilized β-siloxy-vinylcarbenoids have a strong preference for the s-cis conformation where the siloxy group points away from the “wall” of the catalyst.17,18 We postulate that this conformational preference would be consistent with the rhodium carboxylate and the siloxy group adopting an anti-periplanar conformation which would then facilitate the β-elimination to afford the alkynoate product.
Scheme 2.

Proposed mechanism.
The X-ray structure of alkynoate 3 unambiguously shows that both the alkynyl side chain and the siloxy migrating group are on the same face of the cyclohexane. This led us to propose that the siloxy migration could be considered as an intramolecular transfer process that would preferentially occur in a suprafacial manner (path a vs b) as illustrated in Scheme 3.
Scheme 3.

Rationale for the diastereoselectivity.
Having established the basic propargylation method, studies were then conducted to determine the synthetic potential of the transformation. One obvious application would be to use the disiloxyketal as a carbonyl protecting group. Indeed the alkynoate products generated are readily converted into the corresponding ketones in high yield by simple treatment with tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF). As demonstrated in Scheme 4, ketone 16 was isolated in 94% yield.
Scheme 4.

Access to 1,6-dicarbonyl compound 16.
An exploratory study was then conducted to determine whether an enantioselective version of this transformation was feasible. Extensive experimentation revealed that the use of Rh2(S-PTAD)4 at −25 °C in TFT were the optimal conditions (see supporting information for further discussion on the optimized conditions). Representative examples are summarized in Table 2. Moderate levels of enantioinduction were observed, which demonstrated that the chiral catalyst was capable of limited facial selectivity. Interestingly the enantiomeric excess of the products increased proportionally with the size and flexibility of the ring (entries 1, 2 and 4).
Table 2.
Asymmetric alkynoate synthesis.
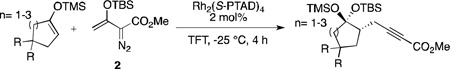 | |||||
|---|---|---|---|---|---|
| Entry | R | n | Yield[a][%] | d.r.[b] | ee[c][%] |
| 1 | H | 1 | 87 | > 20:1 | 50 |
| 2 | H | 2 | 53 | > 20:1 | 60 |
| 3 | O(CH2)2O | 2 | 49 | > 20:1 | 46 |
| 4 | H | 3 | 69 | > 20:1 | 70 |
Isolated yield.
Determined from 1H NMR of the crude reaction mixture.
Determined by chiral HPLC after hydrolysis of the disiloxyketal.
The study was then extended to 3-substituted silyl enol ethers (Scheme 5). The Rh2(esp)2-catalyzed reactions of siloxy-vinyldiazoacetate 2 with 17–19 generated products of type 20–22 bearing three contiguous stereogenic centers as single diastereomers. The relative configuration of 20 was determined by 1D-NOE NMR studies and extended by analogy to compounds 21 to 22. This result is consistent with a nucleophilic addition of the silyl enol ether anti to the 3-substituent, followed by intramolecular siloxy migration syn with respect to the alkyne moiety.
Scheme 5.

Scope of α-substituted silyl enol ethers.
To demonstrate the practicality of this reaction, the above transformation was repeated using the enantioenriched silyl enol ether (−)−19 (Scheme 6, 98% ee). As anticipated, product (+)−22 was isolated as a single diastereomer with complete transfer of stereochemical information. As chiral 3-substituted silyl enol ethers, such as (−)−19, are readily obtained by asymmetric conjugate addition to an unsaturated carbonyl compounds,19 this allows the propargylation procedure to be effectively used for the synthesis of highly enantioenriched products containing three contiguous stereogenic centers.
Scheme 6.
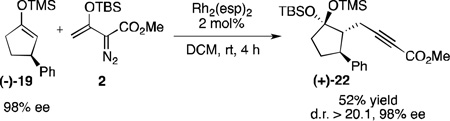
Alkynoate formation with conservation of enantioenrichment.
In summary, we have developed a method for the rapid access to highly functionalized 2-alkynoates in a single step using cyclic silyl enol ethers and siloxyvinylcarbenoids under Rh(II) catalysis in a highly diastereoselective manner. Further studies are in progress to extend this reaction, to acyclic enol ethers and to find systems that will give higher levels of asymmetric induction. These studies underscore the rich chemistry of rhodium-stabilized vinylcarbenoids, which has led to their broad application in a number of novel synthetic transformations.
Experimental Section
General procedure: A solution of siloxyvinyldiazoacetate (1.0 mmol, 2.0 equiv) in 6 mL of dried degassed DCM was added by syringe pump over 3 h at room temperature to a flame-dried 25 mL flask containing Rh2(esp)2 (7.7 mg, 0.02 equiv) and enol ether (0.5 mmol, 1.0 equiv) in 6 mL of dried degassed DCM under an argon atmosphere. The solution was stirred at room temperature for an additional 1 h. The mixture was concentrated under reduced pressure and purified by flash chromatography (pentane/Et2O) on silica gel to yield the product.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (GM099142). The crystallographic data was supplied by the X-ray diffraction laboratory at the Department of Chemistry, Emory University.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org
References
- 1.Jung ME. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 4. Oxford: Pergamon Press; 1991. pp. 1–67. [Google Scholar]
- 2.Selected examples: Prasad JS, Liebeskind LS. Tetrahedron Lett. 1987;28:1857. Lu X, Zhu G. Synlett. 1993:68. Zhu G, Lu X. Tetrahedron Asymmetry. 1995;6:885. Harriman GCB, Chi S, Zhang M, Crowe A, Bennett RA, Parsons I. Tetrahedron Lett. 2003;44:3659. Green JR, Tjeng AA. J. Org. Chem. 2009;74:7411. doi: 10.1021/jo901471b. Taj RA, Green JR. J. Org. Chem. 2010;75:8258. doi: 10.1021/jo102127q. Schaefer C, Miesch M, Miesch L. Org. Biomol. Chem. 2012;10:3253. doi: 10.1039/c2ob07049a. Kitamura T, Otsubo K. J. Org. Chem. 2012;77:2978. doi: 10.1021/jo300021a.
- 3.For a discussion, see: Bello D, Ruiz-Rodriguez J, Albericio F, Ramon R, Lavilla R. Eur. J. Org. Chem. 2010;28:5373.
- 4.For some examples, see: Klein A, Miesch M. Tetrahedron Lett. 2003;44:4483. Mota AJ, Klein A, Wendling F, Dedieu A, Miesch M. Eur. J. Org. Chem. 2005;20:4346. Geoffroy P, Ballet M-P, Finck S, Marchioni E, Marcic C, Miesch M. Synthesis. 2010;1:171.
- 5.For reviews of the Nicholas reaction and its application in natural product synthesis, see: Nicholas KM. Acc. Chem. Res. 1987;20:207. Green JR. Curr. Org. Chem. 2001;5:809. Teobald BJ. Tetrahedron. 2002;58:4133. Diaz DD, Betancort JM, Martin VS. Synlett. 2007;3:343. Kann N. Curr. Org. Chem. 2012;16:322.
- 6.a) Vizniowski CS, Green JR, Breen TL, Dalacu AV. J. Org. Chem. 1995;60:7496. [Google Scholar]; b) Green JR. Chem. Commun. 1998;16:1751. [Google Scholar]; c) Taj RA, Abhayawardhana A, Green JR. Synlett. 2009;2:292. [Google Scholar]
- 7.a) Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; b) Davies HML, Morton D. Chem. Soc. Rev. 2011;40:1857. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]; c) Davies HML, Lian Y. Acc. Chem. Res. ASAP; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Davies HML, Saikali E, Clark TJ, Chee EH. Tetrahedron Lett. 1990;31:6299. [Google Scholar]; b) Davies HML, Saikali E, Young WB. J. Org. Chem. 1991;56:5696. [Google Scholar]; c) Davies HML, Hu B, Saikali E, Bruzinski PR. J. Org. Chem. 1994;59:4535. [Google Scholar]; d) Sevryugina Y, Weaver B, Hansen J, Thompson J, Davies HML, Petrukhina MA. Organometallics. 2008;27:1750. [Google Scholar]; e) Lian Y, Davies HML. Org. Lett. 2010;12:924. doi: 10.1021/ol9028385. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Hansen JH, Davies HML. Chem. Sci. 2011;2:457. [Google Scholar]; g) Lian Y, Davies HML. Org. Lett. 2012;14:1934. doi: 10.1021/ol300632p. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Morton D, Dick AR, Ghosh D, Davies HML. Chem. Commun. 2012;48:5838. doi: 10.1039/c2cc31973j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rh2(esp)2: Bis(rhodium(α,α,α′,α′-tetramethyl-1,3-benzene -dipropionic acid)).. For a leading reference see: Zalatan DN, Bois JD. J. Am. Chem. Soc. 2009;131:7558. doi: 10.1021/ja902893u.
- 10.a) Davies HML, Ahmed G, Calvo RL, Churchill MR, Churchill DG. J. Org. Chem. 1998;63:2641. doi: 10.1021/jo972189b. [DOI] [PubMed] [Google Scholar]; b) Muller P, Bernardinelli G, Allenbach YF, Ferri M, Flack HD. Org. Lett. 2004;6:1725. doi: 10.1021/ol049554n. [DOI] [PubMed] [Google Scholar]
- 11.a) Davies HML, Ahmed G, Churchill MR. J. Am. Chem. Soc. 1996;118:10774. [Google Scholar]; b) Davies HML, Matasi JJ, Hodges LM, Huby NJS, Thornley C, Kong N, Houser JH. J. Org. Chem. 1997;62:1095. [Google Scholar]; c) Reddy RP, Davies HML. J. Am. Chem. Soc. 2007;129:10312. doi: 10.1021/ja072936e. [DOI] [PubMed] [Google Scholar]; d) Schwartz BD, Denton JR, Lian Y, Davies HML, Williams CM. J. Am. Chem. Soc. 2009;131:8329. doi: 10.1021/ja9019484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu X, Hu W-H, Zavalij PY, Doyle MP. Angew. Chem. Int. Ed. 2011;50:11152. doi: 10.1002/anie.201105557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Wang X, Xu X, Zavalij PY, Doyle MP. J. Am. Chem. Soc. 2011;133:16402. doi: 10.1021/ja207664r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xu X, Ratnikov MO, Zavalij PY, Doyle MP. Org. Lett. 2011;13:6122. doi: 10.1021/ol2026125. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang X, Abrahams QM, Zavalij PY, Doyle MP. Angew. Chem. Int. Ed. ASAP; 2012. [DOI] [PubMed] [Google Scholar]; d) Qian Y, Xu X, Wang X, Zavalij PJ, Hu W, Doyle MP. Angew. Chem. Int. Ed. ASAP; 2012. [DOI] [PubMed] [Google Scholar]
- 14.The crystal structure of 3 has been deposited at the Cambridge Crystallographic Data Centre under the deposition number CCDC 882822
- 15.Davies HML, Panaro SA. Tetrahedron. 2000;56:4871. [Google Scholar]
- 16.Davies HML, Ren P. J. Am. Chem. Soc. 2001;123:2070. doi: 10.1021/ja0035607. [DOI] [PubMed] [Google Scholar]
- 17.Lian Y, Hardcastle KI, Davies HML. Angew. Chem. Int. Ed. 2011;50:9370. doi: 10.1002/anie.201103568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen JH, Gregg TM, Ovalles SR, Lian Y, Autschbach J, Davies HML. J. Am. Chem. Soc. 2011;133:5076. doi: 10.1021/ja111408v. [DOI] [PubMed] [Google Scholar]
- 19.Tokunaga N, Yoshida K, Hayashi T. Proc. Natl. Acad. Sci. 2004;101:5445. doi: 10.1073/pnas.0307260101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.