

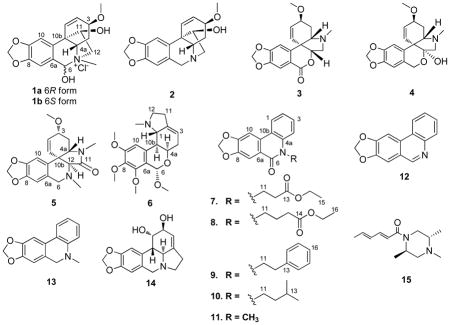
Abstract
Seven new alkaloids, N-methylhaemanthidine chloride (1), N-methyl-5,6-dihydroplicane (5), O-methylnerinine (6), N-ethoxycarbonylethylcrinasiadine (7), N-ethoxycarbonylpropylcrinasiadine (8), N-phenethylcrinasiadine (9) and N-isopentylcrinasiadine (10), together with eight known alkaloids, haemanthamin (2), 3-epimacronine (3), (+)-tazettine (4), N-methylcrinasiadine (11), trisphaeridine (12), 5,6-dihydrobicolorine (13), lycorine (14), and nigragillin (15), were isolated from the whole plants of Zephyranthes candida. The structures of the new compounds were established by spectroscopic data interpretation, with single-crystal X-ray diffraction analysis performed on 1. The absolute configuration of 3-epimacronine (3) was determined by single-crystal X-ray diffraction analysis with CuKα irradiation. Compounds 1–15 were evaluated for their in vitro cytotoxicity against five human cancer cell lines and the Beas-2B immortalized (non-cancerous) human bronchial epithelial cell line. Compounds 1, 2, 9, and 14 exhibited cytotoxicity with IC50 values ranging from 0.81 to 13 μM with selectivity indices as high as 10 when compared to the Beas-2B cell line.
Plants of the family Amaryllidaceae comprise ca. 85 genera and 1100 species that are distributed widely in tropical regions of the world. More than 500 Amaryllidaceae alkaloids representing 18 skeletal types have been isolated and reported to have acetylcholinesterase (AChE) inhibitory, analgesic, antibacterial, antifungal, antimalarial, antitumor, antiviral, and cytotoxic activities.1 Zephyranthes candida (Lindl.) Herb. is an amaryllidaceous bulbous herb native to South America. The plant is widely cultured as an ornamental flower and used as a medicinal plant in mainland China. The whole plants of Z. candida are used to treat infantile convulsions, epilepsy, and tetanus.2 Previous phytochemical studies on Z. candida have focused on the bulbs, leading to reports of four ceramides3,4 and nine alkaloids.5–9 Among these, trans-dihydronarciclasine was obtained as a cytostatic agent with an IC50 value of 3.2 ng/mL.9 Expecting the other plant parts to be chemically distinct from the previously well-studied bulbs, the whole plant extract of Z. candida was investigated, leading to the isolation of seven new alkaloids, compounds 1 and 5–10, and eight known alkaloids (2–4, 11–15). Herein, we report the isolation, structure determination, and cytotoxic activities of compounds 1–15.
RESULTS AND DISCUSSION
The air-dried whole plants of Z. candida were ground and extracted with acidic aqueous EtOH at room temperature. After evaporation of EtOH under reduced pressure, the residue was dissolved in 2% HCl, and extracted with CHCl3 three times. The acidic aqueous phase was adjusted to pH 7 with aqueous NH4OH and then extracted with CHCl3. The resulting CHCl3-soluble fraction was subjected to silica gel, RP (reversed-phase) C18, and Sephadex LH-20 column chromatography, and repeated RP C18 HPLC, to afford compounds 1–15.
Compound 1 was isolated as colorless cubic crystals, mp 237 °C, and exhibited a positive Dragendorff’s test, indicating it to be an alkaloid. The molecular formula for the quaternary base of 1 was determined to be C18H22NO5+ by (+)-HRESIMS of the [M − Cl]+ ion at m/z 332.1488 (calcd 332.1493). The IR spectrum showed the presence of hydroxy group (3239 cm−1), aromatic (3003, 1509 and 1489 cm−1), and methylenedioxy (937 cm−1) absorption. The UV absorption at 245 and 292 nm was consistent with a compound containing a methylenedioxy-substituted benzene ring.10 The NMR spectra (Tables 1 and 2) indicated that compound 1 was isolated as a mixture (ca. 3:2) of two isomers (1a and 1b) at C-6. The 1H NMR spectrum of 1 (Table 1) exhibited signals for an O-methyl (δH 3.40, s), a N-methyl (δH 3.23, s), a methylenedioxy group (δH 6.03, s), two sets of singlet aromatic protons (δH 6.92/6.86, s; 7.03/7.07, s), and two olefinic protons (δH 6.42, m; 6.41, m). In turn, the 13C NMR and DEPT spectra (Table 2) showed the presence of an O-methyl (δC 57.2), a N-methyl (δC 43.8/44.4), a methylenedioxy (δC 103.6), two methylenes (δC 62.9/68.7, 26.2/25.7), four methines bearing heteroatoms (71.9/65.4, 72.5/72.6, 78.1/78.2, 96.2/96.9), four sp2 methines (δC 104.5, 109.2/110.0, 126.8/126.7; 131.3/131.4), a sp3 quaternary carbon (δC 53.4/53.9), and four sp2 quaternary carbons (δC 124.3/125.4, 134.1, 149.3, 150.9/149.3). The NMR data of 1 were similar to those of compound 2, which was identified as haemanthamin,5 and previously isolated from the bulbs of Z. candida.3,5 The major difference found was the chemical shift of C-6 (δC 96.2/96.9), which in 1 was shifted downfield by almost 32 ppm in comparison to that of 2 (δC 64.0), indicating that C-6 in 1 bears two heteroatoms. Furthermore, an additional N-methyl group in 1 was deduced from the HMBC correlations from N-CH3 (δH 3.23, s, 3H) to C-6 (δC 96.2/96.9), C-4a (δC 71.9/65.4), and C-12 (δC 62.9/68.7). The planar structures of 1a and 1b were deduced unambiguously from analysis of the 2D-NMR spectroscopic data, including 1H-1H COSY, HSQC, and HMBC experiments, and the relative configurations of 1a and 1b were determined from the NOESY spectrum. In the NOESY spectrum of 1, the cross peak of OCH3-3 and H-4a indicated the syn-relationship of these groups, which were assigned in a β-orientation, the same as those in 2. The NOESY correlations of H-3/H-4α/H-12 suggested the N-ethyl bridged ring between C-10b and N-5 is α-oriented. The NOESY correlations of H-6 with H-12 in 1a, and H-6 with H-4a in 1b, suggested H-6 in 1a and 1b are in an α and a β-orentation, respectively. This is consistent with the structures of (+)-6-hydroxycrinamine, yemenines B and C,10 and haemanthidine11 which all have a similar epimeric C-6 relationship, while the structure of (+)-6-hydroxycrinamine has been confirmed by single-crystal X-ray diffraction.12 The characteristic Cotton effects [250 nm ([θ], −4686), 289 nm ([θ], +5931) in MeOH] in the circular dichroism (CD) spectrum of 1 were used to determine that the absolute configuration of the 5α,10b-ethanophenanthridine system10 is the same as that of haemanthamin (2). A suitable crystal of 1 was obtained for single-crystal X-ray diffraction analysis, and the result revealed that 1 was obtained in the form of a chloride salt, and confirmed the structure deduced. In the crystal structure (Figure 1), the ratio of the two epimers 1a and 1b was determined as 75:25. Importantly, the presence of the chlorine atom allowed a determination of the chirality of this structure using the Flack coefficient.13 The coefficient obtained [−0.04 (12)] for the given coordinates indicated that 1a and 1b possess the same absolute configurations as both haemanthamin (2), ignoring C-6, and the parent structure haemanthidine, which was isolated from the bulbs of Z. candida.5 It is possible that the chloride atom in 1 was introduced by the addition of HCl during the extraction procedures. Therefore, compound 1 is determined to be N-methylhaemanthidine chloride.
Table 1.
1H NMR [δ, mult, (J in Hz)] Data for Compounds 1, 5, and 6 in CD3OD (400 MHz)
| position | 1a | 1b | 5 | 6 |
|---|---|---|---|---|
| 1 | 6.42, overlap | 6.42, overlap | 5.87, d (10.0) | 2.79, br d (9.6) |
| 2 | 6.41, overlap | 6.41, overlap | 6.13, dd (10.0, 4.9) | |
| 3 | 4.04, overlap | 4.04, overlap | 3.94, ddd (4.9, 4.0, 3.5) | 5.51, overlap |
| 4a | 2.56, ddd (15.6, 13.3, 2.8) | 2.56, ddd (15.6, 13.3, 2.8) | 1.73, ddd (13.4, 10.8, 3.5) | 2.66, m |
| 4β | 2.46, dd (15.6, 4.6) | 2.34, dd (15.6, 4.6) | 2.41, ddd (13.4, 4.4, 4.0) | 2.28, m |
| 4a | 4.16, dd (13.3, 4.6) | 4.16, dd (13.3, 4.6) | 3.79, dd (10.8, 4.4) | 4.38, br d (6.4) |
| 6α | 6.05, s | 3.84, d (15.3) | ||
| 6β | 5.64, s | 3.55, d (15.3) | 5.52, s | |
| 7 | 6.92, s | 6.86, s | 6.54, s | |
| 10 | 7.03, s | 7.07, s | 6.77, s | 6.85, s |
| 10b | 2.41, dd (9.6, 2.0) | |||
| 11 | 4.02, m, overlap | 4.02, m, overlap | 2.50, m | |
| 12α | 3.25, dd (12.7, 2.4) | 3.63, d (13.7) | 3.56, s | 3.15, ddd (10.0, 7.5, 2.9) |
| 12β | 4.58, dd (12.7, 6.3) | 4.20, m, overlap | 2.30, q (10.0) | |
| OCH2O | 6.03, s | 6.03, s | 5.90, dd (4.1, 1.0) | |
| OMe-3 | 3.40, s | 3.40, s | 3.49, s | |
| NMe-5 | 3.23, s | 3.23, s | 2.78, s | |
| OMe-6 | 3.50, s | |||
| OMe-7 | 3.88, s | |||
| OMe-8 | 3.81, s | |||
| OMe-9 | 3.86, s | |||
| NMe-13 | 2.75, s | 2.03, s |
Table 2.
13C NMR Data for Compounds 1, 5, and 6 in CD3OD (100 MHz)
| position | 1a | 1b | 5 | 6 |
|---|---|---|---|---|
| 1 | 126.8 | 126.7 | 136.4 | 69.1 |
| 2 | 131.3 | 131.4 | 126.8 | 141.3 |
| 3 | 72.5 | 72.6 | 72.7 | 117.6 |
| 4 | 26.2 | 25.7 | 30.4 | 32.7 |
| 4a | 71.9 | 65.4 | 63.6 | 67.3 |
| 6 | 96.2 | 96.9 | 53.7 | 98.0 |
| 6a | 124.3 | 125.4 | 129.5 | 134.6 |
| 7 | 109.2 | 110.0 | 107.0 | 153.2 |
| 8 | 149.3 | 149.3 | 148.4 | 142.9 |
| 9 | 150.9 | 151.2 | 148.5 | 154.8 |
| 10 | 104.5 | 104.5 | 109.4 | 110.3 |
| 10a | 134.1 | 134.1 | 131.2 | 121.8 |
| 10b | 53.4 | 53.9 | 45.9 | 44.8 |
| 11 | 78.1 | 78.2 | 173.9 | 29.0 |
| 12 | 62.9 | 68.7 | 68.9 | 57.9 |
| OCH2O | 103.6 | 103.6 | 102.6 | |
| OMe-3 | 57.2 | 57.2 | 57.0 | |
| NMe-5 | 43.8 | 44.4 | 43.7 | |
| OMe-6 | 55.8 | |||
| OMe-7 | 61.6 | |||
| OMe-8 | 61.3 | |||
| OMe-9 | 56.7 | |||
| NMe-13 | 28.1 | 44.4 |
Figure 1.
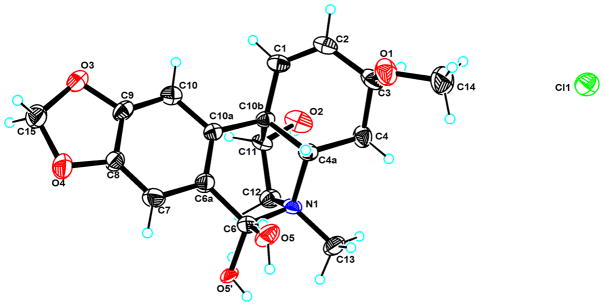
X-ray ORTEP drawing of compound 1 with ellipsoid probability of 30% by MoKα radiation at 298 (2) K.
Compound 5 was obtained as yellow amorphous powder, and its HRESIMS showed quasimolecular ion peaks at m/z 343.1688 [M + H]+ (calcd for C19H23N2O4+, 343.1652), 685.3244 [2M + H]+ (calcd for C38H45N4O8 +, 685.3232), and 707.3064 [2M + Na]+ (calcd for C38H44N4NaO8 +, 707.3051), corresponding to the molecular formula, C19H22N2O4, and indicating ten degrees of unsaturation. The UV spectrum showed absorption maxima at 250 and 290 nm, again consistent with the presence of a methylenedioxy-substituted benzene ring. The NMR data of compound 5 (Tables 1 and 2) were similar to those of tazettine (4).14 However, three differences were evident: (1) the oxygen atom at position C-5 in compound 4 is replaced by a N-Me group (δH 2.78, s, 3H; δC 43.7) in compound 5; (2) C-11 is oxidized from a methylene (δH 3.28, d, J = 10.4 Hz, 1H, H-6α; 2.66, d, J = 10.4 Hz, 1H, H-6β; δC 65.7) in compound 4 to a carbonyl (δC 173.9) in compound 5; (3) the quaternary carbon (δC 102.3) bearing two heteroatoms at C-12 in compound 4 is replaced with a methine (δH 3.56, s, 1H; δC 68.9) bearing a nitrogen atom in compound 5. The planar structure of 5 was deduced from its 1H-1H COSY, HSQC, and HMBC data, and the relative configuration was determined from the NOESY spectrum. The α-orientation of the methoxy group (δH 3.49, s, 3H) at C-3 was inferred from the coupling constant of J2,3 = 4.9 Hz15,16 and the NOE correlation of OCH3-3 to H-10 (δH 6.77, s). The cross peaks between H-12 (δH 3.56, s) and H-1 (δH 5.87, d, J = 10.0 Hz) and H-4α (δH 1.73, ddd, J = 13.4, 10.8, 3.5 Hz) in the NOESY spectrum of 5 indicated H-12, H-1, and H-4α to be in the same orientation. Literature investigation showed that 5 has the same molecular skeleton as plicamine,16 plicane,17 and obliquine,18 where there is a nitrogen atom, instead of an oxygen atom at position C-5 of the basic tazettine nucleus. To our knowledge, compound 5 (N-methyl-5,6-dihydroplicane) is the fourth example of a plicamine-type Amaryllidaceae alkaloid.
Compound 6, a colorless oil, was found to be optically active, [α]25 D +140. The molecular formula of this compound was established as C20H28NO5 by HRESIMS at m/z 362.1965 [M + H]+ (calcd for C20H28NO5 +, 362.1962), in combination with the 1H NMR and 13C NMR data. The 1H NMR spectra showed a N-CH3 signal at δH 2.03 (s, 3H), four O-CH3 signals at δH 3.50 (s, 3H), 3.81 (s, 3H), 3.86 (s, 3H), and 3.88 (s, 3H), an aromatic proton signal at δH 6.85 (s, 1H, H-10), and an olefinic proton signal at 5.51 (m, 1H, H-3). The 13C NMR, DEPT, and HSQC spectra indicated the presence of a N-CH3 (δC 44.4), four O-CH3 (δC 55.8, 56.7, 61.3, 61.6), three methylenes (δC 32.7, C-4; 29.0, C-11; 57.9, C-12), and three methines (δC 98.0, C-6; 67.3, C-4a; 44.8, C-10b). The NMR data (Tables 1 and 2) suggested compound 6 has a similar structure to nerinine,8,19 which was first isolated from the bulbs of Z. candida collected in Japan. The major difference was an additional methoxy group (δH 3.50, s, 3H; δC 55.8) in 6. In the HMBC spectrum of 6, the correlations of the additional methoxy group (δH 3.50, s, 3H) to C-6 (δC 98.0) and H-6 (δH 5.52, s, 1H) to the O-Me (δC 55.8), supported the methoxy group being located at C-6. A series of 2D-NMR experiments including 1H-1H COSY, HSQC, and HMBC allowed assignment of the planar structure of compound 6. The larger coupling constant J1,10b = 9.6 Hz of H-1 (δH 2.79, br d) indicated that H-1 and H-10b are in an anti-relationship, while the smaller coupling constant J4a,10b = 2.0 Hz of H-10b (δH 2.41, dd, J = 9.6, 2.0 Hz) suggested the syn-relationship of H-4a and H-10b. The NOESY correlations of H-4a, H-10b, and OCH3-6 reflected the same orientation. All of the above information suggested compound 6 (O-methylnerinine) had the same configuration as that of nerinine.
Compound 7 was isolated as a gray amorphous powder. The molecular formula was deduced as C19H17NO5 on the basis of the ion peaks at m/z 340.1185 [M + H]+ (calcd for C19H18NO5 +, 340.1180) and 701.2100 [2M + Na]+ (calcd for C38H34N2NaO10 +, 701.2106) in the HRESIMS. The 1H NMR (Table 3) and 13C NMR (Table 4) spectra of 7 resembled those of 11, N-methylcrinasiadine, 20 except that the N-Me in 11 was replaced by a N-ethoxycarbonylethyl in 7. The 1H NMR spectrum showed characteristic signals for O-CH2CH3 at δH 4.16 (q, J = 7.2 Hz, 2H, H-14) and 1.24 (t, J = 7.2 Hz, 3H, H-15), and for N-CH2CH2-at δH 4.67 (t, J = 7.9 Hz, 2H, H-11) and 2.79 (t, J = 7.9 Hz, 2H, H-12). The signals at δC 38.9 (t, C-11), 32.4 (t, C-12), 171.6 (s, C-13), 61.1 (t, C-14), 14.4 (q, C-15) in the 13C NMR and DEPT spectra confirmed the presence of the N-ethoxycarbonylethyl group. In the HMBC spectrum, the correlating peaks between H-11, H-12, and H-14 to C-13 (δC 171.6) further supported the above deduction. In addition, the HMBC correlations of H-11 to C-4a (δC 136.4) and C-6 (δC 160.9) suggested that the N-ethoxycarbonylethyl group was located at N-5. The structure was confirmed by the analysis of the HMBC, HSQC, and 1H-1H COSY spectra. Therefore, compound 7 was assigned to be N-ethoxycarbonylethylcrinasiadine.
Table 3.
1H NMR [δ, mult, (J in Hz)] Data for Compounds 7–10 in CDCl3 (400 MHz)
| position | 7 | 8 | 9 | 10 |
|---|---|---|---|---|
| 1 | 8.10, dd (8.1, 1.1) | 8.08, br d (8.0) | 8.12, br d (8.0) | 8.09, dd (8.0, 1.2) |
| 2 | 7.28, ddd (8.1, 7.0, 1.1) | 7.27, t (7.5) | 7.29, ddd (8.0, 7.0, 1.2) | 7.27, ddd (8.0, 7.1, 1.0) |
| 3 | 7.50, ddd (8.4, 7.0, 1.2) | 7.52, t (7.5) | 7.52, ddd (8.4, 7.0, 1.1) | 7.49, ddd (8.2, 7.1, 1.2) |
| 4 | 7.42, br d (8.4) | 7.58, br d (8.6) | 7.46, br d (8.4) | 7.37, br d (8.2) |
| 7 | 7.87, s | 7.88, s | 7.91, s | 7.89, s |
| 10 | 7.61, s | 7.61, s | 7.63, s | 7.61, s |
| OCH2O | 6.12, s | 6.11, s | 6.12, s | 6.11, s |
| 11 | 4.67, t (7.9) | 4.43, t (7.9) | 4.57, t (8.4) | 4.37, t (8.1) |
| 12 | 2.79, t (7.9) | 2.10, m | 3.06, t (8.4) | 1.66, m |
| 13 | 2.51, t (7.0) | 1.81, m | ||
| 14 | 4.16, q (7.2) | 7.34, m | 1.04, d (6.6) | |
| 15 | 1.24, t (7.2) | 4.15, q (7.1) | 7.38, m | 1.04, d (6.6) |
| 16 | 1.26, t (7.1) | 7.25, m | ||
| 17 | 7.38, m | |||
| 18 | 7.34, m |
Table 4.
13C NMR Data for Compounds 7–10 in CDCl3 (100 MHz)
| position | 7 | 8 | 9 | 10 |
|---|---|---|---|---|
| 1 | 123.6 | 123.3 | 123.5 | 123.4 |
| 2 | 122.7 | 122.5 | 122.5 | 122.3 |
| 3 | 129.3 | 129.3 | 129.2 | 129.1 |
| 4 | 114.8 | 115.5 | 115.0 | 115.2 |
| 4a | 136.4 | 136.7 | 136.6 | 136.8 |
| 6 | 160.9 | 161.0 | 160.8 | 160.8 |
| 6a | 130.8 | 130.8 | 130.7 | 130.7 |
| 7 | 107.1 | 107.1 | 107.1 | 107.1 |
| 8 | 148.7 | 148.6 | 152.5 | 148.6 |
| 9 | 152.6 | 152.5 | 148.7 | 152.4 |
| 10 | 100.7 | 100.6 | 100.7 | 100.6 |
| 10a | 119.7 | 119.6 | 119.7 | 119.7 |
| 10b | 121.3 | 121.3 | 121.5 | 121.5 |
| OCH2O | 102.2 | 102.2 | 102.2 | 102.1 |
| 11 | 38.9 | 42.3 | 44.6 | 41.7 |
| 12 | 32.4 | 22.7 | 33.9 | 36.3 |
| 13 | 171.6 | 31.7 | 138.8 | 26.9 |
| 14 | 61.1 | 173.4 | 129.1 | 22.8 |
| 15 | 14.4 | 60.8 | 128.9 | 22.8 |
| 16 | 14.5 | 126.9 | ||
| 17 | 128.9 | |||
| 18 | 129.1 |
Compound 8 was isolated as a white amorphous powder. Its HRESIMS displayed a quasimolecular ion peak at m/z 354.1340 [M + H]+ (calcd for C20H20NO5 +, 354.1336), corresponding to the molecular formula C20H19NO5. The 1H NMR (Table 3) and 13C NMR (DEPT) (Table 4) spectra of compound 8 exhibited resonances closely related to those of compound 7, except for the appearance of signals for an additional methylene (δH 2.10, m, 2H, H-12; δC 22.7, C-12). The cross peaks of this methylene to H-11 (δH 4.43, t, J = 7.9 Hz, 2H) and H-13 (δH 2.51, t, J = 7.0 Hz, 2H) in the 1H-1H COSY spectrum indicated it to be inserted between C-11 and C-13. This confirmed the presence of a partial structure, -CH2-CH2-CH2-. HMBC correlations of H-12 to C-11 (δC 42.3), C-13 (δC 31.7), and C-14 (173.4), and H-15 (δH 4.15, q, J = 7.1 Hz, 2H) to C-14 suggested the presence of a N-ethoxycarbonylpropyl group. The location of this substituent was concluded from the HMBC cross-peaks of H-11 to C-4a (δC 136.7), C-6 (δC 161.0), C-12, and C-13. The structure of compound 8 was assigned as N-ethoxycarbonylpropylcrinasiadine.
Compound 9 was isolated as a white amorphous powder. Its HRESIMS indicated a molecular formula of C22H17NO3, based on the ion peaks at m/z 344.1282 [M + H]+ (calcd for C22H18NO3 +, 344.1281) and 687.2474 [2M + H]+ (calcd for C44H35N2O6 +, 687.2490). Comparison of their NMR data (Tables 3 and 4) showed that compounds 9 and 7 have the same parent ring system. However, signals for a mono-substituted benzene ring (δH 7.34, m, 2H; 7.38, m, 2H; 7.25, m, 1H; δC 138.8, 129.1, 128.9, 126.9) were apparent in compound 9, but signals for the ethoxycarbonyl group were absent in compound 7. The position of the substituent of the mono-substituted benzene ring was determined from the cross-peaks of H-12 (δH 3.06, t, J = 8.4 Hz) to C-13 (δC 138.8) and C-14 (δC 129.1) in the HMBC spectrum. HMBC correlations of H-11 (δH 4.57, t, J = 8.4 Hz, 2H) with C-4a (δC 136.6), C-6 (δC 160.8), and C-12 (δC 33.9), together with the chemical shifts of H-11 and C-11 suggested the phenethyl is located at position N-5. Accordingly, the structure of 9 was assigned as N-phenylethylcrinasiadine.
The molecular formula of compound 10 was determined as C19H19NO3 by HRESIMS analysis [m/z 310.1434 [M + H]+ (calcd for C19H20NO3 +, 310.1438), 619.2790 [2M + H]+ (calcd for C38H39N2O6 +, 619.2803)]. The 1H NMR, 13C NMR, and DEPT spectra of 10 exhibited signals for two methyls (δH 1.04, d, J = 6.6 Hz, 6H; δC 22.8, C-14/15), two methylenes (δH 1.66, m, 2H, δC 36.3, C-12; δH 4.37, 2H, t, J = 8.1 Hz, δC 41.7, C-11), and a methine (δH 1.81, m, 1H; δC 26.9, C-13). It was concluded from the 1H-1H COSY, HSQC, and HMBC spectra of 10 that the above signals could be attributed to an isopentyl group. In consideration of the chemical shifts of H-11 and C-11, the isopentyl group could be connected to a nitrogen atom. Compound 10 had similar NMR spectra (Tables 3 and 4) compared with compound 11, except that the signals for N-CH3 in 11 were absent and replaced by signals for N-isopentyl group in 10. The cross peaks of H-11 to C-4a (δC 136.8) and C-6 (δC 160.8) in the HMBC spectrum of 10 indicated that the isopentyl group was located at N-5. Detailed 1H-1H COSY, HSQC, and HMBC spectroscopic analysis was used to assign compound 10 as N-isopentylcrinasiadine.
Compounds 2–4, 11–15 were identified as haemanthamin (2),5 3-epimacronine (3),21 (+)-tazettine (4),14 N-methylcrinasiadine (11),20 trisphaeridine (12),22 5,6-dihydrobicolorine (13),21 lycorine (14),23 and nigragillin (15),24 respectively, on the basis of spectroscopic data analysis and comparison with those reported in the literature. The known amaryllidaceous alkaloids 3, 11, and 13 were isolated from Z. candida for the first time. Compound 15 was originally isolated from the fungus Aspergillus niger,24 and is reported from a plant for the first time, although it is possibly biosynthesized by an endophytic fungus of Z. candida. For compounds 7 and 8, it cann’t be ruled out that they are artifacts of isolation, resulting from esterification of the corresponding carboxylic acids, which presently are unknown.
The relative configuration of 3-epimacronine (3) has been confirmed by single-crystal X-ray diffraction analysis irradiated by MoKα at 173 K,25 but the absolute configuration of 3-epimacronine is still unknown. In order to determine its absolute configuration, compound 3 was recrystallized in MeOH containing a small amount of H2O, and the crystal was subjected to single-crystal X-ray diffraction analysis on a Bruker APEX-II diffractometer equipped with graphite-monochromatized CuKα radiation (λ = 1.54178 Å) at 100 (2) K. The results indicated that the molecule crystallized in the enantiomorphic P21 space group (No. 4), and should have optical properties. The Flack parameter, associated with the absolute configuration, was calculated to be −0.01 (12), which confirmed the absolute configuration of compound 3. The crystal structure of compound 3 (3-epimacronine) is shown in Figure 2, and its absolute configuration was determined as 3S, 10bS, 5S, 11R.
Figure 2.

X-ray ORTEP drawing of compound 3 with ellipsoid probability of 30% by CuKα radiation at 100 (2) K.
Since Amaryllidaceae alkaloids are reported to exhibit cytotoxic properties,1,26–33 compounds 1–15 were evaluated for their cytotoxic activities against five human cancer cell lines, namely HL-60 and K562 human myeloid leukemia, A549 lung cancer, HepG2 hepatocellular carcinoma, and HT-29 colon cancer, as well as the immortalized non-cancerous Beas-2B human bronchial epithelial cell line. The results (Supporting Information, Table S1) showed compounds 1, 2, 9, and 14 exhibited more potent cytotoxicity than one of the positive controls (cis-platin), while compounds 3–8, 10–13, and 15 did not show obvious cytotoxicity (IC50 >10 μM). The known compound 2 (haemanthamin) strongly inhibited the proliferation of five cancer cells with IC50 values of 1.4, 2.5, 2.5, 4.8, and 2.1 μM, respectively, which are in agreement with those reported in the literature.26,29 Compound 2 has been reported to be cytostatic, not cytotoxic, and its antiproliferative effects result from its complex formation with RNA.26 The new compound 1 showed more potent cytotoxicity than compound 2 against the same cell lines with IC50 values of 0.91, 1.0, 1.1, 1.5 and 1.2 μM, respectively. The mechanism by which compound 1 exhibits cytotoxicity is likely similar to that of 2, due to their structural similarity. The new compound 9 showed the strongest cytotoxicity against HL-60, K562, A549, HepG2, and HT-29 cells with IC50 values of 0.81, 0.70, 13, 1.4, and 2.3 μM, respectively. Compound 14 (lycorine) strongly inhibited cancer cell proliferation with IC50 values of 1.6, 2.3, 1.9, 3.7, and 3.2 μM, respectively, which was consistent with previous reports,29,30 and it was reported that effects of lycorine (14) on HL-60 cells was via arresting cell cycle and inducing apoptosis,31 due to the down-regulation of Mcl-1 in human leukemia cells.32 Interestingly, compounds 1, 2, 7–9, and 14 exhibited higher IC50 values against the non-cancerous Beas-2B cell line (3.7, 5.0, > 40, 36, 7.3, and 4.9 μM, respectively) than those against the five cancer cell lines, showing these compounds are selective for the cancer cell lines used (see Supporting Information, Table S1). Therefore, understanding the molecular and cellular mechanism of these compounds in affecting cell proliferation or possible apoptosis may be relevant to the design of therapeutic strategies.
EXPERIMENTAL SECTION
General Experimental Procedures
Melting points (uncorrected) were determined on a Beijing Tech X-5 microscopic melting point apparatus. Optical rotations were determined in CHCl3 or MeOH, as indicated, on a Perkin-Elmer 341 polarimeter. The CD spectra were obtained on a JASCO J-810 spectrometer. UV and FT-IR spectra were determined using Varian Cary 50 and Bruker Vertex 70 instrument, respectively. NMR spectra were recorded on a Bruker AM-400 spectrometer, and the 1H and 13C NMR chemical shifts were referenced to the solvent peaks for CDCl3 at δH 7.24 and δC 77.23 or for CD3OD at δH 3.31 and δC 49.15. Electrospray time-of-flight mass spectra (ESI-TOF-MS) (accurate mass) were measured in the positive-ion mode on an Agilent MSD-TOF mass spectrometer, while low-resolution and multidimensional electrospray ion trap mass spectra (ESIMS and ESIMS/MS) were measured in the positive-ion mode on a Thermo Finnigan LCQ Deca XP Max mass spectrometer. The crystallographic data were obtained on a Bruker SMART CCD area-detector diffractometer equipped with graphite-monochromatized MoKα radiation (λ = 0.71073 Å), and a Bruker SMART APEX-II CCD diffractometer equipped with graphite-monochromatized CuKα radiation (λ = 1.54178 Å). HPLC was conducted on an Agilent 1200 instrument with detection at 210 nm using a C18 column (5 μm, 250 × 10 mm, YMC-pack ODS-A) and MeOH–H2O as the mobile phase at 2.0 mL/min flow rate. Column chromatography (CC) was carried out on silica gel (100–200 mesh, 200–300 mesh, and 400 mesh, Qingdao Haiyang Chemical Industry Co. Ltd, P. R. China), RP-C18 silica gel (ODS-A-HG, YMC Co. Ltd, Japan), and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Sweden). Thin-layer chromatography (TLC) was performed with silica gel 60 F254 (Yantai Chemical Industry Research Institute, P. R. China) and RP-C18 F254 plates (Merck, Germany). MPLC was carried out with a EZ Purifier III chromatography system.
Plant Material
The whole plants of Zephyranthes candida were collected at Shiyan, Hubei Province, People’s Republic of China, in June, 2010. The plant material was identified by Professor Changgong Zhang of the School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology. A voucher specimen (No. 20100601) has been deposited at the Hubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology.
Extraction and Isolation
The whole dried plants of Z. candida (10 kg) were extracted four times with 25 L each of 95% aqueous EtOH with 2% HCl at room temperature. The filtrates were combined and concentrated under vacuum to afford 1150 g of crude extract, which was then partitioned between CHCl3 and 2% aqueous HCl (3 L each), followed by re-extracting the aqueous phase three additional times with CHCl3 (3 L). After the aqueous phase was adjusted to pH 7 with NH4OH, it was partitioned between CHCl3 (4 × 1.5 L) for the second time. On evaporation, the CHCl3 phase (18 g) was chromatographed over silica gel by MPLC and eluted with a MeOH–CHCl3 gradient to give five fractions, A–E. Fraction A was separated into three subfractions, A1, A2, and A3, by CC on Sephadex LH–20 eluting with MeOH-CH2Cl2 (50:50). Subfraction A2 was subjected to CC on silica gel using a gradient system of cyclohexane–CHCl3 (33:67) providing two fractions, A2A and A2B. Fraction A2B was further separated by semi-preparative HPLC (70:30 MeOH-H2O mobile phase) to yield compound 11 (5.7 mg, tR 38.4 min). Subfraction A3 was subjected to CC on silica gel using a gradient system of cyclohexane–CHCl3 (25:75) providing two fractions, A3A and A3B. A3B was further separated by semi-preparative HPLC (80:20 MeOH-H2O) to yield compounds 7 (2.0 mg, tR 19.2 min), 8 (3.8 mg, tR 24.5 min), 9 (2.3 mg, tR 45.6 min), and 10 (2.4 mg, tR 48.0 min). Similar to fraction A, fraction B was separated into three subfractions, B1, B2, and B3. B2 was subjected to separation on RP-C18 using a gradient system of MeOH–H2O (50:50–100:0) to provide three subfractions, B2A, B2B, and B2C. Compound 3 (8.6 mg, mp 132 °C) was crystallized from subfraction B2B. Fraction B3 was purified by semi-preparative HPLC (80:20 MeOH-H2O) to yield compounds 13 (6.4 mg, tR 15.3 min) and 12 (5.9 mg, tR 19.4 min). Fraction C was separated into two subfractions (C1 and C2) by Sephadex LH–20 using MeOH-CH2Cl2 (50:50). Subfraction C2 was subjected to separation on RP-C18 using a gradient system of MeOH–H2O (20:80–100:0) to provide three further subfractions, C2A, C2B, and C2C. Fraction C2B was subjected to separation over silica gel using a gradient system of MeOH–CHCl3 (10:90–100:0), to provide three subfractions, C2BA, C2BB, and C2BC. Compounds 5 (16.2 mg, tR 15.9 min), 6 (7.6 mg, tR 36.1 min), and 15 (2.1 mg, tR 14.5 min) were isolated from two subfractions, C2BB and C2BC, using semi-preparative HPLC (70:30 MeOH-H2O), and compound 4 (34.9 mg, mp 210 °C) was crystallized from fraction C2C. Fraction D was separated into two subfractions, D1 and D2, by passage over Sephadex LH–20. Subfraction D2 was subjected to purification on RP-C18 using stepwise gradient elution with MeOH–H2O, to furnish two subfractions, D2A and D2B. Granules of compound 2 (15.7 mg) appeared in fraction D2B. Fraction E was separated on a RP–C18 column using a MeOH-H2O gradient to yield three fractions, E1, E2, and E3. Compound 14 (5.4 mg) was afforded from fraction E2, and compound 1 (63.8 mg, mp 237 °C) was crystallized from fraction E3.
N-Methylhaemanthidine chloride (1)
Colorless cubes; mp 237 °C; [α]25D +5 (c 0.75, CH3OH); UV (MeOH) λmax (log ε) 210 (4.27), 245 (3.47), 292 (3.52) nm ; CD (MeOH) 209 ([θ], +18842), 250 ([θ], −4686), 289 ([θ], +5931) nm; IR (KBr) νmax 3239, 3003, 1509, 1489, 1252, 1132, 1030, 937, 825 cm−1; 1H NMR data, see Table 1; 13C NMR data, see Table 2; (+)-HRESIMS m/z 332.1488 [M − Cl]+ (calcd for C18H22NO5+, 332.1493).
N-Methyl-5,6-dihydroplicane (5)
Yellow amorphous powder; [α]25D +100 (c 0.063, CH3OH); UV (MeOH) λmax (log ε) 210 (4.31), 245 (3.70), 292 (3.51) nm; IR (KBr) νmax 2935, 1607, 1489, 1341, 1048, 1025 cm−1; 1H NMR data, see Table 1; 13C NMR data, see Table 2; HRESIMS m/z 343.1688 [M + H]+ (calcd for C19H23N2O4 +, 343.1652), 685.3244 [2M + H]+ (calcd for C38H45N4O8 +, 685.3232), 707.3064 [2M + Na]+ (calcd for C38H44N4NaO8 +, 707.3051).
O-Methylnerinine (6)
Colorless oil; [α]25D + 140 (c 0.22, CH3OH); UV (MeOH) λmax (log ε) 214 (4.42), 240 (3.90), 284 (3.51) nm; IR νmax 2939, 2909, 1600, 1493, 1460, 1337, 1097, 1051, 1022 cm−1; 1H NMR data, see Table 1; 13C NMR data, see Table 2; HRESIMS m/z 362.1965 [M + H]+ (calcd for C20H28NO5 +, 362.1962).
N-Ethoxycarbonylethylcrinasiadine (7)
Gray amorphous powder; UV (CHCl3) λmax (log ε) 240 (5.72), 311(4.66), 339 (4.62), 368 (4.26), 371 (4.31), 665 (3.66) nm; IR (KBr) νmax 2922, 2853, 1730, 1644, 1627, 1601, 1510, 1484, 1462, 1313, 1037, 751 cm−1; 1H NMR data, see Table 3; 13C NMR data, see Table 4; HRESIMS m/z 340.1185 [M + H]+ (calcd for C19H18NO5 +, 340.1180), 701.2100 [2M + H]+ (calcd for C38H34N2NaO10 +, 701.2106).
N-Ethoxycarbonylpropylcrinasiadine (8)
White amorphous powder; UV (CHCl3) λmax (log ε) 252 (4.58), 257 (4.52), 270 (4.15), 308 (3.14), 318 (3.99), 341 (3.83), 371 (3.17), 668 (2.22) nm; IR (KBr) νmax 2976, 2926, 1892, 1729, 1647, 1626, 1601, 1461, 1199, 1176,1036, 753 cm−1; 1H NMR, see Table 3; 13C NMR, see Table 4; HRESIMS m/z 354.1340 [M + H]+ (calcd for C20H20NO5 +, 354.1336), 729.2395 [2M + Na]+ (calcd for C40H38N2NaO10+, 729.2419).
N-Phenylethylcrinasiadine (9)
White amorphous powder; UV (CHCl3) λmax (log ε) 270 (4.08), 313 (3.91), 341 (3.76), 372 (3.13) nm; IR (KBr) νmax 2957, 2924, 2855, 1747, 1631, 1600, 1462, 1310, 1039, 751 cm−1; 1H NMR, see Table 3; 13C NMR, see Table 4; HRESIMS m/z 344.1282 [M + H]+ (calcd for C22H18NO3 +, 344.1281), 687.2474 [2M + H]+ (calcd for C44H35N2O6 +, 687.2490).
N-Isopentylcrinasiadine (10)
Colorless solid; UV (CHCl3) λmax (log ε) 270 (3.96), 312 (3.80), 341 (3.68), 371 (3.30) nm; IR (KBr) νmax 2924, 2856, 1720, 1639, 1601, 1459, 1312, 1035, 749 cm−1; 1H NMR, see Table 3; 13C NMR, see Table 4; HRESIMS m/z 310.1434 [M + H]+ (calcd for C19H20NO3 +, 310.1438), 619.2790 [2M + H]+ (calcd for C38H39N2O6 +, 619.2803).
Crystallographic Data and X-ray Structure Analysis of 1
A suitable colorless crystal of 1 was obtained by slow evaporation from MeOH at room temperature. Diffraction intensity data were obtained on a Bruker SMART CCD area-detector diffractometer equipped with graphite-monochromatized MoKα radiation (λ = 0.71073 Å) at 298(2) K. Collection was conducted using the multirun procedure in the Bruker SMART software. Data reduction was subsequently performed with Bruker SAINT. Structure solution and refinement were performed with the SHELXTL program package. The final structure model obtained for 1 is shown in Figure 1. Crystal data and experimental details: C18H22ClNO5, formula weight 367.82, crystal size 0.20 × 0.20 × 0.20 mm3, crystal system tetragonal, space group P4 (1), a = 10.0493(12) Å, b = 10.0493(12) Å, c = 17.167(4) Å, α = β = γ = 90°, V = 1733.7(5) Å3, Z = 4, Dc 1.409 Mg/m3, F(000) = 776, absorption coefficient 0.249 mm−1. A total of 11307 reflections was collected in the range 2.03° < θ < 25.99°, with 3406 independent reflections [R(int) = 0.1008], completeness to θmax was 100.0 %; no absorption correction applied; full-matrix least-squares refinement on F2; the number of data/restraints/parameters were 3406/3/241; goodness-of-fit on F2 = 1.065; Final R indices [I > 2σ (I)] R1 = 0.0745, wR2 = 0.1339; R indices (all data) R1 = 0.1170, wR2 = 0.1496; absolute structure parameter −0.04 (12); largest difference peak and hole 0.227 and −0.179 e.Å−3. Crystallographic data for the structure of 1 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC 94570. Copies of these data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk).
Crystallographic Data and Single-Crystal X-ray Absolute Configuration Analysis of 3
A suitable light-yellow crystal of 3 was obtained by slow evaporation from MeOH at room temperature. Diffraction intensity data were obtained on a Bruker APEX-II diffractometer equipped with a graphite-monochromatized CuKα radiation (λ = 1.54178 Å) at 100 (2) K. Collection was conducted using the multirun procedure in the Bruker APEX-II software. Data reduction was subsequently performed with Bruker SAINT. Structure solution and refinement were performed with the SHELXTL program package. The final structure model obtained for 3 is shown in Figure 2. Crystal data and experimental details: empirical formula C18H19NO5·0.13H2O, formula weight 331.68, crystal size 0.80 × 0.60 × 0.25 mm3, crystal system monoclinic, space group P21, a = 8.1308 (2) Å, b = 17.5736 (3) Å, c = 11.0262 (2) Å, α = 90°, β = 94.9400 (10)°, γ = 90°, V = 1569.65 (5) (2) Å3, Z = 4, Dc 1.404 Mg/m3, F(000) = 716, absorption coefficient 0.857 mm−1. A total of 13397 reflections was collected in the range 4.02° < θ < 69.46°, with 5191 independent reflections [R(int) = 0.0274], completeness to θmax was 95.2 %; semi-empirical from equivalents absorption correction with SADABS applied; full-matrix least-squares refinement on F2; the number of data/restraints/parameters were 5191/1/447; goodness-of-fit on F2 = 1.047; final R indices [I > 2σ (I)] R1 = 0.0282, wR2 = 0.0732; R indices (all data) R1 = 0.0282, wR2 = 0.0732; absolute structure parameter 0.01 (10); largest difference peak and hole 0.238 and −0.191 e.Å−3. Crystallographic data for the structure of 3 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication CCDC 94569. Copies of these data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk).
Cytotoxicity Assays
Five human cancer cell lines were used, namely, HL-60 and K562 human myeloid leukemias, A549 lung cancer, HepG2 hepatocellular carcinoma, and HT-29 colon cancer, together with one non-cancerous cell line, Beas-2B human bronchial epithelial. Cells were cultured in RPMI-1640 or in DMEM medium (Hyclone, Logan, UT, USA), supplemented with 10% fetal bovine serum (Hyclone) in 5% CO2 at 37 °C. The cytotoxicity assay was performed using a MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) method in 96-well microplates, as reported previously, with slight modification.34,35 Briefly, 100 μL of adherent cells were seeded into each well of the 96-well culture plates and allowed to adhere for 12 h before adding the test compounds, while suspended cells were seeded into wells at a density of 1 × 105 cells/mL just prior to the addition of the test compounds. Each tumor cell line was exposed to the test compound at concentrations of 0.0625, 0.32, 1.6, 8, and 40 μM in triplicates for 48 h. Wells with DMSO were used as negative controls, and DDP (cis-platin, Sigma, St. Louis, MO, USA) was used as a positive control. After compound treatment, cell viability was detected by a Bio-Rad 680 at λ = 595 nm and a cell growth curve was graphed. IC50 values were calculated by the method described by Reed and Muench.36
Supplementary Material
Acknowledgments
We are grateful to Prof. C.-G. Zhang at Huazhong University of Science and Technology for the authentification of this plant material, and Dr. X.-G. Meng at Central China Normal University and Dr. X.-N. Li at Kunming Institute of Botany, Chinese Academy of Science, for the single-crystal X-ray data collection and analysis. This work was supported financially by the National Natural Science Foundation of China (No. 31170323, to G. Yao), Scientific Research Foundation for the Returned Oversea Chinese Scholars, State Education Ministry of China (2010-1561-40th, to G. Yao), Wuhan Youth Chenguang Program of Science and Technology, China (No. 201271031389, to G. Yao), Program for Youth Chutian Scholar of Hubei Province of China (to G. Yao), Program for New Century Excellent Talents in University, State Education Ministry of China (No. NCET-2008-0224, to Y. Zhang), Program of Recruited Top Talent of Sciences and Technology of Yunnan Province (No. 2009c1120, to Y. Li), and U.S. National Institutes of Health grant (No. P20 RR-016467, to F. D. Horgen).
Footnotes
The authors declare no competing financial interest.
Supporting Information Available. (+)-HR-ESIMS, 1D and 2D NMR spectra for compounds 1, 5–10, CD for compound 1, crystal packing of compounds 1 and 3, X-ray crystallographic data (CIF files) for compounds 1 and 3, as well as cytotoxicity data of compounds 1–15 against five cancer cell lines and one non-cancerous human Beas-2B cell line. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Jin Z. Nat Prod Rep. 2009;26:363–381. doi: 10.1039/b718044f. [DOI] [PubMed] [Google Scholar]; (b) Jin Z. Nat Prod Rep. 2011;28:1126–1142. doi: 10.1039/c0np00073f. [DOI] [PubMed] [Google Scholar]
- 2.State Administration of Traditional Chinese Medicine. Zhonghuabencao. Shanghai Science and Technology Publishing Company; Shanghai: 1999. pp. 7268–7268. [Google Scholar]
- 3.Wu ZP, Chen Y, Xia B, Wang M, Dong YF, Feng X. Lipids. 2009;44:63–70. doi: 10.1007/s11745-008-3246-6. [DOI] [PubMed] [Google Scholar]
- 4.Wu ZP, Chen Y, Feng X, Xia B, Wang M, Dong YF. Chem Nat Compd. 2010;46:187–191. [Google Scholar]
- 5.Yang JS, Feng X, Chen Y, Zhao XZ, Wang M. J Chin Med Mater. 2010;33:1730–1732. [PubMed] [Google Scholar]
- 6.Boit H, Horst E. Chem Ber. 1955;88:1590–1594. [Google Scholar]
- 7.Ozeki S. Chem Pharm Bull. 1964;12:253–254. [PubMed] [Google Scholar]
- 8.Ozeki S. Yakugaku Zasshi. 1964;84:1194–1197. [PubMed] [Google Scholar]
- 9.Pettit GR, Cragg GM, Singh SB, Duke JA, Doubek DL. J Nat Prod. 1990;53:176–178. doi: 10.1021/np50067a026. [DOI] [PubMed] [Google Scholar]
- 10.Abdel-Halim OB, Morikawa T, Ando S, Matsuda H, Yoshikawa M. J Nat Prod. 2004;67:1119–1124. doi: 10.1021/np030529k. [DOI] [PubMed] [Google Scholar]
- 11.Hohmann J, Forgo P, Molnar J, Wolfard K, Molnar A, Thalhammer T, Mathe I, Sharples D. Planta Med. 2002;68:454–457. doi: 10.1055/s-2002-32068. [DOI] [PubMed] [Google Scholar]
- 12.Karle J, Estlin JA, Karle IL. J Am Chem Soc. 1967;89:6510–6515. [Google Scholar]
- 13.Flack HD. Acta Crystallogr Sect A. 1983;39:876–881. [Google Scholar]
- 14.Ling YQ, Feng X, Zhao XZ, Chen Y, Dong YF, Wang M. Nat Prod Res Dev. 2010;22:241–244. [Google Scholar]
- 15.Haugwitz RD, Jeffs PW, Wenkert E. J Chem Soc. 1965:2001–2009. [Google Scholar]
- 16.Ünver N, Gözler T, Walch N, Gözler B, Hesse M. Phytochemistry. 1999;50:1255–1261. [Google Scholar]
- 17.Unver N, Noyan S, Gözler B, Gözler T, Werner C, Hesse M. Heterocycles. 2001;55:641–652. [Google Scholar]
- 18.Brine ND, Campbell WE, Bastida J, Herrera MR, Viladomat F, Codina C, Smith PJ. Phytochemistry. 2002;61:443–447. doi: 10.1016/s0031-9422(02)00206-6. [DOI] [PubMed] [Google Scholar]
- 19.Ozeki S. Yakugaku Zasshi. 1965;85:699–702. [PubMed] [Google Scholar]
- 20.Suau R, Gómez AI, Rico R. Phytochemistry. 1990;29:1710–1712. [Google Scholar]
- 21.Viladomat F, Bastida J, Tribo G, Codina C, Rubiralta M. Phytochemistry. 1990;29:1307–1310. [Google Scholar]
- 22.Ghosal S, Saini KS, Razdan S. Phytochemistry. 1985;24:2141–2156. [Google Scholar]
- 23.Evidente A, Cicala MR, Giudicianni I. Phytochemistry. 1983;22:581–584. [Google Scholar]
- 24.Isogai A, Horii T, Suzuki A, Murakoshi S, Ikeda K, Sato S, Tamura S. Agric Biol Chem. 1975;39:739–740. [Google Scholar]
- 25.Linden A, Akineri G, Noyan S, Gözler T, Hesse M. Acta Crystallogr Sect C. 1998;54:1653–1659. [Google Scholar]
- 26.Van Goietsenoven G, Andolfi A, Lallemand B, Cimmino A, Lamoral-Theys D, Gras T, Abou-Donia A, Dubois J, Lefranc F, Mathieu V, Kornienko A, Kiss R, Evidente A. J Nat Prod. 2010;73:1223–1227. doi: 10.1021/np9008255. [DOI] [PubMed] [Google Scholar]
- 27.Zupkó I, Réthy B, Hohmann J, Molnár J, Ocsovszki I, Falkay G. In Vivo. 2009;23:41–48. [PubMed] [Google Scholar]
- 28.Evidente A, Kireev AS, Jenkins AR, Romero AE, Steelant WF, Van Slambrouck S, Kornienko A. Planta Med. 2009;75:501–507. doi: 10.1055/s-0029-1185340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hohmann J, Forgo P, Molnár J, Wolfard K, Molnár A, Thalhammer T, Máthé I, Sharples D. Planta Med. 2002;68:454–457. doi: 10.1055/s-2002-32068. [DOI] [PubMed] [Google Scholar]
- 30.Lamoral-Theys D, Andolfi A, Van Goietsenoven G, Cimmino A, Le Calvé B, Wauthoz N, Mégalizzi V, Gras T, Bruyère C, Dubois J, Mathieu V, Kornienko A, Kiss R, Evidente A. J Med Chem. 2009;52:6244–6256. doi: 10.1021/jm901031h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Hu WX, He LF, Ye M, Li Y. FEBS Lett. 2004;578:245–250. doi: 10.1016/j.febslet.2004.10.095. [DOI] [PubMed] [Google Scholar]
- 32.Liu XS, Jiang J, Jiao XY, Wu YE, Lin JH, Cai YM. Cancer Lett. 2009;274:16–24. doi: 10.1016/j.canlet.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 33.Weniger B, Italiano L, Beck JP, Bastida J, Bergonon S, Codina C, Lobstein A, Anton R. Planta Med. 1995;61:77–79. doi: 10.1055/s-2006-958007. [DOI] [PubMed] [Google Scholar]
- 34.Mosmann T. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 35.Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemarker RH, Boyd MR. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- 36.Reed LJ, Muench H. Am J Hyg. 1938;27:493–497. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.