

Abstract
Abundant neurochemical, neuropathological, and genetic evidence suggests that a critical number of proinflammatory and innate immune system-associated factors are involved in the underlying pathological pathways that drive the sporadic Alzheimer's disease (AD) process. Most recently, a series of epigenetic factors - including a select family of inducible, proinflammatory, NF-κB-regulated small noncoding RNAs called miRNAs - have been shown to be significantly elevated in abundance in AD brain. These upregulated miRNAs appear to be instrumental in reshaping the human brain transcriptome. This reorganization of mRNA speciation and complexity in turn drives proinflammatory and pathogenic gene expression programs. The ensuing, progressively altered immune and inflammatory signaling patterns in AD brain support immunopathogenetic events and proinflammatory features of the AD phenotype. This report will briefly review what is known concerning NF-κB-inducible miRNAs that are significantly upregulated in AD-targeted anatomical regions of degenerating human brain cells and tissues. Quenching of NF-κB-sensitive inflammatory miRNA signaling using NF-κB-inhibitors such as the polyphenolic resveratrol analog trans-3,5,4'-trihydroxystilbene (CAY10512) may have some therapeutic value in reducing inflammatory neurodegeneration. Antagonism of NF-κB-inducing, and hence proinflammatory, epigenetic and environmental factors, such as the neurotrophic herpes simplex virus-1 and exposure to the potent neurotoxin aluminum, are briefly discussed. Early reports further indicate that miRNA neutralization employing anti-miRNA (antagomir) strategies may hold future promise in the clinical management of this insidious neurological disorder and expanding healthcare concern.
Introduction
Although intensively studied for well over 100 years, the biological factors that initiate and drive the Alzheimer's disease (AD) process remain incompletely understood [1-3]. Anti-AD therapies directed solely against amyloid beta (Aβ) peptides have generally proved extremely disappointing, although therapeutic strategies targeted against multiple AD biomarkers - such as amyloid and tau abundance and processing dysfunction and neuroinflammation - have more recently shown greater promise [1-4].
As one recent example, the experimental drug posiphen, a chirally pure positive enantiomer of phenserine and β-amyloid precursor protein (βAPP) synthesis inhibitor, has shown a significantly improved efficacy against multiple AD-relevant targets, at least in proof-of-principal phase I testing [4]. Interestingly, this drug has been shown not only to attenuate Aβ42 peptide levels but also to lower the inflammatory biomarkers complement factor C3 and monocyte chemotactic protein in the cerebrospinal fluid of patients suffering from mild cognitive impairment [4]. Indeed, significant increases in inflammatory biomarkers such as cytokines, chemokines, complement factors, chemotactic proteins and C-reactive protein, mitochondrial-mediated upregulation of reactive oxygen species (ROS), and the proinflammatory actions of Aβ peptides have long been thought to be involved in a brain-specific inflammatory process as AD initiates and progresses throughout the limbic system of the brain [4-13].
One neurogenetic consequence of increased inflammatory signaling in AD brain is the upregulation of the inducible, proinflammatory transcription factor NF-κB, and NF-κB-driven miRNA expression; hence a self-sustaining, self-reinforcing proinflammatory signaling loop is generated [2,3,7-18]. Whether some of these proinflammatory signaling systems are neuroprotective or beneficial to homeostatic brain cell structure and function remains to be clarified [5-9]. Extrinsic and environmental factors such as herpes simplex virus-1 (HSV-1) infection and aluminum exposure from the environment, as two exceptionally strong inducers of NF-κB and proinflammatory miRNA upregulation, are considered potential contributors to the development of AD pathology. Major points regarding the potential pathogenic role for each of these factors and processes are further discussed in the following sections.
Inflammation and Alzheimer's disease
Inflammation constitutes an intrinsic, physiological defense mechanism aimed at protecting healthy tissues from infection, injury and trauma. As such, inflammation represents an essential, evolutionarily ancient process that normally ceases to function once the physiological insult has been eliminated, and cellular homeostasis has been restored [1-12]. On the contrary, chronic or sustained inflammatory signaling contributes to dys-homeostasis, culminating in progressive cellular damage as is observed in many pathological and progressive degenerative conditions ranging from cancer to AD [4,11-18].
In the central nervous system (CNS), macrophages and glial cells - as the primary immune cells in the brain's privileged immune compartment - function primarily, by a variety of phagocytic and digestive mechanisms, to promote host defense by maintaining tissue homeostasis through the destruction of invading pathogens, through sequestering and eliminating deleterious debris via the cytoplasmic multi-protein inflammasome complex, and by promoting tissue repair [12-39]. On the contrary, sustained, uncontrolled activation of brain macrophages and glial cells can lead to excess production of various pathogenic factors that contribute to neuronal injury, including the significant and dramatic upregulation of proinflammatory chemokines, cytokines and ROS. These in turn are capable of activating inflammatory transcription factors such as NF-κB and proinflammatory gene expression programs that drive cellular fate towards CNS dys-homeostasis, compromised neuronal function and, ultimately, apoptosis and brain cell death [2,3,38-48].
A strong association between inflammation and AD has been suggested for almost 50 years, and to date at least 2,750 peer-reviewed papers have appeared on the contribution of inflammation to the AD process [11-14]. Some of these inflammatory processes may be necessary in an attempt to regain brain cell homeostasis in early AD, but the integration of these processes into AD proliferation and the progression to late-stage AD is not well understood [15-18]. Over the last year there have been at least half-a-dozen excellent reviews on this area of research on the AD-inflammation connection so this topic will not be covered in depth here [5,15-20].
Briefly, AD is characterized neuropathologically by at least five heterogeneous features, all of which support the progressive generation of abnormal tau and amyloid, neural and synaptic deficits and proinflammatory signaling to various degrees. These features include: the appearance of hyperphosphorylated tau-protein containing intracellular neurofibrillary tangles; amyloidogenesis - the progressive, age-related generation, aggregation and accumulation of Aβ peptides into dense, insoluble, proinflammatory and pathogenic deposits of senile plaque; reduced synaptic densities and synaptic protein assemblies; significant neuronal loss in the temporal lobe and hippocampal regions that, as AD progresses, radiates into the more distal parietal, frontal and occipital poles of the brain; and a unique, chronic and progressive smoldering inflammation of the neocortex and limbic system of the brain, especially in the middle to late stages of AD [11-18,20-25].
Until recently, the density of neurofibrillary tangle and senile plaque lesions required extensive postmortem histopathological confirmation for an accurate diagnosis of AD; however, current autoradiographic, nuclear magnetic resonance, tomographic and related electronic digitization and quantification technologies are capable of non-invasively and effectively resolving these insoluble lesions in the aging brains of patients with AD, and in transgenic animal models of AD (Tg-AD) [24-31]. Indeed, the initial aberrant phosphorylation of tau, the generation of Aβ peptides, the progressive aggregation from soluble Aβ peptide monomers into higher-order structures, and ultimately into insoluble deposits, and their unusual protease-resistant biophysical properties have been widely suggested to be the most significant markers for early cognitive disturbances, mild cognitive impairment and early AD onset [12-18,24-32]. These markers may typically precede, by decades, the appearance of fully mature senile plaque and tangle lesions in the AD brain [28-32]. Aβ40 and Aβ42 peptides themselves, and innate immune system interaction and attack of the mature senile plaque and tangle lesions mediated by CNS macrophages and microglia, may represent one of the earliest manifestations of increased immune system activation and inflammatory signaling in AD, and of the ensuing upregulation of chemokines, cytokines IL-1β and TNFα and others, chemotactic proteins and complement factor proteins such as complement factor H (CFH) [7-10,32-38]. That Aβ40 and Aβ42 peptides directly activate microglia and monocytes to progressively generate these endogenous neurotoxins may signify that Aβ peptides or Aβ peptide-containing lesions may be critical for the initial seeding of inflammatory neurodegeneration, as is observed in the AD-affected brain and in amyloid-overexpressing Tg-AD models [33-39].
Proinflammatory transcription factor NF-κB
As previously indicated, neuroinflammatory processes appear, against a background of brain aging, to significantly contribute to a cascade of deleterious events that culminates in progressive synaptic loss and neuronal signaling dysfunction - pathogenic events that critically underlie a number of inflammatory neurodegenerative disorders including AD, age-related macular degeneration (a common AD-like inflammatory degeneration of the human retina) and also human prion disease [16,38-48].
During upregulation of inflammatory processes in the CNS and retina there appears to be a significant parallel upregulation of the dimeric DNA-binding protein NF-κB (as the p50/p65 complex) [40-48]. Indeed, originally described in 1986, NF-κB has emerged as a ubiquitous transcription factor that controls diverse biological functions including inflammatory and immune functions in both the central and peripheral nervous systems [40-45]. NF-κB may be singularly important in regulating genetic responses to nervous system stress through the innate immune response because it belongs to the category of pre-existing primary transcription factors that are already present in cells in an inactive-sensory state and do not require new protein synthesis to be activated [40-45]. That the NF-κB p50 and p65 subunits belong to an expanding family of more than 25 NF-κB subunits indicates that the subunit composition of NF-κB is variable and may be tailored by the cell to accommodate various inflammatory signaling needs [40,41,44-49]. Interestingly, compared with interleukin-1 receptor-associated kinase (IRAK)-1, the more chronic and persistent activation of the NF-κB p50/p65 complex via the IRAK-2 signaling pathway in AD has recently been described [49]. Importantly, NF-κB activation and binding in the promoters of NF-κB-sensitive genes, including miRNA precursors (see below), leads to the facilitated transcription of many hundreds of potentially pathogenic genes, and therefore has the capacity to completely overwhelm the cell's anti-oxidant and anti-inflammatory defenses while at the same time altering the functional properties of nervous system cells [40-49].
Speciation, bioactivity and complexity of miRNA in the human brain
The potential contribution of small, noncoding RNA to human brain genetic function has been known for at least 20 years [50], but more recently there has been a virtual explosion into molecular-genetic research involving the neurobiological function of small, noncoding RNA and miRNA in brain development, injury, aging, health and disease [38,49,51-59]. Indeed, both small, noncoding RNAs and miRNAs are acquiring increasingly important roles in modulating the pathogenesis of progressive human neurologic disorders including inflammatory neurodegeneration, AD, Down's syndrome, epileptogenesis, glioma and glioblastoma, human prion diseases such as Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker syndrome, viral infection and aluminum intoxication of the brain, as well in murine Tg-AD and other transgenic models for progressive human neuro-degenerative disorders [52-60].
The miRNAs represent an evolutionarily conserved class of single-stranded small, noncoding RNAs averaging approximately 21 to 24 ribonucleotides in length. The major mode of action of miRNA is to bind to complimentary RNA sequences in the 3'-UTR of mRNA, and to thereby act as a repressor of that mRNA's expression [7,8,10,33,38,41,51-56]. Upregulated miRNAs are now generally accepted to predominantly act to decrease their target mRNA levels, and hence downregulate the genetic information encoded by that target mRNA [41,50-56]. Upregulated miRNAs and downregulated mRNAs may help explain the general downregulation of gene expression as is observed in the AD brain [9,39,42,51,59]. Of the approximately 2,000 human miRNAs currently known, only about 30 or 40 miRNAs are abundantly expressed in either the brain or the retina [51-56,59]. Figure 1 describes the expression of a small family of potentially pathogenic, NF-κB-regulated miRNAs that are significantly upregulated by a combinatorial cocktail of [IL-1β + Aβ42] in human neuronal-glial (HNG) cells in a primary co-culture [38,55,59]. This represents a physiologically relevant induction as both IL-1β and Aβ42 peptides are increased in abundance in AD brain [1-3,12-15,17,18]. The upregulated miRNA results in Figure 1 have been independently confirmed using RT-PCR and/or Northern and or LED-Northern dot-blot techniques [6,7,55-57,59]. These same miRNAs have been observed to be upregulated in AD and in age-related macular degeneration, but not in unaected anatomical regions of these same brain and retinal tissues [7,8,38,43].
Figure 1.
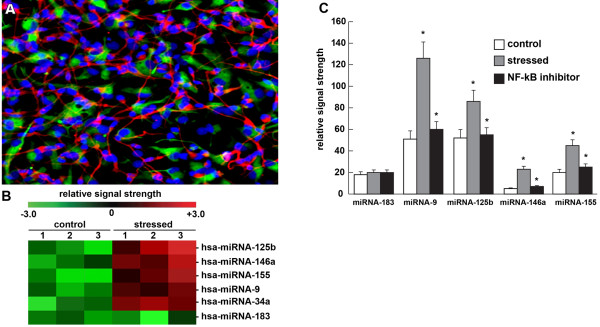
Expression of a small family of potentially pathogenic, NF-κB-regulated miRNAs. (A) Light microscopic photograph of human neuronal-glial (HNG) cells in primary culture stained with: antibody to glial fibrillary acidic protein, a glial-specific cytoplasmic marker (green fluorescence; λmax = 556 nm); antibody to βTUBIII, a neuron-specific cytoplasmic marker (red; λmax = 702 nm); and Hoescht 33258 to highlight the morphological features of both glial cell and neuronal cell nuclei (blue; λmax = 461 nm). Note: large nuclear area, relative to both glial or neuronal cytoplasmic area, is indicative of high levels of transcriptional activity [10,52,55,102] (18 days in culture; 20× magnification) [6,38,78]. (B) miRNA array cluster analysis and heatmap data from [IL-1β+Aβ42]-induced HNG cells in primary culture; relative induction of homo sapien (hsa)-miRNA-125b, hsa-miRNA-146a, hsa-miRNA-155, hsa-miRNA-9, hsa-miRNA-34a and hsa-miRNA-183 in control (n = 3) and stressed (n = 3) HNG cells by a cocktail of [IL-1β+Aβ42] in combination [38,52,78]. Note: twofold to sixfold upregulation in miRNA-125b, miRNA-146a, miRNA-155, miRNA-9 and hsa-miRNA-34a and no significant change in hsa-miRNA-183 (control miRNA in the same sample). (C) [IL-1β+Aβ42]-induced HNG cells in primary culture [38,78] show significant upregulation of hsa-miRNA-9, hsa-miRNA-125b, hsa-miRNA-146a, and miRNA-155 and quenching of their induction using the polyphenolic resveratrol analog CAY10512 (trans-3,5,4'-trihydroxystilbene). Aβ42+IL-1β has been previously shown to induce NF-κB and proinflammatory miRNA expression in several different human brain primary cell types [38,53,72,78]; other classes of NF-κB inhibitors, including the polyphenolic free radical scavenger curcumin and pyrrolidine dithiocarbamate, have also been shown to significantly quench the upregulation of inducible brain-enriched miRNAs indicating their NF-κB sensitivity [38,72,78]. n = 3 to 5; *P <0.01 (analysis of variance), gray bars over white bars (upregulation) or black bars over gray bars (downregulation).
Common to aged, degenerating brain and retina are significant upregulation of miRNA-125b and miRNA-146a, and their increases positively correlate with AD progression [38,59]. As discussed further below, upregulation of these miRNAs has been shown to be involved with a deficit in synaptic and neurotrophic signaling, synaptogenesis and the induction of amyloidogenesis and inflammatory signaling due to their selective targeting of several brain mRNA 3'-UTRs, including a critical down-regulation of 15-lipoxygenase (15-LOX), synapsin-2 (SYN-2), IRAK-1, CFH and tetraspanin-12 (TSPAN12) gene expression [38,55,56,61-82]. Interestingly, the miRNA-mediated downregulation of certain brain mRNAs, and hence the impairment in their expression, contributes downstream to AD-relevant deficits. For example, the miRNA-146a-mediated downregulation of TSPAN12 impairs the disintegrin and metalloproteinase-10 activity, thus shunting βAPP processing activities into more amyloidogenic and proinflammatory Aβ42-generating pathways (Table 1) [1-5,80-82].
Table 1.
An NF-κB-activated miRNA-mediated proinflammatory genetic network in Alzheimer's disease
| Human miRNA | mRNA target | mRNA function | Result of mRNA or gene expression deficit | References |
|---|---|---|---|---|
| miRNA-125b | CDKN2A | Cyclin-dependent kinase inhibitor 2A cell cycle inhibitor; induces cell cycle arrest | Downregulation of cell cycle control: glial cell proliferation | [57-60] |
| miRNA-125b | SYN-2 | Synapsin-2: neuronal synaptic phosphor-protein; coats synaptic vesicles; functions in the regulation of neurotransmitter release | Impairment of neurotransmitter release; synaptic signaling deficits | [2,3,61-65] |
| miRNA-125b | 15-LOX-1 | ALOX15; arachidonate 15-lipoxygenase; essential in the conversion of docosahexaenoic acid to neuroprotectin D1 (NPD1) | Deficit in neurotrophic omega-3 fatty acid derivatives in the brain | [3,66-69] |
| miRNA-146a | CFH | Complement factor H; repressor of activation of the innate immune response in brain and retina at the C3 to C3b transition; deficits in disease are proinflammatory | Defect in control of the innate immune response; chronic stimulation of the innate immune response and proinflammatory signaling | [2,3,8,70-75] |
| miRNA-146a | IRAK-1 | Interleukin-1 receptor-associated kinase 1; initiation of the innate immune response and NF-κB signaling | Compensatory surge in IRAK-2 and chronic stimulation of NF-κB signaling in the brain | [3,76-79] |
| miRNA-146a | TSPAN12 | Transmembrane 4 superfamily member 12; regulator of cell surface receptor signal transduction; activates ADAM10-dependent cleavage activity of βAPP | Results in a shift from neurotrophic (sAPPα) to amyloidogenic (Aβ42 peptide) processing of βAPP | [3,80-83] |
Both miRNA-125b and miRNA-146a target the 3'-UTR of several Alzheimer's disease (AD)-relevant mRNAs; these have been predicted using bioinformatics and confirmed experimentally using multiple analytical approaches including DNA arrays, RT-PCR, Northern and LED-Northern dot blots, and western and ELISA analysis [2,3,6-10,38,51-57,78]. Additional and original references are provided here and in the text. Factors that induce NF-κB such as HSV-1 and aluminum also induce the expression of proinflammatory miRNAs such as miRNA-125b and miRNA-146a [73,83,86,88,101,102,106,107,110]. Overexpression of just two NF-κB-regulated miRNAs (miRNA-125b and miRNA-146a) may in part explain many of the observed pathogenic features of AD including glial cell proliferation, synaptic signaling and neurotrophic deficits, chronic overstimulation of NF-κB and innate immune signaling and proinflammatory amyloidogenesis [8,59]. The mRNA targets for miRNA-9, miRNA-34a and miRNA-155 (Figure 1) and other inducible miRNAs, and their possible contribution to alterations in gene expression in AD, are currently under intensive research investigation by multiple research laboratories. ADAM10, a disintegrin and metalloproteinase-10; βAPP, β-amyloid precursor protein; TSPAN12, tetraspanin-12.
AD-relevant effects of two NF-κB-regulated proinflammatory miRNAs
Table 1 displays some of the integrated neurobiological effects of just two of the most consistently upregulated NF-κB-induced miRNAs in AD brain and in stressed HNG cells in primary culture: miRNA-125b and miRNA-146a. Several of their multiple AD-relevant mRNA targets, the function of those mRNAs and consequences of their deficits, and original key references are shown.
As indicated, inducible miRNA-125b and miRNA-146a have experimentally verified mRNA targets including the glial cell cycle and glial cell proliferation inhibitor cyclin-dependent kinase 2A (CDKN2A), the neurotransmitter release and synaptic protein SYN-2, the essential docosahexaenoic acid-to-neuroprotectin D1 (NPD1) conversion enzyme 15-LOX (also known as ALOX15), the innate immune system regulator CFH, IRAK-1 and the βAPP-disintegrin and metalloproteinase-10 regulatory protein TSPAN12 [3,57-82]. These combined data suggest a complex and highly interactive role for NF-κB, miRNA-125b and miRNA-146a in physiologically stressed HNG cells in primary co-culture. Remarkably, the misregulation of just two NF-κB-regulated, proinflammatory miRNAs has the potential to contribute to the deregulation of several key features of AD neuropathology, including neurotrophic support, synaptogenesis, neuroinflammation, innate immune signaling, and amyloidogenesis in primary human neural cells. Importantly, upregulation of miRNA-125b and miRNA-146a has been observed in anatomical areas of the brain targeted by the AD process, but neither in unaffected regions of the same brain, such as the brain stem or thalamus, nor in the same anatomical areas in healthy age-matched controls [57,74,83].
More recently, interrelated and independent studies further suggest the sensitivity of human miRNA-9, miRNA-34a and miRNA-155 to AD-relevant stress and neuropathology as NF-κB-mediated miRNAs (Figure 1) [38,40,45,59,74,83]. While the neurological activities of miRNA-9, miRNA-34a and miRNA-155 are currently under active investigation by multiple laboratories, miRNA-125b and miRNA-146a and several of their mRNA targets, and the implications, are further discussed in the following sections.
miRNA-125b
One of the most human brain cell-abundant miRNAs, if not the most abundant CNS miRNA, is inducible miRNA-125b [23,45,49,51,55,57,58,83-87]. This extensively studied 22-nucleotide miRNA (encoded at human chromosome 11q24.1: 5'-ucccugagacccuaacuuguga-3' [Genbank:NR_029671.1]) was first shown to be upregulated in both stressed and differentiating mouse and human neurons, and has since been implicated in mammalian neuronal development, brain cell signaling functions and degenerative disease [45,49].
NF-κB-regulated proinflammatory miRNA-125b has been further shown to be induced by human neurotrophic viruses and by neurotoxic metal sulfates, such as aluminum sulfate, that generate robust oxidative stress and ROS in human brain cells [83-101]. The high-abundance miRNA-125b is also associated with brain cancers, where it apparently also targets CDKN2A, a negative regulator of astroglial cell growth and proliferation [57-59]. Consistent upregulation of miRNA-125b, and CDKN2A downregulation, thus associates with deregulated astroglial cell proliferation, and is thereby linked to the proliferation of astroglia in several diverse neurodegenerative conditions including AD, Down's syndrome and epileptogenesis, and in inflammatory glial cell proliferation in glioma and glioblastoma multiforme [57,58,83,86,87,97-110]. Indeed, the capability of miRNA-125b in simultaneously regulating multiple downstream pathogenic gene targets may play a key role in explaining the complex multigenetic mechanisms underlying glioblastoma multiforme, an aggressive grade IV astrocytoma with a 1-year median survival rate and dismal prognosis despite current treatment modalities [57,58].
Interestingly, the pathogenic upregulation of miRNA-125b can be effectively quenched using both anti-NF-κB and anti-miRNA-125b intervention strategies (Figure 1) [51,57,74,83].
miRNA-146a
miRNA-146a (chromosome 5q34; 5'-gagaacugaauucca-uggguu-3' [Genbank:NR_029701.1]) was first described as an NF-κB-regulated proinflammatory miRNA that was found to target signaling proteins of innate immune responses, and more specifically the 3'-UTR of CFH in murine monocytes [70-72,75]. Subsequently, elevated miRNA-146a has also been shown to target human CFH and IRAK-1 in AD brain, and the role of miRNA-146a in altered innate immune responses and neuroinflammation signaling in progressively degenerating human brain cells and tissues is well documented [10,38,53,56,72,101]. Interestingly, although CFH is a highly abundant human serum protein of hepatic origin, abundant CFH presence in brain and retinal tissues suggests CFH involvement in the innate immune response and inflammatory regulation within the privileged immunology of these tissues [71-79].
Although miRNA-146a is a much less basally abundant miRNA when compared with miRNA-125b, it has been found to be the most inducible and upregulated miRNA in AD brain compared with all other NF-κB-regulated species so far indentified (Figure 1 and Table 1). The reason why miRNA-146a is one of the most rapidly induced of all brain miRNAs may be due to the presence of three cannonical tandem NF-κB binding sites in the pre-miRNA-146a promoter located at chromosome 5q34 [38,70,78]. Disease-related upregulation of miRNA-146a has also been observed in human prion disease and in inflammatory processes associated with epilepsy, but no increase in miRNA-146a has been associated with multiple sclerosis, Huntington's disease, schizophrenia, and in certain grades of glioblastoma where the actions of other upregulated miRNAs may predominate [84-86].
miRNA-125b and miRNA-146a mRNA targets in the brain
As indicated in Table 1, upregulation in brain-abundant miRNA-125b is associated with downregulation of the cell cycle inhibitor CDKN2A and glial cell proliferation, a pathological feature of AD gliosis, glioma and glio-blastoma [57,58,72]. Upregulated miRNA-126b also downregulates the synaptic vesicle-associated neuronal-enriched phosphoprotein (which associates with the cytoplasmic surface of synaptic vesicles) and neuro-transmitter release regulator SYN-2 [61-65], as well as the 15-LOX enzyme essential for the conversion of the essential omega-3 fatty acid docosahexaenoic acid into the potent docosahexaenoic acid derivative and neuro-protectant NPD1 [66-69]. Deficits in 15-LOX correlate with NPD1 deficits in AD brain [66-68]. Similarly, a miRNA-146a-regulated CFH is a key negative regulator of the innate immune system, and miRNA-146a upregulation associates with decreased CFH and a chronic inflammatory neural degeneration [38,53,56,87].
Similarly, the mRNA encoding the four-time membrane spanning integral membrane protein TSPAN12 is also a target for miRNA-146a, and upregulated miRNA-146a contributes to the downregulation of TSPAN12 as is observed in AD brain and in cytokine and Aβ peptide-stressed human brain cells [8,80,81]. Just as sufficient TSPAN12 appears to be required for the neurotrophic cleavage of the βAPP, insufficient TSPAN12 is associated with the induction of amyloidogenesis [8,80,81].
The integrated miRNA-mRNA interactions of as few as two human brain miRNAs (miRNA-125b and miRNA-146a) may hence in part explain not only the observed downregulation of CDKN2A, 15-LOX, SYN-2, CFH, IRAK-1 and TSPAN12, but also progressive, pathogenic deficiencies in innate and immune signaling, neurotrophic support, and synaptogenesis and amyloidogenesis in the AD brain.
Extraneural and environmental factors that are strong inducers of NF-κB-mediated and miRNA-mediated proinflammatory signaling
Herpes simplex virus 1
While only about 5% of all AD cases are genetic and 95% of all AD cases are of sporadic (idiopathic, or unknown) origin, a significant epigenetic contributor to sporadic AD may well be of extraneural or environmental origin [1-3]. Two independent factors that have long been thought to contribute to inflammatory aspects of AD are neurotropic viral infection, specifically by HSV-1, and the abundant neurotoxin aluminum in the environment [88-108].
About 95% of all humans harbor HSV-1 in various CNS compartments, and normally HSV-1 remains latent until activated by a number of factors including stress, radiation, trauma or ancillary neurological disease [88-92]. For at least 30 years, HSV-1 activation or previous HSV-1 infection of the human CNS has been associated with increased risk for AD, and the appearance of AD-relevant neuropathological lesions [88-96]. Interestingly, HSV-1 particles are associated with mature senile plaques in AD brain. HSV-1 and experimental infection of HNG cells in primary culture with HSV-1 significantly upregulate both NF-κB and miRNA-146a and a proinflammatory gene expression program. This upregulation culminates in neuronal blebbing and swelling, inflammation and ultimately brain cell death [88-95].
Treatment of AD with antiviral agents - such as the already US Food and Drug Administration-approved acyclovir (brand name Zovirax®; GlaxoSmithKline, London, UK), penciclovir, valacyclovir (brand name Valtrex®; GlaxoSmithKline) or foscarnet - has been suggested as a possible efficacious or adjunct treatment for AD [94-96] (unpublished observations). The pharmacological strategy here is that HSV-1 infection in the brain induces the accumulation of key pathogenic proteins, such as Aβ42 peptides, abnormally phosphorylated tau, and proinflammatory miRNAs, and that these antiviral agents have been shown to greatly reduce the abundance of Aβ42 peptides, phosphorylated tau and proinflammatory miRNA-146a accumulation in human brain cells previously infected with HSV-1 [73,88,94-96].
Aluminum
Aluminum exists in the biosphere as the third most abundant element (after oxygen and silicon) and the first most abundant metal, and hence environmental exposure to aluminum is naturally quite extensive [97-109]. Additional biologically-relevant sources of aluminum come from drinking water, vaccines, medicines, beverages and food [98,100]. A considerable amount of work has been done on studying the effect of environmental toxins such as aluminum hydroxide and aluminum sulfate on NF-κB induction, on miRNA generation, speciation and complexity, and on the effects of aluminum on the pathogenic regulation of AD-relevant gene expression [98,102-107].
Interestingly, aluminum potassium sulfate, or alum (hydrated potassium alum is AlK(SO4)2·12H2O), which is added to water-purification systems worldwide to clarify turbid drinking water, or aluminum hydroxide, used as an adjuvant to stimulate a local inflammatory response during vaccine injection, also strongly induce NF-κB, miRNA-146a, and a proinflammatory gene expression program in human primary brain cell models [103,106,107]. In fact, the capability of aluminum - an extremely high charge-density trivalent cation (Z2 /r = 18, where Z is an unchanging charge of +3 and r is the ionic radius of 0.5 nm) - to crosslink and aggregate biological material is second to none in the realm of biosphere-available neurological metallotoxins [98-108].
Aluminum has also been shown to aggregate Aβ42 peptides into a much more neurotoxic, immunogenic and proinflammatory fibrillar form, as observed within the end-stage senile plaques in advanced AD brain [98,100]. For example, when tested for the ability to induce ROS and NF-κB activation in vitro, comparison of aluminum, cadmium, copper, iron, mercury, gallium, magnesium, manganese, nickel, lead, tin or zinc (as sulfates) at 50 nmol concentrations in HNG cell co-cultures (using the novel, mixed isomer, fluorescent indicator 5-(and-6)-carboxy-2',7'-dichlorofluorescein diacetate) found aluminum to have by far the strongest ROS-inducing, NF-κB-inducing and inflammatory gene expression-inducing capacity of any trace metal tested [10,109,110].
While antiviral therapeutic strategies have been advocated for the clinical treatment of AD [94,95], a single clinical trial using the actinobacterial siderophore desferrioxamine (mesylate) as an anti-oxidant, ROS scavenger and aluminum chelator has proved to be one of the most efficacious treatments yet for mild-to-severe AD [105-107]. This is also in line with the idea that drugs such as desferrioxamine (mesylate) and posiphen that target multiple pathogenic molecules or processes in AD brain may hold the best promise in the clinical management of this complex and multifactorial neurological disorder [1-5,105,108].
Anti-miRNA (antagomir) strategies
Using perfectly complimentary ribonucleotide anti-sense (anti-miRNA; antagomir) sequences to lower the ambient abundance of upregulated miRNA in the brain is a logical approach to neutralizing the pathogenic gene expression eects of some overly expressed miRNAs, and attenuating their effects on selective mRNA abundance. This neutralization has been demonstrated in primary human brain cell tissue co-culture for both miRNA-125b and miRNA-146a [38,75,78,79,83,84]. The structure of these small, single-stranded therapeutic anti-miRNAs can be chemically modified to increase their stability within the cell in vitro, and as little as 5 nM locked nucleic acid-stabilized anti-miRNA per million human brain cells in primary tissue culture has been shown to have a dramatic quenching effect on both the target miRNA and proinflammatory gene expression induction patterns when analyzed using DNA and miRNA arrays and LED-Northern analytical techniques [6,7,55,75,79].
While it is not at the present time clear whether these anti-miRNA strategies can be translated into human therapies for inflammatory degeneration, these kinds of RNA silencing approaches have shown recent promise in the treatment of glioblastoma, the most lethal form of primary malignant tumor in the human CNS [58,83-85].
Conclusion
The six main conclusions from the research work presented in this review are as follows: the miRNA-mediated downregulated expression of several bio-informatics and experimentally confirmed mRNAs are targeted by increases in AD brain-relevant miRNA; stressors known to induce NF-κB also transactivate specific NF-κB-sensitive brain cell miRNAs; single miRNAs, such as miRNA-125b and miRNA-146a, have the potential to regulate multiple mRNA abundances relevant to the AD process; epigenetic and environmental factors such as HSV-1 infection and bioavailable aluminum may be highly relevant to the AD process, as they are both exceedingly strong inducers of NF-κB and proinflammatory miRNAs; specific antiviral, trivalent metal chelation, NF-κB inhibitors, or anti-miRNA strategies may be able to quench pathogenic miRNA overabundance and restore homeostasis to the AD brain, as is seen in models of AD in vitro; and HNG cells in primary co-culture are a proven, reliable, and human brain disease-relevant in vitro cell model to study the mechanism of transcription factor-mediated miRNA activation and speciation, and inflammatory signaling under normal aging, and physiologically relevant stress conditions.
Whether these mechanisms are operative in Tg-AD murine models, whether antiviral, anti-aluminum, anti-NF-κB or anti-miRNA strategies operate mechanistically in the same way in Tg-AD or cell culture models, or whether Tg-AD results can be extrapolated into human clinical trials are currently not known, and are all very active areas of independent research investigation. Since multiple mRNA targets are known to associate with neurodegenerative disease, and participate in complex positive or negative NF-κB-mediated feedback and signaling loops, these miRNA-mRNA linkage studies and their functional interpretations in disease may be more complex than initially anticipated, especially when multiple epigenetic or environmental factors are involved [87-111]. Importantly, the significant overabundance of NF-κB and miRNA in specific anatomical regions in AD neocortex and hippocampus strongly implicates an NF-κB-mediated, miRNA-regulated inflammatory disease mechanism that appears to selectively down-regulate different pathology-associated brain gene transcripts during the sporadic AD process, including those AD-relevant miRNA-mRNA pairings and the pathogenic consequences depicted in Table 1[38,78].
In summary, AD is a complex neurodegenerative disease caused by the dysregulation of numerous brain cell functions and multiple neurobiological networks [1-3,5,34-37,109-114]. A wiser therapeutic strategy may therefore be to consider the use of drugs or drug combinations that have multiple pathogenic targets, with minimal o-target and negligible peripheral toxic effects [1-7,32,109-114]. These effects include the implementation of novel drug delivery systems [111-114]. As an important step to achieve this goal we currently need to better understand the role of brain chromatin-mediated transcription mechanisms in AD and how these compare with normally aging brain, to better understand the role of ancillary DNA-binding proteins and proinflammatory transcription factors such as NF-κB in these processes, and to better understand features of other related epigenetic mechanisms on specific miRNA-mRNA recognition, activation, and signaling pathways. Yet another layer of miRNA-mediated genetic complexity in the brain appears to be the role of miRNA nucleases and the relatively rapid turnover of specific miRNAs, which ultimately modulates the ability of miRNAs to impact pathogenic signaling [38,78,87,115,116]. Paradoxically, certain inflammatory responses may prove to be neuroprotective or beneficial, so it will be important to quantify both the individual contribution, and integration, of each of these proinflammatory signaling pathways to the AD process. Eventually, their net impact on the neurogenetics of brain cell function in healthy aging and in inflammatory neurodegenerative disease will be elucidated, yielding advanced therapeutic strategies and combinatorial approaches that have not yet been considered.
Abbreviations
Aβ: β-amyloid; AD: Alzheimer's disease; βAPP: β-amyloid precursor protein; CDKN2A: cyclin-dependent kinase inhibitor 2A; CFH: complement factor H; CNS: central nervous system; ELISA: enzyme-linked immunosorbent assay; HNG: human neuronal-glial; HSV-1: herpes simplex virus 1; IL: interleukin; IRAK: IL-1 receptor-associated kinase; 15-LOX: 15-lipoxygenase; miRNA: microRNA; NF: nuclear factor; NPD1: neuroprotectin D1 PCR: polymerase chain reaction; ROS: reactive oxygen species; RT: reverse transcription; SYN-2: synapsin-2; Tg-AD: transgenic AD (murine model for disease); TNF: tissue necrosis factor; TSPAN12: tetraspanin-12; UTR: untranslated region.
Competing interests
The author declares that they have no competing interests.
Acknowledgements
This research was presented in part at the 12th annual Alzheimer's Association International Conference (AAIC12) in Vancouver, British Columbia, Canada, 14-19 July 2012. Thanks are extended to Dr Yuhai Zhao, Dr Surjyadipta Bhattacharjee, Dr Brandon M Jones and Dr Darlene Guillot for expert technical assistance, the co-culture of primary HNG cells, unpublished data, recent publications in this research area and helpful interpretative discussions, and to Dr C Eicken, Dr C Hebel and Dr P Dua for the miRNA array work and initial data interpretation. Human brain tissues were provided in part by the Harvard Brain Tissue Bank, the Oregon State University Health Science Centre, the Louisiana State University Health Sciences Center Brain Bank, and by the Memory Impairments and Neurological Disorders Institute at the University of California, Irvine Alzheimer's Disease Research Center (funded in part though NIA P50 AG16573). Thanks are also extended to the physicians, neuropathologists and families who have kindly provided human brain and retinal tissues for research purposes. Research on the structure and function of NF-κB and miRNA expression, speciation and complexity in AD brain and related neurological disorders in the Lukiw laboratory were supported through Grant Number P20RR016456 from the National Center for Research Resources, Translational Research Initiative Grants from LSU Health Sciences Center New Orleans (WJL), Alzheimer Association Investigator-Initiated Research Grant IIRG-09-131729 (WJL), and NIH NIA Grants AG18031 and AG038834 (WJL). The content of this manuscript is solely the responsibility of the author and does not necessarily represent the official views of the National Institute on Aging, National Center for Research Resources, or the National Institutes of Health.
References
- Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ. 100 years of AD research; are we any closer to a cure? Aging Health. 2007;3:279–282. doi: 10.2217/1745509X.3.3.279. [DOI] [Google Scholar]
- Lukiw WJ. Amyloid beta (Aβ) peptide modulators and other current treatment strategies for Alzheimer's disease (AD) Expert Opin Emerg Drugs. 2012. in press . [DOI] [PMC free article] [PubMed]
- Maccecchini ML, Chang MY, Pan C, John V, Zetterberg H, Greig NH. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-β peptide and s levels: target engagement, tolerability and pharmacokinetics in humans. J Neurol Neurosurg Psychiatry. 2012;83:894–902. doi: 10.1136/jnnp-2012-302589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelenboom P, Hoozemans JJ, Veerhuis R, van Exel E, Rozemuller AJ, van Gool WA. Whether, when and how chronic inflammation increases the risk of developing late-onset Alzheimer's disease. Alzheimers Res Ther. 2012;4:15–26. doi: 10.1186/alzrt118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Cui JG, Lukiw WJ. Natural secretory products of human neural and microvessel endothelial cells: implications in pathogenic 'spreading' and Alzheimer's disease. Mol Neurobiol. 2006;34:181–192. doi: 10.1385/MN:34:3:181. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Alexandrov PN, Zhao Y, Hill JM, Bhattacharjee S. Spreading of Alzheimer's disease inflammatory signaling through soluble micro-RNA. Neuroreport. 2012;23:621–626. doi: 10.1097/WNR.0b013e32835542b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Alexandrov PN. Regulation of complement factor H (CFH) by multiple miRNAs in Alzheimer's dsease (AD) brain. Mol Neurobiol. 2012;46:11–19. doi: 10.1007/s12035-012-8234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Bazan NG. Inflammatory, apoptotic, and survival gene signaling in Alzheimer's disease. A review on the bioactivity of neuroprotectin D1 and apoptosis. Mol Neurobiol. 2010;42:10–16. doi: 10.1007/s12035-010-8126-4. [DOI] [PubMed] [Google Scholar]
- Pogue AI, Li YY, Cui JG, Zhao Y, Kruck TP, Percy ME, Tarr MA, Lukiw WJ. Characterization of an NF-κB-regulated, miRNA-146a-mediated downregulation of complement factor H (CFH) in metal-sulfate-stressed human brain cells. J Inorg Biochem. 2009;103:1591–1595. doi: 10.1016/j.jinorgbio.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Katselson EN. Alzheimer's disease simulating an abscess of the left temporal lobe of the brain in a patient with chronic suppurative inflammation of the left middle ear. Zh Ushn Nos Gorl Bolezn. 1968;28:95–96. [PubMed] [Google Scholar]
- Eikelenboom P, Zhan SS, van Gool WA, Allsop D. Inflammatory mechanisms in Alzheimer's disease. Trends Pharmacol Sci. 1994;15:447–450. doi: 10.1016/0165-6147(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Aisen PS, Davis KL. Inflammatory mechanisms in Alzheimer's disease: implications for therapy. Am J Psychiatry. 1994;151:1105–1113. doi: 10.1176/ajp.151.8.1105. [DOI] [PubMed] [Google Scholar]
- NIH Medline Search; Keywords 'Inflammation, Alzheimer's Disease'. http://www.ncbi.nlm.nih.gov/gquery/?term=inflammation%20alzheimer%27s%20disease
- Verri M, Pastoris O, Dossena M, Aquilani R, Guerriero F, Cuzzoni G, Venturini L, Ricevuti G, Bongiorno AI. Mitochondrial alterations, oxidative stress and neuroinflammation in Alzheimer's disease. Int J Immunopathol Pharmacol. 2012;25:345–353. doi: 10.1177/039463201202500204. [DOI] [PubMed] [Google Scholar]
- Wojtera M, Sobow T, Kłoszewska I, Liberski PP, Brown DR, Sikorska B. Expression of immunohistochemical markers on microglia in Creutzfeldt-Jakob disease and Alzheimer's disease: morphometric study and review of the literature. Folia Neuropathol. 2012;50:74–84. [PubMed] [Google Scholar]
- Wilcock DM. Neuroinflammation in the aging down syndrome brain; lessons from Alzheimer's disease. Curr Gerontol Geriatr Res. 2012;2012:170276. doi: 10.1155/2012/170276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povova J, Ambroz P, Bar M, Pavukova V, Sery O, Tomaskova H, Janout V. Epidemiological of and risk factors for Alzheimer's disease. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2012;156:108–114. doi: 10.5507/bp.2012.055. [DOI] [PubMed] [Google Scholar]
- Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer's 1907 paper 'Uber eine eigenartige Erkankung der Hirnrinde'. Clin Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- Hardy J. Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis. 2006;9:151–153. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- Esiri MM, Chance SA. Vulnerability to Alzheimer's pathology in neocortex: the roles of plasticity and columnar organization. J Alzheimers Dis. 2006;9:79–89. doi: 10.3233/jad-2006-9s310. [DOI] [PubMed] [Google Scholar]
- Cui JG, Hill JM, Zhao Y, Lukiw WJ. Expression of inflammatory genes in the primary visual cortex of late-stage Alzheimer's disease. Neuroreport. 2007;18:115–119. doi: 10.1097/WNR.0b013e32801198bc. [DOI] [PubMed] [Google Scholar]
- van de Pol LA, Korf ES, van der Flier WM, Brashear HR, Fox NC, Barkhof F, Scheltens P. Magnetic resonance imaging predictors of cognition in mild cognitive impairment. Arch Neurol. 2007;64:1023–1028. doi: 10.1001/archneur.64.7.1023. [DOI] [PubMed] [Google Scholar]
- Marshall GA, Monserratt L, Harwood D, Mandelkern M, Cummings JL, Sultzer DL, Laforce R Jr, Rabinovici GD. Amyloid imaging in the differential diagnosis of dementia: review and potential clinical applications. Alzheimers Res Ther. 2011;3:31–34. doi: 10.1186/alzrt93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velliquette RA, O'Connor T, Vassar R. Energy inhibition elevates betasecretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J Neurosci. 2005;25:10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers M, Sperling RA, Klunk WE, Weiner MW, Hampel H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer's disease dementia. Trends Neurosci. 2011;34:430–442. doi: 10.1016/j.tins.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemers ER, Quinn JF, Kaye J, Farlow MR, Porsteinsson A, Tariot P, Zoulnouni P, Galvin JE, Holtzman DM, Knopman DS, Satterwhite J, Gonzales C, Dean RA, May PC. Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology. 2006;66:602–604. doi: 10.1212/01.WNL.0000198762.41312.E1. [DOI] [PubMed] [Google Scholar]
- Tomita T. At the frontline of Alzheimer's disease treatment: gamma-secretase inhibitor/modulator mechanism. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:295–300. doi: 10.1007/s00210-007-0206-2. [DOI] [PubMed] [Google Scholar]
- Siemers ER, Dean RA, Friedrich S, Ferguson-Sells L, Gonzales C, Farlow MR, May PC. Safety, tolerability, and effects on plasma and cerebrospinal fluid amyloid-beta after inhibition of gamma-secretase. Clin Neuropharmacol. 2007;30:317–325. doi: 10.1097/WNF.0b013e31805b7660. [DOI] [PubMed] [Google Scholar]
- Epis R, Marcello E, Gardoni F, Luca MD. Alpha, beta-and gamma-secretases in Alzheimer's disease. Front Biosci (Schol Ed) 2012;4:1126–1150. doi: 10.2741/s322. [DOI] [PubMed] [Google Scholar]
- Van Broeck B, Van Broeckhoven C, Kumar-Singh S. Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches. Neurodegener Dis. 2007;4:349–365. doi: 10.1159/000105156. [DOI] [PubMed] [Google Scholar]
- Veremeyko T, Starossom SC, Weiner HL, Ponomarev ED. Detection of miRNAs in microglia by real-time PCR in normal CNS and during neuroinflammation. J Vis Exp. 2012;23:65–71. doi: 10.3791/4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, El Khoury J. The neuroimmune system in Alzheimer's disease: the glass is half full. J Alzheimers Dis. 2012. in press . [DOI] [PMC free article] [PubMed]
- Lai AY, McLaurin J. Clearance of amyloid-β peptides by microglia and macrophages: the issue of what, when and where. Future Neurol. 2012;7:165–176. doi: 10.2217/fnl.12.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp (Warsz) 2012;60:251–266. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B, Choi RH, Chin TJ, Kaur C, Ling EA. Manipulation of microglial activity as a therapy for Alzheimer's disease. Front Biosci (Schol Ed) 2012;4:1402–1412. doi: 10.2741/s342. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Zhao Y, Cui JG. An NF-κB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem. 2008;283:31315–31322. doi: 10.1074/jbc.M805371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer's disease, role of cytokines. Sci World J. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R. The origins of NF-κB. Nat Immunol. 2011;12:686–688. doi: 10.1038/ni.2071. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Baltimore D. MicroRNAs, new effectors and regulators of NF-κB. Immunol Rev. 2012;246:205–220. doi: 10.1111/j.1600-065X.2011.01089.x. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Bazan NG. Strong NF-κB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer's disease superior temporal lobe neocortex. J Neurosci Res. 1998;53:583–592. doi: 10.1002/(SICI)1097-4547(19980901)53:5<583::AID-JNR8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Ottlecz A, Lambrou G, Grueninger M, Finley J, Thompson HW, Bazan NG. Coordinate activation of HIF-1 and NF-κB DNA binding and COX-2 and VEGF expression in retinal cells by hypoxia. Invest Ophthalmol Vis Sci. 2003;44:4163–4170. doi: 10.1167/iovs.02-0655. [DOI] [PubMed] [Google Scholar]
- Cai D, Liu T. Inflammatory cause of metabolic syndrome via brain stress and NF-κB. Aging (Albany NY) 2012;4:98–115. doi: 10.18632/aging.100431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale ST. Hierarchies of NF-κB target-gene regulation. Nat Immunol. 2011;12:689–694. doi: 10.1038/ni.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari OA, Liao JK. NF-κB and innate immunity in ischemic stroke. Ann N Y Acad Sci. 2010;1207:32–40. doi: 10.1111/j.1749-6632.2010.05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D. NF-κB-mediated metabolic inflammation in peripheral tissues versus central nervous system. Cell Cycle. 2009;8:2542–2548. doi: 10.4161/cc.8.16.9386. [DOI] [PubMed] [Google Scholar]
- Granic I, Dolga AM, Nijholt IM, van Dijk G, Eisel UL. Inflammation and NF-κB in Alzheimer's disease and diabetes. J Alzheimers Dis. 2009;16:809–821. doi: 10.3233/JAD-2009-0976. [DOI] [PubMed] [Google Scholar]
- Cui JG, Li YY, Zhao Y, Bhattacharjee S, Lukiw WJ. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-κB in stressed human astroglial cells and in Alzheimer disease. J Biol Chem. 2010;285:38951–38960. doi: 10.1074/jbc.M110.178848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Handley P, Wong L, McLachlan DRC. BC200 and other small RNAs in normal human neocortex, non-Alzheimer dementia (NAD), and senile dementia of the Alzheimer type (AD) Neurochem Res. 1992;17:591–597. doi: 10.1007/BF00968788. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer's disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chen XP, Li YJ. MicroRNA-146a and human disease. Scand J Immunol. 2010;71:227–231. doi: 10.1111/j.1365-3083.2010.02383.x. [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian miRNAs act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui JG, Hill JM, Zhao Y, Lukiw WJ. Expression of inflammatory genes in the primary visual cortex of late-stage Alzheimer's disease. Neuroreport. 2007;18:115–119. doi: 10.1097/WNR.0b013e32801198bc. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Dua P, Pogue AI, Eicken C, Hill JM. Up-regulation of micro RNA-146a (miRNA-146a), a marker for inflammatory neurodegeneration, in sporadic Creutzfeldt-Jakob disease (sCJD) and Gerstmann-Straussler Scheinker (GSS) syndrome. J Toxicol Environ Health. 2011;74:1460–1468. doi: 10.1080/15287394.2011.618973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogue AI, Cui JG, Li YY, Zhao Y, Culicchia F, Lukiw WJ. miRNA-125b (miRNA-125b) function in astrogliosis and glial cell proliferation. Neurosci Lett. 2010;476:18–22. doi: 10.1016/j.neulet.2010.03.054. [DOI] [PubMed] [Google Scholar]
- Nikaki A, Piperi C, Papavassiliou AG. Role of microRNAs in gliomagenesis: targeting miRNAs in glioblastoma multiforme therapy. Expert Opin Investig Drugs. 2012;21:1475–1488. doi: 10.1517/13543784.2012.710199. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ. NF-κB-regulated micro RNAs (miRNAs) in primary human brain cells. Exp Neurol. 2012;235:484–490. doi: 10.1016/j.expneurol.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GeneCards: CDKN2A. http://www.genecards.org/cgi-bin/carddisp.pl?gene=CDKN2A
- Bykhovskaia M. Synapsin regulation of vesicle organization and functional pools. Semin Cell Dev Biol. 2011;22:387–392. doi: 10.1016/j.semcdb.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Fassio A, Raimondi A, Lignani G, Benfenati F, Baldelli P. Synapsins: from synapse to network hyperexcitability and epilepsy. Semin Cell Dev Biol. 2011;22:408–415. doi: 10.1016/j.semcdb.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Valtorta F, Pozzi D, Benfenati F, Fornasiero EF. The synapsins: multitask modulators of neuronal development. Semin Cell Dev Biol. 2011;22:378–386. doi: 10.1016/j.semcdb.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE, Coleman PD. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer's disease. Neurobiol Dis. 2003;12:97–109. doi: 10.1016/S0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
- GeneCards: SYN2. http://www.genecards.org/cgi-bin/carddisp.pl?gene=SYN2
- Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Bazan NG. Docosahexaenoic acid and the aging brain. J Nutr. 2008;138:2510–2514. doi: 10.3945/jn.108.096016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Calon F, Julien C, Winkler JW, Petasis NA, Lukiw WJ, Bazan NG. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARγ-mediated mechanisms in Alzheimer's disease models. PLoS One. 2011;6:e15816. doi: 10.1371/journal.pone.0015816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GeneCards: ALOX15. http://www.genecards.org/cgi-bin/carddisp.pl?gene=ALOX15
- Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of miRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Han SB, Nam SY, Oh KW, Hong JT. Inflammation and Alzheimer's disease. Arch Pharm Res. 2010;33:1539–1556. doi: 10.1007/s12272-010-1006-7. [DOI] [PubMed] [Google Scholar]
- Li YY, Cui JG, Dua P, Pogue AI, Bhattacharjee S, Lukiw WJ. Differential expression of miRNA-146a-regulated inflammatory genes in human primary neural, astroglial and microglial cells. Neurosci Lett. 2011;499:109–113. doi: 10.1016/j.neulet.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Zhao Y, Clement C, Neumann DM, Lukiw WJ. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport. 2009;20:1500–1505. doi: 10.1097/WNR.0b013e3283329c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Pogue AI. Induction of specific micro RNA (miRNA) species by ROS-generating metal sulfates in primary human brain cells. J Inorg Biochem. 2007;101:1265–1269. doi: 10.1016/j.jinorgbio.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GeneCards: CFH. http://www.genecards.org/cgi-bin/carddisp.pl?gene=CFH
- Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. 2010;80:1981–1991. doi: 10.1016/j.bcp.2010.06.020. [DOI] [PubMed] [Google Scholar]
- Gan L, Li L. Regulations and roles of the interleukin-1 receptor associated kinases (IRAKs) in innate and adaptive immunity. Immunol Res. 2006;35:295–302. doi: 10.1385/IR:35:3:295. [DOI] [PubMed] [Google Scholar]
- Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian miRNAs uncovers a subset of brainexpressed miRNAs with possible roles in murine and human neuronal diff erentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GeneCards: IRAK1. http://www.genecards.org/cgi-bin/carddisp.pl?gene=IRAK1
- Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM, Costa M, Rice DS, Ye W. TSPAN12 regulates retinal vascular development by promoting Norrinbut not Wnt-induced FZD4/beta-catenin signaling. Cell. 2009;139:299–311. doi: 10.1016/j.cell.2009.07.048. [DOI] [PubMed] [Google Scholar]
- Xu D, Sharma C, Hemler ME. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 2009;23:3674–3681. doi: 10.1096/fj.09-133462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GeneCards: TSPAN12. http://www.genecards.org/cgi-bin/carddisp.pl?gene=TSPAN12
- Pogue AI, Percy ME, Cui JG, Li YY, Bhattacharjee S, Hill JM, Kruck TPA, Zhao Y, Lukiw WJ. Up-regulation of NF-κB-sensitive miRNA-125b and miRNA-146a in metal sulfate-stressed human astroglial (HAG) primary cell cultures. J Inorganic Biochem. 2011;105:1434–1437. doi: 10.1016/j.jinorgbio.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YY, Alexandrov PN, Pogue AI, Zhao Y, Bhattacharjee S, Lukiw WJ. miRNA-155 up-regulation and complement factor H (CFH) deficits in Down's syndrome. Neuroreport. 2012;23:168–173. doi: 10.1097/WNR.0b013e32834f4eb4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltronieri P, D'Urso PI, Mezzolla V, D'Urso OF. Potential of anti-cancer therapy based on anti-miR-155 oligonucleotides in glioma and brain tumours. Chem Biol Drug Des. 2012. doi: 10.1111/cbdd.12002. [DOI] [PubMed]
- Aronica E, Fluiter K, Iyer A, Zurolo E, Vreijling J, van Vliet EA, Baayen JC, Gorter JA. Expression pattern of miRNA-146a, an inflammation-associated microRNA, in experimental and human temporal lobe epilepsy. Eur J Neurosci. 2010;31:1100–1107. doi: 10.1111/j.1460-9568.2010.07122.x. [DOI] [PubMed] [Google Scholar]
- Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer's disease temporal lobe neocortex. Neurosci Lett. 2009;459:100–104. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Cui JG, Yuan LY, Bhattacharjee PS, Corkern M, Clement C, Kammerman EM, Ball MJ, Zhao Y, Sullivan PM, Hill JM. Acyclovir or Aβ42 peptides attenuate HSV-1-induced miRNA-146a levels in human primary brain cells. Neuroreport. 2010;21:922–927. doi: 10.1097/WNR.0b013e32833da51a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Ball MJ, Neumann DM, Azcuy AM, Bhattacharjee PS, Bouhanik S, Clement C, Lukiw WJ, Foster TP, Kumar M, Kaufman HE, Thompson HW. The high prevalence of herpes simplex virus type 1 DNA in human trigeminal ganglia is not a function of age or gender. J Virol. 2008;82:8230–8234. doi: 10.1128/JVI.00686-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerman EM, Neumann DM, Ball MJ, Lukiw W, Hill JM. Senile plaques in Alzheimer's disease brains: possible association of beta-amyloid with herpes simplex virus type 1 (HSV-1) L-particles. Med Hypotheses. 2006;66:294–299. doi: 10.1016/j.mehy.2005.07.033. [DOI] [PubMed] [Google Scholar]
- Higaki S, Gebhardt B, Lukiw W, Thompson H, Hill J. Gene expression profiling in the HSV-1 latently infected mouse trigeminal ganglia following hyperthermic stress. Curr Eye Res. 2003;26:231–238. doi: 10.1076/ceyr.26.3.231.14892. [DOI] [PubMed] [Google Scholar]
- Hill JM, Lukiw WJ, Gebhardt BM, Higaki S, Loutsch JM, Myles ME, Thompson HW, Kwon BS, Bazan NG, Kaufman HE. Gene expression analyzed by microarrays in HSV-1 latent mouse trigeminal ganglion following heat stress. Virus Genes. 2001;23:273–280. doi: 10.1023/A:1012517221937. [DOI] [PubMed] [Google Scholar]
- Higaki S, Gebhardt BM, Lukiw WJ, Thompson HW, Hill JM. Effect of immunosuppression on gene expression in the HSV-1 latently infected mouse trigeminal ganglion. Invest Ophthalmol Vis Sci. 2002;43:1862–1869. [PubMed] [Google Scholar]
- Wozniak MA, Frost AL, Preston CM, Itzhaki RF. Antivirals reduce the formation of key Alzheimer's disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS One. 2011;6:e25152. doi: 10.1371/journal.pone.0025152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki RF, Wozniak MA. Could antivirals be used to treat Alzheimer's disease? Future Microbiol. 2012;7:307–309. doi: 10.2217/fmb.12.10. [DOI] [PubMed] [Google Scholar]
- Ball MJ, Lukiw WJ, Kammerman E, Hill JM. Intracerebral propagation of Alzheimer's disease confirms its herpes virus etiology. Alzheimer's Dementia. 2012. in press . [DOI] [PMC free article] [PubMed]
- Campbell A, Yang EY, Tsai-Turton M, Bondy SC. Pro-inflammatory effects of aluminum in human glioblastoma cells. Brain Res. 2002;933:60–65. doi: 10.1016/S0006-8993(02)02305-3. [DOI] [PubMed] [Google Scholar]
- Exley C. Aluminium, tau and Alzheimer's disease. J Alzheimers Dis. 2007;12:313–315. doi: 10.3233/jad-2007-12403. [DOI] [PubMed] [Google Scholar]
- Walton JR. Aluminum disruption of calcium homeostasis and signal transduction resembles change that occurs in aging and Alzheimer's disease. J Alzheimers Dis. 2012;29:255–273. doi: 10.3233/JAD-2011-111712. [DOI] [PubMed] [Google Scholar]
- Percy ME, Kruck TP, Pogue AI, Lukiw WJ. Towards the prevention of potential aluminum toxic effects and an effective treatment for Alzheimer's disease. J Inorg Biochem. 2011;105:1505–1512. doi: 10.1016/j.jinorgbio.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara M, Kato-Negishi M. Link between aluminum and the pathogenesis of Alzheimer's disease: the integration of the aluminum and amyloid cascade hypotheses. Int J Alzheimers Dis. 2011;2011:276393. doi: 10.4061/2011/276393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogue AI, Li YY, Cui JG, Zhao Y, Kruck TP, Percy ME, Tarr MA, Lukiw WJ. Characterization of an NF-κB-regulated, miRNA-146a-mediated downregulation of complement factor H (CFH) in metal-sulfate-stressed human brain cells. J Inorg Biochem. 2009;103:1591–1595. doi: 10.1016/j.jinorgbio.2009.05.012. [DOI] [PubMed] [Google Scholar]
- Zhang QL, Jia L, Jiao X, Guo WL, Ji JW, Yang HL, Niu Q. APP/PS1 transgenic mice treated with aluminum: an update of Alzheimer's disease model. Int J Immunopathol Pharmacol. 2012;25:49–58. doi: 10.1177/039463201202500107. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ. Evidence supporting a biological role for aluminum in brain chromatin compaction and epigenetics. J Inorg Biochem. 2010;104:1010–1012. doi: 10.1016/j.jinorgbio.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Percy ME, Kruck TP, Pogue AI, Lukiw WJ. Towards the prevention of potential aluminum toxic effects and an effective treatment for Alzheimer's disease. J Inorg Biochem. 2011;105:1505–1512. doi: 10.1016/j.jinorgbio.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov PN, Zhao Y, Pogue AI, Tarr MA, Kruck TP, Percy ME, Cui JG, Lukiw WJ. Synergistic effects of iron and aluminum on stress-related gene expression in primary human neural cells. J Alzheimers Dis. 2005;8:117–127. doi: 10.3233/jad-2005-8204. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Percy ME, Kruck TP. Nanomolar aluminum induces proinflammatory and pro-apoptotic gene expression in human brain cells in primary culture. J Inorg Biochem. 2005;99:1895–1898. doi: 10.1016/j.jinorgbio.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Pogue AI, Jones BM, Bhattacharjee S, Percy ME, Zhao Y, Lukiw WJ. Metalsulfate induced generation of ROS in human brain cells: detection using an isomeric mixture of 5- and 6-carboxy-2',7'-dichlorofluorescein diacetate (carboxy-DCFDA) as a cell permeant tracer. Int J Mol Sci. 2012;13:9615–9626. doi: 10.3390/ijms13089615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruck TP, Cui JG, Percy ME, Lukiw WJ. Molecular shuttle chelation: the use of ascorbate, desferrioxamine and Feralex-G in combination to remove nuclear bound aluminum. Cell Mol Neurobiol. 2004;24:443–459. doi: 10.1023/B:CEMN.0000022773.70722.b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov PN, Zhao Y, Pogue AI, Tarr MA, Kruck TP, Percy ME, Cui JG, Lukiw WJ. Synergistic effects of iron and aluminum on stress-related gene expression in primary human neural cells. J Alzheimers Dis. 2005;8:117–127. doi: 10.3233/jad-2005-8204. [DOI] [PubMed] [Google Scholar]
- Lane RF, Shineman DW, Steele JW, Lee LB, Fillit HM. Beyond amyloid: the future of therapeutics for Alzheimer's disease. Adv Pharmacol. 2012;64:213–271. doi: 10.1016/B978-0-12-394816-8.00007-6. [DOI] [PubMed] [Google Scholar]
- Ansari N, Khodagholi F. Molecular mechanism aspect of ER stress in Alzheimer's disease: current approaches and future strategies. Curr Drug Targets. 2012. in press . [DOI] [PubMed]
- Schenk D, Basi GS, Pangalos MN. Treatment strategies targeting amyloid β-protein. Cold Spring Harb Perspect Med. 2012;2:a006387. doi: 10.1101/cshperspect.a006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma HS, Castellani RJ, Smith MA, Sharma A. The blood-brain barrier in Alzheimer's disease: novel therapeutic targets and nanodrug delivery. Int Rev Neurobiol. 2012;102:47–90. doi: 10.1016/B978-0-12-386986-9.00003-X. [DOI] [PubMed] [Google Scholar]
- Rüegger S, Großhans H. MicroRNA turnover: when, how, and why. Trends Biochem Sci. 2012;37:436–446. doi: 10.1016/j.tibs.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Qin YW, Brewer G, Jing Q. MicroRNA degradation and turnover: regulating the regulators. Wiley Interdiscip Rev RNA. 2012;3:593–600. doi: 10.1002/wrna.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]