

Abstract
The use of self-assembly for the construction of functional biomaterials is a highly promising and exciting area of research, with great potential for the treatment of injury or disease. By using multiple noncovalent interactions, coded into the molecular design of the constituent components, self-assembly allows for the construction of complex, adaptable, and highly tunable materials with potent biological effects. This review describes some of the seminal advances in the use of self-assembly to make novel systems for regenerative medicine and biology. Materials based on peptides, proteins, DNA, or hybrids thereof have found application in the treatment of a wide range of injuries and diseases, and this review outlines the design principles and practical applications of these systems. Most of the examples covered focus on the synthesis of hydrogels for the scaffolding or transplantation of cells, with an emphasis on the biological, mechanical, and structural properties of the resulting materials. In addition, we will discuss the distinct advantages conferred by self-assembly (compared with traditional covalent materials), and present some of the challenges and opportunities for the next generation of self-assembled biomaterials.
1. Introduction
Over the past three decades, the synthesis and application of biomaterials has become one of the most active and promising areas of research, positioned at the intersection of chemistry, materials science, bioengineering, and medicine. Broadly speaking, a biomaterial is any construct made specifically to interact with biological systems, usually for the treatment of injury or disease. As a result, a biomaterial must have properties (e.g. stiffness, chemical composition, biological signals) compatible with the tissue it will interact with in order to elicit the desired effect. In particular, biomaterials have found broad application as scaffolds for presenting the necessary physical, chemical, and biological cues to either regenerate damaged endogenous cells, or aid in the survival and/or differentiation of exogenously transplanted cells.
A number of organic chemical systems have been explored as biomaterials, including synthetic polymers, materials based on naturally occurring components such as biopolymers, and more recently molecularly designed materials based on biological structural units such as peptides and proteins. These materials generally fall into two categories: (1) systems based on covalent bonds, and (2) materials that self-assemble from molecular components into supramolecular architectures. Of the two, the second category represents the newer approach pioneered by the Stupp laboratory in the form of bioactive and biomimetic structures and is now emerging into a diverse and controllable platform for the synthesis of functional biomaterials.[1–3]
Self-assembly is the spontaneous association of an ensemble of molecules into one or more supramolecular structures, driven by multiple noncovalent interactions. These interactions include hydrogen bonding, electrostatic association, van der Waals forces, and are often mediated by the hydrophobic effect. In most examples, the assembling subunits used in biomaterials form thermodynamically stable structures that reflect a balance between the enthalpic and entropic contributions of both the molecular species as well as the solvent (usually water).
A broad range of self-assembling systems have been explored as biomaterials for physical scaffolds, biological signals, or both. Most of the applications of these systems have been geared towards regenerative medicine, either for healing existing damaged cells in the body, or providing a favorable milieu for externally transplanted components like stem cells. In addition to materials designed for biological applications, there also exists a rich body of literature on self-assembly strategies in bioinspired materials engineering, drug delivery, and a range of pharmaceutical application. However, only biomaterials that have been directly applied in a biological setting, either in vitro or in vivo, will be discussed herein.
The most elegant, complex, and adaptable examples of self-assembly are found in biological systems, and careful studies of proteins, peptides, lipids, and nucleic acids have provided chemists and materials scientists with a rich lexicon of self-assembly motifs and rules. Most biomaterials are designed to mimic the natural extracellular environment, as well as to impart biological signals to guide cell behavior. As a result, the majority of successful self-assembled biomaterials possess one or more biological components (e.g. peptides, proteins), often in hybrid structures to further guide their assembly and function. Materials with biological components have the additional benefit of high compatibility and low cellular toxicity, and are usually biodegradable and easily cleared from the body, allowing for “traceless” scaffolds. As a result, the chemical constituents of self-assembled biomaterials nearly always incorporate at least one biological component for guiding assembly, imbuing signaling, or both.
While examples of materials composed of covalently associated molecules, often based on hydrogels made from polymers like polyethers, polyesters, or polyacrylates, have historically been proven successful, self-assembly affords a number of intrinsic advantages not possible with covalent biomaterials. Because self-assembled structures are composed of many copies of simpler molecular components, they can be highly modular, allowing the material composition to be readily tuned by changing the mixture of the molecules that make it up. Furthermore, the multivalency inherent in the assembly of supramolecular structures often allows for a more potent biological effect, and a higher local concentration of a given chemical signal. Self-assembly usually results in structures with features at the nanometer length-scale, and in many cases the nanostructure geometries formed can be rationally designed by selecting the shapes and noncovalent forces encoded in the constituent molecules. Thus, with the combination of intentional molecular architecture and modularity of the resulting supramolecular structures, great control of length scales, aspect ratios, and higher-order hierarchical self-assembly may be achieved. Since cells interact with their external environment via receptors organized at similar length-scales, self-assembly is a natural approach towards arranging a material’s components for optimal interaction with biological systems.
Because self-assembled structures form due to the aggregation of multiple noncovalent intermolecular interactions, materials incorporating them can often be dynamic (e.g. switching between two or more self-assembled states depending on external condition) and self-healing if the nanostructures are temporarily disrupted, for example by external shear forces. Judiciously selecting the identity of the molecular components also allows for biodegradability and cytocompatibility. Finally, because most self-assembled materials are made up of single molecules, their constituents are monodisperse (especially compared with covalent polymers), and often highly scalable.
2. Peptide-based Biomaterials
2.1 Introduction
Peptides represent the most commonly used biological moiety for constructing self-assembling biomaterials, and possess a number of advantages that make them particularly attractive for this purpose. Unlike more complex biomolecules like proteins, peptides are more synthetically tractable and reasonably scalable. Using solid-phase peptide synthesis, it is possible to quickly construct sequences of up to ~50 amino acids with high purity and good yield (>1 g in some cases). In addition, unnatural amino acids or additional chemical groups can be facilely incorporated, allowing for a greater complexity and flexibility in both structure and function. As biological molecules, peptides can be readily degraded in vivo to harmless byproducts, resulting in an ultimately traceless treatment. Finally, there exists a rich scope of short peptide epitopes for biological function (such as cell adhesion, signaling, or differentiation), so peptide-based materials may elicit specificity without having to use more complex (and often more expensive) molecules like full-size proteins.
The self-assembly behavior of peptides represents the most comprehensively studied of the various classes of self-assembled biomaterials.[4, 5] The two most common “natural” motifs used for peptide-based materials are the α-helix and the β-sheet. However, a number of novel supramolecular interactions (e.g. based on ionic charge, aromatic interactions, or amphiphilicity) can be engineered through the judicious choice of the amino acid sequence. Furthermore, additional chemical functionality can be appended to the peptide epitope to dictate the self-assembly properties and direct the formation of a specific nanostructure.
Peptide-based self-assembled materials may be divided into two general categories. In the first case, systems are composed primarily of amino acids linked by native peptide bonds, in which self-assembly is mostly determined by the primary sequence. In the second case, peptidic systems are designed to incorporate other chemical moieties (e.g. lipids, small molecules) to create hybrid materials.
2.2 β-sheet peptides
2.2.1 Ionic self-complementary peptides
Peptides composed of alternating hydrophobic and hydrophilic amino acids often assemble into β-sheets due to the amphiphilic nature of the molecule. In the early 1990’s, Zhang and coworkers developed this motif for the synthesis of self-assembling biomaterials, synthesizing peptides with alternating hydrophobic and charged amino acids based on the sequence of zuotin, a yeast protein known to bind to Z-DNA.[6] Tuning the position of the positive and negative charges on the hydrophilic face of the molecule (e.g. + + − − + + − − for a peptide with the sequence (RARADADA)2, termed the “RAD” motif) allowed for the synthesis of peptides that self-assembled into staggered β-sheets and then formed entangled nanofibers and membranes (Figure 1A).[7, 8] The exact charge patterns allowed for efficient self-pairing, much like Lego building blocks. These materials were also able to form biocompatible and stable hydrogels at very low concentrations (< 1 wt%) when shielding the charges with physiologically relevant salt concentrations. Interestingly, nanofibers disrupted by mechanical agitation (e.g. sonication), readily reformed and were indistinguishable from the original structures.[9] This feature highlights the self-healing potential afforded by the noncovalent, reversible bonds that hold most self-assembled structures together.
Figure 1. Self-assembling peptides based on the β-sheet.

(A) Schematic of the ionic self-complementary peptides developed by Zhang and coworkers. The peptides pack into sheets and fibers based on hydrophobic interactions on one face of the molecule, and complementary ionic interaction on the other. These fibers then form a highly porous hydrogel. (B) Peptides with alternating hydrophilic and hydrophobic residues assemble into β-sheet structures (left) that form twisted ribbons (right) and bundle into larger fibers. (C) The Q11 peptide (blue) developed by the Collier lab assembles into nanofibers due to β-sheet formation. Appending a peptide epitope (red) results in a high density of the signal on the fibril surface. (D) Amphiphilic tri-block peptides developed by the Hartgerink group. The central hydrophobic block forces self-assembly via hydrophobic between molecules and hydrogen bonding along the fiber axis. Charged lysine residues provide solubility, and balance the hydrophobic forces. (E) The β-hairpin molecule MAX1, which can fold under appropriate conditions and form intramolecular hydrogen bonds. (F) Upon charge screening in cell culture media, the β-hairpin molecule can fold and then subsequently assemble into long nanofibers due to intermolecular interactions. [(A): ref. 7; (B): ref. 21; (C): ref. 28; (D): ref. 29; (E): ref. 32; (F): ref. 38]
The Zhang laboratory has applied these ionic β-sheet peptides to a number of regenerative medicine applications, primarily as matrices for cell growth and transplantation. Peptides based on the RAD self-assembling motif were shown to be effective scaffolds for neuronal culture, demonstrating cell viability, efficient neurite outgrowth, and formation of active synapses.[10] Matrices made from similar peptide gels were able to encapsulate and support the growth of several different cells important in regenerative medicine. For example, encapsulating chondrocytes resulted in the production of extracellular matrix similar to cartilage,[11] liver progenitor differentiated into hepatocyte spheroid bodies in the material,[12] and neural stem cells were able to migrate out of the gels and into organotypic hippocampus brain slices.[13] Gels based on the RAD motif were also able to facilitate the repair of the severed optic nerve in a hamster in vivo, serving as a permissible matrix for axon regeneration through the lesion, and resulting in partial restoration of vision.[14]
Appending a peptide epitope to the architecture of these β-sheet driven assemblies afforded additional biological information and influenced cell phenotype. Zhang and coworkers synthesized RAD peptides with epitopes for cell adhesion (like RGDS from fibronectin, or IKVAV from laminin), or from bone marrow homing motifs, and found that these materials promoted neural stem cell attachment and differentiation more effectively than materials lacking the epitopes.[15] In another example, cell-adhesive nanofiber gels facilitated collagen production from human periodontal ligament fibroblasts, providing a potential matrix for repair of this tissue.[16]
Although the peptides described above are fairly short, much longer alternating charged sequences, not possible with solid-phase synthesis, can be produced by recombinant DNA technology. The Muller group described the synthesis of the (AEAEAKAKAEAEAKAK)9 peptide, possessing the hallmark alternating hydrophobic-hydrophilic pattern of many β-sheet peptides.[17] This polypeptide (which approaches the size of small proteins) formed fibrils 10–20 nm in diameter, which could produce self-supporting hydrogels at physiological pH when the charges were screened with electrolytes. Interestingly, the storage modulus of gels formed from this polypeptide was significantly higher than gels formed from a shorter peptide with a similar sequence, demonstrating peptide length as another parameter by which the properties of self-assembled biomaterials may be tuned.
2.2.2 β-sheet tapes
In addition to these self-complementary, ionic peptides, a number of groups have focused on alternative self-assembling motifs based on the β-sheet. For example, certain sequences of alternating hydrophilic and hydrophobic amino acids (e.g. QQRFQWQFEQQ) can assemble into β-sheet tapes and ribbons (Figure 1B), which subsequently bundle into larger fibers that can be gelled with triggers such as pH or added ions.[18–21] These properties allow a solution of initially monomeric peptides to form a gel after injection in vivo. The Fischer lab used this “viscosity switch” to create injectable lubricants with similar rheological properties to hyaluronic acid (a natural joint lubricant) for the treatment of osteoarthritis.[22] This peptide-based material performed well in cartilage-on-cartilage friction measurements (though it did not outperform hyalouronic acid itself) and, importantly, highlighted the advantage of materials that could assemble in situ. A similar β-sheet fibrillar material was used for the regeneration of enamel, employing an anionic sequence to promote the mineralization of the fibers.[23] Once again, the ability to inject a monomeric solution of peptide that then gelled in vivo was critical to the success of the material.
Collier and Messersmith developed a similar β-sheet motif, based on the related sequence QQKFQFQFEQQ (termed the Q11 peptide), to create self-assembled hydrogels that could be subsequently modified with cell adhesion peptides by using the transglutaminase enzyme.[24] Collier and coworkers further elaborated this platform, tuning the stiffness by crosslinking the nanofibers using native chemical ligation with cysteine and thioester-modified sequences.[25] In addition to changing the stiffness, sequences like RGD could be grafted onto the Q11 sequence, and the amount of epitope could be tuned by simply co-assembling two different molecules, highlighting the modularity inherent in self-assembly. Human umbilical vein endothelial cells (HUVECs) cultured in the stiffer, RGD-functionalized hydrogels demonstrated increased proliferation and expression of CD31 (a cell-cell adhesion protein). Several other epitopes (such as IKVAV, from laminin) could be incorporated into Q11 gels, and the materials were found to be biocompatible and non-immunogenic.[26] Furthermore, mixing two populations of pre-formed Q11 fibers (each with a different ligand) resulted in the spatial segregation of two epitopes in solution due to the high kinetic barrier for peptide exchange between fibers.[27] This heterogeneity in fiber composition allowed for more effective control of stiffness (by biasing inter- vs. intra-fiber crosslinking), whereas biological activity (e.g. cell survival) was higher when an RGD epitope was distributed throughout all the fibers rather than segregated on a subset of them. This last example further highlights the flexibility of self-assembly: multiple distinct nanostructures can be accessed from a common pool of monomers by carefully setting up the self-assembly conditions.
Although most of the works covered thus far have focused on synthesizing hydrogels for tissue engineering and regenerative medicine, the Collier lab extended the Q11 peptide for use as an immune system adjuvant (Figure 1C).[28] Attaching a 17-amino acid peptide (termed OVA) derived from chicken ovalbumin to the Q11 sequence resulted in nanofibers that displayed the antigenic epitope at high density. This multivalency endowed the material with a potent immunostimulatory effect, producing equal amounts of IgG1, IgG2, and IgG3 and higher amounts of IgM as compared to complete Freund’s adjuvant. Nanofibers lacking the OVA epitope did not elicit any immunogenic response, nor did the free OVA peptide itself, indicating that the self-assembled structure was critical for the potent biological effect observed.
2.2.3 Triblock peptides
Another approach to using β-sheet self-assembly has been developed by the Hartgerink group, based on the concept of “molecular frustration”.[29] Tri-block peptides were synthesized according to an ABA motif, where A represents a positively charged segment of two to four lysine residues. The B block was a (QL)n repeat (with n varying from 2–6), resulting in facially amphiphilic molecules. These peptides formed dimers (due to hydrophobic interactions between leucine residues) and the dimers aligned perpendicular to the peptide backbone axis as a result of β-sheet hydrogen bonds (Figure 1D). The supramolecular structures formed represented a balance between the associative forces of the hydrophobic packing and the hydrogen bonds, and the charge repulsion between the flanking A blocks. When the A block contained two lysines, and the B block was composed of five or fewer (QL) repeats, electrostatic repulsion between the lysines resulted in primarily micellar structures. When these counteracting forces were appropriately balanced, however, in the K2(QL)6K2 peptide, nanofibers were observed, and these fibers formed shear-thinning, self-healing gels upon ionic screening of the charges.[30] Increasing the A block to three or more lysines once again tilted the balance in favor of charge repulsion, and micelles were again observed. This work thus showed the delicate interplay between multiple noncovalent forces, and demonstrated the potential for nanostructure control by appropriate engineering of molecular interactions.
Subsequent studies by the Hartgerink group employed these ABA peptides for the construction of hydrogels containing cell adhesive epitopes (RGD; appended to the end of the charged A block) and a cleavage site for the matrix metalloprotinase-2, an enzyme produced by cells and that plays an important role in tissue remodeling, deposition of new ECM, and by extension cell behavior.[31] Mesenchymal stem cells showed optimal viability and spreading when both the adhesive and cleavable epitopes were included (as opposed to either epitope in isolation), once again demonstrating the advantage of self-assembly for the construction of multi-component biomaterials.
2.2.4 β-hairpin peptides
A variation on the β-sheet designs described above was pioneered by Schneider and Pochan in 2002, and utilized a β-hairpin structure as the self-assembling motif.[32] The authors synthesized a peptide with the sequence VKVKVKVKVDPPTKVKVKV (where DP denotes the D enantiomer of proline), which was unstructured at pH 5.5 due to charge repulsion between lysine residues, but upon increasing the pH to 9 folded into a hairpin structure due to formation of an intramolecular β-sheet (Figure 1E). These hairpins aggregated into higher-order fibrillar nanostructures and formed shear-thinning, self-healing hydrogels. Because this transition was based on supramolecular forces, it was highly reversible, with the hairpin reverting to a random coil structure (and concomitantly dissolving the gel) when the pH was lowered. Modifying this peptide sequence slightly to VKVKVKTKVDPPTKVKTKVKV resulted in a molecule that was unstructured at pH 9, and assembled into the β-hairpin when heated to ~60 °C.[33] This conformational change was driven by the entropic liberation of water molecules from the hydrophobic residues in a mechanism similar to the lower critical solution temperature (LCST) phenomenon seen with polymers like poly(N-alkyl acrylamide),[34] and represents another dynamic material trigger dependent on self-assembly. Systematically replacing one or two threonine residues with valine reduced this transition temperature to 40 °C and 25 °C, respectively, by changing the relative hydrophobicity of the molecule, once again due to the balance of multiple supramolecular forces. Further molecular engineering of the peptide structure allowed for stiffer gels by mixing enantiomeric peptides,[35] for gelation to be triggered by zinc ions,[36] or for degradation by matrix metalloproteinase-13.[37]
Hydrogels constructed from these self-assembling β-hairpin based materials proved to be attractive scaffolds for cell encapsulation and scaffolding. Mixing the peptide with cell culture medium sufficiently screened the positive charges to induce folding into the hairpin structure, even at physiological pH (Figure 1F).[38] Fibroblasts cultured on the surface of these gels were able to attach and spread (even in the absence of serum proteins or cell-adhesive epitopes), and proliferate, indicating the biocompatibility and lack of cytotoxicity of the material. These hydrogels could be further engineered to increase the gelation kinetics and allow for three-dimensional cell culture, and the material did not induce any inflammatory effect.[37] Gels made from these materials also demonstrated shear-thinning behavior due to the noncovalent cross-linking forces holding the gel together; upon removal of the force, the gel quickly regained its original stiffness due to re-formation of the disrupted crosslinks.[39] This property (which would likely not be possible with covalent crosslinks) allowed for cells to be encapsulated in a gel, and then delivered through a syringe to a desired delivery site, at which the gel state of the material would re-form. Interestingly, β-hairpin hydrogels were found to be innately toxic to both Gram-positive and Gram-negative bacteria, yet not harmful to mammalian cells.[40] Bacterial membranes were lysed upon contact with the gel, whereas fibroblasts and erythrocytes were not affected, an effect that the authors surmised could be due to the differential interaction of bacterial membranes with the cationic peptide.
2.2.5 Peptides based on amyloid proteins
One of the starkest examples of β-sheet peptide aggregates comes from naturally-occurring amyloid fibers, which are responsible for the degenerative effects of diseases like Alzheimer’s.[41, 42] However, the self-assembly inherent in amyloidogenic sequences has also been adapted to the synthesis of biomaterials. Nomizu and coworkers discovered that a peptide termed A208 with sequence AASIKVAVSADR (containing the IKVAV epitope), from the mouse laminin α-1 chain, formed amyloid-like fibrils.[43] In addition to the inherent activity of the IKVAV sequence, the authors introduced an RGD sequence into the material to further promote cell adhesion. Interestingly, scrambling the A208 amino acid sequence abolished any self-assembly, indicating the importance of the natural protein’s motif for nanostructure formation. Hydrogels formed using this peptide were able to mediate fibroblast adhesion through laminin receptors (via the IKVAV epitope) or integrins (via the RGD epitope). In a separate report, Gras and coworkers used an 11-amino acid sequence (YTIAALLSPYS) found in human transthyretin, a protein known to form amyloid fibers,[44] and appended an RGD sequence in order to facilitate cell adhesion. The peptides self-assembled into twisted ribbon nanostructures, around 30 nm in width, and the density of the epitope could be adjusted through co-assembly with the RGD-lacking base sequence. As with the previous example, fibroblasts attached to the material and formed integrin-mediated focal adhesion complexes. These examples demonstrate the potential for mining the rich diversity of protein structures for novel self-assembling motifs beyond the simpler sequences described previously. An outstanding challenge of these approaches, however, is that it is harder to rationally engineer such sequences until computational models more effectively predict the design rules for more complicated self-assembling peptide sequences.
2.3 α-helical peptides
Although the β-sheet is the most commonly used secondary structure motif in peptide-based materials, self-assembly based on the α-helix has been extensively studied as well, and is just now beginning to be applied to the biological setting. Ubiquitous structural motifs in most natural proteins, a-helices result from the hydrogen bonding of a peptide backbone amide hydrogen to a backbone carbonyl four residues away. This motif results in a right-handed helical twist configuration with a periodicity of 3.6 amino acids per turn. The formation of α-helices is determined by the amino acid sequence, and is dependent on the inclusion of both hydrophilic and hydrophobic amino acid residues such that the resulting helix has a hydrophobic and a hydrophilic face.
Synthetic peptides can be designed (usually with inspiration from naturally occurring helices in proteins) to assemble into α-helical motifs. The design of these systems can be further tuned to elicit higher-order self-assembly into a coiled-coil conformation[45] by taking advantage of the amphiphilic nature of the helix’s faces (Figure 2A,B).[45] In this coiled-coil structure, the α-helices are distorted such that the periodicity of their turns is reduced to 3.5 amino acids, and heptad repeats (corresponding to two such 3.5-residue turns) with the pattern HPPHPP (where H denotes a hydrophobic and P a hydrophilic residue) tend to favor the coiled-coil configuration. A number of design principles to promote the coiled-coil interaction have been elucidated in the past two decades and have been used to promote the design of novel α-helix-based materials. In particular, residues with positive and negative charges at certain positions can help enforce salt bridge interactions, further stabilizing the coiled-coil architecture.
Figure 2. Peptide materials using the coiled-coil motif.
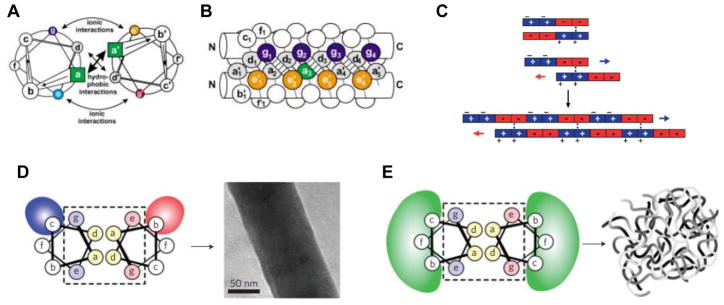
(A) Top-down view of two α-helices interacting via coiled-coil interaction. The a and d residues in the heptad repeat associate due to hydrophobic interactions, whereas ionic interactions like salt bridges between the e and g residues further stabilize the structure. (B) Two side-on views of the same coiled-coil structure, further illustrating the intermolecular associations that hold it together. (C) Redesigning the a-helix motif to create “sticky ends” based on charge-charge interactions allowed the assembly of peptides into nanofibers. (D) The coiled-coil interactions originally designed by the Woolfson group in Ref. 47, where ionic interactions between the b and c positions resulted in the formation of fibers, but not hydrogels. (E) Using weaker interactions (e.g. hydrophobic, H-bonding) between residues b, c, and f resulted in hydrogels. [(A, B): ref. 45; (C): ref. 50; (D, E): ref. 48]
The body of literature exploring the molecular design principles to promote α-helical assemblies is quite extensive and has been reviewed elsewhere.[46, 47] One example of a purely α-helical peptide that was used to construct a hydrogel, was reported by the Woolfson group in 2009.[48] In accordance with the coiled-coil rules,[49] each peptide in Woolfson’s design contained heptad repeats with the sequence abcdefg, where a and d represent hydrophobic residues such as leucine and isoleucine (to promote the coiled-coil interaction), whereas b, c, e, f, and g all represent hydrophilic residues. In a previous work,[50] Woolfson and coworkers demonstrated coiled-coil a-helices with “sticky end” overhangs (as opposed to traditional, fully “in-register” coiled-coils) to promote association into fibers (Figure 2C). In that report, the b and c positions were oppositely charged, inducing electrostatic attraction between two coiled-coils, and causing the fibers to thicken and settle out of solution (Figure 2D). However, this effect prevented the fibers from gelling, and thus posed an obstacle for using the peptides for cell culture applications. In order to promote gelation, Woolfson and coworkers replaced the charged amino acids at positions b and c with alanine or glutamine (Figure 2E), which are expected to have weaker interactions (either hydrophobic or hydrogen-bonding). Furthermore, rather than rely on a peptide to self-assemble with itself, complementary pairs of peptides, with matched noncovalent interactions, were synthesized such that gelation would only occur upon mixing.
As a result of the strategy of systematic variation in peptide sequence, hydrogels with different thermophysical properties could be constructed with great control. For example, by building coiled-coils that participated in inter-helix hydrogen bonding, the hydrogels would melt upon heating. When hydrophobic forces were used instead of hydrogen bonds to promote fiber interaction, the gels became stronger upon heating.[48] This ability to control gelation by using heat provides a versatile scaffold for cell culture, especially when gel dissolution at elevated temperatures is desired. The α-helical peptide gels proved highly cytocompatible, and PC12 cells cultured in them were able to proliferate and differentiate similar to cells cultured in collagen and Matrigel, despite the greater complexity of those materials. Thus, although α-helical biomaterials are still at a nascent stage, their precise supramolecular architecture and tunable properties suggest that they will find an increasingly important role in the future.
2.4 Collagen-mimetic peptides
Collagen makes up the majority of protein content in humans and represents a key structural component of both load-bearing tissues and extracellular matrix (ECM).[51] Collagen-mimetic biomaterials are under investigation, where the format of their peptide sequence is exploited in order to produce the desirable structural properties observed in nature or to induce a specific biological function. Collagen is defined by its triple helical structure, in which three linear peptidic chains are supercoiled into high-aspect-ratio supramolecular fibrils, usually of ~ 300 nm in length. Amino acid patterns that promote collagen assembly contain triads, usually of Gly-Pro- X or Gly-X-Hyp where X represents other naturally occurring amino acids and Hyp denotes hydroxyproline. The high incidence of glycine in collagen is necessary for its supramolecular assembly because glycine’s lack of a side chain precludes steric interference of the tight coil at about every third residue.[51]
Collagen’s strength in connective tissues underlies the push toward collagen-mimetic tissue engineering. Collagen itself has been used extensively as a biomaterial (see Section 4 below for reviews that cover this field), but synthesizing short collagen-mimetic peptides that can recapitulate the effects of the full-length protein (which is ~1,000 amino acids in size) has been a long-standing goal of biomaterials engineering due to the greater flexibility it would afford. Pioneering work by the Raines laboratory exemplifies the design of short synthetic “sticky-end” collagen triple helices. This motif is made up of covalently linked collagen peptides that enforce the correct register, resulting in nanofibers 1 nm in width, and approaching 1 μm in length.[52]
The Chmielewski group described the first collagen-mimetic hydrogel for cell culture applications in 2009.[53] In this work, the authors synthesized a peptide with a central Pro-Lys-Gly peptide segment flanked on either side by four Pro-Hyp-Gly repeats in order to promote triple helix formation analogous to collagen itself. In order to promote gelation, three synthetic groups capable of binding to metal ions were appended to the peptide: (1) a nitriloacetic acid (NTA) moiety at the N-terminus; (2) a His2 sequence at the C-terminus; and (3) a bipyridyl (bpy) moiety on the lysine in the center of the peptide (Figure 3A). The peptides first self-assembled into collagen-like triple helices, and subsequent addition of Ni2+ resulted in these triple helices further assembling into a cross-linked hydrogel network due to metal coordination between triple helices. Other divalent metals similarly induced gelation, and addition of EDTA to chelate the metal ions resulted in dissolution of the formed gel. Furthermore, HeLa cells could be incorporated into the gels and remained alive and proliferative over several days, with no decrease in viability compared to tissue-culture plates. This example demonstrated the power of combining a self-assembly motif culled from nature (that of the collagen triple helix) with the functional flexibility offered by chemical synthesis of small peptides, and illustrates a novel avenue for engineering biomaterials.
Figure 3. Hydrogels constructed from collagen-mimetic peptides.

(A) The structure of a peptide synthesized by the Chmielewski group, with metal-binding groups at the ends and middle of a peptide bearing collagen-mimetic repeats. The peptides self-assemble into triple helices, and addition of metal ions crosslinks them into a hydrogel. (B) Three peptides with collagen-like repeats, developed by the Hartgerink laboratory, self-assemble into staggered, heterotrimeric helices. Interactions between the ends and middle of these helices result in the formation of bundled nanofibers that form a hydrogel. (C) Schematic of the sticky-end interactions that result in formation of the nanofibers observed. Arrows indicate charged residues that promote additional inter-helical bundling. [(A): ref. 53; (B, C): ref. 54]
The Hartgerink group reported the first purely peptide-based (without any additional chemical functionality) collagen-mimetic hydrogel in 2011.[54] In previous work synthesizing heterotrimeric collagen-mimetic helices (where three different strands formed a single ABC complex), Hartgerink and coworkers described the importance of inter-strand hydrogen bonds between cationic and anionic amino acids.[55, 56] Using a similar rationale, the authors synthesized a peptide with the sequence (Pro-Lys-Gly)4(Pro-Hyp-Gly)4(Asp-Hyp-Gly)4 to form staggered triple helices with lysine and aspartate resides available to promote hydrogen bonding interactions between the “sticky ends” (Figure 3B,C). The peptides self-assembled into remarkably monodisperse (as observed by AFM) fibers composed of several bundled triple helices; no widespread and nonspecific aggregation was seen, as with many previously reported collagen-mimetic systems. At concentrations of 0.5 wt%, these fibers formed hydrogels, as confirmed by SEM, within a few hours. These gels were remarkably similar to native collagen in their properties, despite being comprised of peptides with only 36 amino acids (as opposed to 1,000 for collagen). The storage modulus (G’) was similar to that of native collagen hydrogels, the gel dissolved above 40 °C, similar to the temperature at which the collagen triple helix unfolds. In addition, the synthetic gels could be enzymatically broken down by collagenase type IV. This elegant report highlighted the benefits and great potential inherent in understanding the self-assembly behavior of peptides and rational design of supramolecular interactions to bias the structure of the materials formed. These collagen-mimetic hydrogels will undoubtedly find wide applicability in a range of cell culture and biomaterials applications in the future.
Unlike the above examples, which have all relied on purely synthetic peptides, the Yu laboratory developed a different approach, designing peptides that could integrate with natural collagen. Yu and coworkers synthesized short (~30 amino acids) collagen-mimetic peptides (CMPs) with the canonical proline-hydroxyproline-glycine triplet repeats found in collagen, hypothesizing that these “helicogenic” molecules would be able to associate collagen that had been partially denatured and integrate into it via self-assembly.[57] Indeed, collagen films treated with fluorescently modified peptides showed integration of the dye into the film; using a peptide that lacked the appropriate collagen-mimetic sequence did not. This approach allowed for the functionalization of native collagen by employing the specific self-assembly mechanism that forms the triple helix, without relying on chemical modification. As a proof of principle of this method, collagen films could be modified with a collagen-mimetic peptide appended to a 2 kDa PEG chain, a polymer known to prevent cell adhesion. As expected, modified films prevented cell adhesion and migration of both fibroblasts and breast epithelial cells. The Yu laboratory extended these CMPs to a number of self-assembled biomaterials. Synthesizing a CMP with a branched, poly-glutamate section at the N terminus, they were able to create a molecule that could not only bind to collagen films, but also associate (via the multiple negative charges) to growth factors like VEGF.[58] The modified films were able to stimulate HUVEC cells to form cell-cell contacts and tube structures seen in angiogenesis. Extending this idea, the Yu laboratory synthesized a different CMP bearing a peptide sequence derived from VEGF, and known to stimulate angiogenic activity.[59] Incorporating this hybrid peptide into collagen films and gels induced endothelial morphologies indicative of angiogenesis, but without having to use full-length growth factor. Patterning the distribution of the VEGF-mimetic CMP within the collagen scaffold allowed for spatial control of the desired biological effect, paving the way for more precise 3D manipulation of angiogenesis within a biomaterial.
3. Peptide hybrid materials
3.1 Introduction
As demonstrated in the previous section, peptide-based materials can be designed to self-assemble through their amino acid sequence, using design rules derived from natural proteins as well as fundamental physicochemical studies of peptide secondary structure. And alternative way to guide self-assembly, however, is through the incorporation of additional chemical functionality with known assembly properties. This functionality can determine the nanostructure formation on its own, or (as in most cases) complements the peptide’s natural, “built-in” self-assembly tendencies. The majority of such hybrid peptide materials are amphiphilic in nature, attaching a hydrophobic or aromatic moiety onto a hydrophilic peptide. The hydrophobic effect and aromatic π-stacking interactions are well understood, relatively straightforward to design (compared with, for example, more complex peptide or protein folding), and can employ simple and biocompatible components like lipids. Even within the “like dissolves like” paradigm, however, a large variety of supramolecular nanostructures can be accessed by appropriately designing the domains that make up a hybrid material.
This section will cover two broad classes of peptide hybrid materials. The first, developed and widely employed by the Stupp laboratory, are peptide amphiphiles comprised of a linear alkyl tail appended onto a peptide sequence, driving the formation nanofibers through hydrophobic collapse and intermolecular β-sheet hydrogen bonding. The second category involves very short peptides with aromatic moieties that, through a combination of hydrophobic and π-stacking effects, drive their assembly into fibrillar morphologies. A third category of hybrid materials involves peptide/protein-polymer conjugates, whereby the self-assembly of the biological component drives a sol-gel transition in the material. A number of elegant examples exist that employ these peptide-polymer hybrids, especially in the context of responsive/dynamic materials driven by the biological component, though few have been used directly in biomaterials applications as covered in this review. Nonetheless, these systems represent an attractive and fertile area for future research and have been reviewed in depth elsewhere.[60]
3.2 Peptide amphiphiles
Peptide amphiphiles (PA) are characterized by a peptide sequence with an appended alkyl tail. The first example of a PA was reported in 1995 by Tirrell and coworkers, in which a dialkyl ester tail was appended onto a peptide sequence from collagen, resulting in the assembly of a monolayer at the air-water interface.[61] In 2001, the Stupp laboratory reported a novel PA structure, whereby an unbranched palmitic acid tail was appended to the N terminus of a peptide sequence (Figure 4A).[1] Driven by the hydrophobic collapse of the alkyl tail, as well as the conical shape of the molecule, these PAs self-assembled in aqueous solution into high aspect ratio cylindrical nanofibers, ~6 nm in diameter and often several microns in length (Figure 4B). The PA in this report was designed to possess several cysteine residues to covalently trap the supramolecular structure (which formed only at low pH) through disulfide bridge formation. In addition, a phosphoserine residue was included to promote nanofiber mineralization, as well as the cell-adhesive RGD epitope. The close packing of the PAs into the nanofiber structure resulted in an extremely high density of the peptide epitope, and allowed for the mineralization of hydroxyapatite crystals on the repeating surface of phosphoserine residues presented, with the c axis of the crystals aligned along the fiber. In a follow-up report from the Stupp laboratory, the design parameters for PAs was extensively explored, and revealed a wide range of potential molecules with different amino acid sequences and self-assembly triggers.[62]
Figure 4. Peptide amphiphiles and their applications.

(A) Chemical structure of a representative peptide amphiphile (PA), and its assembly into nanofibers, with the four primary regions highlighted. Region I: unbranched alkyl (usually C16) tail; region II: a β-sheet forming segment to promote H-bonding along the fiber axis; region III: charged amino acids for solubility; region IV: a peptide epitope to imbue the material with biological signaling. (B) TEM image demonstrating the self-assembly of PAs into high aspect ratio nanofibers, often several microns in length. (C) Injection of a PA solution containing the IKVAV epitope into the damaged spinal cord of a mouse resulted in regeneration of both ascending and descending axons (ii), whereas a control saline injection did not (i). (D) A VEGF-mimetic PA was able to stimulate angiogenesis in a mouse hind-limb ischemia model, restoring more blood flow to the injured limb after 28 days (top image) compared with the peptide alone, or a PA with a non-bioactive sequence. (E) Schematic of self-assembled sac formation upon injection of a polyelectrolyte solution into an oppositely charged PA solution. (F) SEM demonstrating the hierarchical structure of the sac membrane, including the dense arrangement of fibers perpendicular to the membrane (region 3). (G) SEM images of a PA-based monodomain gel, with fibers aligned in the direction of shear (left); a PA solution that was not heat-annealed shows randomly oriented fibers (right). [(A, B): ref. 79; (C): ref. 64; (D): ref. 67; (E, F): ref. 68; (G): ref. 70]
The highly modular nature of PA molecules has made them well-suited as biomaterials for regenerative medicine. The nanofibers that result from PA self-assembly can often be gelled by screening their surface charges, resulting in a material that can be readily injected and form a gel in situ. Additionally, the high density of peptide epitopes present on their surface can result in an acutely potent cellular response. As with other peptidic biomaterials, PAs are highly biocompatible and biodegradable because they are composed of naturally occurring amino acids and lipids. In 2004, the Stupp and Kessler laboratories cultured neural progenitor cells (NPCs) in a gel comprised of PAs bearing the IKVAV epitope from laminin, relying on simple charge screening to gel the soluble nanofiber-cell mixture.[63] Intriguingly, the NPCs cultured in the gel selectively differentiated into neurons, while suppressing their differentiation into astrocytes, compared with either laminin or poly(D-lysine) coated surfaces. This remarkable capability of the PA gel was attributed to the extremely high density of IKVAV epitope presented on the nanofiber surface (~1015/cm2), around three orders of magnitude greater compared with the laminin-coated surface. Further supporting this theory, this effect was found to be highly dependent on the concentration of the IKVAV PA in the nanofiber; incorporating the IKVAV PA below ~50 mol% (relative to a non-bioactive “filler” PA) resulted in loss of this biological activity. The soluble IKVAV peptide alone also did not promote this dramatic effect, demonstrating that epitope density and presentation on the nanofiber was critical for bioactivity. In a separate report, the Stupp and Kessler groups showed that PA gels could be used for the treatment of acute spinal cord injury.[64] A solution of PA bearing the IKVAV epitope was injected into the crushed spinal cord of a mouse, resulting in gelation in situ due to charge screening by naturally present ions. The nanofiber gel reduced the formation of a glial scar at the site of injury, promoted the regeneration of both descending motor and ascending sensory axons through the material (Figure 4C), and resulted in a marked functional improvement in the mice compared with a saline injection.
In addition to neural regeneration, PAs have been used in a wide range of other applications. Growth factors are often critical for regeneration, but their short-lived nature in vivo requires a prolonged release strategy aided by a biomaterial implant. The Stupp laboratory synthesized a PA bearing an LRKKLGKA epitope derived from a consensus sequence for binding heparin in order to synthesize gels that could slowly release growth factors with heparin-binding domains.[65] Gels made from this PA-heparin mixture were able to sustain delivery of VEGF and FGF-2, and were more effective at promoting angiogenesis in a rat cornea model than collagen gels containing heparin, or heparin itself. Importantly, the PA-based materials were more effective than collagen gels or heparin without the PA matrix at sustained delivery of these therapeutic proteins. The Stupp laboratory has also demonstrated a different approach in which PAs are endowed with affinity for TGFβ-1, a growth factor important in cartilage regeneration.[66] Here, phage display was used to find a peptide epitope that bound TGFβ-1; appending this sequence onto a PA resulted in gels that could slow the release of the protein for controlled delivery. When injected into a chondral defect in a rabbit model, the PA gel was able to promote cartilage regeneration even in the absence of externally applied growth factors, acting instead to concentrate endogenous growth factors at the site of injury.
PAs have also been used to present growth factor mimetic epitopes in order to replace the use of the expensive natural proteins. In 2011, the Stupp laboratory synthesized a PA bearing an epitope that mimicked the receptor-binding domain of VEGF, and had been previously shown to activate VEGF signaling.[67] PA nanofibers bearing this sequence were able to activate both VEGF receptors, to a greater degree than the peptide or even VEGF itself, most likely due to the extremely high density of the peptide signal afforded by the self-assembly process. The PA was also able to stimulate angiogenesis in a chicken chorioallantoic membrane assay, once again showing a marked increase over the free peptide. This result was specific for the epitope; changing the amino acid sequence to mutate several key residues resulted in a non-functional PA. The PA material was also highly functional in vivo in a mouse hind-limb ischemia model, restoring blood flow following excision of the femoral artery (Figure 4D), and promoting significant functional recovery of the injured limb. Furthermore, due to the nanofiber morphology of the PA, the material injected was still present in the limb at least four weeks after the injection, whereas free growth factors are cleared on the order of minutes or hours. The success of this synthetic approach towards growth factor therapy indicated the great potential for PAs as protein mimics and replacements, obviating the need to use expensive growth factors in conjunction with controlled release strategies.
All of the PA-based therapies described thus far relied on randomly entangled networks of PA fibers. One of the distinct advantages of self-assembly, however, is the ability to form hierarchically ordered structures by employing multiple supramolecular forces that operate across different length scales. In 2008, the Stupp laboratory reported that mixing a solution of a polyelectrolyte, like alginate or hyaluronic acid, with a PA solution of opposite charge resulted in the formation of free-standing sacs and membranes at the interface of the two solutions (Figure 4E).[68] The membranes resulted from a dynamic, hierarchical self-assembly process between the polymer and the PA nanofibers: a dense barrier formed initially, preventing chaotic mixing of the two solutions, followed by reptation of the electrolyte through the barrier as a result of an osmotic pressure difference between its two sides. The result was a dense layer of PA-coated fibers perpendicular to the interface between the solutions, creating a membrane with a highly ordered structure over several length scales (Figure 4F). In a subsequent report, membranes were synthesized using hyaluronic acid in conjunction with the heparin-binding PA described previously, resulting in a material that could release growth factors like VEGF for applications like angiogenesis.[69] Yet another elegant example of hierarchical self-assembly was reported by Stupp and coworkers in 2010, when they discovered that heating certain PA molecules to 80 °C for a short time resulted in the formation of two-dimensional plaques that broke into bundles of nanofibers upon cooling.[70] Upon application of a gentle shearing force (such as that of manual pipetting) and gelation with divalent ions, these fibers organized into a birefringent, monodomain gels with a remarkable degree of alignment over arbitrary macroscopic (e.g. centimeter) length scales (Figure 4G). These highly hydrated (>99%) “noodle” gels served as attractive scaffolds for cell encapsulation, given that very low shear forces were necessary for alignment. Mesenchymal stem cells were not only highly viable in the material, but also aligned with the long axis of the noodle, demonstrating the great potential for these materials to serve as scaffolds for intrinsically anisotropic cell types, like neurons or cardiomyocytes. These two examples highlight the great potential inherent in self-assembly to organize materials across multiple length scales, a feat which is significantly more difficult with covalent interactions.
The examples highlighted above represent some of the most promising applications of PAs as biomaterials for regenerative medicine. Indeed, PAs have been applied successfully to a range of other medical applications, including bone and enamel regeneration,[71–74] cancer therapy,[75] islet transplantation,[76] and others covered in a variety of review articles.[77–79] In the specific systems designed for bone regeneration, PAs were designed first to nucleate hydroxyapatite crystals in biomimetic fashion as a potential source of bioactivity.[1, 71] This approach was inspired by previous work in the Stupp laboratory involving the nucleation of hydroxyapatite by polyelectrolytes and polyamino acids to create materials known as organoapatites.[80] [81]
3.3 Short aromatic peptide amphiphiles
A conceptually different approach to self-assembling amphiphilic hybrid materials involves the use of short (e.g. 2–5) peptides with an amphiphilic nature due to either the side chains, an N-terminal modification, or both. Reches and Gazit first reported the ability of aromatic dipeptides to spontaneously assemble into nanotubes and vesicles for materials applications.[82, 83] Soon thereafter, Xu and coworkers reported that di-peptides modified with an N-terminal fluorenylmethyloxycarbonyl (Fmoc) moiety (often used as a protecting group in solid-phase peptide synthesis) were able to self-assemble into nanofibers and hydrogels in water. The Fmoc groups drove the supramolecular association of the molecules through hydrophobic and π-π stacking effects, and inter-molecular β-sheet hydrogen bonds between the dipeptides further enforced the structures. The simplicity of these molecules makes them a particularly elegant and potentially scalable approach towards peptide-based materials; being only 2–3 amino acids in length also allows for a more extensive exploration of sequence space and its influence on nanoscale morphology.
The Ulijn group explored the use of Fmoc-dipeptide hydrogels as biomaterials for cell culture, synthesizing a library of seven molecules incorporating glycine, alanine, leucine, and phenylalanine in order to tune their relative hydrophobicity.[84] All but one of the molecules tested did indeed form hydrogels in water, at concentrations around 0.5 wt%, and at pH values ranging from acidic (< 4) to neutral, depending on the exact sequence. These dipeptides could also be mixed with other dipeptides, or even Fmoc-lysine to form heterogeneous systems, once again highlighting the modularity afforded by self-assembly. Fiber-like morphologies (with diameters ~ 50 nm) were visible by cryogenic scanning electron microscopy (SEM), indicating that multiple supramolecular stacks were bundled together into the observed nanostructures (Figure 5A). The gels formed from the dipeptides that could gel at physiological pH (e.g. Fmoc-FF) were cytocompatible, and allowed for the culture of chondrocytes either on top of a pre-formed gel, or within the three-dimensional gel matrix. The cells were viable and able to proliferate, especially in gels that included 50 mol% Fmoc-lysine, which the authors speculated was due to the preference of chondrocytes for positive charges. In two follow-up studies, the Ulijn group explored the construction of multi-component biomaterials based on this platform. Mixing their Fmoc-FF dipeptide with Fmoc-modified amino acids (e.g. lysine, glutamic acid, arginine, or serine) allowed them to tune the gel stiffness, and certain cell types were found to be more compatible with certain hybrid materials.[85] In a separate report, an Fmoc-RGD peptide was incorporated with the Fmoc-FF sequence in order to endow the material with cell adhesive functionality; once again, the self-assembled nature of the material allowed for a range of compositions to be quickly synthesized by simply mixing the two components in a desired ratio.[86] Human adult dermal fibroblasts could be cultured in the material, attaching and spreading in the RGD-modified material in an integrin-dependent manner; blocking the cell integrins or using a control Fmoc-RGE sequence (which is known not to bind cell receptors) abolished cell adhesion and spreading.
Figure 5. Self-assembling short aromatic peptides.
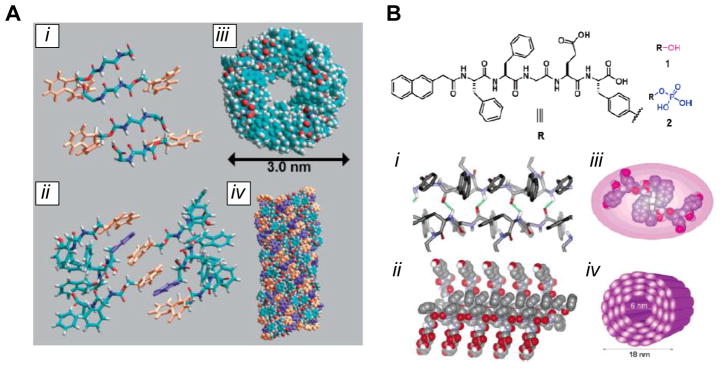
(A) Self-assembly of the Fmoc-FF peptide, as determined by computational modeling. The peptide forms antiparallel β-sheet hydrogen bonds (i); different β-sheets are brought together by π-π stacking between Fmoc groups (orange), with interdigitated phenyl side groups (purple) from the phenylalanine residues (ii); the twist of the β-sheets results in a cylindrical arrangement, shown in top (iii) and side (iv) views. (B) Structure of the Nap-FFGEY peptide, showing the site of phosphorylation that drives the structural transition. The peptide forms a β-sheet structure (i, ii), resulting in further association into nanotube-like structures (iii, iv). [(A): ref. 4; (B): ref. 87]
The Xu group—which first discovered the self-assembly of the Fmoc-FF peptide—has also extensively employed short aromatic peptides for materials construction. Synthesizing the Nap-FFGEY peptide (where Nap denotes naphthalene) resulted in a hydrogel with nanofiber morphology.[87] Adding a kinase to phosphorylate the terminal tyrosine residue, however, increased the hydrophilicity of the molecule and disrupted the balance of supramolecular forces holding the fibers together, resulting in dissolution of the gel (Figure 5B). Interestingly, injecting the soluble phosphorylated material into a mouse resulted in conversion to a gel due to the action of naturally occurring phosphatase enzymes in vivo. This supramolecular switch could thus potentially be used to deliver encapsulated cells or to create a scaffold in situ by relying on natural enzymatic activity coupled to the self-assembly of the material. In a separate report, the Xu laboratory employed Nap-modified dipeptides to create hydrogels in vivo when injected into a rat.[88] By using D-amino acids to construct the peptides, the gels formed were resistant to protease activity, and could prolong the release of radioactive tracers as model drug compounds. Extending this molecular design to append the Nap group at the C-terminus resulted in a molecule that only formed nanofibers and gels within a narrow pH window of 5–6, due to the protonation state of the now-free N-terminus, demonstrating how molecular design can dramatically influence self-assembly behavior.[89] Interestingly, this material was quite toxic to cancerous HeLa cells, while leaving healthy cells unharmed, a result that might be due to the cancerous cells’ acidification of their local environment, resulting in a higher local concentration of cytotoxic cationic fibers.
4. Biomaterials from proteins and large polypeptides
4.1 Introduction
Proteins represent another broad category of biological molecules that have been employed to generate self-assembling biomaterials. Compared to peptides, proteins have the disadvantage that they cannot be made on solid support, and thus incorporating unnatural functionality is usually more difficult. However, recombinant protein production (using E. coli or some other host), combined with genetic engineering can often afford the requisite quantities of proteins in reasonable yield and cost. Like peptides, proteins are usually biocompatible and degradable, but they often possess greater functionality, both for self-assembly as well as for biological effect, due to their greater complexity. The extracellular matrix itself is a collection of self-assembled proteins (e.g. collagen, fibronectin, laminin), so proteins are a natural choice for many biomaterials applications. In addition, their properties and geometries can vary greatly due to their complex structure (as opposed to primarily linear peptides), and a wide range of proteins exists in nature and can serve a source of both inspiration and raw material.
A number of naturally-occurring proteins—including collagen, elastin, or spider silk—self-assembly spontaneously in their natural settings, and a number of laboratories have used these molecules (often in conjunction with genetic engineering to introduce adhesion or differentiation epitopes) for the construction of biomaterials.[60, 90, 91] This area represents a vibrant and highly promising area of research for biomaterials too large to effectively cover in this review, so only proteins-based materials that are engineered de novo will be discussed herein.
4.2 Coiled-coil protein materials
One of the earliest examples of protein-based biomaterials came from Tirrell’s group in 1998, and used alpha-helical coiled-coil interactions to assemble a supramolecular hydrogel.[92] Seeking to rationally engineer a hydrogel material, Tirrell and coworkers designed a triblock polypeptide, with leucine zipper motifs separated by an unstructured ((AG)3PEG)10 amino acid sequence (Figure 6A). Leucine zippers are alpha helical sections with a heptad repeating sequence, and form a supramolecular association complex based on both hydrophobic and charged interactions between amino acid side chains (Figure 6B). The authors constructed a gene for the desired polypeptide and expressed it recombinantly in E. coli, and the material formed a hydrogel at higher concentrations. As expected, the gel could be dissolved to a viscous liquid by increasing the temperature, and thus denaturing the leucine zipper structure, or decreasing the pH, which protonated acidic residues necessary to stabilize the alpha helix. By engineering the exact leucine zipper sequence to promote inter- vs. intra-molecular association, the degradation rates of these gels could be tuned over two orders of magnitude, demonstrating the utility of genetic engineering to adjust protein-based material properties.[93] A number of other examples have been reported in hybrid materials whereby coiled-coil associations serve as the crosslinks between synthetic polymers.[94–96]
Figure 6. Protein-based self-assembled materials.

(A) Structure of the telechelic, coiled-coil based polypeptide designed by the Tirrell group. (B) Association of the end helices into coiled-coils results in hydrogel formation. (C) Chemical structure of the amphiphilic KxLy block co-polypeptide designed by the Deming laboratory. (D) The leucine α-helices self-assemble due to the hydrophobic effect, forming extended tapes and fibers. (E) Naturally-derived WW domains form a noncovalent complex with PPxY domains. The strength of the interaction between the two can be tuned by changing the specific motif; in this example, the Nedd4.3 variant of the WW domain has a weaker interaction with PPxY than the CC43 variant. (F) Incorporating repeating WW and PPxY domains into separate protein polymers allows for the construction of a hydrogel through protein association interactions. [(A): ref. 92; (B): ref. 97; (C, D): ref. 106; (E,F): ref. 107]
The Tirrell and Kornfield groups also applied these coiled-coil telechelic proteins to cellular applications, by synthesizing a shear-thinning hydrogel to serve as a cell delivery system.[97] Applying a shearing force (e.g. injection of the material through a syringe) resulted in a transition from an elastic solid to a liquid, with a decrease in storage modulus by three orders of magnitude. Upon removal of the shearing force, the material regained 90% of its modulus, allowing it to reset into a gel. Because of the reversibility of the self-assembly interactions, cross-links disrupted by shear could readily re-form once the force was removed, once again highlighting the benefit of self-assembly in these materials. The gels were highly biocompatible, and encapsulated CHO cells showed > 95% survival in the material, both before and after applying the shear force, indicating the suitability of the polymer for delivering cells. Interestingly, the length of the unstructured chain between the self-assembling motifs did not affect this shear-thinning behavior, allowing for a potentially diverse set of materials to be constructed in a highly modular fashion.
Although we have limited our description of peptide-based triblock copolymer materials to the coiled-coil examples described above due to their direct application to cell scaffolding and transplantation studies, we would like to point out several other telechelic protein polymers that have been developed, based on elastin association or collagen self-assembly, and which demonstrate the rich scope of self-assembly motifs for the construction of novel materials.[98–102]
4.3 Amphiphilic block co-polypeptides
A conceptually different approach for the synthesis of self-assembled polypeptide biomaterials was pioneered by the Deming lab, and employs amphiphilic block copolymers comprised of alternating sections of charged and hydrophobic amino acids.[103] Deming and coworkers used transition metal catalyzed α-amino-acid N-carboxyanhydride polymerizations to synthesize polymers with sequences like KxLy, KxVy, or ExLy, with x = 160–180 and y = 20–40 (Figure 6C). The hydrophobic segments formed α-helices (for leucine) or β-sheets (for valine). When dissolved in water, the copolymers did not form micelles (as the authors had expected, given the behavior of other block copolymers), but self-assembled via hydrophobic packing into twisted ribbon-like tapes that formed hydrogels (Figure 6D). Unlike the Tirrell hydrogels described above, these materials did not rely on naturally-occurring protein motifs (like the coiled-coil) for the self-assembly, nor did they use recombinant production to make the polymers, given that precise control over sequence was not necessary.
Despite this simpler design, the resulting hydrogels were remarkably stable, even at 90 °C, and the stability could be tuned by the relative length of the hydrophobic section (shorter segments resulted in weaker gels), or by adding salt to screen the charged sections (which allowed gel dissolution at lower temperatures). The hydrophobic packing of the leucine/valine blocks was highly dependent on the structure (helical or sheet) of these segments; using a racemic leucine mixture (which could not form an α-helix), resulted in weaker gels. Similar to previous examples of self-assembled hydrogels, these materials exhibited both shear-thinning and rapid self-healing abilities due to the reversible nature of the supramolecular cross-links holding them together. In subsequent studies, the Deming group expanded this approach to tri-block[104] and penta-block[105] copolymers (e.g. KxLyKz or KxLyKzKxLy, respectively) in order to further tune the morphology and properties of the hydrogels.
In a follow up report, the Deming group studied the biocompatibility of these amphiphilic block copolypeptide gels in the central nervous system, for eventual use as scaffolds for cell transplantation or nerve repair.[106] The stiffness of a biomaterial is one of the most important properties for influencing cellular behavior, and matching the stiffness of the native tissue is often important to ensure the desired biological effect. A number of di-block gels (containing K, R, or E residues for the charged block, and L for the hydrophobic block) possessed storage moduli (G’) similar to that of brain tissue (~200 Pa), and were injected into the caudate putamen nucleus of the brain for evaluation. As shown previously, the gel properties could be tuned by the relative identity and length of the two blocks, and ultimately the authors focused on the KxLy gels due to their favorable properties, as well as high compatibility with neurons (which are routinely cultured on poly-lysine). The gels were highly biocompatible, with minimal inflammation, gliosis, toxicity, or immune response due to their implantation, and the mice that received the injections showed no behavior changes as a result of the implanted material. Over the course of several weeks, new blood vessels were formed in the material, along with some migration of glia and nerve fibers into the gel. The dense vascularization, presence of myelinating oligodendrocyte progenitors, and in-growth of nerve fibers suggested that these materials could serve as attractive scaffolds for neural regeneration applications, especially as scaffolds or growth factor depots.
4.4 Two-component protein hydrogels
A third method for the construction of self-assembling protein biomaterials was developed by the Heilshorn group, and employed a fundamentally different protein-protein association mechanism compared with the works highlighted above. Their approach employed two naturally occurring protein motifs—the WW domain, and the proline-rich peptide (PPxY)—which can associate via noncovalent interactions, with tunable interaction strength depending on the exact motif variant (Figure 6E). Heilshorn and coworkers synthesized synthetic proteins consisting of repeats of either the WW or PPxY domains, separated by short peptide spacers.[107] Upon mixing, the two synthetic proteins, the two domains were able to interact and form a highly cross-linked network (Figure 6F), with the strength of the resulting material dependent on both the number of repeats in the components, as well as their interaction strength. One particular system (consisting of seven WW repeats, nine PPxY repeats, and a strong WW-PPxY association) formed a hydrogel with a G’ value of 50 Pa (similar to Matrigel); reducing the WW-PPxY interaction strength by using a different PPxY motif resulted in a fivefold reduction in G’.
This system, termed a mixing-induced two-component hydrogel (MITCH), formed gels solely as a result of the intermolecular interactions afforded by self-assembly. No external trigger (e.g. pH, temperature, or ionic strength) was necessary to promote gelation as with other materials, and unlike the other protein materials described in this section, the individual protein components did not self-associate, allowing for greater temporal control over the gelling process. Furthermore, cell adhesion epitopes like RGDS could be incorporated into the linkers separating the WW domains (Figure 6F), endowing the gels with biological functionality. Similar to many of the other biomaterials covered in this review, the MITCH system was shear-thinning due to the noncovalent crosslinks, with the strength of the WW-PPxY interaction dictating the gelation time following disruption. The gels were also highly cytocompatible, supporting the culture of PC-12 (a neuronal-like cell line) on their surface. In addition, the authors examined the encapsulation of both HUVEC cells and primary murine neural stem cells in 3D culture. Both cell types were viable in the gels, and the neural stem cells were able to differentiate as expected and grow neurites in the material upon exposure to the appropriate conditions. Finally, because of the predictable and tunable nature of the protein-protein interactions that made up the crosslinks, the gel mechanical properties (e.g. stiffness, gelation time) could be rationally designed with a level of control not possible in many other self-assembled biomaterials.[108]
5. DNA-based Biomaterials
5.1 Introduction
Unlike peptides and proteins, only a few examples exist of DNA-based materials, perhaps due to the higher cost of synthesizing synthetic DNA compared with peptides or proteins. However, DNA possesses great potential for the construction of self-assembled biomaterials due to its highly programmable nature: by designing an appropriate sequence, any two DNA strands can be coaxed to assemble via Watson-Crick base pairing, allowing for a much greater flexibility to construct arbitrary nanostructures. Indeed, the field of DNA nanotechnology has exploited this versatility to construct nanoscale constructs and devices with exquisite precision and complexity.[109–111] Furthermore, the strength of the supramolecular interaction can be tuned by the length and identity of the sequence, and a variety of additional modifications can be carried out by using enzymes like nucleases or ligases. DNA is also inherently biocompatible and biodegradable, two key attributes for a successful biomaterial. Despite these advantages, DNA has yet to be broadly applied in biomaterials synthesis, most likely due to the higher relative cost of custom DNA synthesis compared with solid-phase peptide synthesis or recombinant protein expression. Some examples do exist, however, and DNA-based materials—with their flexible and highly “designable” structure due to the specificity of base-pairing—represent a field with great potential in the future of self-assembling biomaterials.
5.2 DNA-based hydrogels
The Luo laboratory pioneered the use of DNA for the construction of hydrogels, describing a novel material constructed from branched DNA structures.[112] Taking advantage of the aforementioned high degree of specificity allowed by base-pairing, they designed three small DNA structures, in the shape of an X, Y, or T. These branched structures were designed to have a short, palindromic sticky end at the end of each “arm”, allowing them to hybridize with each other: each unit simultaneously serving as both monomer and crosslinker (Figure 7A). Treatment of the solution with T4 DNA ligase sealed the nicks at each sticky and resulted in a cross-linked gel network with entangled fibers and a highly porous structure. The degree of swelling, as well as the mechanical properties, of these gels could be adjusted by changing the concentration of branched DNA molecules. Further, mixing model drug compounds (camptothecin and insulin, in this case) with the DNA prior to gelation resulted in almost 100% encapsulation, with no post-gelation loading necessary, and the molecules could be released over several weeks in a controlled fashion with a rate depending on the geometry (X, Y, or T) of the gel monomers. As expected, these gels were highly biocompatible: CHO cells could be effectively encapsulated prior to gelation with the ligase, and remained viable for several days in the material. Though not demonstrated in this work, the authors pointed out that the cells could potentially be extracted from the gel by digestion with nucleases after gelation.
Figure 7. Hydrogels constructed from DNA.

(A) DNA structures in the shape of X, Y, or T can form extended networks due to complementary sticky ends; addition of a ligase enzyme covalently fixes these links into a hydrogel. (B) By incorporating plasmid DNA into this cross-linked gel, the resulting “P-gel” pads can be used in a cell-free translation system to produce protein more efficiently than if the plasmid was free in solution. (C) Y-shaped DNA monomers can form a hydrogel by association of cytosine-rich i-motifs; the magnification shows one such i-motif at the junction of two DNA arms. (D) A Y-shaped DNA scaffold can be brought together by the addition of a dsDNA linker to form a hydrogel without the need for enzymatic linking. Heating the material can disrupt the sticky-end interactions and melt the gel; alternatively, an enzyme that cleaves a sequence in the middle of the crosslinking strands can be used. [(A): ref. 102; (B): ref. 113; (C): ref. 114; (D): ref. 115]
Although the majority of biological applications for the self-assembled materials described in this review involve cell scaffolding and delivery, DNA-based hydrogels allowed for a novel application possible only with a nucleic acid scaffold. The Luo laboratory used the X-DNA gels described above to encapsulate plasmids coding for proteins, creating a “P-gel” that could be transcribed to RNA and then translated to protein in a cell-free expression system (Figure 7B).[113] The X-shaped branched DNA monomers and T4 ligase were cast in a PDMS mold to produce many small gel pads (1 mm x 1mm x 20 μm); mixing these pads with a coupled transcription-translation system (containing polymerase enzymes, ribosomes, amino acids, ATP, etc.) resulted in efficient protein production of the gene encoded in the plasmid. Remarkably, the P-gels proved far more efficient at producing protein than solution-phase cell-free synthesis (i.e. with the plasmid not in a gel). The amount of protein, the rate of its production, the fraction of functional protein, and the duration of protein synthesis were all increased, and the system proved quite flexible: 16 different proteins, ranging widely in size and physical properties, could be produced from the gels. The increased efficiency of the P-gels was attributed to an increase in transcription, with a greater than 50-fold increase in mRNA produced compared to the soluble system. The authors surmised that this increase was due to three effects promoted by the gel: (1) greater protection of the plasmid DNA due to entrapment in the gel, (2) a higher local concentration of DNA, and (3) more efficient enzymatic turnover due.
The Liu group has explored a different all-DNA approach for the construction of self-assembled gels that avoids the enzymatic ligation step described above.[114] The authors designed a Y-shaped DNA nanostructure, where the “arms” of the Y were comprised of double-stranded DNA ending in a single-stranded sequence rich in cytosine. At low pH values, the protonated C residues formed noncovalent interlocking “i-motif” domains, effectively crosslinking the Y-DNA monomers and resulting in a hydrogel (Figure 7C). Increasing the pH deprotonated the cytosines, broke the i-motif, and resulted in dissolution of the gel. These gels could subsequently be used to reversible trap and then release gold nanoparticles (as a model cargo) by switching the pH. Although this material demonstrated an interesting new way to create supramolecular DNA-based materials, the low pH values (~5) necessary for the i-motif formation prevented their use as cell scaffolds. In a follow-up work, Liu and coworkers adapted a design principle similar to the work by Luo described above. Once again, Y-shaped DNA structures were employed, but rather than allowing them to self-assemble with each other directly, linker strands were used to link the Y-DNA sticky ends (Figure 7D).[115] By using an 8-bp sticky end to link the DNA, the authors created stable hydrogels without the need for subsequent enzymatic ligation; the Luo lab’s work, by comparison, had used less stable 4-bp sticky end that did not afford a gel on its own. Incorporating a nuclease digestion sequence in the linker duplexes allowed for digestion and dissolution of the gel by addition of the enzyme. Alternatively, due to the relatively low melting temperature of the 8-bp sticky ends on the linkers, the gels could be dissolved upon heating to 47 °C. Increasing the sticky ends to 12-bp increased this temperature by about ten degrees, and introducing a single base mismatch lowered the temperature by about the same amount.
The gels described above were comprised entirely of DNA. In addition, DNA has been used as a crosslinking element to control the gelation of synthetic polymers such as polyacrylamide.[116] Although outside the scope of this review, such constructs have been used to create gels that shrink when cross-linked with DNA,[117] or that can capture and release quantum dots.[118] In addition, the ability to incorporate DNA aptamers—ssDNA sequences that are able to bind to specific analytes with high specificity—has allowed for the synthesis of dynamic gels for protein release[119, 120] or small molecule sensing.[121] Although none of these systems have been applied yet in the context of biomaterials, the ability to design crosslinking interactions via sequence design—combined with the dynamic potential afforded by DNA strand exchange, thermal and enzymatic manipulation of DNA, and aptamer binding—will likely prove to be promising for novel biomedical applications in the future.
6. Conclusions and future outlook
The examples highlighted in this review demonstrate the great potential of self-assembly for the preparation of biomimetic and highly functional biomaterials for a wide range of applications in medicine. Whether constructed from peptides, proteins, DNA, synthetic molecules, or hybrid structures, self-assembled biomaterials will continue to evolve to meet biomedical challenges. Biological systems are complex, and treating injury or disease will in many cases necessitate a material that can mimic the complex spatiotemporal dynamics of the tissues they are used to treat. Some of the classes of biomaterials—such as peptide amphiphiles or self-assembling β-sheet peptides—have been explored for a decade or more, and have shown remarkable promise in a number of applications.
Other systems—such as α-helical peptides, collagen mimetics, DNA-based materials, or protein-polymer hybrids—have a rich literature studying their fundamental physicochemical properties, and are now beginning to be applied in a biomedical context. It is likely that hybrid systems that combine multiple chemical elements will be necessary; some components may provide structure, others biological signaling, and yet others a switchable element to imbue dynamic ability to the material.
One of the future challenges in this area will be to develop materials that can respond to external stimuli and reconfigure their physical or chemical properties accordingly. Biological systems are highly dynamic (indeed, this is a prerequisite for life), thus materials that can adapt their properties in response to external information will be extremely useful. For example, biomaterials that can change biological signals, or modulate their stiffness in response to regenerative stages, or systems that can release multiple growth factors depending on when they are most needed by the damaged or transplanted tissue, will have greater efficacy relative to static constructs. Self-assembly is one of the most powerful ways to realize such dynamic materials, precisely because supramolecular structure is dependent on multiple, weak, and thus potentially reconfigurable interactions. Outlined by several examples in this review are instances of minor molecular changes that have dramatic effects on the structure of a system. These instances will likely be extrapolated in the future to create increasingly complex constructs that can integrate multiple external cues will afford truly “smart” behavior.
A second challenge, and opportunity, for self-assembly in the future lies in creating hierarchical structures that can organize material across multiple length scales. Biological systems themselves are hierarchically organized: from molecules, to cells, tissues, and up to entire organisms. Materials that can similarly traverse these scales will be able to meet the needs of a healing organism on multiple levels. Once again, self-assembly provides a natural way to achieve this goal, allowing for the organization of matter on increasingly greater size scales by integrating specific noncovalent interactions with external forces. Most of the systems described in this review show a high degree of organization on the scale of single molecules, or nanostructures of hundreds or thousands of molecules (such as nanofibers or membranes). On a much larger, bulk scale, the macroscopic properties of a material—such as its stiffness or self-healing behavior—can be engineered as well. The future of self-assembled materials lies in bridging these endpoints, and maintaining order from the nano- to the micro- to the macro-scale through the judicious choice of molecular design and external conditions.
The third great challenge in the development of self-assembled biomaterials is their translation to clinical applications. To this end, biocompatibility and scalablity are both required. Many of the examples of self-assembled biomaterials presented are reasonably scalable. For example, the majority of peptide-based molecules are constructed by solid-phase peptide synthesis, a process that can facilely yield multiple millimoles of pure material. Considering that low concentrations (~ tens of mM or less) are generally required for peptide assemblies to form biomaterials (including gels), such yields are often sufficient for clinical applications. In contrast, materials based on DNA, particularly long nucleotides with additional chemical functionality, are less scalable than peptidic systems. As in this example, scalability remains a challenge for select self-assembled systems, but research into the synthetic chemistry of such motifs is resulting in significant technological advancements and increasing the feasibility of applying such systems for practical applications. In addition, biocompatibility of biomimetic and bioinspired self-assemblies will also need to be investigated thoroughly in order to move these materials towards clinical trials; as highlighted in many of the examples herein, however, using naturally occurring molecules such as peptides or proteins represents a powerful way to ensure that the materials remain compatible with the biological milieu. With efforts to address scalability and biocompatibility, the vast array of self-assembling motifs available for designing functional biomaterials provides engineers and scientists with a diverse toolkit with which to confront biomaterials challenges. This highly active field of research provides great scientific and technology opportunities and prospects for the future progress in medical technologies.
Acknowledgments
Research in the authors’ laboratory described in this paper was supported by grants from the National Institutes of Health under Award #’s (5R01DE015920-07; 5R01EB003806-07), the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering under Award # (DE-FG02-00ER45810), and by the National Science Foundation under Award # (DMR1006713). NS gratefully acknowledges support from the IIN Postdoctoral Fellowship and the Northwestern International Institute for Nanotechnology, and from a NIH Ruth L. Kirschstein NRSA postdoctoral fellowship under Award # (1F32NS077728-01A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–8. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 2.Hwang JJ, Iyer SN, Li L-S, Claussen R, Harrington DA, Stupp SI. Proceedings of the National Academy of Sciences. 2002;99:9662–7. doi: 10.1073/pnas.152667399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aida T, Meijer E, Stupp S. Science. 2012;335:813–7. doi: 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ulijn RV, Smith AM. Chem Soc Rev. 2008;37:664–75. doi: 10.1039/b609047h. [DOI] [PubMed] [Google Scholar]
- 5.Zhang S, Marini DM, Hwang W, Santoso S. Current opinion in chemical biology. 2002;6:865–71. doi: 10.1016/s1367-5931(02)00391-5. [DOI] [PubMed] [Google Scholar]
- 6.Zhang S, Holmes T, Lockshin C, Rich A. Proceedings of the National Academy of Sciences. 1993;90:3334. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loo Y, Zhang S, Hauser CAE. Biotechnology Advances. 2011 doi: 10.1016/j.biotechadv.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Zhang S, Holmes TC, DiPersio CM, Hynes RO, Su X, Rich A. Biomaterials. 1995;16:1385–93. doi: 10.1016/0142-9612(95)96874-y. [DOI] [PubMed] [Google Scholar]
- 9.Yokoi H, Kinoshita T, Zhang S. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8414. doi: 10.1073/pnas.0407843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes TC, De Lacalle S, Su X, Liu G, Rich A, Zhang S. Proceedings of the National Academy of Sciences. 2000;97:6728. doi: 10.1073/pnas.97.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kisiday J, Jin M, Kurz B, Hung H, Semino C, Zhang S, et al. Proceedings of the National Academy of Sciences. 2002;99:9996. doi: 10.1073/pnas.142309999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semino CE, Merok JR, Crane GG, Panagiotakos G, Zhang S. Differentiation. 2003;71:262–70. doi: 10.1046/j.1432-0436.2003.7104503.x. [DOI] [PubMed] [Google Scholar]
- 13.Semino CE, Kasahara J, Hayashi Y, Zhang S. Tissue engineering. 2004;10:643–55. doi: 10.1089/107632704323061997. [DOI] [PubMed] [Google Scholar]
- 14.Ellis-Behnke RG, Liang YX, You SW, Tay DKC, Zhang S, So KF, et al. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5054–9. doi: 10.1073/pnas.0600559103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gelain F, Bottai D, Vescovi A, Zhang S. PLoS One. 2006;1:e119. doi: 10.1371/journal.pone.0000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumada Y, Zhang S. PLoS One. 2010;5:e10305. doi: 10.1371/journal.pone.0010305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goeden-Wood NL, Keasling JD, Muller SJ. Macromolecules. 2003;36:2932–8. [Google Scholar]
- 18.Aggeli A, Bell M, Boden N, Keen J, Knowles P, McLeish T, et al. Nature. 1997 doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 19.Aggeli A, Nyrkova I, Bell M, Harding R, Carrick L, McLeish T, et al. Proceedings of the National Academy of Sciences. 2001;98:11857. doi: 10.1073/pnas.191250198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aggeli A, Bell M, Carrick LM, Fishwick CWG, Harding R, Mawer PJ, et al. Journal of the American Chemical Society. 2003;125:9619–28. doi: 10.1021/ja021047i. [DOI] [PubMed] [Google Scholar]
- 21.Fishwick CWG, Beevers AJ, Carrick LM, Whitehouse CD, Aggeli A, Boden N. Nano letters. 2003;3:1475–9. [Google Scholar]
- 22.Bell CJ, Carrick LM, Katta J, Jin Z, Ingham E, Aggeli A, et al. Journal of Biomedical Materials Research Part A. 2006;78:236–46. doi: 10.1002/jbm.a.30672. [DOI] [PubMed] [Google Scholar]
- 23.Kirkham J, Firth A, Vernals D, Boden N, Robinson C, Shore R, et al. Journal of dental research. 2007;86:426–30. doi: 10.1177/154405910708600507. [DOI] [PubMed] [Google Scholar]
- 24.Collier JH, Messersmith PB. Bioconjugate chemistry. 2003;14:748–55. doi: 10.1021/bc034017t. [DOI] [PubMed] [Google Scholar]
- 25.Jung JP, Jones JL, Cronier SA, Collier JH. Biomaterials. 2008;29:2143–51. doi: 10.1016/j.biomaterials.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung JP, Nagaraj AK, Fox EK, Rudra JS, Devgun JM, Collier JH. Biomaterials. 2009;30:2400–10. doi: 10.1016/j.biomaterials.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gasiorowski JZ, Collier JH. Biomacromolecules. 2011 doi: 10.1021/bm200763y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudra JS, Tian YF, Jung JP, Collier JH. Proceedings of the National Academy of Sciences. 2010;107:622–7. doi: 10.1073/pnas.0912124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong H, Paramonov SE, Aulisa L, Bakota EL, Hartgerink JD. Journal of the American Chemical Society. 2007;129:12468–72. doi: 10.1021/ja072536r. [DOI] [PubMed] [Google Scholar]
- 30.Aulisa L, Dong H, Hartgerink JD. Biomacromolecules. 2009;10:2694–8. doi: 10.1021/bm900634x. [DOI] [PubMed] [Google Scholar]
- 31.Galler KM, Aulisa L, Regan KR, D’Souza RN, Hartgerink JD. Journal of the American Chemical Society. 2010;132:3217–23. doi: 10.1021/ja910481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider JP, Pochan DJ, Ozbas B, Rajagopal K, Pakstis L, Kretsinger J. Journal of the American Chemical Society. 2002;124:15030–7. doi: 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- 33.Pochan DJ, Schneider JP, Kretsinger J, Ozbas B, Rajagopal K, Haines L. Journal of the American Chemical Society. 2003;125:11802–3. doi: 10.1021/ja0353154. [DOI] [PubMed] [Google Scholar]
- 34.Heskins M, Guillet JE. Journal of Macromolecular Science—Chemistry. 1968;2:1441–55. [Google Scholar]
- 35.Nagy KJ, Giano MC, Jin A, Pochan DJ, Schneider JP. Journal of the American Chemical Societ. 2011 doi: 10.1021/ja206742m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Micklitsch CM, Knerr PJ, Branco MC, Nagarkar R, Pochan DJ, Schneider JP. Angewandte Chemie. 2011;123:1615–7. doi: 10.1002/anie.201006652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giano MC, Pochan DJ, Schneider JP. Biomaterials. 2011 doi: 10.1016/j.biomaterials.2011.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kretsinger JK, Haines LA, Ozbas B, Pochan DJ, Schneider JP. Biomaterials. 2005;26:5177–86. doi: 10.1016/j.biomaterials.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 39.Haines-Butterick L, Rajagopal K, Branco M, Salick D, Rughani R, Pilarz M, et al. Proceedings of the National Academy of Sciences. 2007;104:7791. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salick DA, Kretsinger JK, Pochan DJ, Schneider JP. Journal of the American Chemical Society. 2007;129:14793–9. doi: 10.1021/ja076300z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selkoe DJ. Neuron. 1991;6:487. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 42.Serpell LC. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2000;1502:16–30. doi: 10.1016/s0925-4439(00)00029-6. [DOI] [PubMed] [Google Scholar]
- 43.Kasai S, Ohga Y, Mochizuki M, Nishi N, Kadoya Y, Nomizu M. Peptide Science. 2004;76:27–33. doi: 10.1002/bip.10565. [DOI] [PubMed] [Google Scholar]
- 44.Gras SL, Tickler AK, Squires AM, Devlin GL, Horton MA, Dobson CM, et al. Biomaterials. 2008;29:1553–62. doi: 10.1016/j.biomaterials.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 45.Mason JM, Arndt KM. ChemBioChem. 2004;5:170–6. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- 46.Woolfson DN, Ryadnov MG. Current opinion in chemical biology. 2006;10:559–67. doi: 10.1016/j.cbpa.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 47.Woolfson DN. Peptide Science. 2010;94:118–27. doi: 10.1002/bip.21345. [DOI] [PubMed] [Google Scholar]
- 48.Banwell EF, Abelardo ES, Adams DJ, Birchall MA, Corrigan A, Donald AM, et al. Nature materials. 2009;8:596–600. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim PS, Berger B, Wolf E. Protein Science. 1997;6:1179–89. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Papapostolou D, Smith AM, Atkins EDT, Oliver SJ, Ryadnov MG, Serpell LC, et al. Proceedings of the National Academy of Sciences. 2007;104:10853. doi: 10.1073/pnas.0700801104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Lullo GA, Sweeney SM, Körkkö J, Ala-Kokko L, San Antonio JD. Journal of Biological Chemistry. 2002;277:4223. doi: 10.1074/jbc.M110709200. [DOI] [PubMed] [Google Scholar]
- 52.Kotch FW, Raines RT. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3028–33. doi: 10.1073/pnas.0508783103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pires MM, Przybyla DE, Chmielewski J. Angewandte Chemie International Edition. 2009;48:7813–7. doi: 10.1002/anie.200902375. [DOI] [PubMed] [Google Scholar]
- 54.O'Leary LER, Fallas JA, Bakota EL, Kang MK, Hartgerink JD. Nature Chemistry. 2011;3:821–8. doi: 10.1038/nchem.1123. [DOI] [PubMed] [Google Scholar]
- 55.Fallas JA, Gauba V, Hartgerink JD. Journal of Biological Chemistry. 2009;284:26851–9. doi: 10.1074/jbc.M109.014753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Russell LE, Fallas JA, Hartgerink JD. Journal of the American Chemical Society. 2010;132:3242–3. doi: 10.1021/ja909720g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang AY, Mo X, Chen CS, Yu SM. Journal of the American Chemical Society. 2005;127:4130–1. doi: 10.1021/ja0431915. [DOI] [PubMed] [Google Scholar]
- 58.Wang AY, Leong S, Liang YC, Huang RCC, Chen CS, Yu SM. Biomacromolecules. 2008;9:2929–36. doi: 10.1021/bm800727z. [DOI] [PubMed] [Google Scholar]
- 59.Chan TR, Stahl PJ, Yu SM. Advanced functional materials. 2011 doi: 10.1002/adfm.201101163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kopecek J, Yang J. Acta biomaterialia. 2009;5:805–16. doi: 10.1016/j.actbio.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berndt P, Fields GB, Tirrell M. Journal of the American Chemical Society. 1995;117:9515–22. [Google Scholar]
- 62.Hartgerink JD, Beniash E, Stupp SI. Proceedings of the National Academy of Sciences. 2002;99:5133. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, et al. Science. 2004;303:1352–5. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 64.Tysseling-Mattiace VM, Sahni V, Niece KL, Birch D, Czeisler C, Fehlings MG, et al. The Journal of Neuroscience. 2008;28:3814–23. doi: 10.1523/JNEUROSCI.0143-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rajangam K, Behanna HA, Hui MJ, Han X, Hulvat JF, Lomasney JW, et al. Nano letters. 2006;6:2086–90. doi: 10.1021/nl0613555. [DOI] [PubMed] [Google Scholar]
- 66.Shah RN, Shah NA, Lim MMDR, Hsieh C, Nuber G, Stupp SI. Proceedings of the National Academy of Sciences. 2010;107:3293–8. doi: 10.1073/pnas.0906501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Webber MJ, Tongers J, Newcomb CJ, Marquardt KT, Bauersachs J, Losordo DW, et al. Proceedings of the National Academy of Sciences. 2011;108:13438–43. doi: 10.1073/pnas.1016546108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Capito RM, Azevedo HS, Velichko YS, Mata A, Stupp SI. Science. 2008;319:1812–6. doi: 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- 69.Chow LW, Bitton R, Webber MJ, Carvajal D, Shull KR, Sharma AK, et al. Biomaterials. 2011;32:1574–82. doi: 10.1016/j.biomaterials.2010.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang S, Greenfield MA, Mata A, Palmer LC, Bitton R, Mantei JR, et al. Nature materials. 2010;9:594–601. doi: 10.1038/nmat2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mata A, Geng Y, Henrikson KJ, Aparicio C, Stock SR, Satcher RL, et al. Biomaterials. 2010;31:6004–12. doi: 10.1016/j.biomaterials.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sargeant TD, Oppenheimer SM, Dunand DC, Stupp SI. Journal of tissue engineering and regenerative medicine. 2008;2:455–62. doi: 10.1002/term.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang Z, Sargeant TD, Hulvat JF, Mata A, Bringas P, Koh C-Y, et al. Journal of Bone and Mineral Research. 2008;23:1995–2006. doi: 10.1359/JBMR.080705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang Z, Newcomb CJ, Bringas P, Jr, Stupp SI, Snead ML. Biomaterials. 2010;31:9202–11. doi: 10.1016/j.biomaterials.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Standley SM, Toft DJ, Cheng H, Soukasene S, Chen J, Raja SM, et al. Cancer research. 2010;70:3020. doi: 10.1158/0008-5472.CAN-09-3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stendahl JC, Wang LJ, Chow LW, Kaufman DB, Stupp SI. Transplantation. 2008;86:478. doi: 10.1097/TP.0b013e3181806d9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matson JB, Stupp SI. Chem Commun. 2011;48:26–33. doi: 10.1039/c1cc15551b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cui H, Webber MJ, Stupp SI. Peptide Science. 2010;94:1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matson JB, Zha RH, Stupp SI. Current Opinion in Solid State and Materials Science. 2011 doi: 10.1016/j.cossms.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stupp SI, Ciegler GW. Journal of Biomedical Materials Research. 1992;26:169–83. doi: 10.1002/jbm.820260204. [DOI] [PubMed] [Google Scholar]
- 81.Müller-Mai CM, Stupp SI, Voigt C, Gross U. Journal of Biomedical Materials Research. 1995;29:9–18. doi: 10.1002/jbm.820290103. [DOI] [PubMed] [Google Scholar]
- 82.Reches M, Gazit E. Science. 2003;300:625–7. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- 83.Reches M, Gazit E. Nano letters. 2004;4:581–5. doi: 10.1021/nl0484189. [DOI] [PubMed] [Google Scholar]
- 84.Jayawarna V, Ali M, Jowitt TA, Miller AF, Saiani A, Gough JE, et al. Advanced Materials. 2006;18:611–4. [Google Scholar]
- 85.Jayawarna V, Richardson SM, Hirst AR, Hodson NW, Saiani A, Gough JE, et al. Acta biomaterialia. 2009;5:934–43. doi: 10.1016/j.actbio.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 86.Zhou M, Smith AM, Das AK, Hodson NW, Collins RF, Ulijn RV, et al. Biomaterials. 2009;30:2523–30. doi: 10.1016/j.biomaterials.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 87.Yang Z, Liang G, Wang L, Xu B. Journal of the American Chemical Society. 2006;128:3038–43. doi: 10.1021/ja057412y. [DOI] [PubMed] [Google Scholar]
- 88.Liang G, Yang Z, Zhang R, Li L, Fan Y, Kuang Y, et al. Langmuir. 2009;25:8419–22. doi: 10.1021/la804271d. [DOI] [PubMed] [Google Scholar]
- 89.Kuang Y, Gao Y, Xu B. Chemical Communications. 2011 doi: 10.1039/c1cc15577f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rodriguez-Cabello JC, Prieto S, Reguera J, Arias FJ, Ribeiro A. Journal of Biomaterials Science, Polymer Edition. 2007;18:269–86. doi: 10.1163/156856207779996904. [DOI] [PubMed] [Google Scholar]
- 91.Kyle S, Aggeli A, Ingham E, McPherson MJ. Trends in biotechnology. 2009;27:423–33. doi: 10.1016/j.tibtech.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281:389–92. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 93.Shen W, Zhang K, Kornfield JA, Tirrell DA. Nature materials. 2006;5:153–8. doi: 10.1038/nmat1573. [DOI] [PubMed] [Google Scholar]
- 94.Wang C, Stewart RJ, KopeCek JR. Nature. 1999;397:417–20. doi: 10.1038/17092. [DOI] [PubMed] [Google Scholar]
- 95.Wang C, Kopecek J, Stewart RJ. Biomacromolecules. 2001;2:912–20. doi: 10.1021/bm0155322. [DOI] [PubMed] [Google Scholar]
- 96.Yang J, Xu C, Wang C, Kopecek J. Biomacromolecules. 2006;7:1187–95. doi: 10.1021/bm051002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Olsen BD, Kornfield JA, Tirrell DA. Macromolecules. 2010 doi: 10.1021/ma101434a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wright ER, McMillan RA, Cooper A, Apkarian RP, Conticello VP. Advanced functional materials. 2002;12:149. [Google Scholar]
- 99.Nagapudi K, Brinkman WT, Thomas BS, Park JO, Srinivasarao M, Wright E, et al. Biomaterials. 2005;26:4695–706. doi: 10.1016/j.biomaterials.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 100.Nagapudi K, Brinkman WT, Leisen J, Thomas BS, Wright ER, Haller C, et al. Macromolecules. 2005;38:345–54. [Google Scholar]
- 101.Martens AA, Portale G, Werten MWT, de Vries RJ, Eggink G, Cohen Stuart MA, et al. Macromolecules. 2009;42:1002–9. [Google Scholar]
- 102.Werten MWT, Teles H, Moers APHA, Wolbert EJH, Sprakel J, Eggink G, et al. Biomacromolecules. 2009;10:1106–13. doi: 10.1021/bm801299u. [DOI] [PubMed] [Google Scholar]
- 103.Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, et al. Nature. 2002;417:424–8. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 104.Breedveld V, Nowak AP, Sato J, Deming TJ, David J. Macromolecules. 2004;37:3943–53. [Google Scholar]
- 105.Li Z, Deming TJ. Soft Matter. 2010;6:2546–51. [Google Scholar]
- 106.Yang CY, Song B, Ao Y, Nowak AP, Abelowitz RB, Korsak RA, et al. Biomaterials. 2009;30:2881–98. doi: 10.1016/j.biomaterials.2009.01.056. [DOI] [PubMed] [Google Scholar]
- 107.Foo CTSWP, Lee JS, Mulyasasmita W, Parisi-Amon A, Heilshorn SC. Proceedings of the National Academy of Sciences. 2009;106:22067–72. doi: 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mulyasasmita W, Lee JS, Heilshorn SC. Biomacromolecules. 2011 doi: 10.1021/bm200959e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pinheiro AV, Han D, Shih WM, Yan H. Nature Nanotechnology. 2011;6:763–72. doi: 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chhabra R, Sharma J, Liu Y, Rinker S, Yan H. Advanced drug delivery reviews. 2010;62:617–25. doi: 10.1016/j.addr.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 111.Seeman NC. Trends in biotechnology. 1999;17:437–43. doi: 10.1016/s0167-7799(99)01360-8. [DOI] [PubMed] [Google Scholar]
- 112.Um SH, Lee JB, Park N, Kwon SY, Umbach CC, Luo D. Nature materials. 2006;5:797–801. doi: 10.1038/nmat1741. [DOI] [PubMed] [Google Scholar]
- 113.Park N, Um SH, Funabashi H, Xu J, Luo D. Nature materials. 2009;8:432–7. doi: 10.1038/nmat2419. [DOI] [PubMed] [Google Scholar]
- 114.Cheng E, Xing Y, Chen P, Yang Y, Sun Y, Zhou D, et al. Angewandte Chemie. 2009;121:7796–9. [Google Scholar]
- 115.Xing Y, Cheng E, Yang Y, Chen P, Zhang T, Sun Y, et al. Advanced Materials. 2011;23:1117–21. doi: 10.1002/adma.201003343. [DOI] [PubMed] [Google Scholar]
- 116.Lin DC, Yurke B, Langrana NA. Journal of biomechanical engineering. 2004;126:104. doi: 10.1115/1.1645529. [DOI] [PubMed] [Google Scholar]
- 117.Murakami Y, Maeda M. Macromolecules. 2005;38:1535–7. [Google Scholar]
- 118.Liedl T, Dietz H, Yurke B, Simmel F. Small. 2007;3:1688–93. doi: 10.1002/smll.200700366. [DOI] [PubMed] [Google Scholar]
- 119.Wei B, Cheng I, Luo KQ, Mi Y. Angewandte Chemie International Edition. 2008;47:331–3. doi: 10.1002/anie.200704143. [DOI] [PubMed] [Google Scholar]
- 120.Zhu Z, Wu C, Liu H, Zou Y, Zhang X, Kang H, et al. Angewandte Chemie. 2010;122:1070–4. [Google Scholar]
- 121.Yang H, Liu H, Kang H, Tan W. Journal of the American Chemical Society. 2008;130:6320–1. doi: 10.1021/ja801339w. [DOI] [PMC free article] [PubMed] [Google Scholar]