

Abstract
Over the last decade, accumulating evidence has suggested a causative link between mitochondrial dysfunction and major phenotypes associated with aging. Somatic mitochondrial DNA (mtDNA) mutations and respiratory chain dysfunction accompany normal aging, but the first direct experimental evidence that increased mtDNA mutation levels contribute to progeroid phenotypes came from the mtDNA mutator mouse. Recent evidence suggests that increases in aging-associated mtDNA mutations are not caused by damage accumulation, but rather are due to clonal expansion of mtDNA replication errors that occur during development. Here we discuss the caveats of the traditional mitochondrial free radical theory of aging and highlight other possible mechanisms, including insulin/IGF-1 signaling (IIS) and the target of rapamycin pathways, that underlie the central role of mitochondria in the aging process.
Introduction
The exact reasons why we age are poorly understood. Aging is thought to be a degenerative process caused by accumulated damage that leads to cellular dysfunction, tissue failure, and death. A number of aging theories have been proposed (1–5), and the mitochondrial free radical theory of aging (MFRTA) has taken center stage for several decades (1). According to this theory, ROS are considered to be unwanted toxic byproducts of aerobic metabolism that induce oxidative damage to various cellular macromolecules due to their high chemical reactivity. The respiratory chain (RC), located in the inner mitochondrial membrane, is a main production site of superoxide, an abundant ROS in the cell formed at the level of complexes I and III during electron transport (Figure 1). The superoxide anion is converted to hydrogen peroxide by SOD. Although hydrogen peroxide itself is not a free radical, it can be converted to the highly reactive hydroxyl radical in the presence of transition metals through the Fenton reaction (Figure 1). The hydroxyl radical is considered to be the most damaging form of ROS, as it is highly reactive and causes oxidative damage to virtually every molecule type in the cell, including lipids, proteins, and nucleic acids.
Figure 1. Schematic model of the oxidative phosphorylation system and the production of ROS.

(A) ATP is generated by oxidative phosphorylation conducted by the four RC complexes (CI–CIV) and ATP synthase (CV) located in the inner mitochondrial membrane. Energy released by the electron transfer from NADH and FADH2 to O2 is used to pump protons (H+) via CI, CIII, and CIV. The proton gradient across the inner mitochondrial membrane drives ATP production via ATP synthase. The proton gradient can be dissipated by re-entry of protons to the mitochondrial matrix through uncoupling proteins (UCPs), which leads to uncoupling of respiration and ATP synthesis. (B) ROS are formed as a byproduct of oxidative phosphorylation. Superoxide is an abundant ROS in the cell and is generated by CI and CIII. Cells protect themselves from oxidative damage by expressing a variety of antioxidant enzymes that convert ROS into less harmful byproducts. Superoxide anion is converted to hydrogen peroxide by manganese SOD. Hydrogen peroxide is then converted to water by glutathione peroxidase, the most abundant peroxidase in the cytosol and mitochondria. Even though hydrogen peroxide is not substantially harmful for the cell, it can be converted to the highly reactive hydroxyl radical (OH•) in the presence of transition metals via the Fenton reaction. CytC, cytochrome C; CoQ, coenzyme Q10.
The MFRTA theory is based on several observations: (a) mitochondrial ROS production increases with age because of a decline in mitochondrial function, (b) activity of several ROS-scavenging enzymes declines with age, (c) mutations of mitochondrial DNA (mtDNA) accumulate during aging, and (d) a vicious cycle occurs because somatic mtDNA mutations impair RC function, which in turn results in a further increase in ROS production and accumulated oxidative damage to proteins, lipids, and DNA (6–8). According to the MFRTA, mitochondria play a crucial role in mediating and amplifying the oxidative stress that drives the aging process.
Although a wealth of correlative data support the MFRTA and a role for increased ROS production and oxidative damage in different types of age-associated diseases (9–12), more recent data cast doubt on this model of aging. Here we discuss the role of mitochondria in aging, with specific emphasis on: (a) the role for mtDNA mutations as a driving force in aging, (b) the role for mitochondrial ROS in aging, and (c) the link between mitochondrial function and signaling pathways reported to regulate longevity.
Mitochondrial function declines with age
Mitochondria regulate a multitude of different metabolic and signaling pathways and also play an important role in programmed cell death. The primary function of mitochondria is to produce ATP through the process of oxidative phosphorylation, which is conducted by the four RC complexes (complexes I–IV) and the ATP synthase (complex V), all located in the inner mitochondrial membrane (Figure 1). Mitochondria are unique among the cellular organelles, as they contain their own genetic information, mtDNA, a double-stranded circular molecule of 16.5 kb encoding 13 proteins, 22 transfer RNAs (tRNAs), and 2 ribosomal RNAs in mammals. The 13 mtDNA-encoded proteins are all components of the RC or the ATP synthase, and oxidative phosphorylation collapses in the absence of mtDNA expression (13).
Mitochondrial function has long been recognized to decline during aging concomitant with the appearance of mitochondrial morphological alterations, e.g., abnormally rounded mitochondria in aged mammals (14). The number of mitochondria decreases with age in liver cells of mice (15), rats (16), and humans (17, 18), concurrent with a decrease in mtDNA copy number and mitochondrial protein levels (19). Additionally, RC capacity is reduced up to 40% in rat liver mitochondria of old animals (24 months) in comparison with juvenile animals (3–4 months) (19). A similar decline of RC function has been reported in the spleen, whereas mitochondrial function in the brain was not reported to change with age in rats (19). The RC capacity has also been reported to decline with age in human liver, heart, and skeletal muscle (20, 21). Some of these changes may be secondary, as RC function is inducible, e.g., in response to hormones, and may depend on physical activity (22). In addition to a general decline of mitochondrial mass and overall function, the activity of specific RC complexes and certain nuclear-encoded mitochondrial proteins, including acylcarnitine transferase and adenine nucleotide translocase, declines as mammals age (23–27). The activity of complexes I and IV decreases with age in liver, brain, heart, and kidney of mice and rats (25–27), whereas, interestingly, the activity of complexes II, III, and V remains mostly unchanged (28). The reasons for these partially contradictory differences in age-related decline in RC function are currently unknown, and reduced expression of mtDNA as well as elevated levels of mtDNA mutations have been suggested as potential causes (27). However, it is important to note that the cell type composition likely changes in aging organs, e.g., due to fibrosis or changes in vascularization, and therefore a difference in mitochondrial function in tissue homogenates from young and old individuals may be hard to interpret. Additional factors, such as differences in the applied methodology (29) and difficulties in finding suitable controls to aged patient cohorts (22), further complicate this type of comparison.
While aging is associated with a decline in mitochondrial function, this observation alone does not imply causality because age-associated changes in mitochondrial function might be secondary to other mechanisms (30). More recent genetic models suggest that mtDNA mutations promote aging phenotypes and create RC dysfunction (see further discussion below). However, when considering the contribution of mitochondrial dysfunction to aging, it is important to note that mitochondrial biogenesis is controlled simultaneously at many different levels. For instance, hormones such as thyroid hormones, estrogens, and glucocorticoids not only play important roles in cell growth and differentiation, but are also important regulators of mitochondrial biogenesis (31–35). Therefore, age-related decline in RC function may at least partly be caused by other age-related changes, e.g., decline in hormonal levels or peripheral insulin resistance. In agreement with this suggestion, physical activity and caloric restriction (CR) can reduce oxidative damage and improve mitochondrial function (36, 37).
Somatic mtDNA mutations increase with age
There is solid evidence that the amount of mtDNA mutations increases with age in humans; for example, deletions in mtDNA have been observed in the aged human central nervous system, skeletal muscle, and hepatocytes (38–40), whereas mtDNA point mutations accumulate in aging colonic crypts (41). In general, mutations of mtDNA may arise as a consequence of unrepaired DNA damage, for example, damage caused by ROS, or by replication errors during normal mtDNA synthesis (42). It is intuitive to assume that the de novo mtDNA mutations observed during aging are due to accumulated, unrepaired damage, but some evidence actually suggests that replication errors may be the more important culprit (43–45). A human somatic cell typically contains thousands of copies of mtDNA, whereas an oocyte contains approximately 105 copies. The replication of mtDNA occurs independently of the cell cycle, and a particular mtDNA molecule may therefore be replicated many times or not at all as a cell divides (46). Furthermore, a single cell can contain mtDNA of more than one genotype, a condition termed heteroplasmy. In the case of heteroplasmy for a pathogenic mutation, the levels of mutated mtDNA will determine whether or not RC dysfunction occurs. The threshold level when mutated mtDNA impairs RC function is typically high but also depends on the type of mutation. Large single deletions of mtDNA cause RC dysfunction if the level of mutated mtDNA exceeds approximately 60% (47), whereas certain pathogenic mitochondrial tRNA mutations only cause dysfunction if present at levels above 95% (48, 49). The relaxed replication of mtDNA creates mitotic segregation with uneven distribution of wild-type and mutated mtDNA molecules between different tissues and even between different cells of the same tissue (50, 51). This apparently random segregation can create a mosaic RC dysfunction (42) even if the absolute levels of mutated mtDNA are low within the affected tissue. The mitotic segregation explains how the low levels of mtDNA deletions and point mutations found in aging frequently accumulate in individual cells to cause mosaic RC deficiency, as seen in brain, heart, skeletal muscle, and colon (40, 41, 50, 52–57). Although it has been known for more than two decades that mutations of mtDNA can cause disease and that somatic mtDNA mutations increase with age, experimental data supporting a role for mtDNA mutations in aging were only recently obtained by the generation of mtDNA mutator mice.
Are mtDNA mutations a driving force in aging?
The first experimental evidence that accumulation of mtDNA mutations can lead to a premature aging phenotype was obtained by the creation of mtDNA mutator mice (58, 59). The mtDNA mutator mice are homozygous for a knock-in mutation that leads to the expression of a proofreading-deficient catalytic subunit of mtDNA polymerase (PolgAmut). The expression of PolgAmut causes extensive mtDNA mutagenesis with the formation of three different forms of mtDNA mutations: random point mutations, linear molecules with large deletions, and, in certain tissues, molecules containing multimers of the control region (an area of the mitochondrial genome that contains noncoding mtDNA) (58, 60, 61). Furthermore, a single report has argued that a fourth type of mtDNA mutation, consisting of circular mtDNA molecules with large deletions, is prevalent and even drives the premature aging phenotype (60). However, a number of studies have subsequently refuted this claim (reviewed in ref. 62), indicating that the very low levels of circular mtDNA molecules with large deletions are unlikely to have any phenotypic consequences in mtDNA mutator mice.
The mtDNA mutator mice display a range of phenotypes reminiscent of naturally occurring aging, including kyphosis, anemia, hair loss, alopecia, graying of the hair, hearing loss, cardiomyopathy, reduced fertility, and weight loss, and have a shortened life span. Experimental evidence strongly suggests that the high number of mtDNA point mutations in mtDNA mutator mice leads to the synthesis of RC subunits with amino acid substitutions that cause instability of the RC complexes (63). The mtDNA deleter mouse, which expresses a mutant-dominant version of twinkle, the replicative DNA helicase in mitochondria, display an accumulation of low levels of large-scale mtDNA deletions in postmitotic tissues. Although the mtDNA deleter mice show progressive RC dysfunction and late-onset mitochondrial myopathy, they do not display a progeroid phenotype and have a normal life span, thus suggesting that accumulation of mtDNA deletions and progressive RC dysfunction may not be sufficient to accelerate aging (64).
Recently, the progeroid phenotype of the mtDNA mutator mice has been, at least partly, attributed to embryonic-onset dysfunction of somatic stem cells (65). Ahlqvist and coworkers demonstrated that development of neural and hematopoietic progenitor cells of the mtDNA mutator mice is already affected during fetal development, and that neural stem cells showed decreased abundance in vivo as well as reduced self-renewal in vitro (65). Their data suggest that mtDNA mutagenesis affects the quality and quantity of stem cells and interferes with the maintenance of the quiescent state, which is important for reconstitution capacity and long-term sustenance of somatic stem cells (Figure 2). Although RC dysfunction was observed in postmitotic tissues of the mtDNA mutator mice, it appeared late in life, when the progeroid phenotypes already had started to be evident. The early onset of dysfunction of somatic stem cells is thus an early event in the mtDNA mutator mice and may drive important premature aging phenotypes.
Figure 2. Model for mtDNA mutations in the stem cell hypothesis of aging.
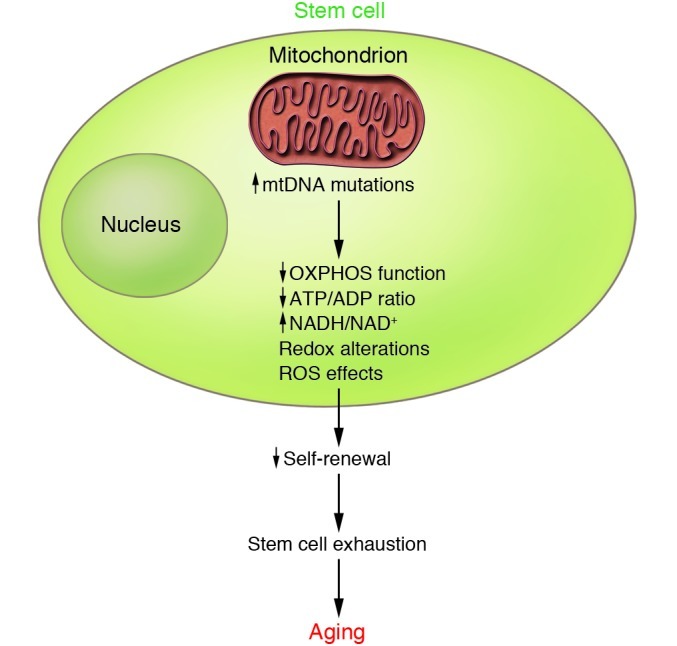
Data from the mtDNA mutator mice suggest that increased mtDNA mutations that arise during development may, by affecting mitochondrial bioenergetic capacity, ROS production, or redox status of the cell, contribute to deregulated stem cell homeostasis and premature aging phenotypes. OXPHOS, oxidative phosphorylation.
The observation that most somatic mutations in mtDNA mutator mice occur as replication errors during development and do not result from damage accumulation during adult life (45) lends further support to the stem cell hypothesis. It is currently unclear how the increased mutation rate in mtDNA mutator mice results in early-onset dysfunction of somatic stem cells. The observation that treatment with the antioxidant N-acetyl cysteine restores the self-renewal ability of neural progenitor cells in mtDNA mutator mice (65) implies that subtle changes in cellular redox state or ROS levels are important for the regulation of somatic stem cell function (Figure 2). The mtDNA mutator mice have little or no increase in levels of ROS production and oxidative damage even though oxidative phosphorylation capacity is severely affected (59, 66). This finding argues against the concept of a vicious cycle in which decline of RC function leads to increased ROS production, which in turn would supposedly increase mtDNA mutation levels to cause further deterioration of RC function and accelerated ROS production.
Free radical theory of aging: dead or alive?
Another relevant piece of information that casts a doubt on the causal role of oxidative damage in the aging process is the absence of a clear correlation between efficacy of antioxidant defenses and longevity. According to the MFRTA, life span is determined by the rate of free radical damage at the cellular and tissue levels. Cells protect themselves from oxidative damage by expressing a variety of non-enzymatic and enzymatic antioxidant defenses that convert ROS into less harmful byproducts (Figure 1). Thus, equilibrium between oxidants and antioxidants is essential to prevent cellular functional impairment. From this perspective, lowering ROS levels by increasing cellular antioxidant defenses should slow the progression of age-related alterations and eventually result in the life span prolongation. In contrast to expectations, genetic manipulations of antioxidant defense genes in animal models show no clear correlation between oxidative damage and life span regulation (67–77). For instance, overexpression of mitochondrial antioxidant enzymes did not extend life spans in different species and even resulted in the shortening of life spans in some cases (68–72, 78). While there are reports that dietary supplementation with antioxidants may improve cellular functions, decrease oxidative stress, and reverse age-related decline in antioxidant defenses in rodents (79, 80), numerous human intervention studies suggest that dietary antioxidant supplementation with vitamin E, beta carotene, or vitamin A has no beneficial effect in prevention of age-related diseases and may even lead to an overall increase of mortality (81–84). Thus, in contrast to the popular notion that increased intake of antioxidants like vitamin C and vitamin E will have beneficial effects on health and life span, these studies suggest that antioxidants should be used with care as therapeutic tools. However, it is important to note that the discrepancies between animal and human studies may be explained by differences in the timing of antioxidant treatment, as some human studies were begun only after subjects had clinical symptoms. In addition, the efficacy of a given antioxidant might also depend on the presence of the other antioxidants or on certain physiological conditions. Certainly the use of mitochondrial-targeted antioxidants is a step toward understanding whether antioxidants can be efficient preventive or therapeutic tools against age-related diseases (reviewed in ref. 85).
Similar to what has been reported concerning antioxidant defense status, the rate of ROS production also does not clearly correlate with species-specific life span. For instance, interspecies comparisons between insect, avian, and mammalian species with very different life spans suggests a positive correlation between mitochondrial hydrogen peroxide production and longevity, potentially implicating ROS levels as a determining factor for life span regulation (86, 87). However, important exceptions cast doubt on the generality of this rule. For example, naked-mole rats are the longest-living rodent species, with a maximum life span of approximately 25–30 years, but they have similar mitochondrial ROS production as mice, which show a maximum life span of about 3–4 years. In addition, naked-mole rats do not show age-dependent variations in terms of antioxidant enzyme expression (88), whereas they have strongly increased levels of oxidative damage with age (89). The findings in naked-mole rats are consistent with the recent observation that increased ROS levels even may result in life span extension in worms, flies, and mice (90–92), suggesting that ROS are not simply unwanted byproducts of oxygen metabolism but that they also act as important signaling molecules to promote longevity (refs. 93–95 and Figure 3). This possibility is further emphasized by a number of recent studies suggesting that ROS are important regulators of cell cycle progression, cell signaling, and apoptosis, among other processes (reviewed in refs. 96–98). Thus, physiological levels of ROS are likely essential for the maintenance of cellular homeostasis, while an increase in ROS production above a certain level has a detrimental effect on cellular physiology. In this respect, it is interesting to note that exercise-induced increase of ROS production and oxidative stress in human skeletal muscle ameliorate insulin resistance and improve glucose metabolism (99), thus suggesting that increased ROS levels might not be the sole determinant of age-related functional alterations.
Figure 3. Model for the effects of impaired IIS and CR on mitochondrial function and aging.
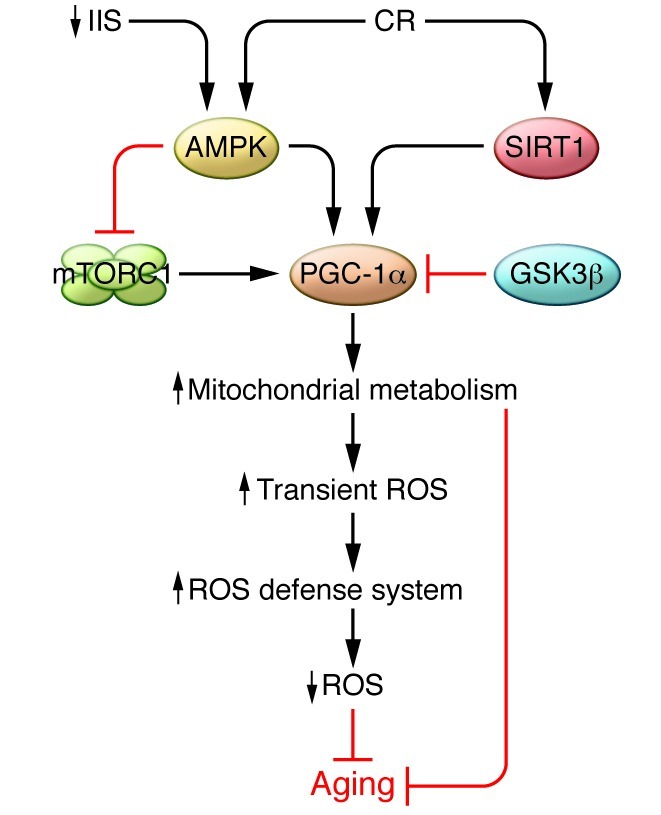
Mitochondrial metabolism plays an important role in mediating longevity promoted by nutrient-sensing pathways like the IIS, the TOR pathway, and CR. Impaired IIS leads to reduced availability of intracellular glucose and consequently to an elevated cellular AMP/ATP ratio that activates AMPK. In its turn, AMPK activates PGC-1α and increases mitochondrial metabolism and respiration rate, which consequently results in transient ROS production. The transient increase in ROS levels activates the expression of scavenging enzymes, e.g., SOD and catalase, resulting in decreased ROS levels, increased stress resistance, and extended life span. Furthermore, current findings suggest that the beneficial effect of CR on longevity is at least partly mediated by improved mitochondrial function mediated through activation of SIRT1 and PGC-1α. The activity of PGC-1α is tightly controlled via glycogen synthase kinase-3β (GSK3β), which acts via phosphorylation to prime PGC-1α for ubiquitinylation and degradation.
In summary, the age-related increases in oxidative damage and ROS production are relatively small and may not explain the rather severe physiological alterations occurring during aging. Consistent with this hypothesis, the absence of a clear correlation between oxidative stress and longevity also suggests that oxidative damage does not play an important role in age-related diseases (e.g., cardiovascular diseases, neurodegenerative diseases, diabetes mellitus) and aging. Experimental results from mtDNA mutator mice suggest that mtDNA mutations in somatic stem cells may drive progeroid phenotypes without increasing oxidative stress, thus indicating that mtDNA mutations that lead to a bioenergetic deficiency may drive the aging process. There is as yet no firm evidence that the overall low levels of mtDNA mutations found in mammals drive the normal aging process. One way to address this experimentally would be to generate anti-mutator animal models to determine whether decreased mtDNA mutation rates prolong their life span.
The signaling role of mitochondria in aging
Though primary mitochondrial dysfunction affects aging, different cellular and metabolic alterations also contribute to the aging process by promoting secondary changes in mitochondrial energy production or mitochondrial biogenesis (Figure 3). Therefore, aging-associated phenotypes have been linked not only to mitochondrial dysfunction but also to aberrant mitochondrial biogenesis caused by impaired retrograde signaling regulated by nuclear genes and factors dependent on mitochondrial metabolism (e.g., ATP, Ca2+, ROS, NO, NAD+/NADH) (100). Numerous studies have shown that mitochondrial metabolism is important in mediating longevity through nutrient-sensing pathways and dietary restriction (101–104). Insulin/IGF-1 signaling (IIS) and target of rapamycin (TOR) signaling pathways are the two main nutrient-sensing pathways that have been linked to the regulation of life span. Impaired IIS and inhibition of TOR activity extend life span in worms, flies, and mammals (101, 103, 105–110). Reduced nutrient availability, also termed CR, extends life span in species ranging from yeast to mammals and improves the health status of rodents and primates (111–113). The effects of CR on longevity are very complex and include many organs and different pathways. Still, the exact underlying mechanisms are unknown. For instance, CR decreases the incidence of cardiovascular diseases in animals, and it has been suggested that the anti-aging effect of CR is propagated through a reduction of metabolic rate and oxidative damage, which consequently inhibits signaling pathways regulated by mitochondria-derived ROS (114). Additionally, a number of studies also show that CR in mice and rats increases mitochondrial biogenesis and respiration through activation of sirtuin 1 (SIRT1), which further activates its downstream effector, PPARγ coactivator-1α (PGC-1α) (refs. 115, 116, and Figure 3). There is evidence that PGC-1α (117–119) and SIRT1 (120) are involved in the regulation of mitochondrial metabolism and life span (refs. 121–123 and Figure 3). However, it should be noted that PGC-1α does not regulate basal mitochondrial biogenesis, but rather is involved in increasing mitochondrial function on demand by activating the expression of certain nuclear genes in different tissues such as heart, brain, and skeletal muscle (124). In summary, current findings suggest that the beneficial effects of CR on longevity are, at least partly, mediated by improved mitochondrial function.
It is important to note that both nutrient-sensitive pathways TOR and IIS activate the common downstream effector ribosomal protein S6 kinase 1 (S6K1), which plays a key role in the regulation of aging in worms and mammals (110). S6K1 knockout mice display ameliorated age-related pathology, life span extension, and gene expression changes similar to those observed under CR. Interestingly, the loss of S6K1 increases AMPK activity (110), which further controls the activity of the mammalian homolog TOR complex 1 (TORC1) (125). In addition, it has been shown that impaired IIS in C. elegans causes an elevated cellular AMP/ATP ratio that activates AMPK, which in turn induces a metabolic shift characterized by increased respiration and transiently increased ROS production, resulting in life span extension (93). Though this mechanism has not been addressed in other model organisms besides worms, it is tempting to speculate that impaired IIS and CR share a common mechanism through AMPK, in which ROS at physiological levels act as signaling molecules to trigger a health-promoting metabolic state by inducing mitochondrial metabolism (ref. 93 and Figure 3). Thus, TOR kinase and AMPK, which are major upstream regulators of mitochondrial metabolism, and the RC itself may all have crucial roles in mediating longevity (Figure 3). However, there is a clear need for future experimental studies to determine whether the contribution of mitochondrial dysfunction to different age-related diseases is explained by a cellular bioenergetic deficiency or by changes in mitochondrial ROS production affecting oxidative damage and signaling. There is also a need to develop novel experimental strategies to interfere with ROS production in a selective way, for example, to reduce oxidative damage without affecting cell signaling. An increased basic understanding of the role for ROS in different cellular processes should make it possible to substantially improve the design of human intervention studies aimed at reducing the oxidative damage associated with disease and aging.
Concluding remarks
A number of recent studies have significantly advanced our understanding of mitochondrial dysfunction in disease and aging. These studies link mitochondrial function to signaling pathways that regulate life span and to the aging process. The MFRTA has largely been refuted, and new efforts have been made to reconcile a unifying aging theory. It is apparent that ROS cannot be the initial cause of the aging process and that mitochondria can cope with physiological levels of oxidative damage. In contrast, genetic mouse models have shown that somatic mtDNA mutations can cause progeroid phenotypes in mammals without increasing oxidative stress. However, it remains unclear how mtDNA mutations accumulate and how they are relevant to aging, as mtDNA mutations are present at relatively low overall levels in normal aging tissues. Numerous studies have shown that clonal expansion of somatic mtDNA mutations creates mosaic RC dysfunction in aging tissues and thereby leads to functional impairment. Thus, an important future goal in understanding the role of mtDNA mutations in the aging process is to investigate whether decreased mtDNA mutation levels have beneficial effects on health and life span. Furthermore, the beneficial effects of CR on longevity are at least partly mediated by improved mitochondrial function. Therefore, important challenges in the future include the development of therapeutic strategies to improve mitochondrial function as a possible means to delay the onset of age-related diseases.
Acknowledgments
The authors thank S. Grönke for critical reading of the manuscript.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(3):951–957. doi:10.1172/JCI64125.
References
- 1.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.Villeponteau B. The heterochromatin loss model of aging. Exp Gerontol. 1997;32(4–5):383–394. doi: 10.1016/S0531-5565(96)00155-6. [DOI] [PubMed] [Google Scholar]
- 3.Kirkwood TB. Evolution of ageing. Nature. 1977;270(5635):301–304. doi: 10.1038/270301a0. [DOI] [PubMed] [Google Scholar]
- 4.Campisi J. The biology of replicative senescence. Eur J Cancer. 1997;33(5):703–709. doi: 10.1016/S0959-8049(96)00058-5. [DOI] [PubMed] [Google Scholar]
- 5.Hamilton WD. The moulding of senescence by natural selection. J Theor Biol. 1966;12(1):12–45. doi: 10.1016/0022-5193(66)90184-6. [DOI] [PubMed] [Google Scholar]
- 6.Fraga CG, Shigenaga MK, Park JW, Degan P, Ames BN. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc Natl Acad Sci U S A. 1990;87(12):4533–4537. doi: 10.1073/pnas.87.12.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stadtman ER. Protein oxidation and aging. Science. 1992;257(5074):1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 8.Marnett LJ, et al. Naturally occurring carbonyl compounds are mutagens in Salmonella tester strain TA104. Mutat Res. 1985;148(1–2):25–34. doi: 10.1016/0027-5107(85)90204-0. [DOI] [PubMed] [Google Scholar]
- 9.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Schapira AH. Mitochondrial diseases. Lancet. 2012;379(9828):1825–1834. doi: 10.1016/S0140-6736(11)61305-6. [DOI] [PubMed] [Google Scholar]
- 11.Müftüoglu M, et al. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord. 2004;19(5):544–548. doi: 10.1002/mds.10695. [DOI] [PubMed] [Google Scholar]
- 12.Grünewald A, et al. Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS One. 2010;5(9):e12962. doi: 10.1371/journal.pone.0012962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larsson NG, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18(3):231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 14.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91(23):10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herbener G. A morphometric study of age-dependent changes in mitochondrial populations of mouse liver and heart. J Gerontol. 1976;31(1):8–12. doi: 10.1093/geronj/31.1.8. [DOI] [PubMed] [Google Scholar]
- 16.Stocco DM, Hutson JC. Quantitation of mitochondrial DNA and protein in the liver of Fischer 344 rats during aging. J Gerontol. 1978;33(6):802–809. doi: 10.1093/geronj/33.6.802. [DOI] [PubMed] [Google Scholar]
- 17.Yen T-C, Chen Y-S, King K-L, Yeh S-H, Wei Y-H. Liver mitochondrial respiratory functions decline with age. Biochem Biophys Res Commun. 1989;165(3):994–1003. doi: 10.1016/0006-291X(89)92701-0. [DOI] [PubMed] [Google Scholar]
- 18.Tauchi H, Sato T. Age changes in size and number of mitochondria of human hepatic cells. J Gerontol. 1968;23(4):454–461. doi: 10.1093/geronj/23.4.454. [DOI] [PubMed] [Google Scholar]
- 19.Stocco DM, Cascarano J, Wilson MA. Quantitation of mitochondrial DNA, RNA, and protein in starved and starved-refed rat liver. J Cell Physiol. 1977;90(2):295–306. doi: 10.1002/jcp.1040900215. [DOI] [PubMed] [Google Scholar]
- 20.Short KR, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102(15):5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ojaimi J, Masters CL, Opeskin K, McKelvie P, Byrne E. Mitochondrial respiratory chain activity in the human brain as a function of age. Mech Ageing Dev. 1999;111(1):39–47. doi: 10.1016/S0047-6374(99)00071-8. [DOI] [PubMed] [Google Scholar]
- 22.Brierley EJ, Johnson MA, James OF, Turnbull DM. Mitochondrial involvement in the ageing process. Facts and controversies. Mol Cell Biochem. 1997;174(1–2):325–328. doi: 10.1023/A:1006847319162. [DOI] [PubMed] [Google Scholar]
- 23.Yan LJ, Sohal RS. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc Natl Acad Sci U S A. 1998;95(22):12896–12901. doi: 10.1073/pnas.95.22.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Killilea DW, Ames BN. Age-associated mitochondrial oxidative decay: improvement of carnitine acetyltransferase substrate-binding affinity and activity in brain by feeding old rats acetyl-L- carnitine and/or R-alpha -lipoic acid. Proc Natl Acad Sci U S A. 2002;99(4):1876–1881. doi: 10.1073/pnas.261709098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benzi G, et al. The mitochondrial electron transfer alteration as a factor involved in the brain aging. Neurobiol Aging. 1992;13(3):361–368. doi: 10.1016/0197-4580(92)90109-b. [DOI] [PubMed] [Google Scholar]
- 26.Lenaz G, et al. Mitochondrial complex I defects in aging. Mol Cell Biochem. 1997;174(1–2):329–333. doi: 10.1023/A:1006854619336. [DOI] [PubMed] [Google Scholar]
- 27.Manczak M, Jung Y, Park BS, Partovi D, Reddy PH. Time-course of mitochondrial gene expressions in mice brains: implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J Neurochem. 2005;92(3):494–504. doi: 10.1111/j.1471-4159.2004.02884.x. [DOI] [PubMed] [Google Scholar]
- 28.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292(2):C670–C686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 29.Picard M, et al. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell. 2010;9(6):1032–1046. doi: 10.1111/j.1474-9726.2010.00628.x. [DOI] [PubMed] [Google Scholar]
- 30.Evans WJ. Protein nutrition, exercise and aging. J Am Coll Nutr. 2004;23(6 suppl):601S–609S. doi: 10.1080/07315724.2004.10719430. [DOI] [PubMed] [Google Scholar]
- 31.Knuever J, et al. Thyrotropin-releasing hormone controls mitochondrial biology in human epidermis. J Clin Endocrinol Metab. 2012;97(3):978–986. doi: 10.1210/jc.2011-1096. [DOI] [PubMed] [Google Scholar]
- 32.Weitzel J, Iwen K, Seitz H. Regulation of mitochondrial biogenesis by thyroid hormone. Exp Physiol. 2003;88(1):121–128. doi: 10.1113/eph8802506. [DOI] [PubMed] [Google Scholar]
- 33.Scheller K, Sekeris CE. The effects of steroid hormones on the transcription of genes encoding enzymes of oxidative phosphorylation. Exp Physiol. 2003;88(1):129–140. doi: 10.1113/eph8802507. [DOI] [PubMed] [Google Scholar]
- 34.Chen J-Q, Cammarata PR, Baines CP, Yager JD. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta. 2009;1793(10):1540–1570. doi: 10.1016/j.bbamcr.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez-Vizarra E, Enriquez JA, Pérez-Martos A, Montoya J, Fernandez-Silva P. Mitochondrial gene expression is regulated at multiple levels and differentially in the heart and liver by thyroid hormones. Curr Genet. 2008;54(1):13–22. doi: 10.1007/s00294-008-0194-x. [DOI] [PubMed] [Google Scholar]
- 36.Radák Z, Zhao Z, Koltai E, Ohno H, Atalay M. Oxygen consumption and usage during physical exercise: the balance between oxidative stress and ROS-dependent adaptive signaling [published online ahead of print: November 16, 2012]. Antioxid Redox Signal. doi: 10.1089/ars.2011.4498. doi: 10.1089/ars.2011 .4498 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nisoli E. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310(5746):314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 38.Corral-Debrinski M, et al. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992;2(4):324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- 39.Yen T-C, Su J-H, King K-L, Wei Y-H. Ageing-associated 5 kb deletion in human liver mitochondrial DNA. Biochem Biophys Res Commun. 1991;178(1):124–131. doi: 10.1016/0006-291X(91)91788-E. [DOI] [PubMed] [Google Scholar]
- 40.Fayet G, et al. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord. 2002;12(5):484–493. doi: 10.1016/S0960-8966(01)00332-7. [DOI] [PubMed] [Google Scholar]
- 41.Taylor RW, et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest. 2003;112(9):1351–1360. doi: 10.1172/JCI19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larsson N-G. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. doi: 10.1146/annurev-biochem-060408-093701. [DOI] [PubMed] [Google Scholar]
- 43.Stewart JB, et al. Strong purifying selection in transmission of mammalian mitochondrial DNA. PloS Biol. 2008;6(1):e10. doi: 10.1371/journal.pbio.0060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart JB, Freyer C, Elson JL, Larsson N-G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat Rev Genet. 2008;9(9):657–662. doi: 10.1038/nrg2396. [DOI] [PubMed] [Google Scholar]
- 45.Ameur A, et al. Ultra-deep sequencing of mouse mitochondrial DNA: mutational patterns and their origins. PLoS Genet. 2011;7(3):e1002028. doi: 10.1371/journal.pgen.1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28(4):693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi J, et al. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc Natl Acad Sci U S A. 1991;88(23):10614–10618. doi: 10.1073/pnas.88.23.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chomyn A, et al. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc Natl Acad Sci U S A. 1992;89(10):4221–4225. doi: 10.1073/pnas.89.10.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holme E, et al. Multiple symmetric lipomas with high levels of mtDNA with the tRNA(Lys) A-->G(8344) mutation as the only manifestation of disease in a carrier of myoclonus epilepsy and ragged-red fibers (MERRF) syndrome. Am J Hum Genet. 1993;52(3):551–556. [PMC free article] [PubMed] [Google Scholar]
- 50.Durham SE, Samuels DC, Chinnery PF. Is selection required for the accumulation of somatic mitochondrial DNA mutations in post-mitotic cells? Neuromuscul Disord. 2006;16(6):381–386. doi: 10.1016/j.nmd.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 51.Elson JL, Samuels DC, Turnbull DM, Chinnery PF. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68(3):802–806. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bender A, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38(5):515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 53.Schwarze SR, Lee CM, Chung SS, Roecker EB, Weindruch R, Aiken JM. High levels of mitochondrial DNA deletions in skeletal muscle of old rhesus monkeys. Mech Ageing Dev. 1995;83(2):91–101. doi: 10.1016/0047-6374(95)01611-3. [DOI] [PubMed] [Google Scholar]
- 54.Kraytsberg Y, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38(5):518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 55.Müller-Höcker J. Cytochrome-c-oxidase deficient cardiomyocytes in the human heart — an age-related phenomenon. A histochemical ultracytochemical study. Am J Pathol. 1989;134(5):1167–1173. [PMC free article] [PubMed] [Google Scholar]
- 56.Müller-Höcker J. Cytochrome c oxidase deficient fibres in the limb muscle and diaphragm of man without muscular disease: An age-related alteration. J Neurol Sci. 1990;100(1–2):14–21. doi: 10.1016/0022-510X(90)90006-9. [DOI] [PubMed] [Google Scholar]
- 57.Cottrell DA, et al. Cytochrome c oxidase deficient cells accumulate in the hippocampus and choroid plexus with age. Neurobiol Aging. 2001;22(2):265–272. doi: 10.1016/S0197-4580(00)00234-7. [DOI] [PubMed] [Google Scholar]
- 58.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 59.Kujoth GC. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 60.Vermulst M, et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40(4):392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- 61.Williams SL, et al. The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab. 2010;12(6):675–682. doi: 10.1016/j.cmet.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park CB, Larsson N-G. Mitochondrial DNA mutations in disease and aging. J Cell Biol. 2011;193(5):809–818. doi: 10.1083/jcb.201010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Edgar D, et al. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009;10(2):131–138. doi: 10.1016/j.cmet.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 64.Tyynismaa H, et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102(49):17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ahlqvist KJ, et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012;15(1):100–109. doi: 10.1016/j.cmet.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 66.Trifunovic A, et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A. 2005;102(50):17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 1994;263(5150):1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- 68.Mockett RJ, Bayne A-CV, Kwong LK, Orr WC, Sohal RS. Ectopic expression of catalase in Drosophila mitochondria increases stress resistance but not longevity. Free Radic Biol Med. 2003;34(2):207–217. doi: 10.1016/S0891-5849(02)01190-5. [DOI] [PubMed] [Google Scholar]
- 69.Orr WC, Mockett RJ, Benes JJ, Sohal RS. Effects of overexpression of copper-zinc and manganese superoxide dismutases, catalase, and thioredoxin reductase genes on longevity in Drosophila melanogaster. J Biol Chem. 2003;278(29):26418–26422. doi: 10.1074/jbc.M303095200. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, et al. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J Gerontol A Biol Sci Med Sci. 2009;64A(12):1212–1220. doi: 10.1093/gerona/glp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5(2):e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Doonan R, et al. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008;22(23):3236–3241. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kayser EB, Morgan PG, Hoppel CL, Sedensky MM. Mitochondrial expression and function of GAS-1 in Caenorhabditis elegans. J Biol Chem. 2001;276(23):20551–20558. doi: 10.1074/jbc.M011066200. [DOI] [PubMed] [Google Scholar]
- 74.Duttaroy A, Paul A, Kundu M, Belton A. A Sod2 null mutation confers severely reduced adult life span in Drosophila. Genetics. 2003;165(4):2295–2299. doi: 10.1093/genetics/165.4.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li Y, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 76.Lebovitz RM, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93(18):9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun J, Folk D, Bradley TJ, Tower J. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics. 2002;161(2):661–672. doi: 10.1093/genetics/161.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mockett RJ, Sohal BH, Sohal RS. Expression of multiple copies of mitochondrially targeted catalase or genomic Mn superoxide dismutase transgenes does not extend the life span of Drosophila melanogaster. Free Radic Biol Med. 2010;49(12):2028–2031. doi: 10.1016/j.freeradbiomed.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O’donnell E, Lynch M. Dietary antioxidant supplementation reverses age-related neuronal changes. Neurobiol Aging. 1998;19(5):461–467. doi: 10.1016/S0197-4580(98)00082-7. [DOI] [PubMed] [Google Scholar]
- 80.Alvarado C, et al. Dietary supplementation with antioxidants improves functions and decreases oxidative stress of leukocytes from prematurely aging mice. Nutrition. 2006;22(7–8):767–777. doi: 10.1016/j.nut.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 81.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2012;3:CD007176. doi: 10.1002/14651858.CD007176.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walker C. Antioxidant supplements do not improve mortality and may cause harm. Am Fam Physician. 2008;78(9):1079–1080. [PubMed] [Google Scholar]
- 83.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297(8):842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 84.Bjelakovic G, Nikolova D, Simonetti R, Gluud C. Antioxidant supplements for prevention of gastrointestinal cancers: a systematic review and meta-analysis. Lancet. 2004;364(9441):1219–1228. doi: 10.1016/S0140-6736(04)17138-9. [DOI] [PubMed] [Google Scholar]
- 85.Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110(8):1109–1124. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lambert AJ, et al. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell. 2007;6(5):607–618. doi: 10.1111/j.1474-9726.2007.00312.x. [DOI] [PubMed] [Google Scholar]
- 87.Cochemé HM, et al. Measurement of H2O2 within living Drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. . Cell Metab. 2011;13(3):340–350. doi: 10.1016/j.cmet.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andziak B, O’Connor TP, Buffenstein R. Antioxidants do not explain the disparate longevity between mice and the longest-living rodent, the naked mole-rat. Mech Ageing Dev. 2005;126(11):1206–1212. doi: 10.1016/j.mad.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 89.Andziak B, et al. High oxidative damage levels in the longest-living rodent, the naked mole-rat. Aging Cell. 2006;5(6):463–471. doi: 10.1111/j.1474-9726.2006.00237.x. [DOI] [PubMed] [Google Scholar]
- 90.Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PloS Biol. 2010;8(12):e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Copeland JM, et al. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19(19):1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 92.Csiszar A, et al. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. Am J Physiol Heart Circ Physiol. 2008;295(5):H1882–H1894. doi: 10.1152/ajpheart.412.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zarse K, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15(4):451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011;21(10):569–576. doi: 10.1016/j.tcb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PloS Biol. 2007;5(10):e259. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Verbon EH, Post JA, Boonstra J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene. 2012;511(1):1–6. doi: 10.1016/j.gene.2012.08.038. [DOI] [PubMed] [Google Scholar]
- 97.Burhans WC, Heintz NH. The cell cycle is a redox cycle: Linking phase-specific targets to cell fate. Free Radic Biol Med. 2009;47(9):1282–1293. doi: 10.1016/j.freeradbiomed.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 98.Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Radic Biol Med. 2012;52(3):539–555. doi: 10.1016/j.freeradbiomed.2011.10.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ristow M, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106(21):8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76(1):701–722. doi: 10.1146/annurev.biochem.76.052305.091720. [DOI] [PubMed] [Google Scholar]
- 101.Holzenberger M, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421(6919):182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 102.Choi J-S, Choi K-M, Lee C-K. Caloric restriction improves efficiency and capacity of the mitochondrial electron transport chain in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2011;409(2):308–314. doi: 10.1016/j.bbrc.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 103.Kapahi P, et al. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14(10):885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007;5(4):265–277. doi: 10.1016/j.cmet.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Powers RW. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20(2):174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vellai T, et al. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426(6967):620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- 107.Kaeberlein M. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310(5751):1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- 108.Murphy CT, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424(6946):277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 109.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6(4):280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 110.Selman C, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326(5949):140. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Weindruch R, Walford RL.The Retardation of Aging and Disease by Dietary Restriction. St. Louis, Missouri, USA: Charles C Thomas Pub Ltd.; 1988. [Google Scholar]
- 112.Kaeberlein M, Burtner C. Recent developments in yeast aging. PLoS Genet. 2007;3(5):e84. doi: 10.1371/journal.pgen.0030084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Colman RJ, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110(8):1109–1124. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cohen HY. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305(5682):390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 116.López-Lluch G, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006;103(6):1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 118.Anderson RM, et al. Dynamic regulation of PGC-1α localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7(1):101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Choi CS, et al. Paradoxical effects of increased expression of PGC-1α on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc Natl Acad Sci U S A. 2008;105(50):19926–19931. doi: 10.1073/pnas.0810339105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Finley LWS, Haigis MC. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res Rev. 2009;8(3):173–188. doi: 10.1016/j.arr.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baker DJ, Betik AC, Krause DJ, Hepple RT. No decline in skeletal muscle oxidative capacity with aging in long-term calorically restricted rats: effects are independent of mitochondrial DNA integrity. J Gerontol A Biol Sci Med Sci. 2006;61(7):675–684. doi: 10.1093/gerona/61.7.675. [DOI] [PubMed] [Google Scholar]
- 122.Katic M, et al. Mitochondrial gene expression and increased oxidative metabolism: role in increased lifespan of fat-specific insulin receptor knock-out mice. Aging Cell. 2007;6(6):827–839. doi: 10.1111/j.1474-9726.2007.00346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rera M, et al. Modulation of longevity and tissue homeostasis by the Drosophila PGC-1 homolog. Cell Metab. 2011;14(5):623–634. doi: 10.1016/j.cmet.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee C-K, Allison DB, Brand J, Weindruch R, Prolla TA. Transcriptional profiles associated with aging and middle age-onset caloric restriction in mouse hearts. Proc Natl Acad Sci U S A. 2002;99(23):14988–14993. doi: 10.1073/pnas.232308999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]