

Abstract
Aging is like the weather: everyone talks about it, but no one seems to do anything about it. We believe this may soon change, as an improved understanding of the molecular and genetic pathways underlying aging suggests it is possible to therapeutically target the aging process and increase health span. This Review series focuses on fundamental cellular mechanisms of aging and their relationship to human disease. These pathways include telomere dysfunction in cellular senescence and induction of the senescence-associated secretory phenotype (SASP) in systemic aging, sirtuin family regulation of metabolism and aging-associated diseases, mitochondrial metabolism in aging, the mechanistic target of rapamycin (mTOR) signaling pathway and the use of mTOR inhibitors to increase longevity, the progressive decline of the immune system with age, and aging-associated changes to pancreatic islet β cells that may contribute to diabetes. Together, these articles explore pathways affecting aging and possible interventional targets to slow or delay the onset of age-related pathologies.
Introduction
Diverse factors, including the sharp peak in births following World War II (the “baby boom”), a rise in worldwide life expectancy (from 48 years in the 1950s to a projected 76 years by 2050), and a decline in fertility, are contributing to a dramatic increase in the percentage of humans over the age of 60 years (1). Currently, approximately 800 million people are at least 60 years old, which represents about 11% of the world’s population; by 2050, this is expected to increase to more than 2 billion people, representing 22% of the population. On a positive note, we can expect a longer period of work productivity and life experiences, in many cases stretching well beyond the age of 60 and into the 70s and 80s. Less positive is the specter of an ever-expanding pool of people experiencing inevitable age-related deterioration in health and onset of frailty, and the impact of such a surge on health care systems and national economies already under severe duress. Despite an expectation that a greater proportion of our society will enjoy a sustained period of “healthy aging,” aging remains a strong risk factor for all of our most prevalent and life-threatening chronic diseases, including cardiovascular disease, cancer, type 2 diabetes, and skeletal/muscular diseases.
In light of the profound effects of aging on human health, the JCI has commissioned a series of Review articles highlighting the fundamental cellular mechanisms of aging and their relationship to human diseases and conditions. The series is not intended to be exhaustive, but rather is focused on selected mechanisms that affect several aspects of aging biology. In particular, we have asked the authors to focus on near-term therapeutic and clinical implications of an enhanced understanding of aging biology.
Historical background
Classic broad symptoms of aging in mammalian species include increased curvature of the spine (kyphosis), reduced fertility, loss of hearing and eyesight, graying and loss of hair, anemia and immune failure, weight loss, frailty, and loss of cognition. These systemic changes are driven by a variety of molecular, biochemical, and metabolic alterations that occur at the cellular level. The field of aging research has grappled for years to develop unifying mechanisms to explain the broad biological effects of the aging process, and to identify pathways that can be targeted to delay or reverse it. This work is complicated by the fact that a major outcome measure in aging research is life span, which is of course affected by lethal diseases such as cancer and cardiovascular disease that develop either as a consequence of or independently from the aging process per se. Though aging remains an inexorable fact of human life, this series highlights important insights that have been gained in the mechanistic understanding of this fundamental process (Figure 1).
Figure 1. Molecular pathways implicated in aging.
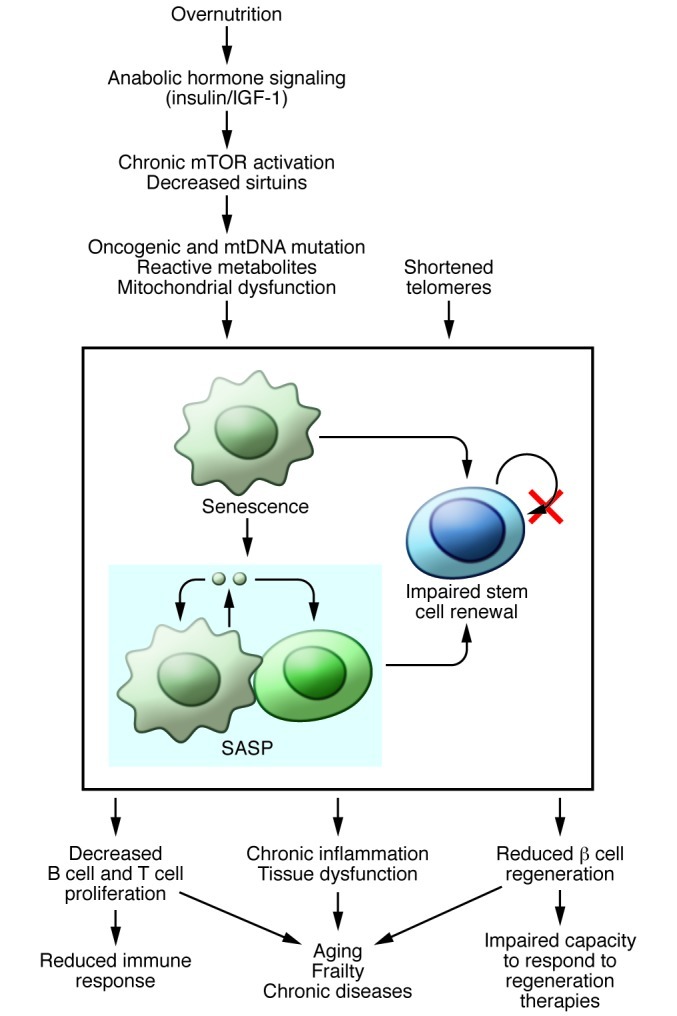
Molecular pathways that drive cellular senescence may contribute to aging and age-related diseases by disrupting tissue homeostasis through impaired stem cell renewal and by promoting the spread of senescence and inducing chronic inflammation through the SASP. Reviews in this series highlight several of these pathways, including the contribution of telomere dysfunction to age-associated disease (24), the mTOR signaling pathway and mechanisms underlying mTOR inhibitors in increasing longevity (13), the role of the sirtuin family of proteins in regulating metabolism and aging-associated diseases (16), the role of mitochondrial dysfunction in aging, including caveats of the long-held free radical hypothesis of aging (8), and the role of cellular senescence and the induction of the SASP in systemic aging (23). The SASP may promote increased tissue inflammation and dysfunction, which leads to aging phenotypes such as the decline in immune system function (26). Aging also decreased β cell regeneration in pancreatic islets (25).
Several major theories have been advanced to explain the biology of aging that are not mutually exclusive. The free radical theory of aging postulates that gradual aging-associated dysfunction of the mitochondrial electron transport chain results in a higher rate of production of superoxide and related oxygen radicals. The accumulation of these radicals in turn leads to oxidative damage of multiple cellular elements, including structural proteins and membrane lipids, hastening the overall phenotype of cellular senescence and functional decline. In addition, alterations in nutrient-sensing pathways, most notably the insulin/IGF-1 signaling pathway and the target of rapamycin (TOR) pathways, are thought to underlie the aging process. Higher activity of these pathways is promoted by nutrient abundance (the “anabolic state”) and is associated with more rapid aging for reasons that remain unclear. The senescence theory of aging suggests that replication-competent cells become exhausted and enter a permanent state of growth arrest called “cellular senescence” as a result of a variety of insults including short telomeres and other forms of DNA damage, oxidative stress, and/or changes in chromatin architecture. This decline in replicative capacity can lead to functional compromise in organs that require homeostatic cellular replication, such as the bone marrow, thymus, and endocrine pancreas. Moreover, senescent cells elaborate a host of potent cytokines (which compose the senescence-associated secretory phenotype [SASP]), and therefore the accumulation of senescent cells with aging is also associated with the production of proinflammatory factors. Chronic inflammation, whether as a consequence of the accumulation of senescent cells or from immune dysfunction, is a defining characteristic of mammalian aging, is thought to promote several age-related phenotypes, and has been dubbed “inflammaging.”
A limited number of interventions for enhancing life span have also emerged that are linked to the foregoing core pathways, including caloric restriction (CR) and inhibition of the mechanistic TOR (mTOR) pathway. CR clearly results in increased life span in a broad array of species, including yeast, C. elegans, flies, and rodents. Interestingly, two long-term studies in primates (rhesus monkeys) have resulted in different outcomes: one study demonstrated a benefit of CR on longevity, but the other showed no effect; the reasons for this difference are currently unknown but are being investigated (2, 3). The benefits of CR for humans have been challenged by several large observational studies and a recent meta-analysis suggesting that lower BMI in humans is associated with increased mortality relative to subjects who are mildly overweight (4–6). Critics of this work suggest that BMI is a poor surrogate for adiposity and effective CR, but the data seem clear that the extreme states of underweight sought by humans attempting CR are not associated with reduced mortality on a population basis. The precise mechanism by which CR enhances longevity in experimentally housed cohorts of model organisms remains uncertain, although CR suppresses TOR and insulin/IGF-1 signaling while inducing sirtuin 1 (SIRT1) and other sirtuins. Pharmacologic inhibition of TOR — primarily the mTOR complex 1 (mTORC1)— with rapamycin and its analogs increases life span in a similarly broad array of species as reported for CR. Importantly, the ability of CR and TOR inhibition to increase longevity in lower organisms suggests an intrinsic effect of these agents on life span that is independent of disease modifiers of longevity. For example, yeast, flies, and worms do not develop cancer, indicating that the effects of rapamycin to promote longevity are not simply explained by the drug’s known antitumor effects. Whether the ability of rapamycin to promote longevity in rodents reflects a true anti-aging effect, or merely its known anticancer activity, is unsettled. Similarly, in model organisms, perturbation of insulin and IGF-1 signaling increases longevity independent of disease modifiers.
The Review articles presented here expand upon these core concepts, in some cases criticizing underlying assumptions and developing a context for gaining deeper understanding. All of the articles explore the potential utility of different pathways for therapeutic intervention.
Role of mitochondria
The “free radical hypothesis” of aging described by Harman in 1956 (7) is perhaps the oldest theory of aging and still has many firm adherents. However, as pointed out in the Review by Bratic and Larsson in this series (8), and by others (9, 10), the free radical hypothesis of aging has become increasingly controversial. What is troubling for this model is the reproducible finding that pharmacologic or genetic neutralization of toxic oxygen radicals or augmentation of antioxidant defenses, while sufficient to reduce measures of damaging oxidative stress, are often not effective in enhancing life span. Furthermore, there is a growing appreciation that acute production of ROS has normal and beneficial effects on signal transduction, control of gene expression, and other biological functions, indicating that chronic or complete suppression of ROS production could be deleterious. Bratic and Larsson provide evidence for an alternative pathway of aging-associated mitochondrial dysfunction that can lead to functional decline of cells independent of ROS production (8). Aging is known to be associated with mutations in genes of the mitochondrial genome, which encodes key proteins of the respiratory complex, including components of the electron transport chain and ATP synthase complexes. Because the mitochondrial genome replicates independently of the nuclear genome, mutations generated during replication of the mitochondrial genome can result in uneven distribution of wild-type and mutated mitochondrial DNA (mtDNA) between different cells and tissues. Thus, some cells may have a preponderance of mutated mtDNA even when the absolute levels of mutated mtDNA are low on an organism-wide basis. This can lead to mosaic deficiencies in mitochondrial function among different cells and tissues.
Recent support for this hypothesis has been gained by the development of mtDNA mutator mice (11), which lack expression of the proofreading enzyme mtDNA polymerase, leading to accelerated and extensive mtDNA mutagenesis. These mice develop a progeroid, or early-aging syndrome. In contrast, a different mouse strain, the mtDNA deletor mouse, expresses a dominant-negative version of twinkle, the mtDNA helicase, resulting in accumulation of large-scale deletions in mtDNA in postmitotic tissues (12). Although these animals exhibit late-onset mitochondrial myopathy and a decline in respiratory function, they have no obvious progeroid syndrome, which suggests that accumulation of mtDNA mutations and mitochondrial dysfunction are not sufficient to accelerate aging. Instead, recent work has suggested that the absence of a fully functional pool of somatic stem cells contributes to the premature aging phenotype of the mtDNA mutator mouse strain (8). While interesting, it remains to be determined whether accrual of mtDNA mutations contributes to the normal aging process, given that the high rate of such mutations in murine models will not be matched in terms of timing or frequency in normal human aging.
Rapalogs and TOR
Lamming et al. focus on the TOR pathway in aging and on TOR inhibitors as a potential interventional tool (13). The most well-known TOR inhibitor, rapamycin, has been used in humans for many years to inhibit graft rejection in tissue transplant subjects and has garnered recent attention as a potential anticancer therapeutic. Interest in rapamycin as an anti-aging drug stems from multiple lines of genetic evidence implicating TOR and its substrate S6 kinase 1 (S6K1) in aging in yeast, flies, worms, and rodents. Rapamycin extends life span in all of these species and is thus touted as the most widely tested and efficacious pharmacotherapy for aging that is currently known. However, the mechanisms involved in this effect remain uncertain. mTORC1 and S6K1 regulate general protein translation via the eukaryotic initiation factor eIF4E binding protein (4E-BP). Inhibition of mTOR also leads to increased degradation and recycling of proteins via activation of the process of autophagy. Thus, rapamycin may reduce cell stress via a dual-edged mechanism for controlling cellular protein content, with salutary effects such as reducing flux of protein through the endoplasmic reticulum. Yet the TOR pathway also has life span–extending activity independent of S6K1 or translation initiation factors. The possibility remains that selective inhibition of the translation of certain transcripts may play a key role in the anti-aging effects of rapamycin, a topic that is gaining increasing attention. In an interesting convergence with the mtDNA mutation hypothesis, rapamycin has potent effects on stem cell function, with the general property of promoting self-renewal capacity.
Although rapamycin is used clinically to prevent organ rejection, treat renal cell carcinoma, and coat coronary stents, and is being tested in various other clinical trials, it is unlikely to be of use in increasing longevity in healthy subjects due to its diverse and still-emerging side effects. The most prominent among these are related to its immunosuppressive activities, including increased incidence of viral and fungal infections. Rapamycin also has been shown to cause edema, ulcers, rash, and hair and nail disorders, as well as a host of metabolic changes reminiscent of metabolic syndrome, including hyperlipidemia, insulin resistance, and glucose intolerance. Lamming et al. suggest that some, if not all, of these side effects could be averted by development of new forms of TOR inhibitors that modestly suppress mTORC1, with no chronic effect on mTORC2 (13). Interestingly, the most widely used diabetes drug in the world today, metformin, was recently discovered to be an inhibitor of mTORC1, and is capable of decreasing phosphorylation of the mTORC1 substrates S6K1 and 4E-BP1 (14). Unlike the effects of chronic rapamycin treatment, metformin reverses several of the symptoms of metabolic syndrome and is very well tolerated over long periods of time in humans. It also increases longevity in species ranging from yeast to mice and was even found to decrease mortality from all causes in humans in a long-term diabetes study (15). The mechanism of action of metformin in diabetes treatment is not fully understood. Interestingly, the drug decreases the activity of complex I of the electron transport chain, and also increases the activity of AMPK. AMPK inhibits mTORC1 complex activity, providing a link between the rapamycin and metformin mechanisms for promoting longevity.
The role of sirtuins in aging
Hall et al. discuss the possible role of the sirtuin family of enzymes in the control of aging (16), a field that had its origins with the report that overexpression of the yeast sirtuin Sir2 resulted in increased life span in that simple organism (17). Since then, the field has been remarkably controversial, and some uncertainty remains as to the extent to which sirtuins influence fundamental aging processes, as opposed to control of activities that can contribute to life span such as energy metabolism, development of cancer, and gene transcription (18, 19). Further complexity is introduced by the fact that mammals express 7 sirtuin gene family members localized to diverse subcellular compartments, with different tissue distribution patterns and still-emerging differences in catalytic and biologic functions. Finally, many of the claims related to sirtuins and age-related pathologies come from studies relying on putative sirtuin activators (e.g., resveratrol), but several groups have challenged the pharmacologic effects of these agents (20–22). The Review by Hall et al. highlights some of the controversies concerning sirtuins and aging, but also focuses on the role of each of the sirtuin family members in important areas of biology (16). One connection that emerges between sirtuins and the core aging process in mammals is the induction of several family members in response to CR. Increased expression of the nuclear sirtuin SIRT1 and the mitochondrial sirtuin SIRT3 contributes to metabolic adaptations to CR. SIRT1 catalyzes the deacetylation of the key transcriptional coregulator PGC-1α, enhancing its activity and contributing to transcriptional upregulation of multiple genes encoding enzymes of the fatty acid β-oxidation pathway. Coinduction of SIRT3 results in deacetylation of some of the same enzymes that are upregulated by SIRT1/PGC-1α, increasing their enzymatic activity. However, it remains unclear why selective activation of oxidation of fatty acids relative to glucose is a strategy to promote longevity. The confusion is heightened by studies that show mitigation of metabolic dysfunction and insulin resistance in response to either overexpression or knockdown of SIRT1. Nevertheless, emerging evidence for cardioprotective, antitumoral, and neuroprotective effects of the mammalian sirtuins, as summarized in the Review by Hall et al. (13), argue that further investigation of the connections between sirtuins and aging could well be fruitful.
Cellular replication and aging
The series includes four articles that discuss cellular replication and aging. Two Reviews focus on the role of cellular senescence in aging (23, 24), whereas the others describe the consequences of a decline in age-associated tissue regeneration in the endocrine pancreas (25) and cellular immune system (26).
“Senescence” in common parlance refers to any functional decline with aging, but “cellular senescence” in cell biology refers to a specific phenomenon of permanent cellular growth arrest, first described by Hayflick and Moorehead in vitro in the 1960s (27). Senescent cells (in the Hayflickian sense) exhibit several phenotypic features including changes in gene expression and morphology, and remain growth arrested in culture for years or longer. Senescent cells cannot generally be coaxed to reenter the cell cycle by ordinary means, such as expression of oncogenes or addition of mitogens. Several lines of recent evidence have compellingly demonstrated that senescent cells accumulate in vivo with aging in mammals and that the activation of the senescence program is a key component of mammalian anticancer defenses. Therefore, the senescence theory of aging holds that this mechanism is antagonistically pleiotropic, preventing cancer in young age, but promoting aging in older organisms. The loss of replicative capacity and accumulation of senescent cells is thought to promote aging in at least two ways: through the loss of the beneficial, replicative function of certain cell types and through the production of proinflammatory cytokines (i.e., the SASP). The Reviews by Kushner (25) and the Dorshkind group (26) predominantly discuss the consequences of reduced replication, while Tchkonia et al. address the age-promoting effects of the SASP (23). The Review by Armanios (24) describes the in vivo consequences of telomere dysfunction, which is one of the most important organismal mediators of senescence. However, it is clear that not all replicative failure with aging results from cellular senescence (also discussed in refs. 25, 26), and it is not generally known, even for the endocrine pancreas and lymphoid organ, to what degree the failure of cellular replication that occurs with aging in these tissues is the result of senescence activation versus other forms of nonpermanent growth arrest.
Telomeres are complex nucleoprotein structures that serve to protect chromosomal ends from activating DNA damage response and end-to-end fusions. Telomere length shortens slightly with each cell division, and when telomere shortening occurs beyond a certain point, telomere dysfunction ensues, which is associated with activation of a DNA damage response and the induction of cellular senescence. Counteracting the effects of cellular replication on telomere length is the telomerase holoenzyme complex, which is active in germ cells and certain self-renewing somatic cells to preserve replicative capacity throughout life. As described by Armanios (24), humans and mice lacking normal telomerase activity have been shown to exhibit some features of premature aging, and in particular, human telomerase deficiency is associated with pulmonary fibrosis, bone marrow failure, cirrhosis, and other age-associated conditions. The Review by Armanios describes the use of telomere-based tests for the diagnosis and management of human conditions, as well as the role of therapies aimed at altering telomere metabolism to combat certain age-related conditions (24).
Cellular senescence is activated by a number of cellular stresses, including telomere dysfunction, with the beneficial effect of preventing cancer. In particular, senescence is associated with activation of two classic tumor suppressor pathways, p53 and p16INK4a-Rb, to induce durable growth arrest. In contrast to cellular quiescence (i.e., reversible growth arrest), senescence is also associated with specific global changes in chromatin architecture as well as durable changes in gene expression. Importantly, senescent cells become a veritable factory of inflammatory cytokines, producing high levels of potent, pleiotropic molecules such as IL-6, IL-8, TNF-α, and VEGF-a, with profound effects on local inflammation, angiogenesis, fibrosis, and wound healing. Tchkonia et al. (23) describe the effects of the SASP in aging mammals, as well as the therapeutic potential of reducing the burden of senescent cells as a treatment for aging. This model of senescence and aging has particular appeal, as one can envision several approaches to counteract the adverse effects of SASP-associated cytokines or to clear senescent cells through an immune-mediated approach.
A decline in cellular immunity is one of the most clinically familiar and morbid features of human aging. Montecino-Rodriguez et al. (26) carefully describe the features of aging of the various components of the cellular immune system including HSCs, thymus, and B and T lymphocytes. HSC function appears to decline with aging in the absence of senescence induction, whereas the function of lymphoid progenitors may exhibit senescence-dependent replicative failure. The Review discusses important recent findings regarding the role of milieu in cellular immune aging, summarizing data showing how “inflammaging” and a decline in thymic function can both induce an age-related decline in lymphocyte function. Additionally, the Review includes a discussion of provocative approaches to “rejuvenate” immune aging in humans through the use of IL-7 and other factors to counteract the loss in thymic function and naive T cell production that occurs with age.
The response of pancreatic islet β cells to aging provides an interesting context for describing the contribution of molecular mechanisms to reduced proliferative and regenerative capacity of tissues over time, as reviewed by Kushner in this series (25). The loss of regenerative capacity in β cells that occurs with age is relevant because inadequate β cell mass and function are at the heart of development of both major forms of diabetes, and no reliable strategies are currently available to reverse this problem. Proliferation of β cells slows markedly with age in rodents and humans, and islets from aged rodents are poorly responsive to proliferative stimuli such as glucagon-like peptide 1 and high-fat feeding. In β cells and other tissues, aging-related decreases in proliferation are linked to increased expression of cyclin-dependent kinase inhibitors (CDKIs). In islets, p16INK4a plays a particularly prominent role, as supported by several lines of evidence. First, knockout of p16INK4a increases, and overexpression of p16INK4a decreases, β cell proliferation (28). Second, manipulation of multiple pathways and factors that control p16INK4a, including the polycomb proteins BMI1 and EZH2, and the PDGF and p38 MAPK signaling pathways, alter β cell replication in rodent models of aging in a p16INK4a-associated fashion. One recent study demonstrated that islets from aged rodents and humans have reduced expression of the PDGFR (29). Restoration of PDGFR expression in older mice enhanced β cell proliferation via induction of the p16INK4a suppressive factor EZH2. Despite these advances in understanding age-related changes in control of the cell cycle in islet cells, no methods have yet been reported to induce proliferation of human islet β cells from older donors in vitro. Interestingly, there is one report showing that transplantation of pancreatic islets into young but not old mice enables β cell proliferation in islets from older rodent or human donors (30). These findings implicate a factor or factors present in young mice that can activate proliferation in otherwise quiescent islets from aged donors. Further studies will be required to identify such putative factors and to test their utility for expansion of functional islet cell mass in post-adolescent diabetic patients.
Summary and conclusions
This collection of Reviews reinforces the view that our basic understanding of the cellular and molecular biology of aging is beginning to lead to therapies to address this fundamental process in humans. Novel aging diagnostics related to telomeres and other markers of senescence are being validated for use in certain clinical scenarios. Human trials of agents including metformin, rapalogs, PDGF, and IL-7, thought to counter aging and aging sequelae, are being planned or are already started. Molecular events associated with CR and anabolic signaling are increasingly understood and may eventually become anti-aging targets. These Reviews suggest even more definitive (and presently more theoretical) approaches such as the generation of replacement cells using induced pluripotent stem cell technology or stimulating immune clearance of damaged or senescent cells through a vaccination approach. While still in the conception stage, such ideas are supported by preclinical efficacy studies in rodents and, we believe, have the potential to change the arc of physiological aging in humans in our lifetime.
Footnotes
Conflict of interest: Christopher Newgard has received financial support from Pfizer and research support from Pfizer and Eli Lilly. Norman Sharpless has received financial support from MolecularMD and has a financial stake in G1 Therapeutics.
Citation for this article: J Clin Invest. 2013;123(3):946–950. doi:10.1172/JCI68833.
References
- 1. Bloom DE, Boersch-Supan A, McGee P, Seike A. Population aging: facts, challenges and responses. PGDA working paper No. 71, Harvard. http://www.hsph.harvard.edu/pgda/WorkingPapers/2011/PGDA_WP_71.pdf . Updated May 2011. Accessed January 18, 2013.
- 2.Colman RJ, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mattison JA, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489(7415):318–322. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303(3):235–41. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 5.Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta-analysis. JAMA. 2013;309(1):71–82. doi: 10.1001/jama.2012.113905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jee SH, et al. Body-mass index and mortality in Korean men and women. N Engl J Med. 2006;355(8):779–87. doi: 10.1056/NEJMoa054017. [DOI] [PubMed] [Google Scholar]
- 7.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 8.Bratic A, Larsson N-G. The role of mitochondria in aging. J Clin Invest. 2013;123(3):951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Radic Biol Med. 2012;52(3):539–555. doi: 10.1016/j.freeradbiomed.2011.10.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pérez VI, et al. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790(10):1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 12.Tyynismaa H, et al. Mutant mitochondrial helicase twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci U S A. 2005;102(49):17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 2013;123(3):980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67(22):10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 15.Scarpello JH. Improving survival with metformin: the evidence base today. Diabetes Metab. 2003;29(4 pt 2):6S36–6S43. doi: 10.1016/S1262-3636(03)72786-4. [DOI] [PubMed] [Google Scholar]
- 16.Hall JA, Dominy JE, Lee Y, Puigserver P. The sirtuin family’s role in aging and age-associated pathologies. J Clin Invest. 2013;123(3):973–979. doi: 10.1172/JCI64094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13(19):2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burnett C, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011;477(7365):482–485. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinclair D, Verdin E. The longevity of sirtuins. Cell Rep. 2012;2(6):1473–1474. doi: 10.1016/j.celrep.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Kaeberlein M, Kennedy BK. Does resveratrol activate yeast Sir2 in vivo? Aging Cell. . 2007;6(4):415–416. doi: 10.1111/j.1474-9726.2007.00314.x. [DOI] [PubMed] [Google Scholar]
- 21.Beher D, et al. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem Biol Drug Des. 2009;74(6):619–624. doi: 10.1111/j.1747-0285.2009.00901.x. [DOI] [PubMed] [Google Scholar]
- 22.Pacholec M, et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem. 2010;285(11):8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Armanios M. Telomeres and age-related disease: how telomere biology informs clinical paradigms. J Clin Invest. 2013;123(3):996–1002. doi: 10.1172/JCI66370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kushner JA. The role of aging upon β cell turnover. J Clin Invest. 2013;123(3):990–995. doi: 10.1172/JCI64095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K. Causes, consequences, and reversal of immune system aging. J Clin Invest. 2013;123(3):958–965. doi: 10.1172/JCI64096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 28.Krishnamurthy J, et al. P16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 29.Chen H, et al. PDGF signalling controls age-dependent proliferation in pancreatic beta-cells. Nature. 2011;478(7369):349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian L, et al. Comparison of exendin-4 on beta-cell replication in mouse and human islet grafts. Transpl Int. 2011;24(8):856–864. doi: 10.1111/j.1432-2277.2011.01275.x. [DOI] [PubMed] [Google Scholar]