

Abstract

Although mu opioid (MOP) receptor agonists are the most commonly used analgesics for the treatment of moderate to severe pain in the clinic, the side effects of MOP agonists such as abuse liability limit their value as a medication. Research to identify novel analgesics without adverse effects is pivotal to advance the health care of humans. The nociceptin/orphanin FQ peptide (NOP) receptor, the fourth opioid receptor subtype, mediates distinctive actions in nonhuman primates which suggests the possibility that activity at this receptor may result in strong analgesia in the absence of virtually all of the side effects associated with MOP agonists. The present review highlights the recent progress of pharmacological studies of NOP-related ligands in primates. Selective NOP agonists, either peptidic or nonpeptidic, produce full analgesia in various assays in primates, when delivered systemically or intrathecally. Yet small molecule NOP agonists do not serve as reinforcers, indicating a lack of abuse liability. Given that NOP agonists have low abuse liability and that coactivation of NOP and MOP receptors produces synergistic antinociception, it is worth developing bifunctional NOP/MOP ligands. The outcomes of these studies and recent developments provide new perspectives to establish a translational bridge for understanding the biobehavioral functions of NOP receptors in primates and for facilitating the development of NOP-related ligands as a new generation of analgesics without abuse liability in humans.
Keywords: Nociceptin/orphanin FQ, NOP receptors, analgesia, abuse liability, macaques, spinal cord
Pain is a symptom of many clinical disorders that afflict a large population of humans and is often treated with pharmacological agents. There have been remarkable advances in our understanding of some pharmacological bases of pain and analgesia in the past decade. However, opioid analgesics remain the most effective and widely used drugs for pain management, and the most clinically used opioids are mu opioid (MOP) receptor agonists (Table 1).1−3 There are several adverse effects associated with the use of MOP agonists. These side effects include abuse liability, respiratory depression, constipation, and itch/pruritus. The side effect profile of MOP agonists has been and remains a serious public health concern and limits the value of opioid analgesics for pain management.1−3 Therefore, research to identify potential analgesics with fewer side effects and reduced abuse liability is pivotal to advances in health care of humans.
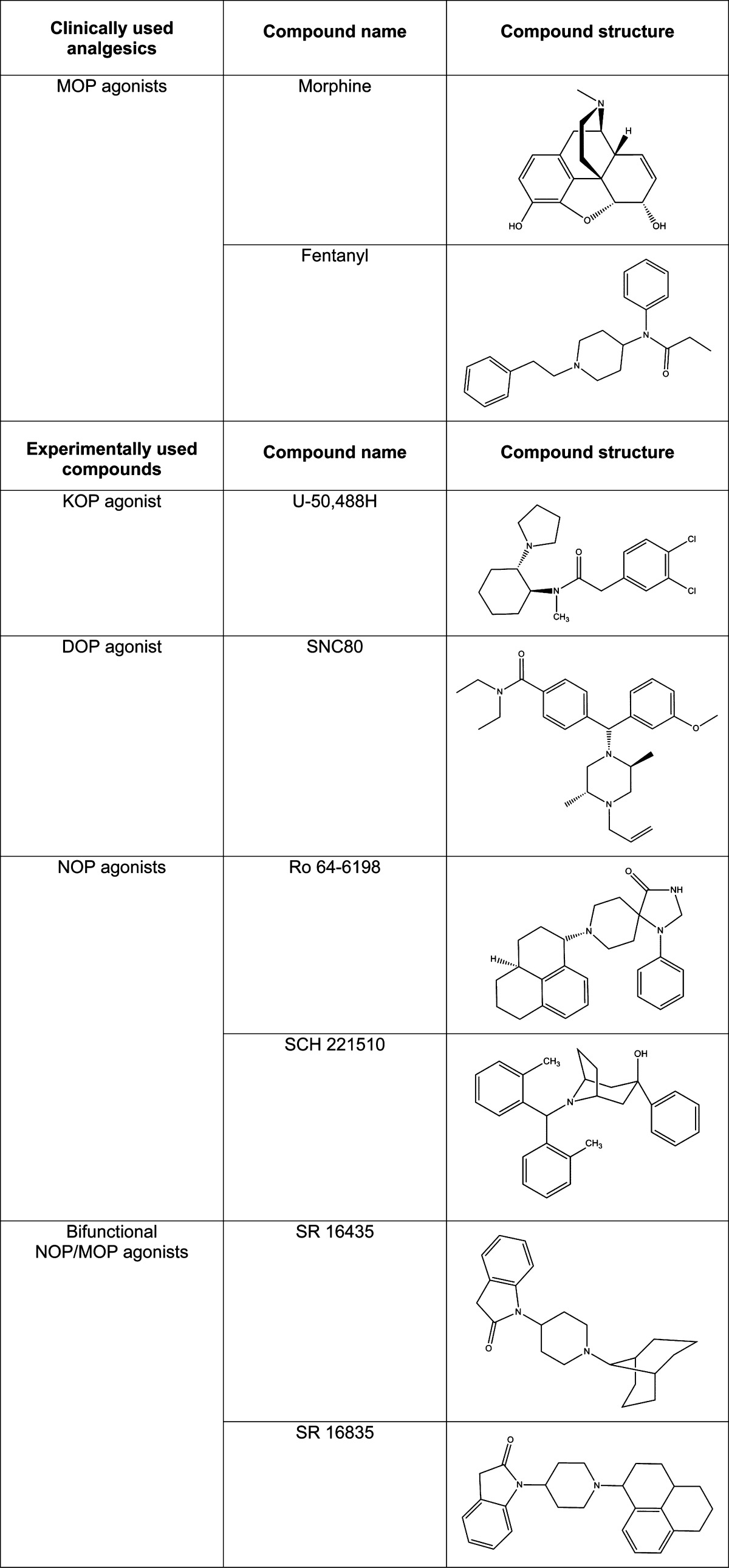
Table 1. Structures of Clinically Used Analgesics (MOP agonists) and Experimental Compounds (KOP, DOP, NOP, and bifunctional NOP/MOP agonists).
Limitations of Using MOP Agonists
Abuse liability is the most devastating side effect of MOP agonists as analgesics. Unpleasant sensations experienced after withdrawing from MOP agonists compel continuing utilization which results in physical dependence that can ultimately become abuse. Psychological dependence might also be factored into the development of opioid abuse due to the euphoric feelings induced by the administration of MOP agonists. Opioid abuse is mainly observed in self-medication, use for experiencing euphoria, compulsive-addictive use, and diversion for profit.2,3 Owing to its powerful and potent nature in alleviating pain and inducing euphoria, opioid abuse has been a growing problem that poses serious threats to society. A world drug report issued by the United Nations Office on Drugs and Crimes estimated that 10–25% of the 24–35 million people who used opioids in 2008 became problem drug users; up to 26% of out-patients on chronic opioid therapy have opioid abuse problems.4 These reports indicate that pain is no longer a one-dimensional problem; it has become a multidimensional problem5 due to the use of MOP agonists, which creates significant difficulties for both the patients and society. The total cost of nonmedical use of prescription opioids was estimated at $53.4 billion in 2006.6 In the health care system, the mean annual direct cost for patients abusing opioids is 8.7 times higher than that for nonabusers.3 Furthermore, overdose potentials give rise to significant mortality rates.5 Given the staggering outcomes related to the abuse of opioid analgesics, both the risks and benefits of MOP agonists as analgesics have been debated.
In addition to the most deleterious side effect, abuse liability, MOP agonists also produces other side effects such as respiratory depression/arrest, constipation, and itch/pruritus. Opioid therapy is most often employed in treating patients with progressive and far-advanced illnesses. One of the most common side effects seen in these patients utilizing opioid analgesics is chronic constipation. Current treatments for opioid-induced constipation involve stimulant laxatives, known to cause negative physiological effects such as dehydration and abdominal cramping.7,8 Peripherally restricted MOP antagonists are available, and they appear to block the constipating effects of MOP agonists with no effect on analgesia. These compounds, such as methylnaltrexone (Relistor) and alvimopan (Entereg), have been approved by the FDA for restricted populations (e.g., postoperative ileus for Entereg), but a treatment for opioid bowel dysfunction associated with a long-term use of MOP agonists has not yet been approved.7,8 Opioid-induced constipation remains a crucial problem because it impacts the opioid use pattern, resource utilization, and post an additional medical burden to the health care system.8,9
Activation of MOP receptors can reduce breathing frequency and tidal volume in patients, as well as the respiratory response to carbon dioxide-stimulated chemoreceptors, leaving a window of decreasing respiratory rate and increasing unresponsiveness.10,11 This problem directly relates to patient mortality rates in hospitals and necessitates vigilant patient care. While systemic administration of morphine is used in opioid therapy targeting chronic pain, intrathecal administration of MOP agonists has been one major breakthrough in pain management during the past three decades and has since become the most common way of inducing analgesia in obstetrics and for surgery.10,12 However, spinal administration of MOP agonists is most often accompanied by the unwanted itch sensation (pruritus) in patients.12,13 In obstetrics, pruritus has been a long-term concern that requires the use of antipruritic drugs. Antipruritic drugs, however, have varied efficacies that may contribute to hormonal changes in obstetric patients that might expose patients to both psychological and physiological downsides.13,14 MOP agonist-induced itch/pruritus is a common “symptom” seen in postoperative patients and, therefore, becomes an issue worthy of attention because it limits the value of spinal MOP agonists as analgesics.12−14
For the reason that the majority of analgesics are MOP agonist-derived,15,16 quest for a comparable analgesic with reduced abuse liability and fewer side effects is pivotal to substantially advance health care for humans. Search of alternative pathways of pain medication is warranted under the circumstance, as the MOP pathway does not provide a well-rounded treatment for patients. Clearly, there is an unmet need to identify and develop strong analgesics that would act on different receptor mechanisms potentially with fewer side effects.
Research and Development of KOP and DOP Agonists
There are two other opioid receptor subtypes, and each has been evaluated for their ability to mediate analgesia. There has, for example, been a tremendous effort to develop kappa opioid (KOP) receptor and delta opioid (DOP) receptor agonists (Table 1) as pain therapeutic agents in the last two decades. Although neither KOP nor DOP agonists significantly produce reinforcing effects, drugs in both categories do not have promising pharmacological profiles as strong analgesics comparable to MOP agonists. To date, none of KOP and DOP agonists are approved by the FDA. Centrally penetrating KOP agonists’ antinociceptive effects were compromised by sedation, inhibited food consumption, dysphoria, and psychotomimetic effects.15,17−19 Nevertheless, a KOP agonist (nalfurafine) is available in Japan for the treatment of uremic pruritus in patients undergoing hemodialysis and is being investigated for the treatment of pruritus in patients with chronic liver disease.20 DOP agonists produced only moderate antinociception associated with the potential for convulsant effects in nonhuman primates.21−23 However, recent development of novel DOP agonists suggests that potent antinociceptive effects can be produced without convulsions in rodents, particularly under chronic pain models.24 These recent findings indicate that the DOP still represents a promising target for the development of innovative analgesics.
Comparing the antinociceptive effects of KOP and DOP agonists with MOP agonists, MOP agonists are still the predominant pain reliever in health care. For example, Corbett and colleagues published an article15 entitled, 75 years of opioid research: the exciting but vain quest for the Holy Grail, which itself conveys a similar message to the notion of Rang and colleagues in the textbook16 they wrote: “Morphine is, as expected of ‘God’s own medicine’, very hard to beat!” Despite the research identifying other opioid receptor subtypes and developing selective agonists and antagonists for each receptor, their analgesic effects are not comparable with those of MOP.15,16 Therefore, there has been a critical need to develop strong analgesics with fewer side effects. Aiming for finding an alternative for MOP agonists, recent studies focus on the exploration of liability-free pharmacological systems/neurotransmission pathways, hoping to introduce a new perspective to the field of pain medication.
Species Differences in Pharmacological Profiles of Opioid-Related Ligands
In the research and development of analgesics, it is often learned that nonhuman primates have distinct pharmacological and behavioral responses from rodents, and are more like humans than rodents when appropriate comparisons are made. There are several examples documenting species differences in the pharmacological actions of drugs and corresponding receptor functions between rodents and primates.
First, for studying MOP-related ligands, a buprenorphine-like compound, BU72, was characterized as a MOP agonist with a wide therapeutic window in rodents, but BU72 was very potent in producing respiratory depression/arrest in nonhuman primates and it displayed a very narrow window between antinociceptive doses and doses producing side effects in nonhuman primates.25 6β-Naltrexol was characterized as a neutral MOP antagonist, but naltrexone displayed an inverse MOP agonist action in morphine-dependent mice.26 In contrast, 6β-naltrexol failed to block naltrexone-precipitated withdrawal in morphine-dependent monkeys; instead, it potentiated the effects of naltrexone when the dose of 6β-naltrexol was increased, indicating that both naltrexone and 6β-naltrexol have the same pharmacological actions only with large potency differences in primates.27 These findings suggest that there might be difference between primates and rodents in terms of the MOP basal signaling activity changed by repeated MOP agonist administration.
Second, for studying KOP-related ligands, naloxone benzoylhydrazone was characterized as a KOP-3 subtype-selective agonist that produced antinociception in mice.28 Later, naloxone benzoylhydrazone was further studied in various primate assays for its discriminative stimulus, analgesic, and respiratory effects. Naloxone benzoylhydrazone clearly displayed MOP antagonist actions without detectable KOP agonist activity in primates.29 Although the receptor mechanisms of naloxone benzoylhydrazone are not clear, the in vivo actions of this compound are different between rodents and primates. Another interesting example is the behavioral responses elicited by a KOP antagonist, nor-binaltorphimine. Subcutaneous administration of nor-binaltorphimine produced profound scratching responses in mice, indicating the tone of endogenous KOP-preferring peptides in regulating itch sensation.30 However, following subcutaneous or intrathecal administration route, nor-binaltorphimine alone did not elicit scratching in primates and it only produced the KOP antagonist effects in primates receiving exogenously administered KOP agonists.31,32 These in vivo findings go along with other in vitro studies demonstrating differences in KOP-regulated desensitization and phosphorylation between human KOP and rat KOP receptors.33
Third, for studying DOP-related ligands, intrathecal administration of a DOP-selective agonist, BW373U86, produced antinociceptive effects in mice.34 However, intrathecal BW373U86 failed to produce antinociceptive effects in nonhuman primates. This functional difference could be contributed potentially by the relatively low DOP receptor density and corresponding receptor-G protein activation in the monkey spinal cord.35,36 In addition to opioid receptor subtypes, rodent and primate species differences in the pharmacological actions and/or receptor functions also exist in nonopioid receptors and channels, including bradykinin B1 receptors, tachykinin receptors, and transient receptor potential ankyrin-1 channel, that regulate pain neurotransmission.37−39
It is the case that humans and monkeys have similar thresholds for detecting noxious stimuli and the neural systems responsible for these sensations in the two species are fundamentally similar.40−43 In the clinic, intrathecal administration of morphine provides patients with pain relief, but it also elicits side effects such as itch/pruritus.12,13 This common clinical issue derived from the spinal delivery of morphine can be observed in monkeys as intrathecal morphine dose-dependently produces antinociception with simultaneous itch/scratching responses in monkeys,44,45 but not in rodents.46 More importantly, the potency and duration of analgesic action of spinal morphine are similar between humans and monkeys.44,47,48 In addition, the relative ratios of opioid receptor subtypes, MOP, KOP, and DOP, in the monkey central nervous system are similar to those of receptor densities observed in the cortex and thalamus of humans.35,42,49 Furthermore, several clinically used MOP agonists in terms of their potencies and therapeutic profiles can be manifested and simulated very well in monkey models.50−52 Therefore, these examples document the necessity and importance of using nonhuman primates to identify, study, and develop analgesics, and more importantly translate these findings into future clinical trials in humans.
Actions of NOP Receptors in Vitro
The nociceptin/orphanin FQ peptide (NOP) receptor, previously called the ORL1/opioid-receptor-like receptor, is a member of the G protein-coupled receptor family. It is also defined as the fourth member within the opioid receptor family by the International Union of Pharmacology.53,54 The sequence of the NOP receptor is closely related to each of the classical, well-characterized MOP, KOP, and DOP receptors.55,56 However, ligands that bind to these classical opioid receptor subtypes do not bind NOP receptors with high affinity. For example, classical opioid agonists and antagonists, such as morphine and naloxone, are not active on the NOP receptor.55−57 The NOP receptor is widely distributed in the central and peripheral nervous system, the cardiovascular system, the airways, the gastrointestinal tract, the urogenital tract, and the immune system.58,59 Due to its widespread locations, NOP receptor may participate in a broad range of physiological and behavioral functions producing pleiotropic effects.58−60
Like most of classical opioids, nociceptin/orphanin FQ (N/OFQ), a heptadecapeptide that is an endogenous ligand for the NOP receptor, inhibits cyclic AMP accumulation,61,62 inhibits Ca2+ entry,61 and increases K+ conductance in neurons.63,64 These actions result in a decreased neuronal excitability and neurotransmitter release.58−60 In addition, N/OFQ inhibits the release of pronociceptive substances from peripheral sensory nerves.65 These in vitro findings suggest an “inhibitory” function of the NOP receptors, which is quite similar to classical opioid receptor subtypes, MOP, DOP, and KOP in the nociception-relevant process.65,66 Although similar in function to the classical opioid agonists, N/OFQ was found to have almost no affinity for the three classical opioid receptors,61,62 indicating that this peptide acts through a different receptor.
In light of the discovery of N/OFQ and NOP receptors, the development and synthesis of selective NOP agonists and antagonists continue to progress in an attempt to define the roles of NOP receptors in diverse physiological functions. Although the actions of N/OFQ and NOP agonists have much in common with those of classical opioids at the cellular level, behavioral studies of NOP agonists in rodents suggest a high degree of complexity.58−60 The present review highlights recent pharmacological studies of NOP agonists in primates, in contrast to what has been known in rodents. Given that both spinal and systemic administration are common routes for delivery of analgesics, sections below specifically discuss the pharmacological profiles of NOP agonists as compared to clinically used MOP agonists following intrathecal and systemic administration.
Effects of Intrathecal NOP-Related Ligands
After N/OFQ was discovered, several laboratories have reported antinociceptive effects of intrathecally administered N/OFQ at the dose range of nmol in rodents under different pain modalities.67−69 However, ultralow doses of intrathecal N/OFQ (i.e., fmol) produced spontaneous agitation and painlike behavior including biting, scratching, and licking responses in mice,70 indicating that biphasic actions of intrathecal N/OFQ were observed in rodents.
In contrast, intrathecal administration of N/OFQ at the dose range of nmol produced antinociceptive effects and N/OFQ(2–17), a major fragment of N/OFQ, was inactive in changing the nociceptive threshold in monkeys.71 As mentioned above, monkey subjects can be used to validate whether newly developed opioid analgesics possess an itch-eliciting property.44,72 The findings that intrathecal N/OFQ produced antinociception without scratching responses in primates strongly support the notion that the spinal NOP receptor is a viable target for research and development to provide spinal analgesia.71−73 More importantly, unlike dual actions (i.e., both pronociceptive and antinociceptive actions) of intrathecal N/OFQ in rodents, intrathecal N/OFQ over a wide dose range from femtomolar to nanomolar only produced antinociception in primates. For direct comparisons in the same group of subjects, intrathecal substance P was used to elicit hyperalgesic responses and intrathecal DAMGO was shown to elicit both antinociception and itch/scratching.73 These primate studies provide a valuable pharmacological basis for identifying and developing spinal analgesics.
In an attempt to develop a long-acting spinal analgesic like morphine, scientists at the University of Ferrara, Italy, have successfully modified N/OFQ and identified UFP-112 by increasing its agonist potency and decreasing its susceptibility to peptidase actions.74−76 In rodent assays investigating a variety of physiological functions, UFP-112 consistently produced N/OFQ-like effects with markedly higher potency and longer duration of actions.76 A recent primate study further demonstrates that intrathecal administration of UFP-112 produced a long-lasting morphine-comparable antinociception against both acute pain and capsaicin-induced allodynia in primates.77 Intrathecal UFP-112 was more potent than intrathecal morphine, but both compounds had the same duration of antinociceptive actions (Figure 1). Likewise, intrathecal UFP-112-induced antinociception was not accompanied by itch scratching responses and its actions were exclusively mediated by spinal NOP receptors. These findings provide direct functional evidence and translational values of NOP agonists as spinal analgesics in primates. In particular, compared to previous findings that intrathecal administration of selective KOP and DOP agonists did not produce morphine-comparable antinociceptive effects in primate models,35,36 promising outcomes derived from primate studies of intrathecal NOP agonists encourage the therapeutic development of NOP-related ligands as spinal analgesics for future clinical trials.
Figure 1.

Comparison of behavioral effects produced by intrathecal administration of UFP-112 and morphine. Abscissas: time in hours after intrathecal administration. Ordinates: latency to withdraw the tail in 50 °C water (top panels) and scratches per 15 min (bottom panels). Each value represents mean ± SEM (n = 6). Symbols represent different dosing conditions in monkeys. The asterisk represents a significant difference from the vehicle condition at corresponding time points (*p < 0.05). [Reprinted with permission from Pain.77]
Effects of Systemic NOP-Related Ligands
Ro 64–6198 is the first reported nonpeptidic selective NOP agonist (Table 1).78 Following systemic administration, Ro 64–6198 significantly reduced the reinforcing effects of ethanol without producing reinforcing or aversive properties of its own in mice and this compound did not induce conditioned place preference in rats.79,80 Systemic administration of Ro 64–6198 did not produce antinociceptive effects in both mouse and rat tail-flick assays,78,81 but it produced antinociception in the mouse hot-plate test.82
Contrary to the results from rodent studies, systemic administration of Ro 64–6198 produced antinociceptive effects against acute noxious stimulus in monkeys.83,84 Importantly, systemic Ro 64–6198 produced antiallodynic effects in the primate capsaicin-induced allodynia model. Capsaicin elicits pain sensation by activating the vanilloid receptor and stimulating the release of pronociceptive neuropeptides.85 Capsaicin-induced allodynia has been used in both monkeys and humans to study the pain mechanisms and pharmacological interventions.86−88 Given that capsaicin-sensitive nerve fibers are involved in diverse nociceptive conditions,85 the effectiveness of Ro 64–6198 in attenuating capsaicin-induced allodynia indicates that NOP agonists may be effective for treating pain derived from different nociceptive origins.
Furthermore, systemic administration of a newly developed nonpeptidic selective NOP agonist, SCH 221510 (Table 1),89 also produced antinociception against acute pain and capsaicin-induced allodynia in primates.52,89 In a newly developed primate model of inflammatory pain, MOP agonists, NSAIDs, and NOP agonists were equally effective in inhibiting carrageenan-induced allodynia/hyperalgesia.37,89 Equal effectiveness of MOP and NOP agonists across different primate pain models are very promising. In particular, cross-examinations of Ro 64-6198 and a MOP agonist, alfentanil, were conducted in monkeys for antinociceptive effects (Figure 2). While a NOP antagonist, J-113397, was able to antagonize the effects of Ro 64-6198, it was not effective to alfentanil. Naltrexone, a MOP antagonist, reversed the antinociceptive effects of alfentanil but not Ro 64-6198. These results clearly demonstrate that the similar degree of antinociceptive effects can be produced by two independent receptor mechanisms in primates.77,84
Figure 2.

Effects of MOP and NOP receptor antagonists on alfentanil- and Ro 64-6198-induced antinociceptive effects in monkeys. A MOP receptor antagonist (naltrexone, 0.03 mg/kg, s.c.) or a NOP receptor antagonist (J-113397, 0.1 mg/kg, s.c.) was administered s.c. 15 min before redetermination of the dose–response curve of alfentanil and Ro 64-6198. Left panel: antagonist effects of s.c. naltrexone and J-113397 on the dose–response curve of Ro 64-6198-induced antinociception in 50 °C water. Right panel: antagonist effects of s.c. naltrexone and J-113397 on the dose–response curve of alfentanil-induced antinociception in 50 °C water. Each data point represents a mean ± SEM (n = 6). [Reprinted with permission from Neuropsychopharmacology.84]
What is more exciting and stimulating is that Ro 64-6198 produced MOP agonist-comparable antinociception without reinforcing effects as compared to other reinforcers including alfentanil, cocaine, and methohexital in monkeys.84 Lack of reinforcing effects by Ro 64-6198 might be expected because several rodent studies have shown that NOP agonists do not have either reinforcing or aversive properties of their own.79,80 Nevertheless, the primate assay of intravenous self-administration of drugs is considered the best validated approach to evaluate and predict the abuse liability of newly developed drugs.91 This primate drug self-administration procedure has been used extensively in drug abuse research for over 40 years and has provided useful information on the behavioral processes associated with drug addiction as well as drug abuse liability in humans.91 As mentioned above one of the biggest concerns with the use of opioids is the abuse liability and the search for an abuse-free, strong analgesic has been ongoing for decades.15,16 Lack of reinforcing effects of Ro 64-6198 in alfentanil-, cocaine- and methohexital-maintained monkeys strongly indicates that NOP agonists will not have abuse liability in humans and opens a new avenue to develop strong analgesics without abuse liability.
Following systemic administration, alfentanil produced antinociception in the presence of respiratory depression and itch scratching in primates. In the same group of subjects, systemic NOP agonists produced antinociception without such side effects, but caused sedation at a dose that was approximately 30-fold higher than the full antinociceptive doses.52,84,92 These results indicate that NOP agonists have a much wider therapeutic window compared with MOP agonists in primates. In particular, lack of respiratory depression of NOP agonists are expected to be much safer than the use of MOP agonists. More importantly, using radiopaque markers, we have recently developed a procedure to study the gastrointestinal transit function in primates. When the equi-analgesic doses of morphine and SCH 221510 were administered, systemic morphine significantly delayed the transit of the markers across the different regions of colons and the excretion time of the markers. In contrast, systemic SCH 221510 produced no change in the bowel motility as compared to the vehicle treatment in the same group of subjects.52,90 Based on the literature of behavioral neuropharmacology in nonhuman primates, NOP agonists are the first series of ligands that provide a proof of concept in producing strong antinociception with fewer side effects in monkeys (Figure 3).52,83,84,90 It is extremely exciting and appealing to document a promising therapeutic profile of NOP agonists in primates, namely they produce MOP agonist-comparable antinociception without abuse liability, constipation potential, respiratory depression, and itch pruritus. Nevertheless, future studies are warranted to investigate and compare the rates and degrees of potential physical dependence and tolerance in primates receiving repeated/chronic administration of NOP agonists as compared to MOP agonists.
Figure 3.

Illustration of the pharmacological effects of MOP and NOP receptor agonists in the preclinical models of primates. Solid arrows indicate the confirmed causality between MOP agonists and the side effects. Dashed lines indicate the weak association between NOP agonists and the side effects.
Development of Bifunctional NOP/MOP Ligands
As indicated above, there are marked differences between rodents and primates with respect to the antinociceptive effects of NOP agonists. There are also differences between rodents and primates with respect to the interactions between NOP and MOP receptors at this endpoint. In rodents following supraspinal or systemic administration, NOP agonists have been shown to have anti-MOP effects.58−60 However, in rodents following spinal administration, NOP agonists potentiated MOP agonist-induced antinociception.58−60 In contrast, systemic or spinal administration of NOP agonists in primates enhanced the analgesic effects of MOP agonists. For example, when N/OFQ was combined with intrathecal morphine, intrathecal N/OFQ dose-dependently enhanced morphine-induced analgesia without compromising the primate’s motor function.73 When an inactive dose of UFP-112 was combined intrathecally with an inactive dose of morphine, such a mixture significantly produced antinociception against capsaicin-induced allodynia.77 These findings strongly indicate that mixed NOP/MOP agonists (i.e., bifunctional NOP/MOP agonists) may represent a novel strategy for developing analgesics.
The potentiated antinociception produced by coactivation of NOP and MOP receptors occurs not only at the spinal level, but also at the systemic level in primates. Buprenorphine is well known as a MOP partial agonist that has been used for both pain management and opioid abuse/addiction and it has less rewarding property compared to other MOP agonists in primates.51,93 When a NOP agonist (either Ro 64-6198 or SCH 221510) is combined with systemic buprenorphine, the NOP agonist dose-dependently potentiated buprenorphine-induced analgesia.52 It is worth noting that NOP antagonists potentiated the antinociceptive effects of buprenorphine in rodents, indicating that buprenorphine’s own NOP agonist activity attenuated its own MOP agonist activity.94,95 Unlike rodent studies, the NOP antagonist J-113397 only blocked NOP agonist-induced antinociception, but could neither enhance nor attenuate buprenorphine-induced antinociception in monkeys.52 By using the isobologram dose-addition analysis, primate studies demonstrated that the NOP agonist interacted with buprenorphine in a synergistic manner to produce antinociceptive effects.52 More importantly, by increasing the relative ratio of the NOP agonist in combination with buprenorphine, the specific mixture potently produced full antinociception without detectable respiratory depression and itch scratching.52 These findings not only provide a new perspective on the interactions between NOP and MOP receptors at the systemic level, but also generate pharmacological evidence that coactivation of NOP and MOP receptors produces synergistic antinociceptive effects without other side effects in primates.
Since NOP agonists have well-documented antirewarding and/or lack of rewarding property,96 bifunctional NOP/MOP ligands may not elicit significant rewarding effects, but they are expected to be more potent and effective in producing antinociceptive effects. Fortunately, several leading medicinal chemists have developed and synthesized bifunctional NOP/MOP agonists that have different efficacies on both receptor sites.96−99 As both NOP and MOP receptor components have been demonstrated to independently produce antinociception at both spinal and systemic levels,77,84 adding the NOP agonist component to MOP agonists such as buprenorphine may generate a much wider therapeutic window (Figure 4). In other words, by reserving most functional receptor reservoirs, simultaneous activation of two receptor components to a small degree will produce desirable therapeutic effects without either receptor-derived side effects. When two receptors are repeatedly activated due to a large reservoir for both receptor populations, bifunctional NOP/MOP ligands may also have a reduced dependence and tolerance liability as compared to MOP- and NOP-selective ligands. To date, however, there has been no effort made to better define and characterize any mixed NOP/MOP agonists systemically in primates. Future pharmacological studies in primates are definitely needed to compare and determine the ideal candidate for bifunctional NOP/MOP ligands (i.e., to provide strong analgesia without rewarding effects). More importantly, future studies are warranted to determine whether such a bifunctional NOP/MOP agonist (Table 1) has an advantage over the selective NOP agonist following acute and chronic administration in primates.
Figure 4.

General hypothetical framework of comparison of the therapeutic windows of MOP, NOP, and bifunctional NOP/MOP agonists based on current literature. Solid lines indicate the doses at which antinocieption/analgesia will occur. Dashed lines indicate the doses at which side effects, especially respiratory depression and sedation, emerge.
Conclusions
Pharmacological studies in primates have been instrumental in evaluating newly developed ligands and comparing their antinociceptive effects with clinically used analgesics. After intrathecal and systemic administration, both peptidic and nonpeptidic NOP agonists display a promising pharmacological profile in primates, indicating that NOP agonists may have therapeutic values as novel analgesics without abuse liability, respiratory depression, constipation potential, and itch pruritus in humans. These pharmacological studies in primates provide a new exciting chapter to the research and development of opioid-related analgesics.
In addition to the promising pharmacological findings in primates, researchers have recently developed PET radioligands such as 11C-NOP-1A to reliably quantify NOP receptors in both monkey and human brains.100,101 It provides a valuable tool to study the NOP receptors by correlating the receptor occupancy with functional evidence. More importantly, the crystal structure of human NOP receptor has been recently discovered102 and it provides a great opportunity to investigate the ligand structure–activity relationships. With additional molecular dynamics stimulations, they offer new insights into the NOP receptor activated by agonists and facilitate the future design of novel NOP-related ligands.102,103
Hopefully, some future candidate NOP-related ligands can be further studied in primate models. In particular, the physiological functions of the NOP receptor should be and can be extensively investigated and elucidated by both using diverse NOP-related ligands with different selectivities and efficacies and by using various behavioral and functional assays in a broader context in primates. From our perspective, it is important to conduct studies in behaving primates as a preclinical framework to establish the therapeutic profiles of NOP-related ligands, to translate these findings into clinical trials in humans, and to facilitate the development of NOP-related ligands as a new generation of analgesics without abuse liability in the global community.
Author Contributions
A.P.L. and M.C.K. contributed equally by drafting, revising, and completing this article.
We are thankful for the strong support by the U.S. Department of Defense, Peer Reviewed Medical Research Program, Grant No. W81XWH-07-1-0162, and National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases, Grant No. R01-AR-059193, and National Institute on Drug Abuse, Grant No. R01-DA-032568.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Cicero T. J.; Dart R. C.; Inciardi J. A.; Woody G. E.; Schnoll S.; Munoz A. (2007) ) The development of a comprehensive risk-management program for prescription opioid analgesics: researched abuse, diversion and addiction-related surveillance (RADARS). Pain Med. 8, 157–170. [DOI] [PubMed] [Google Scholar]
- Sullivan M. D.; Von Korff M.; Banta-Green C.; Merrill J. O.; Saunders K. (2010) Problems and concerns of patients receiving chronic opioid therapy for chronic non-cancer pain. Pain 149, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal N.; Manchikanti L.; Smith H. S. (2012) Prescription opioid abuse in chronic pain: a review of opioid abuse predictors and strategies to curb opioid abuse. Pain Physician 15, ES67–ES92. [PubMed] [Google Scholar]
- Boscarino J. A.; Rukstalis M.; Hoffman S. N.; Han J. J.; Erlich P. M.; Gerhard G. S.; Stewart W. F. (2010) Risk factors for drug dependence among out-patients on opioid therapy in a large US health-care system. Addiction 105, 1776–1782. [DOI] [PubMed] [Google Scholar]
- Nelson L. S.; Perrone J. P. (2012) Curbing the opioid epidemic in the United States: the risk evaluation and mitigation strategy (REMS). JAMA, J. Am. Med. Assoc. 308, 457–458. [DOI] [PubMed] [Google Scholar]
- Hansen R. N.; Oster G.; Edelsburg J.; Woody G. E.; Sullivan S. D. (2011) Economic costs of nonmedical use of prescription opioids. Clin. J. Pain 27, 194–202. [DOI] [PubMed] [Google Scholar]
- Licup N.; Baumrucker S. J. (2010) Methylnaltrexone: treatment for opioid-induced constipation. Am. J. Hosp. Palliative Med. 28, 59–61. [DOI] [PubMed] [Google Scholar]
- Camilleri M. (2011) Opioid-induced constipation: challenges and therapeutic opportunities. Am. J. Gastroenterol. 106, 835–842. [DOI] [PubMed] [Google Scholar]
- Hjalte F.; Berggren A.; Bergendahl H.; Hjortsberg C. (2010) The direct and indirect costs of opioid-induced constipation. J. Pain Symptom Manage. 40, 696–703. [DOI] [PubMed] [Google Scholar]
- DeBalli P.; Breen. T. W. (2003) Intrathecal opioids for combined spinal-epidural analgesia during labour. CNS Drugs 17, 889–904. [DOI] [PubMed] [Google Scholar]
- Macintyre P. E.; Loadsman J. A.; Scott D. A. (2011) Opioids, ventilation and acute pain management. Anaesth. Intensive Care 39, 545–558. [DOI] [PubMed] [Google Scholar]
- Cousins M. J.; Mather L. E. (1984) Intrathecal and epidural administration of opioids. Anesthesiology 61, 276–310. [PubMed] [Google Scholar]
- Ganesh A.; Maxwell L. G. (2007) Pathophysiology and management of opioid-induced pruritus. Drugs 67, 2323–2333. [DOI] [PubMed] [Google Scholar]
- Szarvas S.; Harmon D.; Murphy D. (2003) Neuraxial opioid-induced pruritus: a review. J. Clin. Anesth. 15, 234–239. [DOI] [PubMed] [Google Scholar]
- Corbett A. D.; Henderson G.; McKnight A. T.; Paterson S. J. (2006) 75 years of opioid research: the exciting but vain quest for the Holy Grail. Br. J. Pharmacol. 147, S153–S162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang H. P., Dale M. M., and Ritter J. M. (2007) Rang and Dale’s Pharmacology (6th Ed.), Analgesic Drugs, pp 562–584, Edinburgh, Churchill Livingstone. [Google Scholar]
- Dykstra L. A.; Gmerek D. E.; Winger G.; Woods J. H. (1987) Kappa opioids in rhesus monkeys. I. Diuresis, sedation, analgesia and discriminative stimulus effects. J. Pharmacol. Exp. Ther. 242, 413–420. [PubMed] [Google Scholar]
- Butelman E. R.; Harris T. J.; Kreek M. J. (1999) Effects of E-2078, a stable dynorphin A(1–8) analog, on sedation and serum prolactin levels in rhesus monkeys. Psychopharmacology 147, 73–80. [DOI] [PubMed] [Google Scholar]
- Ko M. C. H.; Johnson M. D.; Butelman E. R.; Willmont K. J.; Mosberg H. I.; Woods J. H. (1999b) Intracisternal nor-binaltorphimine distinguishes central and peripheral kappa-opioid antinociception in rhesus monkeys. J. Pharmacol. Exp. Ther. 291, 1113–1120. [PMC free article] [PubMed] [Google Scholar]
- Nagase H.; Fujii H. (2011) Opioids in preclinical and clinical trials. Top. Curr. Chem. 299, 29–62. [DOI] [PubMed] [Google Scholar]
- Dykstra L. A.; Schoenbaum G. M.; Yarbrough J.; McNutt R.; Chang K. J. (1993) A novel delta opioid agonist, BW373U86, in squirrel monkeys responding under a schedule of shock titration. J. Pharmacol. Exp. Ther. 267, 857–882. [PubMed] [Google Scholar]
- Negus S. S.; Butelman E. R.; Chang K. J.; DeCosta B.; Winger G.; Woods J. H. (1994) Behavioral effects of the systemically active delta opioid agonist BW373U86 in rhesus monkeys. J. Pharmacol. Exp. Ther. 270, 1025–1034. [PubMed] [Google Scholar]
- Negus S. S.; Gatch M. B.; Mello N. K.; Zhang X.; Rice K. (1998) Behavioral effects of the delta-selective opioid agonist SNC80 and related compounds in rhesus monkeys. J. Pharmacol. Exp. Ther. 286, 362–375. [PubMed] [Google Scholar]
- Nozaki C.; Le Bourdonnec B.; Reiss D.; Windh R. T.; Little P. J.; Dolle R. E.; Kieffer B. L.; Gaveriaux-Ruff C. (2012) Delta-opioid mechanisms for ADL5747 and ADL5859 effects in mice: analgesia, locomotion, and receptor internalization. J. Pharmacol. Exp. Ther. 342, 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilan C. L.; Husbands S. M.; Breeden S.; Ko M. C.; Aceto M. D.; Lewis J. W.; Woods J. H.; Traynor J. R. (2004) Characterization of the complex morphinan derivative BU72 as a high efficacy, long-lasting μ-opioid receptor agonist. Eur. J. Pharmacol. 499, 107–116. [DOI] [PubMed] [Google Scholar]
- Raehal K. M.; Lowery J. J.; Bhamidipati C. M.; Paolino R. M.; Blair J. R.; Wang D.; Sadee W.; Bilsky E. J. (2005) In vivo characterization of 6β-naltrexol, an opioid ligand with less inverse agonist activity compared with naltrexone and naloxone in opioid-dependent mice. J. Pharmacol. Exp. Ther. 313, 1150–1162. [DOI] [PubMed] [Google Scholar]
- Ko M. C. H.; Divin M. F.; Lee H.; Woods J. H.; Traynor J. R. (2006a) Differential in vivo potencies of naltrexone and 6beta-naltrexol in the monkey. J. Pharmacol. Exp. Ther. 316, 772–779. [DOI] [PubMed] [Google Scholar]
- Paul D.; Levison J. A.; Howard D. H.; Pick C. G.; Hahn E. F.; Pasternak G. W. (1990) Naloxone benzoylhydrazone (NalBzoH) analgesia. J. Pharmacol. Exp. Ther. 255, 769–774. [PubMed] [Google Scholar]
- France C. P.; Woods J. H. (1992) Naloxone benzoylhydrazone is a mu-selective opioid antagonist without kappa-agonist effects in rhesus monkeys. Behav. Pharmacol. 3, 133–141. [PubMed] [Google Scholar]
- Kamei J.; Nagase H. (2001) Norbinaltorphimine, a selective kappa-opioid receptor antagonist, induces an itch-associated response in mice. Eur. J. Pharmacol. 418, 141–145. [DOI] [PubMed] [Google Scholar]
- Ko M. C. H.; Lee H.; Song M. S.; Sobczyk-Kojiro K.; Mosberg H. I.; Kishioka S.; Woods J. H.; Naughton N. N. (2003a) Activation of kappa Opioid Receptors Inhibits Pruritus Evoked by Subcutaneous or Intrathecal Administration of Morphine in Monkeys. J. Pharmacol. Exp. Ther. 305, 173–179. [DOI] [PubMed] [Google Scholar]
- Lee H.; Naughton N. N.; Woods J. H.; Ko M. C. (2007) Effects of butorphanol on morphine-induced itch and analgesia in primates. Anesthesiology 107, 478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Chen L. Y. (2004) Agonist-induced regulation and trafficking of kappa-opioid receptors. Life Sci. 75, 511–536. [DOI] [PubMed] [Google Scholar]
- Wild K. D.; McCormick J.; Bilsky E. J.; Vanderah T.; McNutt R. W.; Chang K. J.; Porreca F. (1993) Antinociceptive actions of BW373U86 in the mouse. J. Pharmacol. Exp. Ther. 267, 858–865. [PubMed] [Google Scholar]
- Ko M. C. H.; Lee H.; Harrison C.; Clark M. J.; Song H. F.; Naughton N. N.; Woods J. H.; Traynor J. R. (2003b) Studies of μ-, κ-, and δ-opioid receptor density and G protein activation in the cortex and thalamus of monkeys. J. Pharmacol. Exp. Ther. 306, 179–186. [DOI] [PubMed] [Google Scholar]
- Ko M. C. H.; Lee H.; Traynor J. R.; Woods J. H.; Naughton N. N. (2003c) Differential antinociceptive effects of intrathecally administered opioid agonists selective for mu, kappa, and delta opioid receptors in monkeys. Drug Alcohol Depend. 69(Suppl), 87.12536069 [Google Scholar]
- Hawkinson J. E.; Szoke B. G.; Garofalo A. W.; Hom D. S.; Zhang H.; Dreyer M.; Fukuda J. Y.; Chen L.; Samant B.; Simmonds S.; Zeitz K. P.; Wadsworth A.; Liao A.; Chavez R. A.; Zmolek W.; Ruslim L.; Bova M. P.; Holcomb R.; Butelman E. R.; Ko M. C.; Malmberg A. B. (2007) Pharmacological, pharmacokinetic, and primate analgesic efficacy profile of the novel bradykinin B1 receptor antagonist ELN441958. J. Pharmacol. Exp. Ther. 322, 619–630. [DOI] [PubMed] [Google Scholar]
- Leffler A.; Ahlstedt I.; Engberg S.; Svensson A.; Billger M.; Oberg L.; Bjursell M. K.; Lindstrom E.; von Mentzer B. (2009) Characterization of species-related differences in the pharmacology of tachykinin NK receptors 1, 2 and 3. Biochem. Pharmacol. 77, 1522–1530. [DOI] [PubMed] [Google Scholar]
- Bianchi B. R.; Zhang X. F.; Reilly R. M.; Kym P. R.; Yao B. B.; Chen J. (2012) Species comparison and pharmacological characterization of human, monkey, rat, and mouse TRPA1 channels. J. Pharmacol. Exp. Ther. 341, 360–368. [DOI] [PubMed] [Google Scholar]
- LaMotte R. H.; Campbell J. N. (1978) Comparison of responses of warm and nociceptive C-fiber afferents in monkey with human judgments of thermal pain. J. Neurophysiol. 41, 509–528. [DOI] [PubMed] [Google Scholar]
- Rozsa A. J.; Molinari H. H.; Greenspan J. D.; Kenshalo D. R. Sr. (1985) The primate as a model for the human temperature-sensing system: 1. Adapting temperature and intensity of thermal stimuli. Somatosens. Res. 2, 303–314. [DOI] [PubMed] [Google Scholar]
- Mansour A.; Khachaturian H.; Lewis M. E.; Akil H.; Watson S. J. (1988) Anatomy of CNS opioid receptors. Trends Neurosci. 11, 308–314. [DOI] [PubMed] [Google Scholar]
- Kenshalo D. R. Jr.; Anton F.; Dubner R. (1989) The detection and perceived intensity of noxious thermal stimuli in monkey and in human. J. Neurophysiol. 62, 429–436. [DOI] [PubMed] [Google Scholar]
- Ko M. C. H.; Naughton N. N. (2000) An experimental itch model in monkeys: Characterization of intrathecal morphine-induced scratching and antinociception. Anesthesiology 92, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M. C.; Husbands S. M. (2009) Effects of atypical kappa-opioid receptor agonists on intrathecal morphine-induced itch and analgesia in primates. J. Pharmacol. Exp. Ther. 328, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Naughton N. N.; Woods J. H.; Ko M. C. H. (2003) Characterization of scratching responses in rats following centrally administered morphine or bombesin. Behav. Pharmacol. 14, 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P. L.; Rhondeau S.; Schafer P. G.; Lu J. K.; Timmins B. S.; Foster W.; Pace N. L.; Stanley T. H. (1993) Dose-response pharmacology of intrathecal morphine in human volunteers. Anesthesiology 79, 49–59. [DOI] [PubMed] [Google Scholar]
- Palmer C. M.; Emerson S.; Volgoropolous D.; Alves D. (1999) Dose-response relationship of intrathecal morphine for postcesarean analgesia. Anesthesiology 90, 437–444. [DOI] [PubMed] [Google Scholar]
- Kuhar M. J.; Pert C. B.; Snyder S. H. (1973) Regional distribution of opiate receptor binding in monkey and human brain. Nature (London, U.K.) 245, 447–450. [DOI] [PubMed] [Google Scholar]
- Zacny J. P.; Conley K.; Galinkin J. (1997) Comparing the subjective, psychomotor and physiological effects of intravenous buprenorphine and morphine in healthy volunteers. J. Pharmacol. Exp. Ther. 282, 1187–1197. [PubMed] [Google Scholar]
- Pergolizzi J.; Aloisi A. M.; Dahan A.; Filitz J.; Langford R.; Mercadante S.; Morlion B.; Raffa R. B.; Sabatowski R. (2010) Current knowledge of buprenorphine and its unique pharmacological profile. Pain Pract. 10, 428–450. [DOI] [PubMed] [Google Scholar]
- Cremeans C. M.; Gruley E.; Kyle D. J.; Ko M. C. (2012) Roles of μ-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J. Pharmacol. Exp. Ther. 343, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollereau C.; Parmentier M.; Mailleux P.; Butour J. L.; Moisand C.; Chalon P.; Caput D.; Vassart G.; Meunier J. C. (1994) ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 341, 33–38. [DOI] [PubMed] [Google Scholar]
- Foord S. M.; Bonner T. I.; Neubig R. R.; Rosser E. M.; Pin J.-P.; Davenport A. P.; Spedding M.; Harmar A. J. (2005) International Union of Pharmacology. XLVI. G Protein-Coupled Receptor List. Pharmacol. Rev. 57, 279–288. [DOI] [PubMed] [Google Scholar]
- Henderson G.; McKnight A. T. (1997) The orphan opioid receptor and its endogenous ligand—nociceptin/orphanin FQ. Trends Pharmacol. Sci. 18, 293–300. [PubMed] [Google Scholar]
- Calò G.; Guerrini R.; Rizzi A.; Salvadori S.; Regoli D. (2000) Pharmacology of nociceptin and its receptor: a novel therapeutic target. Br. J. Pharmacol. 129, 1261–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelli O.; Nothacker H. P.; Reinscheid R. (1998) Reverse physiology: discovery of the novel neuropeptide, orphanin FQ/nociceptin. Crit. Rev. Neurobiol. 12, 163–176. [DOI] [PubMed] [Google Scholar]
- Mogil J. S.; Pasternak G. W. (2001) The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol. Rev. 53, 381–415. [PubMed] [Google Scholar]
- Lambert D. G. (2008) The nociception/orphanin FQ receptor: a target with broad therapeutic potential. Nat. Rev. Drug Discovery 8, 694–710. [DOI] [PubMed] [Google Scholar]
- Largent-Milnes T. M.; Vanderah T. W. (2010) Recently patented and promising ORL-1 ligands: where have we been and where are we going?. Expert Opin. Ther. Pat. 20, 291–305. [DOI] [PubMed] [Google Scholar]
- Meunier J. C.; Mollereau C.; Toll L.; Suaudeau C.; Moisand C.; Alvinerie P.; Butour J. L.; Guillemot J. C.; Ferrara P.; Monsarrat B.; Mazarguil H.; Vassart G.; parmentier M.; Constantin J. (1995) Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature 377, 532–535. [DOI] [PubMed] [Google Scholar]
- Reinscheid R. K.; Nothacker H. P.; Bourson A.; Ardati A.; Henningsen R. A.; Bunzow J. R.; Grandy D. K.; Langen H.; Monsma F. J. Jr.; Civelli O. (1995) Orphanin FQ: A neuropeptide that activates an opioidlike G protein-coupled receptor. Science 270, 792–794. [DOI] [PubMed] [Google Scholar]
- Connor M.; Yeo A.; Henderson G. (1996) The effect of nociceptin on Ca2+ channel current and intracellular Ca2+ in the SH-SY5Y human neuroblastoma cell line. Br. J. Pharmacol. 118, 205–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan C. W.; Christie M. J. (1996) Increase by the ORL1 receptor (opioid receptor-like-1) ligand, nociceptin, of inwardly rectifying K conductance in dorsal raphe nucleus neurones. Br. J. Pharmacol. 117, 1609–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani S.; Maggi C. A. (1996) Inhibition of tachykinin release from peripheral endings of sensory nerves by nociceptin, a novel opioid peptide. Br. J. Pharmacol. 118, 1567–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helyes Z.; Nemeth J.; Pinter E.; Szolcsanyi J. (1997) Inhibition by nociceptin of neurogenic inflammation and the release of SP and CGRP from sensory nerve terminals. Br. J. Pharmacol. 121, 613–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. J.; Hao J. X.; Wiesenfeld-Hallin Z. (1996) Nociceptin or antinociceptin: potent spinal antinociceptive effect of orphanin FQ/nociceptin in the rat. NeuroReport 7, 2092–2094. [PubMed] [Google Scholar]
- Erb K.; Liebel J. T.; Tegeder I.; Zeilhofer H. U.; Brune K.; Geisslinger G. (1997) Spinally delivered nociceptin/orphanin FQ reduces flinching behaviour in the rat formalin test. NeuroReport 8, 1967–1970. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.; Nozaki-Taguchi N.; Kimura S. (1997) Analgesic effect of intrathecally administered nociceptin, an opioid receptor-like1 receptor agonist, in the rat formalin test. Neuroscience 81, 249–254. [DOI] [PubMed] [Google Scholar]
- Sakurada T.; Katsuyama S.; Sakurada S.; Inoue M.; Tan-No K.; Kisara K.; Sakurada C.; Ueda H.; Sasaki J. (1999) Nociceptin-induced scratching, biting and licking in mice: involvement of spinal NK1 receptors. Br. J. Pharmacol. 127, 1712–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M. C. H.; Wei H.; Woods J. H.; Kennedy R. T. (2006b) Effects of intrathecally administered nociception/orphanin FQ in monkeys: behavioral and mass spectrometric studies. J. Pharmacol. Exp. Ther. 318, 1257–1264. [DOI] [PubMed] [Google Scholar]
- Ko M. C. H.; Song M. S.; Edwards T.; Lee H.; Naughton N. N. (2004) The role of central mu opioid receptors in opioid-induced itch in primates. J. Pharmacol. Exp. Ther. 310, 169–176. [DOI] [PubMed] [Google Scholar]
- Ko M. C.; Naughton N. N. (2009) Antinociceptive effects of nociception/orphanin FQ administered intrathecally in monkeys. J. Pain 10, 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arduin M.; Spagnolo B.; Calo G.; Guerrini R.; Carra G.; Fischetti C.; Trapella C.; Marzola E.; McDonald J.; Lambert D. G.; Regoli D.; Salvadori S. (2007) Synthesis and biological activity of nociceptin/orphanin FQ analogues subsituted in position 7 or 11 with Cα,α-dialkylated amino acids. Bioorg. Med. Chem. 15, 4434–43. [DOI] [PubMed] [Google Scholar]
- Rizzi A.; Spagnolo B.; Wainford R. D.; Fischetti C.; Guerrini R.; Marzola G.; Baldisserotto A.; Salvadori S.; Regoli D.; Kapusta D. R.; Calo G. (2007) In vitro and in vivo studies on UFP-112, a novel potent and long lasting agonist selective for the nociceptin/orphanin FQ receptor. Peptides 28, 1240–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo’ G.; Rizzi A.; Cifani C.; Micioni, Di Bonaventura M. V.; Regoli D.; Massi M.; Salvadori S.; Lambert D. G.; Guerrini R. (2011) UFP-112 a potent and long-lasting agonist selective for the Nociceptin/Orphanin FQ receptor. CNS Neurosci. Ther. 17, 178–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E.; Calo G.; Guerrini R.; Ko M. C. (2010) Long-lasting antinociceptive spinal effects in primates of the novel nociception/orphanin FQ receptor agonist UFP-112. Pain 148, 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenck F.; Wichmann J.; Dautzenberg F. M.; Moreau J. L.; Ouagazzal A. M.; Martin J. R.; Lundstrom K.; Cesura A. M.; Poli S. M.; Roever S.; Kolczewski S.; Adam G.; Kilpatrick G. (2000) A synthetic agonist at the orphanin FQ/nociceptin receptor ORL1: anxiolytic profile in the rat. Proc. Natl. Acad. Sci. U.S.A. 97, 4938–4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pen G.; Wichmann J.; Moreau J. C.; Jenck F. (2002) The orphanin receptor agonist Ro 64-6198 does not induce place conditioning in rats. NeuroReport 13, 451–454. [DOI] [PubMed] [Google Scholar]
- Kuzmin A.; Sandin J.; Terenius L.; Ogren S. O. (2003) Acquisition, Expression, and Reinstatement of Ethanol-Induced Conditioned Place Preference in Mice: Effects of Opioid Receptor-Like 1 Receptor Agonists and Naloxone. J. Pharmacol. Exp. Ther. 304, 310–318. [DOI] [PubMed] [Google Scholar]
- Dautzenberg F. M.; Wichmann J.; Higelin J.; Py-Lang G.; Kratzeisen C.; Malherbe P.; Kilpatrick G. J.; Jenck F. (2001) Pharmacological characterization of the novel nonpeptide orphanin FQ/nociceptin receptor agonist Ro 64-6198: rapid and reversible desensitization of the ORL1 receptor in vitro and lack of tolerance in vivo. J. Pharmacol. Exp. Ther 298, 812–819. [PubMed] [Google Scholar]
- Reiss D.; Wichmann J.; Tekeshima H.; Kieffer B. L.; Ouagazzal A. M. (2008) Effects of nociceptin/orphanin FQ receptor (NOP) agonist, Ro 64–6198, on reactivity to acute pain in mice: Comparison to morphine. Eur. J. Pharmacol. 579, 141–148. [DOI] [PubMed] [Google Scholar]
- Ko M. C. (2004) Antinociceptive effects of ORL1 agonists in monkeys. FASEB J. 18, A961. [Google Scholar]
- Ko M. C.; Woods J. H.; Fantegrossi W. E.; Galuska C. M.; Wichmann J.; Prinssen E. P. (2009) Behavioral effects of a synthetic agonist selective for nociception/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology 34, 2088–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szallasi A.; Cortright D. N.; Blum C. A.; Eid S. R. (2007) The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof of concept. Nat. Rev. Drug Discovery 6, 357–372. [DOI] [PubMed] [Google Scholar]
- Park K. M.; Max M. B.; Robinovitz E.; Gracely R. H.; Bennett G. J. (1995) Effects of intravenous ketamine, alfentanil, or placebo on pain, pinprick hyperalgesia, and allodynia produced by intradermal capsaicin in human subjects. Pain 63, 163–172. [DOI] [PubMed] [Google Scholar]
- Eisenach J. C.; Hood D. D.; Curry R.; Tong C. (1997) Alfentanil, but not amitriptyline, reduces pain, hyperalgesia, and allodynia from intradermal injection of capsaicin in humans. Anesthesiology 86, 1279–1287. [DOI] [PubMed] [Google Scholar]
- Butelman E. R.; Harris T. J.; Kreek M. J. (2004) Antiallodynic effects of loperamide and fentanyl against topical capsaicin-induced allodynia in unanesthetized primates. J. Pharmacol. Exp. Ther. 311, 155–163. [DOI] [PubMed] [Google Scholar]
- Varty G. B.; Lu S. X.; Morgan C. A.; Cohen-Williams M. E.; Hodgson R. A.; Smith-Torhan A.; Zhang H.; Fawzi A. B.; Graziano M. P.; Ho G. D.; Matasi J.; Tulshian D.; Coffin V. L.; Carey G. J. (2008) The anxiolytic-like effects of the novel, orally active nociception opioid receptor agonist SCH 221510. J. Pharmacol. Exp. Ther 326, 672–682. [DOI] [PubMed] [Google Scholar]
- Waldischkin K. A.; Dysko R. C.; Collins G. T.; Ko Y. A.; Winger G.; Ko M. C. (2012) Phamacological characterization of NOP receptor agonists as abuse-free and constipation-free analgesics in monkeys. FASEB J. 26, 1123.3. [Google Scholar]
- Weerts E. M.; Fantegrossi W. E.; Goodwin A. K. (2007) The value of nonhuman primates in drug abuse research. Exp. Clin. Psychopharmacol. 15, 309–327. [DOI] [PubMed] [Google Scholar]
- Podlesnik C. A.; Ko M. C.; Winger G.; Wichmann J.; Prinssen E. P.; Woods J. H. (2011) The effects of nociception/orphanin FQ receptor agonist Ro 64-6198 and diazepam on antinociception and remifentanil self-administration in rhesus monkyes. Psychopharmacology 213, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas S. E.; Griffiths R. R.; Brady J. V. (1983) Buprenorphine self-administration by the baboon: comparison with other opioids. NIDA Res. Monogr. 43, 178–183. [PubMed] [Google Scholar]
- Lutfy K.; Eitan S.; Bryant C. D.; Yang Y. C.; Saliminejad N.; Walayn W.; et al. (2003) Buprenorphine-induced antinociception is mediated by mu opioid receptors and compromised by concomitant activation of opioid receptor-like receptors. J. Neurosci. 23, 10331–10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khroyan T. V.; Polgar W. E.; Jiang F.; Zaveri N. T.; Toll L. (2009) Nociceptin/orphanin FQ receptor activation attenuates antinociception induced by mixed nociception/orphanin FQ/mu opioid receptor agonists. J. Pharmacol. Exp. Ther. 331, 946–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri N. T. (2011) The Nociceptin/Orphanin FQ Receptor (NOP) as a Target for Drug Abuse Medications. Curr. Top. Med. Chem. 11, 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cami-Kobeci G.; Polgar W. E.; Khroyan T. V.; Toll L.; Husbands S. M. (2011) Structural determinants of opioid and NOP receptor activity in derivatives of buprenorphine. J. Med. Chem. 54, 6531–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khroyan T. V; Polgar W. E.; Cami-Kobeci G.; Husbands S. M.; Zaveri N. T.; Toll L. (2011) The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl]-3,3-dimethyl- pentan-2-ol (BU08028): characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. J. Pharmacol. Exp. Ther. 336, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari S.; Camarda V.; Rizzi A.; Marzola G.; Salvadori S.; Marzola E.; Molinari P.; McDonald J.; Ko M. C.; Lambert D. G.; Calo’ G.; Guerrini R. (2012) [Dmt(1) ]N/OFQ(1–13)-NH(2), a potent nociceptin/orphanin FQ and opioid receptor universal agonist. Br. J. Pharmacol. 10.1111/j.1476-5381.2012.02115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y.; Fujita M.; Hong J.; Lohith T. G.; Gladding R. L.; Zoghbi S. S.; Tauscher J. A.; Goebl N.; Rash K. S.; Chen Z.; Pedregal C.; Barth V. N.; Pike V. W.; Innis R. B. (2011) Brain and whole-body imaging in rhesus monkeys of 11C-NOP-1A, a promising PET radioligand for nociception/orphanin FQ peptide receptors. J. Nucl. Med. 52, 1638–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohith T. G.; Zoghbi S. S.; Morse C. L.; Araneta M. F.; Barth V. N.; Goebl N. A.; Tauscher J. T.; Pike V. W.; Innis R. B.; Fujita M. (2012) Brain and whole-body imaging of nociception/orphanin FQ peptide receptor in humans using the PET ligand 11C-NOP-1A. J. Nucl. Med. 53, 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. A.; Liu W.; Chun E.; Katritc V.; Wu H.; Vardy E.; Huang X. P.; Trapella C.; Guerrini R.; Calo G.; Roth B. L.; Cherezov V.; Stevens R. C. (2012) Structure of the nociception/orphanin FQ receptor in complex with a peptide mimetic. Nature 485, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daga P. R.; Zaveri N. T. (2012) Homology modeling and molecular dynamics simulations of the active state of the nociception receptor reveal new insights into agonist binding and activation. Proteins 80, 1948–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]