

Abstract
Polarised tissue elongation during morphogenesis involves cells within epithelial sheets or tubes making and breaking intercellular contacts in an oriented manner. Growing evidence suggests that cell adhesion can be modulated by endocytic trafficking of E-cadherin (E-cad), but how this process can be polarised within individual cells is poorly understood. The Frizzled (Fz)-dependent core planar polarity pathway is a major regulator of polarised cell rearrangements in processes such as gastrulation, and has also been implicated in regulation of cell adhesion through trafficking of E-cad; however, it is not known how these functions are integrated. We report a novel role for the core planar polarity pathway in promoting cell intercalation during tracheal tube morphogenesis in Drosophila embryogenesis, and present evidence that this is due to regulation of turnover and levels of junctional E-cad by the guanine exchange factor RhoGEF2. Furthermore, we show that core pathway activity leads to planar-polarised recruitment of RhoGEF2 and E-cad turnover in the epidermis of both the embryonic germband and the pupal wing. We thus reveal a general mechanism by which the core planar polarity pathway can promote polarised cell rearrangements.
Keywords: Planar polarity, E-cadherin, Morphogenesis
INTRODUCTION
During morphogenesis, elongation of epithelial sheets or tubes can be achieved by planar-polarised cell intercalation, involving the oriented making and breaking of intercellular contacts (reviewed by Keller, 2002; Bertet and Lecuit, 2009; Affolter et al., 2009; Vichas and Zallen, 2011). The mechanisms by which cells can remodel their junctional contacts are still poorly understood, but one possibility is that this occurs via localised endocytic trafficking of E-cad (reviewed by Wirtz-Peitz and Zallen, 2009).
A well-characterised system for studying tissue elongation during development is the Drosophila embryonic tracheal system (Uv et al., 2003; Affolter et al., 2009). It is formed from invaginating groups of epithelial cells that migrate and rearrange to form an interconnected tubular structure with branches projecting dorsally and ventrally from a central trunk. Both the dorsal branches and parts of the ventral branches undergo cell intercalation such that they transform from shorter tubes that have two cells around their circumference to longer tubes with only one cell around their circumference, in the process remodelling their adherens junctions (AJs) from intercellular (joining neighbouring cells) to autocellular (in which they wrap around the tube lumen and adhere to the same cell). This remodelling process requires modulation of E-cad (Shotgun – FlyBase) trafficking, at least in part via regulation of its rate of endocytic recycling by Rab11 (Shaye et al., 2008; Shindo et al., 2008). However, cell intercalation is likely to require selective remodelling of particular junctions within a cell, rather than just a general modulation of cell adhesion, suggesting there might be additional mechanisms that spatially polarise E-cad trafficking at the subcellular level.
The Fz-dependent core planar polarity pathway (hereafter, the ‘core pathway’) is a known regulator of polarised cell rearrangements in processes such as gastrulation (Keller, 2002; Goodrich and Strutt, 2011; Gray et al., 2011), and has also been implicated in regulation of cell adhesion through trafficking of E-cad (Classen et al., 2005; Ulrich et al., 2005). The core pathway specifies planar polarity at the cellular level via the asymmetric subcellular localisation of proteins to cell junctions (Goodrich and Strutt, 2011; Gray et al., 2011). For instance, in Drosophila epithelia, the core proteins Fz, Dishevelled (Dsh) and Diego localise to one cell edge, and Strabismus (Stbm, also known as Van Gogh) and Prickle (Pk) to the other, with Flamingo (Fmi, also known as Starry Night) localising to both edges. The core proteins then recruit tissue-specific ‘effectors’ to specific cell edges to mediate polarised changes in cell morphology and behaviour. Thus, the core pathway is a good candidate for regulating polarised E-cad endocytosis during cell intercalation in contexts such as the tracheal system.
Despite the known function of the core planar polarity pathway in promoting cell intercalation, so far there is little evidence that it might play this role during tubulogenesis, although it is implicated in directing oriented cell divisions in kidney tubules (Saburi et al., 2008). Nevertheless, the core pathway is active during development of the Drosophila embryonic tracheal system, with loss of activity resulting in a convoluted dorsal trunk phenotype via an unknown mechanism (Chung et al., 2009; Nelson et al., 2012). We therefore considered this to be a good context in which to investigate the role of the core pathway in cell intercalation.
MATERIALS AND METHODS
Fly genetics
Alleles and transgenes are described in FlyBase and supplementary material Table S1. Core planar polarity pathway mutant alleles were crossed out to a wild-type stock for multiple generations to reduce the effects of possible background mutations on the strength of embryonic phenotypes. All embryos examined for fz, stbm, pk, fy and mwh were maternal-zygotic mutants for null alleles, for dsh they were maternal-zygotic mutants for the planar polarity-specific allele dsh1, and for fmi and Src42A, embryos are zygotic mutants for a null allele. The antimorphic RhoGEF26.5 allele was examined in heterozygotes. Expression of fz from the transgene UAS-fz was induced in the trachea by crossing to hs-FLP; btl >y+>GAL4 (Ribeiro et al., 2004) and subjecting embryos to a 15-minute heat shock at 38°C 1 hour before imaging. Pupae were aged at 25°C for 28 hours prior to dissection of pupal wings.
Immunolabelling and co-immunoprecipitation
Primary antibodies used were rabbit anti-GFP (Abcam), rabbit anti-Fz (Bastock and Strutt, 2007), rabbit anti-RhoGEF2 (gift from Jörg Grosshans) (Grosshans et al., 2005), rabbit anti-pSrc42A (pY148; Invitrogen), mouse monoclonal anti-βgal (Promega), rabbit anti-Vasa (gift from Ralf Jauch, Alf Herzig and Ralf Pflanz, MPI, Göttingen), mouse monoclonal anti-Crumbs [Developmental Studies Hybridoma Bank (DSHB)] (Tepass et al., 1990), mouse monoclonal anti-Fmi (DSHB) (Usui et al., 1999), mouse anti-Arm (DSHB) (Peifer et al., 1994), rat monoclonal anti-E-cad (DSHB) (Oda et al., 1994), mouse anti-Myc 9E10 (BioServ), rabbit anti-Zipper/MyoII (gift from Thomas Lecuit) (Levayer et al., 2011), guinea pig anti-Bazooka (gift from Jennifer Zallen) (Blankenship et al., 2006). Rabbit anti-Stbm was generated against a His-tagged fusion protein corresponding to amino acids 406-584. Secondary antibodies were purchased from Molecular Probes and Jackson ImmunoResearch.
Embryos were fixed in formaldehyde and devitellinised either by hand or in methanol. For anti-RhoGEF2 embryos were heat fixed and hand devitellinised. Wings were dissected at 28 hours after prepupa formation at 25°C unless otherwise stated, immunolabelled and imaged as previously described (Strutt, 2001). Embryos were mounted in 50% glycerol and wings were mounted in 10% glycerol with 2.5% DABCO.
Confocal images of fixed embryos or pupal wings were either taken using a Nikon A1 LSM confocal microscope with a 40× NA1.2 oil plan apochromatic objective at 2× zoom, or a Leica SP1 confocal microscope with a 40× NA1.32 oil plan apochromatic objective at 2× zoom. For lateral views of tracheal branches stacks of ∼50 slices were taken, the interval between slices was 0.5 μm.
For S2 cell immunoprecipitations, GFP-tagged Dsh and Myc-tagged RhoGEF2 were inserted in the pAc5.1 vector (Invitrogen). S2 cell lysates were made in 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM Na3VO4, 5 mM NaF, 1× protease inhibitor cocktail (Roche). Immunoprecipitations used goat anti-Myc agarose (Abcam), and western blots were probed with mouse anti-Myc 9E10 (BioServe) or rabbit anti-GFP (Abcam).
Junctional intensity measurements
For quantification of junctional intensity in the trachea, embryos were either imaged live as described below or they were fixed and mounted, and images were taken at constant confocal settings. Stacks of images were obtained and the three slices with the strongest staining were averaged for measuring. Mean pixel intensities for junctions were collected in ImageJ using the line measurement tool set to a six-pixel width and ‘laser-off’ background was subtracted. Wild-type and mutant mean intensities were averaged per embryo then statistically compared in Prism (v.5 GraphPad) using an ANOVA and the Dunnett’s multiple comparison post test or an unpaired two-tailed Student’s t-test if only two genotypes were compared.
For epidermal junctional regions, measurements were made in the same way as in the tracheal branches and the angle of each line was also recorded. The mean intensities were normalised for laser-off background and grouped into junctions crossing the dorsoventral axis with an angle of between 0° and 45° or with an angle of between 135° and 180° relative to the anteroposterior axis (horizontal junctions), or crossing the anteroposterior axis (vertical junctions) with an angle of between 45° and 135° relative to the anteroposterior axis. The mean fluorescent intensities of the vertical and horizontal junctions were then statistically compared using an unpaired two-tailed Student’s t-test if two genotypes were compared or an ANOVA and the Dunnett’s multiple comparison test to compare multiple groups to a control genotype. Error bars on the graphs are s.e.m. unless otherwise stated.
Cell packing was analysed using Packing Analyzer v2.0 (Aigouy et al., 2010).
Quantifying tracheal phenotypes
Tracheal dorsal and ventral branches were assayed for the presence of cell intercalations by immunolabelling embryos for Crumbs distribution to highlight the adherens junctions and blind scoring for the percentage of branches in each embryo that show incomplete intercalations. The data were then averaged across embryos. The number of nuclei per dorsal tracheal branch was counted using live embryos expressing btl-GAL4/UAS-α-Cat to mark the junctions and UAS-NLS-red-stinger to mark the nucleus, excluding the branches at the very anterior and posterior of each embryo.
Live imaging and fluorescence recovery after photobleaching (FRAP) analysis
FRAP in the wing was carried out as previously described (Strutt et al., 2011). For FRAP in the embryo, embryos were collected overnight, dechorionated and mounted on glass coverslips (20×50 mm) coated with heptane glue. A gasket of one layer of parafilm was constructed and placed on the coverslip and embryos were then placed in the gasket and covered with Halocarbon 700 oil. Embryos were imaged for a maximum of 3 hours. To check for viability, slides were kept after imaging to confirm that embryos subsequently hatched as larvae.
Samples were imaged on an inverted Zeiss LSM 510 confocal microscope, with a Zeiss 40× NA1.4 oil apochromatic objective lens at 2× zoom with the pinhole open to maximise light detected. For imaging, the 488 nm Argon laser was used at an output of 20%, and a 505-550 nm band-pass filter was used for detection. Single images with no averaging were taken to reduce acquisition bleaching. For FRAP of cellular junctions in the trachea, bleach regions 2 μm2 (106 pixels) in size were selected on vertical branches at stage 14/15. In 28-hour pupal wings and in stage 8 ventral epidermis, 2 μm2 bleach regions were placed on horizontal and vertical junctions. Junctions were bleached using the 488 nm Argon laser at 100% with 20 passes over a region of interest (ROI). Three pre-bleach images were captured, as well as an immediate post-bleach image, and then an image was taken every 15 seconds for up to 30 minutes. For data analysis, Volocity (v.4.4 Improvision) was used. Regions were manually reselected and mean fluorescence was quantified for each bleach region in the field of view, laser-off background was then subtracted. To measure acquisition bleaching, intensity measurements were collected from four non-bleached control regions, of the same size as the bleach regions. Data were then corrected for acquisition bleaching and normalised against the pre-bleached value. Data were plotted on an xy graph in Prism (v.5 GraphPad), and a one-phase exponential association curve was fitted. An extra sum-of-squares F-test was performed to compare curve plateaux (Ymax). Note that, in most cases, the half-life of recovery was less than the acquisition interval (15 seconds), and, thus, the rate of recovery could not be accurately determined.
For FRAP of E-cad-GFP, three different constructs were used. The expression of an UAS-E-cad-GFP insertion located on the second chromosome was driven in the trachea using btl-GAL4; although expression is likely to be higher than endogenous, we did not observe a disrupted tracheal phenotype when expressed in control embryos. We were unable to use ubiquitously expressed E-cad-GFP [using either the ubi-E-cad-GFP construct (Oda and Tsukita, 2001) or the knock-in of GFP to the endogenous locus (Huang et al., 2009)] for FRAP on the tracheal branches owing to fluorescence present in surrounding tissues within the embryo. To control for the presence of another UAS transgene insertion in addition to the UAS-E-cad-GFP, control crosses were carried out using UAS-NLS-red-stinger.
For FRAP in the epidermis and wing, ubi-E-cad-GFP was used to express E-cad in wild-type embryos under the control of a ubiquitous promoter. E-cad-GFP expressed using this construct is reported to behave similarly to endogenous E-cad (Oda and Tsukita, 2001). Although, in our experiments, higher than normal levels of E-cad are present due to expression of both endogenous and GFP-tagged forms, in our FRAP experiments we saw similar results using either this genotype or a fly strain with GFP knocked into the endogenous locus (Huang et al., 2009).
For quantification of germband extension, embryos were imaged from embryonic stage 6 every 10 minutes for up to 5 hours using a Zeiss LSM510 with brightfield illumination, 25× NA1.2 apochromatic objective.
RESULTS
Tracheal branch cell intercalation defects in core pathway mutants
We examined dorsal and ventral branches of the Drosophila tracheal system for cell intercalation defects in embryos either lacking maternal and zygotic expression or with excess core protein activity. The branches appeared normal at stage 12 (supplementary material Fig. S1A′; data not shown) and subsequently showed no difference in the initial pairing or arrangement of the cells compared with wild type (supplementary material Fig. S1L,M). However, by later stages of embryogenesis, when intercalation was largely complete in wild-type embryos, many branches in mutant embryos had failed to complete intercalation to form autocellular junctions (Fig. 1A-D; supplementary material Fig. S1H-J), instead showing characteristic loops of intercellular AJs (Jaźwińska et al., 2003; Ribeiro et al., 2004). This defect was not due to a general delay in embryonic development as other processes progressed at the normal rate (supplementary material Fig. S1A-C). We also observed a convoluted dorsal trunk phenotype in the mutant embryos, as previously reported (supplementary material Fig. S1D-G) (Chung et al., 2009; Nelson et al., 2012).
Fig. 1.

The core planar polarity pathway is required for cell intercalation in the tracheal branches. (A-C) Lateral view of embryonic tracheal branches at stage 14 showing cell intercalation in Drosophila embryos stained for Crumbs in wild type (w1118) (A) and maternal zygotic mutants for fzP21 (B) and stbm6 (C). Insets show magnified regions of indicated dorsal and ventral branches, arrowheads indicate unresolved intercalations. Anterior is to the left and dorsal up in this and following images. (D) Quantification of the proportion of branches per embryo with unresolved intercalations at indicated stages. ANOVAs were used to compare simultaneously the control (w1118) and the mutant conditions at each stage: stage 13, P=0.0075; stage 14-15, P≤0.0001; stage 16-17, P≤0.0001. (E-H) Embryonic tracheal branches imaged live showing α-Catenin-GFP (green) and NLS-red-stinger (red), under the control of btl-GAL4 in wild type stage 13 (E) and stage 17 (G), and dsh1 stage 13 (F) and stage 17 (H). Insets show magnifications of single branches. White asterisks mark individual nuclei in branches, blue asterisks indicate nuclei from different branches, arrowheads indicate the joining point of the dorsal branch to its reciprocal dorsal branch on the other side of the embryo. (I) Quantification of the number of cells per dorsal branch during cell intercalation. ANOVAs were used to compare the control and the mutant conditions for each stage: stage 12, P=0.078; stage 13, P≤0.0001; stage 15, P≤0.0001; stage 16, P≤0.0001; stage 17, P≤0.0001. (J-N) Live images of branches with cells expressing the Apoliner construct under control of btl-GAL4 in wild type stage 13 (J), fzP21 stage 13 (K), fzP21 stage 14 (L), wild type stage 15 (M) and fzP21 stage 15 (N). Arrowheads mark cells with GFP-positive nuclei. Arrow marks a pair of cells both expressing GFP in the nucleus. Error bars represent s.e.m.
Upon further examination, we noticed that, in addition to showing unresolved intercalations, at later stages the branches had reduced cell numbers. Quantifying the dorsal branches at stage 12 showed that about six cells were present (Samakovlis et al., 1996; Baer et al., 2010) as expected in both wild-type and mutant embryos (Fig. 1E-I). By stage 17, wild-type embryos showed a small reduction in cell number, but we observed a greater loss in the mutant embryos (Fig. 1E-I). During normal development, cells are occasionally extruded primarily at the base of branches, and this is believed to be due to stresses induced by junctional remodelling in this region causing cells to activate the apoptotic pathway (Baer et al., 2010). We reasoned that the greater loss of cells we observed could be due to a similar mechanism, triggered by the defects in cell intercalation. To confirm this, we assayed activation of apoptosis using the ‘Apoliner’ construct (Bardet et al., 2008), which causes GFP to translocate to the nucleus upon caspase activation. As predicted, in mutant embryos we saw a large increase in cells with GFP-positive nuclei in tracheal branches (Fig. 1L,N), with this sometimes being observed in pairs of cells yet to intercalate (Fig. 1N, arrow). We surmise that pairs of cells become stressed and activate the apoptosis pathway while attempting to intercalate, resulting in one cell of the pair leaving the branch and thus reducing the stress. Consistent with this, we also see single cells remaining in branches with green nuclei. These cells appear not to leave the branches, as we never see breaks in branches, and so we assume they are able to reverse the apoptotic process once stress is relieved.
The core pathway regulates E-cad levels and turnover in tracheal branches
Similar defects in tracheal branch intercalation have previously been observed in mutant backgrounds in which E-cad levels or turnover are disrupted (Shaye et al., 2008; Shindo et al., 2008). Notably, we found that loss of core protein activity resulted in an increase in levels of endogenous E-cad in the tracheal branches (Fig. 2A), whereas Fz overexpression resulted in a decrease (Fig. 2B), which was also cell-autonomous (Fig. 2C), suggesting a direct effect. We saw no evidence for the core pathway affecting levels of E-cad transcription (Fig. 4M), suggesting that it might be altering E-cad turnover.
Fig. 2.

The core planar polarity pathway controls E-cad levels and turnover in the tracheal branches. (A,B) Intensity measurements of junctional endogenous E-cad in stage 14 tracheal branches in control and core pathway mutant or overexpressing Drosophila embryos. (A) w1118 (control), fzP21, stbm6, dsh1; (B) btl-GAL4 (control), btl-GAL4/UAS-NLS-red-stinger, btl-GAL4/UAS-fz. ANOVAs were used to compare the control and mutant conditions: in A, P≤0.0001; in B, P=0.0002. Asterisks indicate individual results from a Dunnett’s multiple comparison test (NS, not significant; *P≤0.05, **P≤0.01, ***P≤0.0001). (C,C′) Dorsal branches containing clones of cells overexpressing UAS-fz together with UAS-GFP (green) under control of btl>stop>GAL4, labelled for E-cad (magenta in C, or shown as intensity table in C′). E-cad levels are cell-autonomously reduced in cells overexpressing fz (green) compared with non-overexpressing cells (arrowheads). (D) FRAP analysis in dorsal tracheal branches on junctional E-cad-GFP expressed under control of btl-GAL4 in wild-type and core pathway mutant backgrounds. The lower level of fluorescent recovery after bleaching in the mutants indicates the presence of a larger stable fraction of E-cad-GFP compared with wild type (one-way ANOVA comparing stable fractions P≤0.0001). We were unable to determine accurately the half-lives of recovery, as in all genotypes recovery was too rapid compared with the interval between time frames. (E,F) FRAP analysis of E-cad-GFP expressed under control of btl-GAL4 with co-expression of fz (E) or Rab5SN (F). In both cases, there is an increase in the stable fraction of E-cad-GFP compared with the NLS-red-stinger-expressing control (t-test comparing stable fractions P≤0.0001 for both). (G) Quantification of the intercalation phenotype in tracheal branches of dsh1 embryos heterozygous for shgIG27 (the gene coding for E-cad), which is suppressed compared with dsh1 (t-test, **P=0.0016). Error bars represent s.e.m.
Fig. 4.

The core planar polarity pathway regulates planar-polarised distribution of E-cad in the embryonic epidermis. (A-A″) Image of ventrolateral epidermis in a stage 8 Drosophila embryo labelled for E-cad (magenta in A, white in A′) and Fz (green in A, white in A″). (B-B″) Localisation of E-cad (B), Fmi (B′) and Stbm (B″) in stage 15 epidermis. (C-D″) Localisation of RhoGEF2 (red or white) and Arm (green or white) in ventrolateral epidermis of wild-type (C) or fzP21 (D) stage 8 embryos. (E) Quantification of junctional E-cad and Fz levels on vertical and horizontal junctions in the ventrolateral epidermis of stage 8 wild-type embryos (t-test for E-cad, ***P≤0.0001 and for Fz, **P=0.0035). Error bars are s.d. (F) Quantification of junctional asymmetry for RhoGEF2 and Arm in stage 8 epidermis in wild-type embryos (white) and fzP21 embryos (green). t-tests were used to compare horizontal and vertical intensities, RhoGEF2 in w1118 (wild type), ***P≤0.0001 and in fzP21, P=0.26; Arm in w1118, ***P≤0.0001 and in fzP21, **P=0.0035. Error bars are s.d. (G) Quantification of junctional E-cad in stage 8 epidermis in wild-type embryos (white) and fzP21 (green), dsh1 (blue) or stbm6 (orange). Intensity of E-cad increases in polarity mutants and E-cad asymmetry is lost. Asterisks show individual results from a Bonferroni multiple comparison test (NS, not significant; *P≤0.05, **P≤0.01, ***P≤0.0001). Error bars are s.d. An ANOVA comparing just the horizontal intensity values of mutants to wild type gives P≤0.0001. (H-L) FRAP analysis in the epidermis of junctional E-cad-GFP expressed under control of the ubiquitin promoter in w1118 (wild-type; H) embryos (P≤0.0001 comparing stable fractions on vertical and horizontal junctions using t-test), fzP21 (I; P=0.28), stbm6 (J; P=0.13), en-GAL4/UAS-fz (K; P=0.064) and RhoGEF26.5/+ antimorphs (L; P=0.084). (M) Quantification of βgal labelling showing levels of transcription from shg-lacZ (an enhancer trap in the locus encoding E-cad) (Shindo et al., 2008) in wild-type and dsh1 epidermal cells (t-test, P=0.90). Error bars are s.d.
To investigate E-cad turnover, we carried out FRAP experiments on E-cad-GFP to determine the stable fraction present in cell junctions in the tracheal system. In wild-type embryos, 47% of fluorescence was recovered after bleaching, showing that the remaining 53% was in a stable fraction (Fig. 2D). Importantly, the stable fraction of E-cad-GFP increased in branches of mutant embryos (Fig. 2D), consistent with reduced E-cad turnover. Overexpression of Fz also caused a modest increase in the stable fraction of E-cad-GFP at junctions (Fig. 2E). However, taking into account the observation that total levels of E-cad are significantly reduced in this background (Fig. 2B), we infer that this corresponds to a decrease in the overall levels of both stable and unstable E-cad at junctions.
These results suggest that the core pathway normally acts to promote E-cad turnover from junctions, and loss of activity increases both the amount and the stable fraction at junctions. In particular, the core pathway might be promoting E-cad endocytosis, as blocking endocytosis, for instance by reducing Rab5 activity, also causes a tracheal branch intercalation defect accompanied by an increase in E-cad levels (Shaye et al., 2008), and we also find an increase in the size of the stable fraction of E-cad-GFP at junctions (Fig. 2F). In support of the view that the increased levels of E-cad in core pathway mutant backgrounds are responsible for the intercalation defect, we found that the dsh phenotype was suppressed by a reduction in E-cad gene dosage (Fig. 2G).
E-cad turnover in the tracheal branches depends on RhoA activity
We next investigated how the core pathway might regulate E-cad turnover. In hair and bristle formation in the adult cuticle, the core pathway acts through effectors such as Fuzzy and Multiple Wing Hairs (Goodrich and Strutt, 2011); however, loss of their activity had no effect on tracheal branch intercalation (supplementary material Fig. S2A-C). Small GTPases of the Rho family are also implicated as effectors of the core pathway in flies and vertebrates (Goodrich and Strutt, 2011), and both Rac and Rho play roles in E-cad regulation during Drosophila embryogenesis (Chihara et al., 2003; Pirraglia et al., 2006; Levayer et al., 2011). As Rac is not an effector of the core pathway in other contexts in Drosophila (Hakeda-Suzuki et al., 2002), we focused our attention on the single Rho homologue RhoA (Rho1 – FlyBase). Expression of either activated or dominant-negative RhoA caused general disruption of tracheal development (Fig. 3A,B) as previously reported (Lee and Kolodziej, 2002), precluding analysis of a specific defect in tracheal branch intercalation. Nevertheless, we did carry out FRAP on branches with reduced RhoA function and found an increase in the stable fraction of E-cad-GFP (Fig. 3C), suggesting that RhoA is an effector of the core pathway in this tissue. Furthermore, embryos with reduced activity of the RhoA exchange factor RhoGEF2 showed tracheal branch intercalation defects (Fig. 3D,E), and an increase in endogenous E-cad levels (Fig. 3F), and overexpression of RhoGEF2 showed an increase in the stable fraction of E-cad-GFP at junctions (Fig. 3G). The similar FRAP phenotypes of RhoGEF2 overexpression and loss of RhoA activity suggests that excess RhoGEF2 acts as a dominant negative in this context, and that the core proteins normally promote E-cad turnover through activation of RhoA.
Fig. 3.
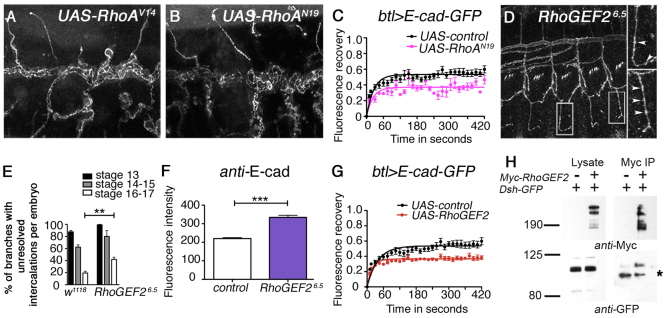
The core planar polarity pathway acts via RhoGEF2 in the tracheal system. (A,B) Lateral view of disrupted tracheal branches stained for Crumbs in Drosophila embryos expressing activated RhoAV14 (A) or dominant-negative RhoAN19 (B) under control of btl-Gal4. (C) FRAP analysis of E-cad-GFP expressed under control of btl-GAL4 with co-expression of RhoN19. There is an increase in the stable fraction of E-cad-GFP compared with the NLS-red-stinger-expressing control (t-test comparing stable fractions, P≤0.0001). (D) Tracheal intercalation defect in embryos heterozygous for the RhoGEF26.5 antimorphic allele stained with Crumbs. Insets show magnifications of single branches, arrowheads indicate unresolved intercalations. (E) Quantification of the intercalation phenotype in embryos heterozygous for the RhoGEF26.5 antimorphic allele. Note similar phenotype to that of the core pathway mutants (see Fig. 1D). A t-test was used to compare w1118 and RhoGEF26.5 at stage 16-17. **P=0.0029. (F) Intensity measurements of junctional E-cad in stage 14 tracheal branches of embryos heterozygous for the RhoGEF26.5 antimorphic allele compared with w1118 control. A t-test was used to determine the statistical significance. ***P≤0.0001. (G) FRAP analysis in dorsal tracheal branches on junctional E-cad-GFP expressed under control of btl-GAL4 with co-expression of UAS-RhoGEF2, showing an increase in the stable fraction of E-cad-GFP compared with the NLS-red-stinger-expressing control (t-test comparing stable fractions, P≤0.0001). (H) Immunoprecipitation of Myc-RhoGEF2 from Drosophila S2 cells, showing co-precipitation of Dsh-GFP. Asterisk indicates non-specific band seen in pull-down. Error bars represent s.e.m.
Recent data from vertebrates indicate that a RhoGEF2-related protein (PDZ-RhoGEF) can be directly recruited by core proteins acting together with the formin DAAM1 (Nishimura et al., 2012). Interestingly, we find that RhoGEF2 can also directly interact with Dsh (Fig. 3H), suggesting that a similar mechanism might operate in Drosophila. Furthermore, it has been reported that asymmetrically distributed RhoGEF2 in the embryonic epidermis regulates planar-polarised E-cad endocytosis via activation of RhoA activity (Levayer et al., 2011). Taken together, this suggests a model in which the core pathway promotes cell rearrangements in the tracheal system by locally regulating E-cad endocytosis through direct recruitment of RhoGEF2 to junctions.
These findings raise the possibility that the core proteins show asymmetric distribution or activity within cells of the tracheal branches, as they do in other epithelia (Goodrich and Strutt, 2011; Gray et al., 2011), and thus promote asymmetric endocytosis of E-cad, perhaps weakening junctional contacts in regions in which cells need to slide over one another. To gather evidence for this, we looked for asymmetric distribution of core proteins within cells of the tracheal branches. As seen by others (Chung et al., 2009; Förster and Luschnig, 2012; Nelson et al., 2012), we observe core proteins localising to junctions, and colocalising with E-cad (supplementary material Fig. S2D), consistent with a local effect on E-cad endocytosis, but did not see any evidence of asymmetric localisation. However, we note that such asymmetry might only be transient in re-arranging cells, and indeed the asymmetry of core protein localisation has proved difficult to see in other dynamic contexts (Goodrich and Strutt, 2011; Gray et al., 2011).
Regulation of E-cad levels and turnover in the embryonic epidermis
To seek further evidence for planar-polarised E-cad turnover controlled by the core pathway, we looked in the embryonic epidermis. Here, ventrolateral cells intercalate along the dorsoventral axis during germband extension (Irvine and Wieschaus, 1994), starting from around stage 7. During this process, E-cad distribution is planar polarised (Blankenship et al., 2006), with more on ‘horizontal’ junctions (i.e. running anteroposteriorly), whereas RhoGEF2 shows the reciprocal distribution with more on ‘vertical’ junctions (Levayer et al., 2011), where it locally promotes E-cad endocytosis. However, the signal leading to polarised RhoGEF2 distribution is unknown.
We examined the distribution of core proteins in the ventrolateral epidermis. During cell intercalation, we found that Fz was enriched on vertical junctions (Fig. 4A,E), consistent with the possibility that it might recruit RhoGEF2 to this location. At later stages of embryonic development, core protein asymmetry became even stronger, with E-cad distribution on horizontal junctions also becoming more prominent (Fig. 4B), consistent with the observation that cell rearrangements are continuing at this stage (Simone and DiNardo, 2010). Notably, in embryos lacking Fz activity, RhoGEF2 appeared to be less tightly localised to junctions (Fig. 4C,D) and its preferential enrichment on vertical junctions was lost (Fig. 4F). Similarly, endogenous E-cad also lost its enrichment on horizontal junctions, and showed increased levels on all junctions in the absence of core protein activity (Fig. 4G). These results support the hypothesis that Fz-dependent recruitment of RhoGEF2 is responsible for increased endocytosis of E-cad from vertical junctions.
Given the loss of RhoGEF2 and E-cad asymmetry upon removal of core pathway activity, we also examined the effects on the localisation of Zipper (MyoII), Bazooka and Armadillo (Arm), which are also asymmetrically distributed at these stages (Zallen and Wieschaus, 2004; Bertet et al., 2004; Blankenship et al., 2006). Interesting, both Zipper and Bazooka also lose asymmetry in core pathway mutants (supplementary material Fig. S4A,B), although Arm does not (Fig. 4F).
If the core pathway is locally promoting E-cad endocytosis, then, by analogy to what we saw in the tracheal branches, horizontal junctions (with less core protein activity and more E-cad) should show a larger stable fraction of E-cad, whereas vertical junctions (with higher core protein activity and less E-cad) should show a smaller stable fraction. Strikingly, this is exactly what we see, carrying out FRAP either on E-cad-GFP expressed under the ubiquitin promoter (Oda and Tsukita, 2001) (Fig. 4H) or its own promoter (Huang et al., 2009) (supplementary material Fig. S3E). As expected, this difference disappears upon removal or overexpression of the core proteins (Fig. 4I-K; supplementary material Fig. S3A-C,F), and also when RhoGEF2 activity is reduced (Fig. 4L), supporting the view that the core proteins act via RhoGEF2. The actual size of the stable fraction varies between different genotypes; however, this observation is hard to interpret as the levels of E-cad-GFP at junctions are reduced when the levels of endogenous E-cad increase in embryos lacking core protein activity (supplementary material Fig. S3D). The key finding is that there is no longer a difference between the vertical and horizontal junctions.
Another factor known to regulate cell intercalation via effects on E-cad, in both tracheal branches and the embryonic epidermis, is the kinase Src (Takahashi et al., 2005; Shindo et al., 2008). Furthermore, in the epidermis, we found activated Src (pSrc) to be preferentially localised to horizontal junctions (supplementary material Fig. S3G), consistent with the possibility that it could act upstream or downstream of the core pathway. However, arguing against Src acting together with the core pathway, Src in part modulates E-cad by altering its transcription (Shindo et al., 2008), whereas in core pathway mutants we see no change in expression of a reporter of E-cad transcription (Fig. 4M). Furthermore, pSrc remains asymmetric in the absence of core pathway activity in the epidermis (supplementary material Fig. S3G) and is also unaltered in the tracheal system (Förster and Luschnig, 2012; Nelson et al., 2012). Finally, reduction in zygotic activity of Src in Src42A mutant embryos results in a striking aggregation of E-cad-GFP at junctions (supplementary material Fig. S3H), unlike what is seen in embryos lacking core pathway activity, but if FRAP is carried out on non-aggregated E-cad-GFP in this background, there is still a larger stable fraction of E-cad-GFP on horizontal junctions than vertical junctions (supplementary material Fig. S3I). We conclude that these pathways act in parallel.
Polarised turnover of E-cad in the pupal wing
It has previously been reported that the core pathway promotes E-cad-dependent junctional remodelling in the Drosophila pupal wing, and that loss of core protein function results in increased junctional E-cad (Classen et al., 2005). Interestingly, a planar-polarised distribution of the exocyst component Sec5 was also observed on proximodistal cell junctions, dependent on activity of the core pathway. This might suggest that the core pathway promotes local exocytosis of E-cad on proximodistal junctions, which in turn might lead to higher levels of E-cad in this position. However, a planar-polarised difference in E-cad distribution in the pupal wing has not been observed, and the hypothesis that the core pathway promotes exocytosis is hard to reconcile with overall higher levels of E-cad when core pathway function is removed.
To investigate this further, we measured junctional levels of endogenous E-cad in pupal wings, at a stage when the core proteins are localised to proximodistal (‘vertical’) junctions. We found increased levels of E-cad on anteroposterior (‘horizontal’) junctions (Fig. 5A,B; supplementary material Fig. S5A-G), and a reciprocal enrichment of RhoGEF2 to vertical junctions (Fig. 5C,D; supplementary material Fig. S5H-N). The asymmetric distribution of both was lost in the absence of core protein activity, and, in addition, loss of RhoGEF2 activity abolished E-cad asymmetry (supplementary material Fig. S4E,F). We obtained additional support for the core proteins recruiting RhoGEF2 to junctions, by examining the effects of overexpressing the core protein Pk. This causes accumulation of all core proteins at high levels at cells junctions (Tree et al., 2002), and, as expected, also resulted in an increase in RhoGEF2 levels (Fig. 5E,F). As we observed in the embryonic epidermis, the horizontal junctions with lower Fz activity and higher E-cad levels also showed a larger stable fraction of E-cad (Fig. 5G) and this difference was lost upon removal of core pathway activity (Fig. 5H,I). These data are consistent with the core pathway normally promoting endocytosis of E-cad from vertical junctions via asymmetric recruitment of RhoGEF2.
Fig. 5.

The core planar polarity pathway regulates planar-polarised distribution of E-cad in the pupal wing. (A-A″) E-cad (red in A, white in A′) and Stbm (blue in A, white in A″) in a fmiE59 pupal wing clone marked by absence of lacZ expression (green in A). Distal to the right. (B) Quantification of endogenous junctional E-cad (blue) in wild type and fmiE59, and of Stbm (orange) in wild-type 28-hour pupal wings. E-cad is increased on the horizontal junctions in wild-type tissue but this is lost in fmiE59 mutant cells (t-tests comparing horizontal and vertical intensities: Stbm in wild type, ***P≤0.0001; E-cad in wild type, ***P=0.0004; E-cad in fmiE59, P=0.3623). Error bars are s.d. (C-C″) RhoGEF2 (red in C, white in C′) and Stbm (blue in C, white in C″) in a fmiE59 pupal wing clone marked by absence of lacZ expression (green in C). Specificity of the RhoGEF2 immunolabelling was confirmed using RNAi knockdown (see supplementary material Fig. S4E). (D) Quantification of endogenous junctional RhoGEF2 (purple) in wild type and fmiE59, and of Stbm (orange) in wild-type 28-hour pupal wings. RhoGEF2 is increased on the vertical junctions in wild-type tissue but this is lost in fmiE59 mutant cells (t-tests comparing horizontal and vertical intensities: Stbm in wild type, ***P≤0.0001; RhoGEF2 in wild type, ***P≤0.0001; RhoGEF2 in fmiE59, P=0.9593). Error bars are s.d. (E-E″) Pupal wing overexpressing Pk under control of the ptc-GAL4 driver between veins 3 and 4 (region indicated by white bar) immunolabelled for Fmi (green in E, white in E′) and RhoGEF2 (magenta in E, white in E″). (F) Quantification of Fmi (red) and RhoGEF2 (purple) in wild-type and Pk-overexpressing tissue. Asterisks correspond to t-tests of mean intensity of a large region in the ptc-Gal4 expression domain compared with a wild-type region in the same wing (Fmi, **P=0.0042; RhoGEF2 *P=0.0388. Error bars are s.d.). (G-I) FRAP analysis of junctional E-cad-GFP expressed under the ubiquitin promoter in 28-hour pupal wings, showing a difference between vertical and horizontal junctions in wild type (G; t-test P≤0.0001), this difference is lost in fzP21 (H; t-test P=0.79) and stbm6 (I; t-test P=0.36). Note that there is also an increase in the half-life of E-cad-GFP fluorescence recovery in the absence of core protein activity, again consistent with a role of core proteins in promoting E-cad turnover. (J-L) Quantification of endogenous junctional E-cad (blue) and Fmi (red) asymmetry in pupal wings at 20 hours (J), 24 hours (K) and 28 hours (L). t-tests comparing horizontal and vertical intensities, *P≤0.05, **P≤0.01, ***P≤0.0001. Error bars are s.d. NS, not significant.
Finally, if planar-polarised distribution of the core proteins is responsible for the asymmetry of E-cad, then we would expect E-cad asymmetry to increase as core protein asymmetry becomes stronger between 20 and 28 hours of pupal life (Aigouy et al., 2010). This is what we observe, with both Fmi and E-cad showing low asymmetry at 20 hours, but by 28 hours both are significantly asymmetric (Fig. 5J-L; supplementary material Fig. S4G-I).
DISCUSSION
Looking in three different tissues, we find that core planar polarity pathway activity promotes E-cad turnover from junctions, most likely via local recruitment and regulation of RhoGEF2 and RhoA activity. In general terms, it is believed that local assembly or disassembly of adherens junctions through trafficking of E-cad is likely to be important for polarised tissue rearrangement (Wirtz-Peitz and Zallen, 2009); however, few specific contexts in which this occurs have been identified.
One process in which regulation of E-cad turnover is strongly linked to cell intercalation is elongation of branches in the Drosophila embryonic tracheal system (Shaye et al., 2008; Shindo et al., 2008). We show that loss of core pathway function and also reduction of RhoGEF2 and RhoA activity give a similar phenotype to blocking endocytosis in this tissue, resulting in increases in both overall levels of and the stable fraction of E-cad at junctions, and a delay in cell intercalation. Consistent with the increase in E-cad levels being the cause of the intercalation defect in core pathway backgrounds, this phenotype can be suppressed by lowering E-cad gene dosage. We speculate that core planar polarity proteins might transiently show polarised distribution or activity in this context, thus selectively weakening junctions and allowing cells to slide over one another. However, consistent with previous studies (Chung et al., 2009; Förster and Luschnig, 2012; Nelson et al., 2012), we failed to detect such asymmetry. We therefore cannot rule out the possibility that core pathway activity is uniform within cells in this tissue, and only plays a role in general modulation of E-cad trafficking.
In the pupal wing, the core pathway has already been linked to regulation of E-cad trafficking, and evidence has been presented that this promotes junctional remodelling that gives rise to a regular hexagonal arrangement of the cells (Classen et al., 2005). The exact mechanism by which the core pathway modulates E-cad trafficking was not defined, although the observation that Sec5 is recruited to proximodistal junctions suggested that there might be a role for local exocytosis of E-cad. Looking at a stage shortly after junctional remodelling, when the core proteins are strongly asymmetrically distributed, we observed planar-polarised localisation of RhoGEF2 to proximodistal junctions, but also a decrease in overall levels and the stable fraction of E-cad in this position. This appears to rule out a role for increased E-cad exocytosis on proximodistal junctions. Interestingly, as previously noted by Classen et al. (Classen et al., 2005), although Sec5 is best characterised as a component of the exocyst, it has also been implicated in endocytosis in the Drosophila oocyte (Sommer et al., 2005) and perhaps this is also true in the wing. It is not clear how planar-polarised E-cad trafficking would contribute to formation of a regular hexagonal array of cells, as removing E-cad from proximodistal distal junctions might be expected to cause shrinkage of these junctions at the expense of anteroposterior junctions. However, during the peak period of junctional rearrangement (from ∼18 hours of pupal life) (Classen et al., 2005), the planar-polarised asymmetric distribution of the core proteins is largely lost (Aigouy et al., 2010), and so it might be that during the crucial stage of morphogenesis the core pathway promotes relatively uniform endocytosis of E-cad.
Our observation of a role for the core pathway in modulating E-cad turnover in the epidermis of the embryonic germband is particularly intriguing, as loss of core pathway activity does not result in a defect in embryonic germband extension (supplementary material Fig. S4C,D) (Zallen and Wieschaus, 2004), even though a planar-polarised distribution of E-cad has been implicated as a key mechanism in promoting cell intercalation in this context (Rauzi et al., 2010; Levayer et al., 2011). We speculate that planar polarisation of E-cad might be only one of a number of mechanisms that operate redundantly during the crucial developmental event of germband extension. Among other mechanisms reported are localised actomyosin contraction at vertical junctions (Bertet et al., 2004; Zallen and Wieschaus, 2004; Blankenship et al., 2006), inhibition of Bazooka localisation on vertical junctions by local Rho kinase (Rok) activity (Simões et al., 2010) and alteration of Arm (β-catenin) dynamics on vertical junctions by localised activity of the Abl kinase (Tamada et al., 2012). Interestingly, we find that loss of core pathway activity also abolishes Zipper and Bazooka asymmetry, but not Arm asymmetry (we have not examined Abl or Rok asymmetry). Additionally, we also observed a planar-polarised distribution of activated Src kinase to horizontal junctions. Although Src kinase is a known modulator of E-cad trafficking in the Drosophila embryo (Takahashi et al., 2005; Shindo et al., 2008), the significance of its planar-polarised distribution is unclear, as loss of core pathway function did not affect the distribution of Src, but did block the planar-polarised distribution of E-cad.
Another context in which the core pathway might modulate E-cad turnover is during ommatidial rotation in the developing Drosophila eye, in which possible involvement of both RhoA and the kinase Nemo have been reported (Mirkovic and Mlodzik, 2006; Mirkovic et al., 2011).
In summary, we present evidence that the core planar polarity pathway acts to locally promote E-cad endocytosis via local recruitment of RhoGEF2 and activation of RhoA activity. This represents a mechanism by which the core pathway can promote planar-polarised cell rearrangements.
Supplementary Material
Acknowledgments
We thank Markus Affolter, Jörg Grosshans, Shigeo Hayashi, Thomas Lecuit, Stefan Luschnig, Ralf Pflanz, Stephen Rogers, Jean-Paul Vincent, Andreas Wodarz, Jennifer Zallen, the Bloomington Drosophila Stock Center, the Kyoto Drosophila Genetic Resource Center, the Bloomington Drosophila Genomics Resource Center, the Developmental Studies Hybridoma Bank and BioServ UK for fly stocks, constructs and antibodies; and Dominique Förster, Stefan Luschnig and Marta Llimargas for comments on the manuscript.
Footnotes
Funding
This work was supported by a Wellcome Trust Senior Fellowship to D.S. Confocal facilities were provided by the Wellcome Trust and Yorkshire Cancer Research. Deposited in PMC for immediate release.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.088724/-/DC1
References
- Affolter M., Zeller R., Caussinus E. (2009). Tissue remodelling through branching morphogenesis. Nat. Rev. Mol. Cell Biol. 10, 831–842 [DOI] [PubMed] [Google Scholar]
- Aigouy B., Farhadifar R., Staple D. B., Sagner A., Röper J.-C., Jülicher F., Eaton S. (2010). Cell flow reorients the axis of planar polarity in the wing epithelium of Drosophila. Cell 142, 773–786 [DOI] [PubMed] [Google Scholar]
- Baer M. M., Bilstein A., Caussinus E., Csiszar A., Affolter M., Leptin M. (2010). The role of apoptosis in shaping the tracheal system in the Drosophila embryo. Mech. Dev. 127, 28–35 [DOI] [PubMed] [Google Scholar]
- Bardet P. L., Kolahgar G., Mynett A., Miguel-Aliaga I., Briscoe J., Meier P., Vincent J. P. (2008). A fluorescent reporter of caspase activity for live imaging. Proc. Natl. Acad. Sci. USA 105, 13901–13905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastock R., Strutt D. (2007). The planar polarity pathway promotes coordinated cell migration during Drosophila oogenesis. Development 134, 3055–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertet C., Lecuit T. (2009). Planar polarity and short-range polarization in Drosophila embryos. Semin. Cell Dev. Biol. 20, 1006–1013 [DOI] [PubMed] [Google Scholar]
- Bertet C., Sulak L., Lecuit T. (2004). Myosin-dependent junction remodelling controls planar cell intercalation and axis elongation. Nature 429, 667–671 [DOI] [PubMed] [Google Scholar]
- Blankenship J. T., Backovic S. T., Sanny J. S., Weitz O., Zallen J. A. (2006). Multicellular rosette formation links planar cell polarity to tissue morphogenesis. Dev. Cell 11, 459–470 [DOI] [PubMed] [Google Scholar]
- Chihara T., Kato K., Taniguchi M., Ng J., Hayashi S. (2003). Rac promotes epithelial cell rearrangement during tracheal tubulogenesis in Drosophila. Development 130, 1419–1428 [DOI] [PubMed] [Google Scholar]
- Chung S., Vining M. S., Bradley P. L., Chan C. C., Wharton K. A., Jr, Andrew D. J. (2009). Serrano (sano) functions with the planar cell polarity genes to control tracheal tube length. PLoS Genet. 5, e1000746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Classen A. K., Anderson K. I., Marois E., Eaton S. (2005). Hexagonal packing of Drosophila wing epithelial cells by the planar cell polarity pathway. Dev. Cell 9, 805–817 [DOI] [PubMed] [Google Scholar]
- Förster D., Luschnig S. (2012). Src42A-dependent polarized cell shape changes mediate epithelial tube elongation in Drosophila. Nat. Cell Biol. 14, 526–534 [DOI] [PubMed] [Google Scholar]
- Goodrich L. V., Strutt D. (2011). Principles of planar polarity in animal development. Development 138, 1877–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R. S., Roszko I., Solnica-Krezel L. (2011). Planar cell polarity: coordinating morphogenetic cell behaviors with embryonic polarity. Dev. Cell 21, 120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans J., Wenzl C., Herz H. M., Bartoszewski S., Schnorrer F., Vogt N., Schwarz H., Müller H. A. (2005). RhoGEF2 and the formin Dia control the formation of the furrow canal by directed actin assembly during Drosophila cellularisation. Development 132, 1009–1020 [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S., Ng J., Tzu J., Dietzl G., Sun Y., Harms M., Nardine T., Luo L., Dickson B. J. (2002). Rac function and regulation during Drosophila development. Nature 416, 438–442 [DOI] [PubMed] [Google Scholar]
- Huang J., Zhou W., Dong W., Watson A. M., Hong Y. (2009). From the cover: directed, efficient, and versatile modifications of the Drosophila genome by genomic engineering. Proc. Natl. Acad. Sci. USA 106, 8284–8289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine K. D., Wieschaus E. (1994). Cell intercalation during Drosophila germband extension and its regulation by pair-rule segmentation genes. Development 120, 827–841 [DOI] [PubMed] [Google Scholar]
- Jaźwińska A., Ribeiro C., Affolter M. (2003). Epithelial tube morphogenesis during Drosophila tracheal development requires Piopio, a luminal ZP protein. Nat. Cell Biol. 5, 895–901 [DOI] [PubMed] [Google Scholar]
- Keller R. (2002). Shaping the vertebrate body plan by polarized embryonic cell movements. Science 298, 1950–1954 [DOI] [PubMed] [Google Scholar]
- Lee S., Kolodziej P. A. (2002). The plakin Short Stop and the RhoA GTPase are required for E-cadherin-dependent apical surface remodeling during tracheal tube fusion. Development 129, 1509–1520 [DOI] [PubMed] [Google Scholar]
- Levayer R., Pelissier-Monier A., Lecuit T. (2011). Spatial regulation of Dia and Myosin-II by RhoGEF2 controls initiation of E-cadherin endocytosis during epithelial morphogenesis. Nat. Cell Biol. 13, 529–540 [DOI] [PubMed] [Google Scholar]
- Mirkovic I., Mlodzik M. (2006). Cooperative activities of drosophila DE-cadherin and DN-cadherin regulate the cell motility process of ommatidial rotation. Development 133, 3283–3293 [DOI] [PubMed] [Google Scholar]
- Mirkovic I., Gault W. J., Rahnama M., Jenny A., Gaengel K., Bessette D., Gottardi C. J., Verheyen E. M., Mlodzik M. (2011). Nemo kinase phosphorylates β-catenin to promote ommatidial rotation and connects core PCP factors to E-cadherin-β-catenin. Nat. Struct. Mol. Biol. 18, 665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K. S., Khan Z., Molnár I., Mihály J., Kaschube M., Beitel G. J. (2012). Drosophila Src regulates anisotropic apical surface growth to control epithelial tube size. Nat. Cell Biol. 14, 518–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T., Honda H., Takeichi M. (2012). Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell 149, 1084–1097 [DOI] [PubMed] [Google Scholar]
- Oda H., Tsukita S. (2001). Real-time imaging of cell-cell adherens junctions reveals that Drosophila mesoderm invagination begins with two phases of apical constriction of cells. J. Cell Sci. 114, 493–501 [DOI] [PubMed] [Google Scholar]
- Oda H., Uemura T., Harada Y., Iwai Y., Takeichi M. (1994). A Drosophila homolog of cadherin associated with armadillo and essential for embryonic cell-cell adhesion. Dev. Biol. 165, 716–726 [DOI] [PubMed] [Google Scholar]
- Peifer M., Sweeton D., Casey M., Wieschaus E. (1994). wingless signal and Zeste-white 3 kinase trigger opposing changes in the intracellular distribution of Armadillo. Development 120, 369–380 [DOI] [PubMed] [Google Scholar]
- Pirraglia C., Jattani R., Myat M. M. (2006). Rac function in epithelial tube morphogenesis. Dev. Biol. 290, 435–446 [DOI] [PubMed] [Google Scholar]
- Rauzi M., Lenne P. F., Lecuit T. (2010). Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature 468, 1110–1114 [DOI] [PubMed] [Google Scholar]
- Ribeiro C., Neumann M., Affolter M. (2004). Genetic control of cell intercalation during tracheal morphogenesis in Drosophila. Curr. Biol. 14, 2197–2207 [DOI] [PubMed] [Google Scholar]
- Saburi S., Hester I., Fischer E., Pontoglio M., Eremina V., Gessler M., Quaggin S. E., Harrison R., Mount R., McNeill H. (2008). Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat. Genet. 40, 1010–1015 [DOI] [PubMed] [Google Scholar]
- Samakovlis C., Hacohen N., Manning G., Sutherland D. C., Guillemin K., Krasnow M. A. (1996). Development of the Drosophila tracheal system occurs by a series of morphologically distinct but genetically coupled branching events. Development 122, 1395–1407 [DOI] [PubMed] [Google Scholar]
- Shaye D. D., Casanova J., Llimargas M. (2008). Modulation of intracellular trafficking regulates cell intercalation in the Drosophila trachea. Nat. Cell Biol. 10, 964–970 [DOI] [PubMed] [Google Scholar]
- Shindo M., Wada H., Kaido M., Tateno M., Aigaki T., Tsuda L., Hayashi S. (2008). Dual function of Src in the maintenance of adherens junctions during tracheal epithelial morphogenesis. Development 135, 1355–1364 [DOI] [PubMed] [Google Scholar]
- Simões S. M., Blankenship J. T., Weitz O., Farrell D. L., Tamada M., Fernandez-Gonzalez R., Zallen J. A. (2010). Rho-kinase directs Bazooka/Par-3 planar polarity during Drosophila axis elongation. Dev. Cell 19, 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone R. P., DiNardo S. (2010). Actomyosin contractility and Discs large contribute to junctional conversion in guiding cell alignment within the Drosophila embryonic epithelium. Development 137, 1385–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer B., Oprins A., Rabouille C., Munro S. (2005). The exocyst component Sec5 is present on endocytic vesicles in the oocyte of Drosophila melanogaster. J. Cell Biol. 169, 953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutt D. I. (2001). Asymmetric localization of frizzled and the establishment of cell polarity in the Drosophila wing. Mol. Cell 7, 367–375 [DOI] [PubMed] [Google Scholar]
- Strutt H., Warrington S. J., Strutt D. (2011). Dynamics of core planar polarity protein turnover and stable assembly into discrete membrane subdomains. Dev. Cell 20, 511–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M., Takahashi F., Ui-Tei K., Kojima T., Saigo K. (2005). Requirements of genetic interactions between Src42A, armadillo and shotgun, a gene encoding E-cadherin, for normal development in Drosophila. Development 132, 2547–2559 [DOI] [PubMed] [Google Scholar]
- Tamada M., Farrell D. L., Zallen J. A. (2012). Abl regulates planar polarized junctional dynamics through β-catenin tyrosine phosphorylation. Dev. Cell 22, 309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepass U., Theres C., Knust E. (1990). crumbs encodes an EGF-like protein expressed on apical membranes of Drosophila epithelial cells and required for organization of epithelia. Cell 61, 787–799 [DOI] [PubMed] [Google Scholar]
- Tree D. R. P., Shulman J. M., Rousset R., Scott M. P., Gubb D., Axelrod J. D. (2002). Prickle mediates feedback amplification to generate asymmetric planar cell polarity signaling. Cell 109, 371–381 [DOI] [PubMed] [Google Scholar]
- Ulrich F., Krieg M., Schötz E. M., Link V., Castanon I., Schnabel V., Taubenberger A., Mueller D., Puech P. H., Heisenberg C. P. (2005). Wnt11 functions in gastrulation by controlling cell cohesion through Rab5c and E-cadherin. Dev. Cell 9, 555–564 [DOI] [PubMed] [Google Scholar]
- Usui T., Shima Y., Shimada Y., Hirano S., Burgess R. W., Schwarz T. L., Takeichi M., Uemura T. (1999). Flamingo, a seven-pass transmembrane cadherin, regulates planar cell polarity under the control of Frizzled. Cell 98, 585–595 [DOI] [PubMed] [Google Scholar]
- Uv A., Cantera R., Samakovlis C. (2003). Drosophila tracheal morphogenesis: intricate cellular solutions to basic plumbing problems. Trends Cell Biol. 13, 301–309 [DOI] [PubMed] [Google Scholar]
- Vichas A., Zallen J. A. (2011). Translating cell polarity into tissue elongation. Semin. Cell Dev. Biol. 22, 858–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz-Peitz F., Zallen J. A. (2009). Junctional trafficking and epithelial morphogenesis. Curr. Opin. Genet. Dev. 19, 350–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen J. A., Wieschaus E. (2004). Patterned gene expression directs bipolar planar polarity in Drosophila. Dev. Cell 6, 343–355 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.