

Abstract
Aim
To test the hypothesis that the clinical efficacy of triptans reflects convergent modulation of ion channels also involved in inflammatory mediator (IM)-induced sensitization of dural afferents.
Methods
Acutely dissociated retrogradely labeled dural afferents were studied with whole cell and perforated patch techniques in the absence and presence of sumatriptan and/or IM (prostaglandin E2, bradykinin, and histamine).
Results
Sumatriptan dose-dependently suppressed voltage-gated Ca2+ currents. Acute (2 min) sumatriptan application increased dural afferent excitability and occluded further IM-induced sensitization. In contrast, pre-incubation (30 min) with sumatriptan had no influence on dural afferent excitability and partially prevented IM-induced sensitization of dural afferents. The sumatriptan-induced suppression of voltage-gated Ca2+ currents, acute sensitization and pre-incubation-induced block of IM-induced sensitization were blocked by the 5-HT1D antagonist, BRL 15572. Pre-incubation failed to suppress the IM-induced decrease in action potential threshold and overshoot (which results from modulation of voltage-gated Na+ currents) and activation of Cl− current, and had no influence on the Cl− reversal potential. However, pre-incubation with sumatriptan caused a dramatic hyperpolarizing shift in the voltage dependence of K+ current activation.
Discussion
These results indicate that while the actions of sumatriptan on dural afferents are complex, at least two distinct mechanisms underlie the antinociceptive actions of this compound. One of these mechanisms, the shift in the voltage-dependence of K+ channel activation may suggest a novel strategy for future development of anti-migraine agents.
Keywords: Nociceptor sensitization, patch clamp, current clamp, in vitro
INTRODUCTION
Migraine is a debilitating neurological disorder that impacts a large percentage of the population (1). Furthermore, the social and economic burden of this disorder remains a major concern despite the prophylactic and abortive agents used to treat migraine pain (2, 3). Previous data indicate that the release of inflammatory mediators in the dura and subsequent dural afferent sensitization are important for initiating migraine pain (4, 5). Furthermore, we have recently demonstrated that inflammatory mediators (IM) not only sensitize the vast majority of dural afferents (6), but that this sensitization reflects the modulation of a number of different ion channels, at least one of which appears to be unique to dural afferents (7).
Triptans, one of the most effective classes of drugs for the treatment of migraine pain, are serotonin 1B/1D (5-HT1B/1D) receptor agonists. While the 5-HT1B receptors appear to be primarily post-synaptic, located on vascular smooth muscle, the 5-HT1D receptors are located on the peripheral and central terminals of dural afferents (8, 9). Interestingly, despite widespread distribution of 5-HT1D receptors in trigeminal (TG) and dorsal root ganglion (DRG) neurons (9), clinical data indicate that these compounds have little, if any utility in the treatment of anything but migraine pain (10). And while pre-clinical data suggest that these compounds may have anti-inflammatory efficacy (11) and may be analgesic when directly applied to the CNS (12, 13), systemic administration of triptans selectively inhibit nociceptive behavior (14), neuropathic pain behavior (15), and evoked activity in trigeminal dorsal horn neurons (16) in response to noxious stimulation of trigeminal targets.
These observations raised the possibility that the clinical selectivity and efficacy of triptans reflect a unique mechanism of action on dural afferents. To begin to assess this possibility, we examined the effect of sumatriptan on the excitability of dural afferents as well as the influence of this compound on IM-induced sensitization of dural afferents.
MATERIALS AND METHODS
Animals
Adult female Sprague Dawley rats (Harlan, Indianapolis, IN) weighing between 180–290 g were used for all experiments. Rats were housed two per cage at the University of Pittsburgh animal facility on a 12:12 light: dark schedule with food and water freely available. Prior to all procedures, animals were deeply anesthetized with an i.p. injection (1 ml/kg) of rat cocktail containing ketamine (55mg/kg), xylazine (5.5 mg/kg) and acepromazine (1.1 mg/kg). Experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with National Institutes of Health guidelines for the use of laboratory animals in research. All efforts were employed to minimize the total number of animals used.
Retrograde labeling
Afferents innervating the dura were identified as previously described following labeling with the retrograde tracer 1,1'-dioctadecyl-3,3,3',3'-tetramethylindocarbocyanine perchlorate (DiI, Invitrogen, Carlsbad, CA) (8). Immediately post-operatively, animals received a single i.m. injection of penicillin G (10,000 units/kg) and a single injection of buprenorphine (0.03 mg/kg) to minimize infection and discomfort. Subsequent administration of ketoprofen was provided if evidence of hypersensitivity persisted over subsequent days post-labeling.
Tissue Preparation
Ten to fourteen days following DiI application, trigeminal ganglia (TG) were removed, enzymatically treated and mechanically dissociated as previously described (7). Acutely dissociated cells were plated on laminin/ornithine coated glass coverslips. Changes in current and excitability were measured 2–8 hours after cells were plated.
Electrophysiology
All whole cell and perforated patch-clamp recordings were performed with a HEKA EPC10 amplifier (HEKA Electronik, Lambrecht/Rhineland-Pfalz, Germany). Data were low-pass filtered at 5–10 kHz with a four-pole Bessel filter and digitally sampled at 25–100 kHz.
Current Clamp
To assess changes in excitability, borosilicate glass electrodes were filled with (mM) K-methanesulfonate 110, KCl 30, NaCl 5, CaCl2 1, MgCl2 2, HEPES 10, EGTA 11, Mg-ATP 2, Li-GTP 1, pH 7.2 (adjusted with Tris-base), 310 mOsm (adjusted with sucrose). Bath solution contained (mM) KCl 3, NaCl 130, CaCl2 2.5, MgCl2 0.6, HEPES 10, glucose 10, pH 7.4 (adjusted with Tris-base), 325mOsm (adjusted with sucrose) and either vehicle (0.01% ETOH and 0.1% acetic acid) or test compounds: IM [(μM) bradykinin 10, histamine 1, and prostaglandin E2 1]; and/or sumatriptan (1μM). Excitability was assessed with three parameters as previously described (6): rheobase, action potential threshold, and the response to suprathreshold stimulation. A neuron was considered sensitized if application of a test solution resulted in a hyperpolarization of action potential threshold, decrease in rheobase, and/or an increase in the response to suprathreshold stimulation greater than 2 SD's from the baseline mean.
Passive properties measured were resting membrane potential (Em), and input resistance (Rin). Rin was assessed with five 750-ms hyperpolarizing current injections (2–5 pA) from Em immediately before and 90 s after the application of sumatriptan alone, IM alone, or sumatriptan and IM. Active electrophysiological properties were assessed with an action potential (AP) evoked with a 4-ms depolarizing current pulse. These included: AP duration at 0 mV, magnitude of AP overshoot, magnitude of the after-hyperpolarization (AHP), AHP decay (τ AHP). The magnitude of the overshoot was measured from 0 mV. The magnitude of the AHP was measured from the Em. Decay of the AHP was estimated by fitting the decay phase of the AHP with a single exponential function.
Voltage Clamp
To isolate Ca2+ currents, electrodes were 1 – 4 MΩ when filled with and electrode solution containing (mM): Cs-methanesulfonate 100, Na-methanesulfonate 5, TEA-Cl 40, CaCl2 1, MgCl2 2, EGTA 11, HEPES 10, pH 7.2 (adjusted with Tris-base), 310 mOsm (adjusted with sucrose). The bath solution contained (mM): choline-Cl 100, TEA-Cl 30, CaCl2 2.5, MgCl2 0.6, NFA 0.1, HEPES 10, glucose 10, pH 7.4 (adjusted with Tris-base), 325 mOsm (adjusted with sucrose).
IM-induced Cl− currents (IIM-Cl) were isolated with electrode solutions containing (mM) Cs-methanesulfonate 100, CsCl 30, CaCl2 1, MgCl2 2, HEPES 10, EGTA 11, Mg-ATP 2, Li-GTP 1, pH 7.2 (adjusted with Tris-base), 310 mOsm (adjusted with sucrose) and bath solution containing (mM) Choline-Cl 130, CaCl2 2.5, MgCl2 0.6, HEPES 10, glucose 10, pH 7.4 (adjusted with Tris-base), 325mOsm (adjusted with sucrose). IIM-Cl was elicited with 100 ms test pulses from −70 to +50mV following a 40ms prepulse to 0 mV to evoke Ca2+ currents in the presence of sumatriptan with and without IM. IIM-Cl was also recorded with Ca2+ artificially buffered to 622nM with an electrode solution containing EGTA (1.2 mM), Ca2+ (1 mM) and Mg2+ (2 mM) and influx via voltage-gated Ca2+ channels was also blocked by the addition of Cd2+ (50 μM) to the bath solution. MaxChelator was used to generate estimates of resting free intracellular Ca2+.
To isolate K+ currents, electrodes were 1–4 MΩ when filled with (mM) K-methanesulfonate 110, KCl 30, NaCl 5, CaCl2 1, MgCl2 2, HEPES 10, EGTA 11, Mg-ATP 2, Li-GTP 1, pH 7.2 (adjusted with Tris-base), 310 mOsm (adjusted with sucrose). Bath solution contained (mM) KCl 3, Choline-Cl 130, CaCl2 2.5, MgCl2 0.6, NFA 0.1, HEPES 10, glucose 10, pH 7.4 (adjusted with Tris-base), 325 mOsm (adjusted with sucrose). Because the bath solution contained Ca2+, total K+ current consisted of both voltage-gated K+ and Ca2+ modulated K+ currents (7).
Drugs
All salts and reagents were obtained from Sigma-Aldrich (St. Louis, MO), unless indicated below. Bradykinin was dissolved in 1% acetic acid (23.58mM stock concentration), PGE2 was dissolved in 100% ETOH (10mM stock concentration), and histamine was dissolved in water (100mM stock concentration). All stock solutions were stored at −20°C until the day of use. IM-vehicle bath containing the final concentration of ETOH (0.01%) and acetic acid (0.001%) was used as a control. Niflumic acid (NFA) was dissolved in 100% ETOH. Sumatriptan was a generous gift from Glaxo Smith Kline. Sumatriptan was dissolved as a 10mM stock solution in water and subsequently diluted in bath solution. The 5-HT1D receptor antagonist BRL 15572 was obtained from Tocris Biosciences (R & D Systems, Minneapolis, MN), was dissolved as a 10 mM stock in 100% ethanol and diluted in bath solution.
Data Analysis
Data were analyzed with PulseFit (HEKA), Sigma Plot and Sigma Stat software (Systat Software Inc., Richmond, CA). Conductance-voltage (G-V) curves were constructed from I–V curves by dividing the evoked current by the driving force on the current, such that G = I/(Vm - Vrev) where Vm is the potential at which current was evoked and Vrev is the reversal potential for the current was measured directly (for K and Ca2+). Instantaneous I–V data was obtained from the tail currents measured following activation of voltage-gated Ca2+ currents.
Statistical Analysis
For comparisons of data collected before and after IM application, either a paired t-test or Repeated Measures ANOVA was used if data were parametric. Otherwise, a Wilcoxin or Friedman test was used for nonparametric analysis. For unpaired comparisons, Student's t-test, one- and two-way ANOVA were used for parametric data and a Mann Whitney U for nonparametric analysis. Data were considered statistically significant when p < 0.05. All data are represented as mean ± standard error.
RESULTS
Data was collected from 78 dural afferents acutely dissociated from 15 female Sprague Dawley rats. Of these, 53 were studied in voltage-clamp and 25 were studied in current clamp.
Sumatriptan dose-dependently inhibits voltage-gated calcium currents (VGCC)
Previous data suggests a primary mechanism of triptan action is a G protein-mediated inhibition of voltage-gated Ca2+ currents (17, 18). To determine if such a mechanism exists in dural afferents and to determine the appropriate concentration of sumatriptan for subsequent experiments, VGCC in dural afferents (n=7) were studied with increasing concentrations (0.001 to 10 μM) of sumatriptan. Currents were evoked with 50ms pulses from −60 to +80mV following a 100ms prepulse to −100mV.
A concentration-dependent inhibition of VGCC was observed in 7 of 7 dural afferents studied (Fig 1A, B). Inhibition of peak current evoked at 10 mV was converted to percent inhibition and data from all 7 neurons were pooled, plotted as a function of the concentration of sumatriptan and fitted with a modified Hill equation (Fig 1C). The IC50 for sumatriptan-induced inhibition of VGCC was 142 nM, with a maximal fractional inhibition of 20 ± 2%. Interestingly, there was no evidence of a low threshold VGCC in any of the dural afferents studied.
Figure 1. Sumatriptan inhibits high threshold voltage gated Ca2+ currents (ICa) in dural afferents.

A. Example of sumatriptan mediated inhibition of ICa. B. Bath application of increasing concentrations of sumatriptan from 0.001 to 10μM suppressed ICa amplitude recorded with a single pulse to 10mV in 7 of 7 dural afferents studied. C. The IC50 for sumatriptan was determined with percent inhibition plotted against sumatriptan concentration. D. To examine the voltage dependence of inhibition, currents were elicited with a test pulse to +10mV following pre-pulses to −60 and +80mV before (Baseline) and after sumatriptan (10 μM Suma) application (n=5). The ratio of the current amplitude following a prepulse to +80 divided by the current amplitude following a prepulse to −60 was determined before and after sumatriptan application. Following sumatriptan application, there was no significant difference in the current ratio. E. Instantaneous I–V data were plotted from tail currents. Sumatriptan decreased the amplitude of the tail currents but did not produce a shift in their voltage dependence of activation.
To determine if a membrane delimited displacement of the N-type Ca2+ channel β-subunit by the G-protein βγ subunits (19) could also be mediating the decrease in VGCC with sumatriptan, Ca2+ currents were elicited with a two-pulse protocol in which a test pulse to +10mV was preceded by a conditioning pulse to either −60 mV or +80 mV (20). A 50 ms step to −60 mV between the conditioning and test pulses was used to enable channel deactivation following the step to +80 mV (n=5). Consistent with the absence of a detectable shift in the VGCC instantaneous I–V curve, there was no evidence of pre-pulse potentiation as the ratio of the currents elicited before sumatriptan application (1.07 ± 0.03) were comparable to that after application (1.12 ± 0.07, Fig 1D). To confirm that sumatriptan-induced inhibition of voltage-gated Ca2+ currents in dural afferents was mediated by the 5-HT1D receptor, sumatriptan (1 μM) was co-applied to 3 dural afferents with the 5-HT1D receptor selective antagonist BRL 15572 (1 μM). Two minutes after the application of the combination of sumatriptan and BRL 15572, the decrease in maximal conductance (7.0 ± 0.1% of baseline) was significantly (p < 0.01, Student's t-test) less than that observed with sumatriptan alone.
Acute sumatriptan increases baseline dural afferent excitability
Acute (2 min) application of 1μM sumatriptan alone produced a significant increase in excitability of dural afferents (n = 7) as evidenced by changes in rheobase (p < 0.01, Fig 2A), action potential threshold (p < 0.01, Fig 2B), and the response to suprathreshold current injection (p < 0.01, Fig 2C). These changes were associated with a significant (p < 0.01, paired t test) depolarization of Em from −71.3 ± 1.6 mV to −54.0 ± 3.6 mV. These sumatriptan-induced changes in excitability were blocked by the co-application of the 5-HT1D receptor antagonist BRL 15572 (1 μM, n = 5, Fig 2A, B and C).
Figure 2. Acute sumatriptan increases dural afferent excitability.

A. Acute bath application of 1μM sumatriptan resulted in a significant reduction in rheobase in dural afferents (n=7). This effect was blocked when sumatriptan was co-applied with the 5-HT1D receptor antagonist BRL 15572 (Antag, 1 μM, n = 6). When IM were applied to dural afferents in the presence of sumatriptan, there was no further decrease in rheobase. B. Acute sumatriptan application also significantly hyperpolarized the AP threshold. This change was also blocked when sumatriptan was co-applied with BRL 15572. There was no further change in AP threshold following IM application. Data in A and B were analyzed with a one way ANOVA with a Holm-Sidak test used for post-hoc analysis. The most relevant comparisons are illustrated for clarity where * is p < 0.05. C. The stimulus response function data in C for neurons treated with sumatriptan alone (Suma) or sumatriptan + BRL 15572 (Suma + Antag) were analyzed with a Fisher Exact test. The proportion of neurons treated with Suma alone (7 of 7) with a left shift in the stimulus response function (relative to baseline) was significantly (p < 0.05) greater than that for the Suma + Antag group (2 of 6). There was no further shift in the stimulus response function in the Suma group (0 of 7) following application of IM. Baseline data are plotted for comparison.
To determine whether sensitization of dural afferents via sumatriptan or IM involve comparable mechanisms, IM were applied following sumatriptan. No further increase in excitability was detected in these neurons (Fig 2). These results suggest that either sumatriptan-induced sensitization shares common mechanisms with those of IM, or this drug has blocked the actions of IM.
Prolonged sumatriptan exposure has no influence on baseline excitability and attenuates IM-induced sensitization of dural afferents
There is evidence that triptan analgesia does not occur immediately after administration. Instead, pain relief is experienced typically 20–30 min after taking the drug (21, 22). Therefore, we examined the possibility that with a longer exposure time, sumatriptan may switch from being excitatory to inhibitory.
Following 30 minute pre-incubation with sumatriptan, in which neurons (n = 8) were incubated in sumatriptan prior to recording, there was no significant (p > 0.05, Student's t test) difference in rheobase compared to that in control (vehicle) neurons (n=8) indicating that the decrease in rheobase following acute sumatriptan application returns to baseline levels with longer incubation times: rheobase normalized to membrane capacitance was 8.4 ± 1.3 pA/pF and 9.5 ± 1.5 pA/pF in neurons from vehicle and sumatriptan treated groups, respectively. Similarly, sumatriptan pre-incubation produced no significant (p > 0.05, Student's t test) changes in AP threshold compared to control: AP threshold was −29.5 ± 1.6 mV and −23.6 ± 4.3mV in neurons from vehicle and sumatriptan treated groups, respectively. There was also no significant (p < 0.05, two-way repeated measure ANOVA) influence of sumatriptan pre-incubation on the response to suprathreshold current injection (Fig 3C). Furthermore, pre-incubation of sumatriptan with BRL 15572 (n = 5) had no detectable influence on rheobase (which was 7.3 ± 2.1 pA/pF), action potential threshold (which was −25.4 ± 4.0), the or the response to suprathreshold current injection (Fig 3C).
Figure 3. Prolonged sumatriptan exposure has no influence on excitability and attenuates IM-induced sensitization of dural afferents.
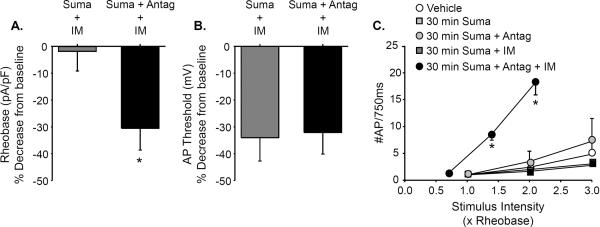
A. Following 30 minute pre-incubation with sumatriptan, IM application (Suma + IM) had little influence on rheobase (expressed as a % of baseline determined prior to the application of IM for each neuron). However, the IM-induced decrease in rheobase in neurons pre-incubated with the combination of sumatriptan and BRL 15572 (Suma + antag + IM) was significantly (Student's t test) greater than the change in observed in neurons treated with sumatriptan alone. B. In contrast to rheobase, application of IM resulted in a decrease in AP threshold in neurons preincubated with sumatriptan alone as well as the combination of sumatriptan and BRL 15572. There was no significant difference between these groups. C. Sumatriptan pre-incubation (Suma) had no significant influence on the baseline response to suprathreshold stimulation. Nor was there an influence of pre-incubating neurons with the combination of sumatriptan and BRL 15572 (Suma + Antag). Data were analyzed with a two-way repeated measures ANOVA and compared to control neurons incubated in vehicle for 30 minutes (Vehicle). Furthermore, the application of IM to neurons pre-incubated for 30 minutes with sumatriptan (Suma + IM) had no significant influence on the stimulus response function as determined with a one-way repeated measures ANOVA. However, application of IM to neurons pre-incubated with the combination of sumatriptan and BRL 15572 (Suma + Antag +IM) resulted in a significant leftward shift in the stimulus response function as determined by both the increase in the number of evoked action potentials at 2× and 3× rheobase (relative to the response prior to the application of IM), as well as the proportion of neurons in which IM produced a change (4 of 4) relative to the Suma + IM group (0 of 7, p < 0.01, Fisher Exact test). * Indicates a significant difference between groups in A and before and after IM application in C where p<0.05.
To determine the effects of sumatriptan pre-incubation on IM-induced sensitization of dural afferents, changes in excitability were recorded with IM in the presence of sumatriptan. In contrast to our previous observations in which application of IM to dural afferents resulted in a significant decrease in rheobase and leftward shift in the response to suprathreshold stimulation (6), IM had no significant influence on rheobase (Fig 3A) or the response to suprathreshold stimulation (Fig 3C) in dural afferents pre-incubated with sumatriptan as compared to vehicle treated dural afferents. However, pre-incubation with sumatriptan did not prevent IM-induced hyperpolarization of AP threshold (Fig 3B). The suppressive effects of sumatriptan pre-incubation on IM-induced changes in rheobase and the response to suprathreshold current injection were blocked by the presence of BRL 15572 during the 30 min pre-incubation (n = 5, Fig 3A and C).
Sumatriptan Modulates Active and Passive Electrophysiological Properties
To begin to determine the basis for the sumatriptan-induced decrease in dural afferent excitability as well as the inhibition of IM-induced sensitization, changes in passive and active electrophysiological properties were examined. Thirty minute sumatriptan pre-incubation had no effect on baseline passive electrophysiological properties as assessed by the resting membrane potential and input resistance as these values, −69.0 ± 1.7 mV and 635 ± 146 MΩ (n = 8), were comparable to values previously reported (i.e., −71.3 ± 1.6 mV and 473 ± 57.3 MΩ (6)).
We previously demonstrated that IM produce significant changes in passive and active electrophysiological properties of dural afferents (6). These IM-induced changes included a ~10 mV membrane depolarization that was accompanied by a decrease in Rin subsequent to activation of IIM-Cl (6). While sumatriptan pre-incubation did not prevent the IM-induced decrease in Rin, it blocked the IM-induced membrane depolarization as the IM-induced depolarization in neurons pre-incubated with the combination of sumatriptan and BRL 15572 (n = 5) was significantly (p < 0.01) larger than that in neurons pre-incubated with sumatriptan alone (n = 8, Table 1). We also previously demonstrated an IM-induced increase the AP overshoot subsequent to modulation of voltage gated Na+ currents (VGSC) in dural afferents (6). Sumatriptan pre-incubation did not prevent the IM-induced increase in AP overshoot, either, suggesting that sumatriptan did not prevent IM modulation of VGSC.
Table 1.
Sumatriptan blocks IM-induced changes in passive and active electrophysiological properties
| Group | N | Δ Em (mV) | % Rin (MΩ) | Δ AP Duration (ms) | Δ AP overshoot (mV) | Δ AHP Magnitude (mV) | Δ τ AHP (ms) |
|---|---|---|---|---|---|---|---|
| 30 min Suma + IM | 8 | 1.0 ± 0.9 | 41.2 ± 14.6 | 0.38 ± 0.4 | 2.9 ± 5.8 | 2.3 ± 1.6 | 17.9 ± 26.5 |
| 30 min Suma + Antag + IM | 5 | 7.5 ± 1.7 | 42.3 ± 9.0 | 0.34 ± 0.5 | 2.6 ± 1.4 | 3.1 ± 1.6 | 24.5 ± 11.4 |
Neurons were pre-incubated with sumatriptan (Suma, 1 μM) for 30 minutes alone or with the 5-HT1D receptor antagonist (Antag) BRL 15572 (1 μM) prior to electrophysiological analysis. The IM-induced change in resting membrane potential (Δ Em) calculated as the difference between Em after IM and Em before IM is significantly (p < 0.01, Student's t test) greater in the antagonist group. All other IM-induced changes were comparable between the two groups. N is the number of neurons studied in each group. Rin is input resistance. AP Duration is the duration of the action potential at 0 mV. AP overshoot is the amplitude of the action potential over 0 mV. AHP magnitude is the magnitude of the afterhyperpolarization following the AP relative to Em. τ AHP is the time constant of decay of the AHP.
Sumatriptan does not Prevent IM-induced Activation of IIM-Cl
While data from the AP waveform suggest that VGSC are probably not a convergent target of sumatriptan, the observation that sumatriptan was able to block the IM-induced depolarization suggests that these drugs may block IM-induced activation of IIM-Cl. To test this possibility, IM-induced changes in IIM-Cl were monitored in dural afferents (n = 7) with a protocol in which IIM-Cl was evoked with 100 ms test pulses from −70mV to +50mV following a 40ms pre-pulse to 0mV to evoke Ca2+ currents (Fig 4A). The currents reversed at −30mV close to the predicted reversal potential for Cl− (−34 mV) based on the composition of our intracellular and extracellular solutions. Pre-incubation with sumatriptan had no detectable influence on the peak density or rectification of IIM-Cl (Fig 4A).
Figure 4. Sumatriptan does not prevent IM-induced activation of IIM-Cl.

IIM-Cl was activated by IM application and elicited with 100 ms test pulses from −70mV to +50mV following a 40ms pre-pulse to 0mV to evoke Ca2+ currents (n=7) and isolated as the difference between current evoked before and after application of IM (IIM-Cl Difference Current). A. Pre-incubation with sumatriptan had no significant (p > 0.05, two-way repeated measures ANOVA) influence on peak IIM-Cl density (at any voltage tested). B. To determine if sumatriptan may change the sensitivity of IIM-Cl to high intracellular Ca2+, IIM-Cl was recorded in the presence of Cd2+ and low intracellular EGTA to buffer intracellular Ca2+ at 622nM (n=5). Sumatriptan had no significant (p > 0.05, two-way repeated measures ANOVA) influence on the amplitude of IIM-Cl at any potential under these conditions. Currents were blocked with 100μM niflumic acid (NFA). C. The reversal potential for Cl- was recorded in response to a ramp voltage protocol from +50mV to −100mV using the gramicidin perforated patch configuration (n=5). Sumatriptan pre-incubation had no significant (p > 0.05, Student's t test) influence on the reversal potential of the IM-induced current.
Given the influence of both sumatriptan and IM on VGCC in dural afferents (7), to rule out a potential interaction between changes in Ca2+ influx and IIM-Cl activation, this experiment was repeated in the presence of Cd2+ to block VGCC and low intracellular EGTA (1.2mM) to buffer intracellular Ca2+ at a high concentration (622nM). IIM-Cl was again recorded with test pulses from −70mV to +50mV. Sumatriptan did not produce any change in the amplitude of IIM-Cl (n=5) at any potential under these conditions (Fig 4B). Furthermore, IIM-Cl recorded in the presence of sumatriptan was blocked by Cl− channel blocker niflumic acid (100μM, Fig 4B) as previously demonstrated (7).
Because the excitatory influence of IM-induced activation of IIM-Cl on dural afferents appears to reflect a depolarized Cl− equilibrium potential (ECl) in these neurons (7), we also examined the effects of sumatriptan on the reversal potential of IIM-Cl. Cl− currents were recorded in response to a ramp voltage protocol from +50mV to −100mV using gramicidin perforated patch to prevent dialysis of intracellular Cl− (23). Sumatriptan pre-incubation (n=5) did not shift the reversal potential of IIM-Cl (Fig 4C).
Sumatriptan Modulates K+ Currents and Inhibits IM-induced Suppression of K+ Currents
The decrease in Rin observed in the absence of an IM-induced depolarization of Em suggests that pre-incubation with sumatriptan may result in the activation of a K+ current (IK) that counters the depolarization driven by the activation of IIM-Cl. To test this possibility, IK was evoked with voltage protocols described in Methods, in the absence (n=7) and presence (n=6) of 30 min pre-incubation with sumatriptan and IM (Fig 5A). From these data, changes in the voltage dependence of activation and maximal conductance (Gmax) were determined. Consistent with the decrease in Rin in the absence of Em depolarization, 30 min sumatriptan pre-incubation resulted in a dramatic leftward shift in the voltage dependence of IK activation. There was a significant (p < 0.01) hyperpolarization of the V0.05 of activation (Fig 5B) following sumatriptan pre-incubation (−27.3 ± 4.7mV) as compared to control (−11.5± 2.4mV). IM application alone produced no change in the voltage dependence of activation of IK (Fig 5B). However, IM (n=7) significantly reduced the maximal conductance (Fig 5C). This effect was completely blocked by sumatriptan (n=8) pre-incubation (Fig 5C).
Figure 5. Sumatriptan both modulates K+ currents and blocks IM-induced suppression of K+ currents.

A. The voltage-dependence of K+ current activation was determined with current-voltage protocols consisting of 10mV, 500ms voltage-steps between −60 and +60mV following a 500ms pre-pulse to −120mV. B. 30 minutes of sumatriptan (Suma) pre-incubation (n=6) resulted in a significant (p < 0.01, Student's t test) left shift in the voltage dependence of K+ current activation compared to vehicle (V) control (n=7): the V0.5 of current activation was shifted from −11.5± 2.4mV to −27.3 ± 4.7mV. IM application produced no significant (p > 0.05, one way ANOVA with Holm-Sidak post-hoc) change in the voltage dependence of K+ current activation in the presence or absence of sumatriptan. C. IM resulted in a significant (p > 0.05, one-way ANOVA with Holm-Sidak post-hoc test) reduction maximal K+ conductance (normalized by membrane capacitance), compared to vehicle treated neurons. However, there was no significant influence of IM on the maximal K+ conductance when applied to neurons pre-incubated with sumatriptan. Inset: When analyzed as a change from baseline, the IM-induced decrease in K+ conductance observed in vehicle treated neurons (V) was significantly greater (p < 0.05, Student's t test) than that observed in neurons pre-incubated with sumatriptan (Suma).
Because the shift in IK activation should have attenuated the initial sumatriptan-induced sensitization of dural afferents, our current clamp results suggested that this shift takes time to develop. To begin to test this suggestion, we recorded IK in dural afferents before and after the application of sumatriptan. Results of this analysis confirmed that this shift takes time to develop as the change in the V0.05 of activation was −2.5 ± 1.8 mV after 5 minutes of incubation and −3.7 ± 3.7 mV after 10 minutes (n = 3).
DISCUSSION
The purpose of this study was to identify the ionic mechanism(s) underlying the actions of sumatriptan on dural afferents. Our results indicate that 1) acute sumatriptan application produces an increase in baseline dural afferent excitability that is blocked by the 5-HT1D receptor antagonist BRL 15572. No further increase in excitability was observed following subsequent application of IM, 2) 30 min sumatriptan pre-incubation has no detectable influence on dural afferent excitability but attenuates IM induced-sensitization in a 5-HT1D receptor antagonist-dependent manner, 3) while sumatriptan produced an expected inhibition of VGCC, pre-incubation with sumatriptan did not attenuate the IM-induced decrease in AP threshold or action potential overshoot (changes that appear to depend on an increase in TTX-R Na+ currents(7)) or IIM-Cl, and 4) Sumatriptan both increases K+ currents in dural afferents via a leftward shift in the voltage-dependence of activation, and attenuates, IM-induced suppression of total K+ current.
Sumatriptan Mediated Inhibition of VGCC
Our data demonstrate that sumatriptan concentration-dependently inhibits VGCC in dural afferents. One of the most dramatic mechanisms of G-protein coupled receptor mediated inhibition of VGCC involves a rapid displacement of the VGCC β subunit via the G-protein βγ subunit (19). A unique feature of this form of inhibition is that it can be overcome with a strong depolarizing pre-pulse (20). However, following a depolarizing pre-pulse to +80mV, Ca2+ currents did not recover from sumatriptan inhibition. Furthermore, a lack of depolarizing shift in the voltage dependence of activation with increasing concentrations of sumatriptan suggests that this inhibition is via an as of yet unidentified intracellular second messenger. These conclusions are consistent with previous results from a study of Xenopus larvae spinal neurons indicating that 5-HT1B/1D receptor agonists (L694 247) reduce high voltage activated N and P/Q type currents by a G-protein activated diffusible second messenger pathway (18). More relevantly, these data are also consistent with previous reports that zolmitriptan can block P/Q and possibly R type currents in dissociated TG neurons. This effect was pertussis toxin sensitive indicating the activation of Gi/Go class of G proteins (17). Such a mechanism was recently suggested to account for the sumatriptan-induced suppression of capsaicin-evoked currents in dural afferents (24).
Acute Sumatriptan Application Increases Dural Afferent Excitability
Acute application of sumatriptan produced an increase in dural afferent excitability. These data may explain the clinical observation that triptans transiently aggravate headache. Within 5–15 min of taking sumatriptan, approximately 50% of patients experience exacerbated pain that lasts for about 10 – 15 min (25) before the onset of pain relief. Our data are also consistent with previous observations that sumatriptan can drive a Ca2+ dependent discharge (26), an increase the firing rate of C and Aδ meningeal nociceptors, and increase their mechanical sensitivity (25).
Multiple mechanisms are likely involved in this sumatriptan-induced transient increase in dural afferent excitability. However, the only change in active or passive electrophysiological properties observed in this study following acute sumatriptan was a significant depolarization in the membrane potential from −71.3mV to −54.0mV. That the depolarization was not accompanied by a significant change in Rin suggests that there was no net change in the number of open channels, only a shift in the proportion of the various types of channels that were open.
Pre-Incubation with Sumatriptan has no Influence on Dural Afferent Excitability
There was no significant influence of pre-incubation with sumatriptan on baseline dural afferent excitability. This is somewhat surprising, in retrospect, given the dramatic leftward shift in the activation of IK. Multiple K+ currents are expressed in sensory neurons and are critically involved in regulating their excitability (27). K+ channels regulate the timing between APs and therefore impact AP frequency. Thus, the dramatic shift in the voltage dependence of activation of K+ currents should have resulted in an increase in rheobase and/or a decrease in the response to suprathreshold current injection. The failure to detect such changes in excitability suggests that the shift in the voltage-dependence of K+ current activation is compensated, at least in part, by excitatory changes that persist following acute application of summatriptan. One such mechanism would include a suppression of Ca2+ dependent K+ channels secondary to the sumatriptan-induced inhibition of VGCC. We have recently demonstrated that such a channel is present in a subpopulation of cutaneous neurons where it plays a significant role in the regulation of afferent excitability (28) and appears to be tightly coupled to the Ca2+ influx via VGCC (29). As we have also demonstrated that a Ca2+ dependent K+ channel is present in dural afferents and suppressed following IM application (7), sumatriptan-induced suppression of such a current could also account for the apparent block of the IM-induced suppression of total K+ current following sumatriptan pre-incubation. Such an explanation would suggest that the shift in the voltage-dependence of K+ current activation is associated with an increase in K+ channel density. That is, an increase in one K+ channel type associated with the shift in the voltage-dependence of activation would compensate for a decrease in Ca2+-dependent K+ channel activity resulting in the observed no net change in peak K+ conductance. K+ channel subunits present in sensory neurons that could undergo such dramatic shifts in the voltage-dependence of activation include Kv2.1 (30), which can undergo a ~26 mV hyperpolarizing shift in the G-V following Ca2+/calcineurin dependent dephosphorylation (31). Future studies will be needed to identify the K+ channel subunit(s) that underlie the actions of sumatriptan in dural afferents.
Sumatriptan Selectivity
The observation that both the sumatriptan-induced acute sensitization and the subsequent inhibition of IM-induced sensitization of dural afferents were blocked by BRL 15572 indicates that both processes are mediated by the 5-HT1D receptor. This is consistent with previous data suggesting that while both 5-HT1B and 5-HT1D receptors are present on trigeminal ganglion neurons (32, 33), the vasoconstrictive effects of triptans are due to the 5-HT1B receptors on the dural vasculature (34), while the selective therapeutic efficacy of triptans for migraine is due to 5-HT1D receptors in dural afferents (35). However, evidence that the 5-HT1D receptor is present on subpopulations of afferents throughout the body (9) and that triptans have analgesic efficacy in other preclinical pain models (12, 13), still begs the question as to the basis of the selective clinical profile of this class of drugs. Our recent observation that the higher density of the 5-HT1DR in nerve fibers preferentially involved in signaling migraine pain may partially explain the selectivity of these drugs (8). However, in light of the fact that a receptor for these drugs is present in other afferent populations, albeit at lower densities, we proposed that other mechanisms likely contribute to efficacy and selectivity. Given evidence that IM-induced activation of IIM-Cl appears to be a relatively unique mechanism underlying the sensitization of dural afferents, sumatriptan-induced inhibition of IIM-Cl would provide another mechanism to account for the therapeutic selectivity of this compound. The observations that sumatriptan neither blocked the activation of IIM-Cl, nor shifted the equilibrium potential for Cl− indicates that this channel cannot account for the therapeutic actions of triptans. However, the modulation of IK could account for the therapeutic selectivity of this compound if data from subsequent studies confirm that this modulation is only observed in dural afferents.
The complex actions of sumatriptan on dural afferents raise at least 3 questions. One question is how could a decrease in VGCC contribute to the antinociceptive efficacy of triptans at the same time triptans have increased excitability of dural afferents. VGCCs are largely responsible for the influx of Ca2+ necessary to enable transmitter release from pre-synaptic terminals. The suppression of VGCC on the central terminals of dural afferents should contribute to the antinociceptive efficacy of triptans and account for the normalization dural stimulation-induced activity in trigeminal dorsal horn neurons following IM-induced sensitization (36). While there is evidence that low threshold or T-type VGCC may contribute to afferent sensitization (37), the high threshold channels described in the present study that mediate transmitter release have a minimal direct contribution to action potential generation (38). However, Ca2+ influx through these channels may contribute to the activation and/or modulation of a number of channels including 2-pore K+ channels (39) and Ca2+ modulated K+ channels (28). Thus, as noted above, while the triptan-induced suppression of VGCC in dural afferents may occur in parallel with the increase in excitability, the two may be causally linked. A second question pertains to the differential time course of the sumatriptan-induced excitation and inhibition of IM-induced sensitization, particularly if both processes are mediated by the same receptor. While pharmacokinetics could explain the relatively slow onset of triptan-induced pain relief observed clinically, the present results suggest an alternative possibility: distinct cellular processes underlie excitatory and inhibitory actions of the drug where those underlying inhibition develop far more slowly than those underlying excitation. Additional work will be needed to tease apart the specific mechanisms underlying the actions of sumatriptan in dural afferents, but the literature is now full of examples of receptor mediated processes, in particular those like the 5-HT1D receptor that are coupled to G-proteins, that develop over very different time scales. For example, the membrane delimited form of G-protein-mediated suppression of VGCC can occur within tens of milliseconds in sensory neurons (40), while there is evidence that metabotropic glutamate receptor-mediated decrease in membrane ionotropic glutamate receptors develops over 10s of minutes (41). A third question is why triptans fail alleviate migraine pain once it is already established. Our results indicating the pre-incubation with sumatriptan blocked IM-induced sensitization are consistent with the evidence that triptans administered prior to the development of migraine pain can abort a migraine. However, the leftward shift in the activation of K+ currents should enable sumatriptan to reverse afferent sensitization, even after it is established. The observation that triptans fail to reverse IM-induced sensitization of dural afferents (36) suggests that the second messenger pathways activated by IM block the actions of sumatriptan, at least those underlying the modulation of K+ currents. Ongoing experiments are designed to identify the point(s) of convergence of the underlying second messenger pathways.
SUMMARY
We have described both excitatory and inhibitory effects of sumatriptan that follow a time course that may explain why some migraineurs experience increases in pain sensitivity before the onset of pain relief. Additional work is needed to identify the ionic mechanisms underlying the excitatory effects of sumatriptan, as the ability to block these effects may ultimately increase the efficacy of these compounds. We have ruled out two important targets for the therapeutic actions of sumatriptan, TTX-R INa and IIM-Cl. The implication of this observation is that there is a balance between the excitatory actions of inflammatory mediators and the inhibitory actions of triptans which appear to be acting on different targets. Relatively more excitation and/or less inhibition in a subpopulation of patients would result in a population unresponsive to triptans. Differences in the relative balance between excitation and inhibition may suggest an explanation for why triptans are only effective in ~70% of migraineurs (3) (although those pain free at 2 hrs may be considerably lower (42)). More importantly, in addition to voltage-gated Ca2+ channels previously identified by others, we have identified a novel target that may account of the therapeutic actions of triptans. Maximizing the hyperpolarizing shift in IK may provide a novel approach for the treatment of migraine.
Acknowledgements
We thank Dr. Bradley Alger, Norman Capra, Amy MacDermott and Danny Weinreich for helpful feedback on the preparation of this manuscript. Sumatriptan used in this study was a generous gift from Glaxo-Smith-Kline. Work was support by NIH grants: NS059153 (AMH) NS41384 (MSG) and DE018252 (MSG).
Footnotes
Conflict of Interest: The Authors declare that there is no conflict of interest with the material in this manuscript.
References
- [1].Lipton RB, Stewart WF, Scher AI. Epidemiology and economic impact of migraine. Curr Med Res Opin. 2001;17(Suppl 1):s4–12. doi: 10.1185/0300799039117005. [DOI] [PubMed] [Google Scholar]
- [2].Galletti F, Cupini LM, Corbelli I, Calabresi P, Sarchielli P. Pathophysiological basis of migraine prophylaxis. Prog Neurobiol. 2009;89:176–92. doi: 10.1016/j.pneurobio.2009.07.005. [DOI] [PubMed] [Google Scholar]
- [3].Tfelt-Hansen P, De Vries P, Saxena PR. Triptans in migraine: a comparative review of pharmacology, pharmacokinetics and efficacy. Drugs. 2000;60:1259–87. doi: 10.2165/00003495-200060060-00003. [DOI] [PubMed] [Google Scholar]
- [4].Sarchielli P, Alberti A, Codini M, Floridi A, Gallai V. Nitric oxide metabolites, prostaglandins and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. Cephalalgia. 2000;20:907–18. doi: 10.1046/j.1468-2982.2000.00146.x. [DOI] [PubMed] [Google Scholar]
- [5].Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–4. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- [6].Harriott AM, Gold MS. Electrophysiological Properties of Dural Afferents in the Absence and Presence of Inflammatory Mediators. J Neurophysiol. 2009;101:3126–34. doi: 10.1152/jn.91339.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vaughn AH, Gold MS. Ionic mechanisms underlying inflammatory mediator-induced sensitization of dural afferents. J Neurosci. 2010;30:7878–88. doi: 10.1523/JNEUROSCI.6053-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Harriott AM, Gold MS. Serotonin type 1D receptors (5HTR) are differentially distributed in nerve fibres innervating craniofacial tissues. Cephalalgia. 2008;28:933–44. doi: 10.1111/j.1468-2982.2008.01635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Potrebic S, Ahn AH, Skinner K, Fields HL, Basbaum AI. Peptidergic nociceptors of both trigeminal and dorsal root ganglia express serotonin 1D receptors: implications for the selective antimigraine action of triptans. J Neurosci. 2003;23:10988–97. doi: 10.1523/JNEUROSCI.23-34-10988.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dao TT, Lund JP, Remillard G, Lavigne GJ. Is myofascial pain of the temporal muscles relieved by oral sumatriptan? A cross-over pilot study. Pain. 1995;62:241–4. doi: 10.1016/0304-3959(95)00025-N. [DOI] [PubMed] [Google Scholar]
- [11].Pierce PA, Xie GX, Peroutka SJ, Levine JD. Dual effect of the serotonin agonist, sumatriptan, on peripheral neurogenic inflammation. Reg Anesth. 1996;21:219–25. [PubMed] [Google Scholar]
- [12].Nikai T, Basbaum AI, Ahn AH. Profound reduction of somatic and visceral pain in mice by intrathecal administration of the anti-migraine drug, sumatriptan. Pain. 2008 doi: 10.1016/j.pain.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vera-Portocarrero LP, Ossipov MH, King T, Porreca F. Reversal of inflammatory and noninflammatory visceral pain by central or peripheral actions of sumatriptan. Gastroenterology. 2008;135:1369–78. doi: 10.1053/j.gastro.2008.06.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ottani A, Ferraris E, Giuliani D, Mioni C, Bertolini A, Sternieri E, Ferrari A. Effect of sumatriptan in different models of pain in rats. Eur J Pharmacol. 2004;497:181–6. doi: 10.1016/j.ejphar.2004.06.053. [DOI] [PubMed] [Google Scholar]
- [15].Kayser V, Aubel B, Hamon M, Bourgoin S. The antimigraine 5-HT 1B/1D receptor agonists, sumatriptan, zolmitriptan and dihydroergotamine, attenuate pain-related behaviour in a rat model of trigeminal neuropathic pain. Br J Pharmacol. 2002;137:1287–97. doi: 10.1038/sj.bjp.0704979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cumberbatch MJ, Hill RG, Hargreaves RJ. Differential effects of the 5HT1B/1D receptor agonist naratriptan on trigeminal versus spinal nociceptive responses. Cephalalgia. 1998;18:659–63. doi: 10.1046/j.1468-2982.1998.1810659.x. [DOI] [PubMed] [Google Scholar]
- [17].Morikawa T, Matsuzawa Y, Makita K, Katayama Y. Antimigraine drug, zolmitriptan, inhibits high-voltage activated calcium currents in a population of acutely dissociated rat trigeminal sensory neurons. Mol Pain. 2006;2:10. doi: 10.1186/1744-8069-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sun QQ, Dale N. G-proteins are involved in 5-HT receptor-mediated modulation of N-and P/Q- but not T-type Ca2+ channels. J Neurosci. 1999;19:890–9. doi: 10.1523/JNEUROSCI.19-03-00890.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature. 1996;380:255–8. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- [20].Jiang M, Gold MS, Boulay G, Spicher K, Peyton M, Brabet P, et al. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc Natl Acad Sci U S A. 1998;95:3269–74. doi: 10.1073/pnas.95.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fox AW. Onset of effect of 5-HT1B/1D agonists: a model with pharmacokinetic validation. Headache. 2004;44:142–7. doi: 10.1111/j.1526-4610.2004.04030.x. [DOI] [PubMed] [Google Scholar]
- [22].Pilgrim AJ. The clinical profile of sumatriptan: efficacy in migraine. Eur Neurol. 1994;34(Suppl 2):26–34. doi: 10.1159/000119529. [DOI] [PubMed] [Google Scholar]
- [23].Akaike N. Gramicidin perforated patch recording and intracellular chloride activity in excitable cells. Prog Biophys Mol Biol. 1996;65:251–64. doi: 10.1016/s0079-6107(96)00013-2. [DOI] [PubMed] [Google Scholar]
- [24].Evans MS, Cheng X, Jeffry JA, Disney KE, Premkumar LS. Sumatriptan Inhibits TRPV1 Channels in Trigeminal Neurons. Headache. 2012 doi: 10.1111/j.1526-4610.2011.02053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Burstein R, Jakubowski M, Levy D. Anti-migraine action of triptans is preceded by transient aggravation of headache caused by activation of meningeal nociceptors. Pain. 2005;115:21–8. doi: 10.1016/j.pain.2005.01.027. [DOI] [PubMed] [Google Scholar]
- [26].Strassman AM, Levy D. The anti-migraine agent sumatriptan induces a calcium-dependent discharge in meningeal sensory neurons. Neuroreport. 2004;15:1409–12. doi: 10.1097/01.wnr.0000132771.64590.42. [DOI] [PubMed] [Google Scholar]
- [27].Gold MS, Shuster MJ, Levine JD. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J Neurophysiol. 1996;75:2629–46. doi: 10.1152/jn.1996.75.6.2629. [DOI] [PubMed] [Google Scholar]
- [28].Zhang XL, Mok LP, Katz EJ, Gold MS. BK(Ca) currents are enriched in a subpopulation of adult rat cutaneous nociceptive dorsal root ganglion neurons. Eur J Neurosci. 2010;31:450–62. doi: 10.1111/j.1460-9568.2009.07060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang X-L, Mok L, Charbonnet M, Lee K-Y, Gold MS. Inflammation-induced changes in BKCa currents in cutaneous dorsal root ganglion neurons from the adult rat. Mol Pain. doi: 10.1186/1744-8069-8-37. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bocksteins E, Raes AL, Van de Vijver G, Bruyns T, Van Bogaert PP, Snyders DJ. Kv2.1 and silent Kv subunits underlie the delayed rectifier K+ current in cultured small mouse DRG neurons. Am J Physiol Cell Physiol. 2009;296:C1271–8. doi: 10.1152/ajpcell.00088.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Park KS, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–9. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- [32].Nicholson R, Small J, Dixon AK, Spanswick D, Lee K. Serotonin receptor mRNA expression in rat dorsal root ganglion neurons. Neurosci Lett. 2003;337:119–22. doi: 10.1016/s0304-3940(02)01256-9. [DOI] [PubMed] [Google Scholar]
- [33].Hou M, Kanje M, Longmore J, Tajti J, Uddman R, Edvinsson L. 5-HT(1B) and 5-HT(1D) receptors in the human trigeminal ganglion: co-localization with calcitonin gene-related peptide, substance P and nitric oxide synthase. Brain Res. 2001;909:112–20. doi: 10.1016/s0006-8993(01)02645-2. [DOI] [PubMed] [Google Scholar]
- [34].Razzaque Z, Pickard JD, Ma QP, Shaw D, Morrison K, Wang T, Longmore J. 5-HT1B-receptors and vascular reactivity in human isolated blood vessels: assessment of the potential craniovascular selectivity of sumatriptan. Br J Clin Pharmacol. 2002;53:266–74. doi: 10.1046/j.0306-5251.2001.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Donaldson C, Boers PM, Hoskin KL, Zagami AS, Lambert GA. The role of 5-HT1B and 5-HT1D receptors in the selective inhibitory effect of naratriptan on trigeminovascular neurons. Neuropharmacology. 2002;42:374–85. doi: 10.1016/s0028-3908(01)00190-3. [DOI] [PubMed] [Google Scholar]
- [36].Levy D, Jakubowski M, Burstein R. Disruption of communication between peripheral and central trigeminovascular neurons mediates the antimigraine action of 5HT 1B/1D receptor agonists. Proc Natl Acad Sci U S A. 2004;101:4274–9. doi: 10.1073/pnas.0306147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zamponi GW, Lewis RJ, Todorovic SM, Arneric SP, Snutch TP. Role of voltage-gated calcium channels in ascending pain pathways. Brain Res Rev. 2009;60:84–9. doi: 10.1016/j.brainresrev.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22:10277–90. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Czirjak G, Enyedi P. TRESK background K(+) channel is inhibited by phosphorylation via two distinct pathways. J Biol Chem. 2010;285:14549–57. doi: 10.1074/jbc.M110.102020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wilding TJ, Womack MD, McCleskey EW. Fast, local signal transduction between the mu opioid receptor and Ca2+ channels. J Neurosci. 1995;15:4124–32. doi: 10.1523/JNEUROSCI.15-05-04124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sanderson TM, Collingridge GL, Fitzjohn SM. Differential trafficking of AMPA receptors following activation of NMDA receptors and mGluRs. Mol Brain. 2011;4:30. doi: 10.1186/1756-6606-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Goadsby PJ, Sprenger T. Current practice and future directions in the prevention and acute management of migraine. Lancet Neurol. 2010;9:285–98. doi: 10.1016/S1474-4422(10)70005-3. [DOI] [PubMed] [Google Scholar]