

Certain transmembrane ERAD substrates are segregated into specialized ER subdomains, termed ER-associated compartments (ERACs), before degradation. COPII components and Hsp40s act in the same pathway to sequester ERAD substrates into ERACs. The findings point to an as-yet-undefined role of COPII proteins in the formation of ERACs.
Abstract
Proteins that fail to fold in the endoplasmic reticulum (ER) are subjected to ER-associated degradation (ERAD). Certain transmembrane ERAD substrates are segregated into specialized ER subdomains, termed ER-associated compartments (ERACs), before targeting to ubiquitin–proteasome degradation. The traffic-independent function of several proteins involved in COPII-mediated ER-to-Golgi transport have been implicated in the segregation of exogenously expressed human cystic fibrosis transmembrane conductance regulator (CFTR) into ERACs in Saccharomyces cerevisiae. Here we focus on the properties of COPII components in the sequestration of enhanced green fluorescent protein (EGFP)–CFTR into ERACs. It has been demonstrated that the temperature-sensitive growth defects in many COPII mutants can be suppressed by overexpressing other genes involved in COPII vesicle formation. However, we show that these suppression abilities are not always correlated with the ability to rescue the ERAC formation defect, suggesting that COPII-mediated EGFP-CFTR entry into ERACs is independent of its ER-to-Golgi trafficking function. In addition to COPII machinery, we find that ER-associated Hsp40s are also involved in the sequestration process by directly interacting with EGFP-CFTR. COPII components and ER-associated Hsp40, Hlj1p, act in the same pathway to sequester EGFP-CFTR into ERACs. Our findings point to an as-yet-undefined role of COPII proteins in the formation of ERACs.
INTRODUCTION
The endoplasmic reticulum (ER) is responsible for the synthesis of secretory, transmembrane, and ER-resident proteins. Newly synthesized proteins are translocated into the ER lumen or ER membrane through the translocation channel, where they undergo folding, assembly, and posttranslational modifications with the aid of a variety of ER chaperones. During the folding process, a cellular mechanism referred to as “ER quality control” continually monitors the protein folding status, and only correctly folded and assembled proteins are segregated from the ER-resident proteins and transported to the Golgi apparatus via membrane-bound vesicles or carrier intermediates for further processing and secretion. At the same time, the ER quality control system also plays important roles in preventing the accumulation of unfolded proteins in the ER. Misfolded or incompletely assembled proteins are retained within the ER to either complete the folding process or be targeted for retrotranslocation into the cytosol and subjected to ER-associated protein degradation (ERAD) by the ubiquitin–proteasome system (Nakatsukasa and Brodsky, 2008; Vembar and Brodsky, 2008; Brodsky and Skach, 2011).
Studies using both yeast and mammalian cells demonstrated that certain transmembrane ERAD substrates are physically segregated into specialized ER subdomains before degradation (Valetti et al., 1991; Kamhi-Nesher et al., 2001; Kiser et al., 2001; Zhang et al., 2001). The human cystic fibrosis transmembrane conductance regulator (CFTR), which normally resides at the plasma membrane, serves as an integral membrane ERAD substrate when expressed in the yeast Saccharomyces cerevisiae (Kiser et al., 2001; Zhang et al., 2001). Human CFTR expressed in yeast was shown to be sequestered into specialized ER subdomains called ER-associated compartments (ERACs), and such sequestration seemed to be a prerequisite for ER retrotranslocation and the subsequent proteasomal degradation (Fu and Sztul, 2003; Huyer et al., 2004). However, the underlying mechanisms that drive the sequestration of transmembrane ERAD substrates into ERACs are unknown.
On the other hand, successfully folded and assembled proteins are exported from the ER in COPII-coated vesicular carriers (Sato and Nakano, 2007; Dancourt and Barlowe, 2010; Schmidt and Stephens, 2010; Gillon et al., 2012). COPII vesicle–mediated protein export is believed to occur at specialized regions of the ER termed ER exit sites (ERES; Orci et al., 1991; Bannykh et al., 1996). In these distinct zones of the ER, a set of cytoplasmic proteins, collectively known as the COPII coat, generates COPII vesicles through a sequence of events under the control of multiple regulatory mechanisms (Aridor and Balch, 2000; Lee and Linstedt, 2000; Blumental-Perry et al., 2006; Yamasaki et al., 2006; Higashio et al., 2008; Rismanchi et al., 2009; Kodera et al., 2011; Yorimitsu and Sato, 2012; Yoshibori et al., 2012). The COPII coat is responsible for the direct or indirect capture of cargo proteins and for the physical deformation of the ER membrane that drives the COPII vesicle formation. In brief, the assembly of the COPII coat is triggered by GDP–GTP exchange on the small GTPase Sar1p, which is catalyzed by the ER-resident Sec12p guanine nucleotide exchange factor (Nakano and Muramatsu, 1989; Barlowe and Schekman, 1993). The activated Sar1p-GTP in turn binds to the ER membranes and recruits the Sec23/24p heterodimer by binding to the Sec23p portion, and the Sec24p subunit captures the cargo protein to form a prebudding complex (Barlowe et al., 1994; Kuehn et al., 1998; Miller et al., 2003; Mossessova et al., 2003). The membrane association of Sec23/24p is stabilized through the interactions with transmembrane cargo proteins and repeated cycles of Sec12p-dependent GTP loading of Sar1p, which facilitate proper and efficient cargo sorting into COPII vesicles (Sato and Nakano, 2005; Tabata et al., 2009). Subsequently the prebudding complex recruits the Sec13/31p heterotetramer onto Sec23/24p, which polymerizes and drives membrane deformation and vesicle budding.
Although much of the attention to COPII function has focused on events associated with vesicular trafficking, an earlier study showed that the sequestration of enhanced green fluorescent protein (EGFP)–CFTR into ERACs in S. cerevisiae requires the COPII components (Fu and Sztul, 2003). In the absence of COPII function, EGFP-CFTR remains distributed throughout the ER rather than being segregated into ERACs, indicating the possibility that COPII machinery participates in the sequestration of transmembrane ERAD substrates to the ERACs by a noncanonical function. However, the exact mode of action of individual COPII components during these processes is very unclear. Here we investigate in detail the contribution of COPII components to the sequestration of EGFP-CFTR into ERACs in yeast cells. In addition to COPII components, we find that the ER-associated Hsp40 cochaperones are also involved in the sequestration process. Our results show that COPII components, along with Hsp40s, participate in the targeting of EGFP-CFTR to ERACs, independent of their role in ER-to-Golgi transport.
RESULTS
EGFP-CFTR–induced ERAC formation in COPII mutant strains
Fu and Sztul (2003) observed that the sequestration of the membrane ERAD substrate EGFP-CFTR into degradative ERACs in S. cerevisiae was impaired by temperature-sensitive mutations in key COPII components, including SEC12, SEC13, and SEC23, but not by a mutation in SEC18 (mutant in transport vesicle fusion). These findings led to the idea that the segregation of EGFP-CFTR into ERACs required COPII function, but the function was independent of ER-to-Golgi trafficking. To further examine the potential involvement of the COPII machinery in the sequestration of EGFP-CFTR in the ER, we quantitatively analyzed the localization of EGFP-CFTR expressed under the control of a copper-inducible promoter in various temperature-sensitive mutants defective in COPII-mediated transport to the Golgi. We examined the mutant alleles sar1D32G, sec12-4, sec23-1, sec13-1, sec16-2, sec17-1, and sec18-1, all of which block ER-to-Golgi transport at restrictive conditions (37ºC). As shown in Figure 1A, 2 h of EGFP-CFTR induction in yeast cells yielded three different localization patterns: the ERAC pattern, the ER pattern, or both the ERAC and ER patterns. We found that >95% of the wild-type cells expressing EGFP-CFTR and >80% of the mutant cells expressing EGFP-CFTR displayed the ERAC pattern at the permissive temperature (23ºC; Figure 1B, top). In contrast, when EGFP-CFTR was expressed at the restrictive temperature in the sar1D32G, sec12-4, sec23-1, sec13-1, and sec16-2 mutant strains, all of which were defective in COPII vesicle formation near the point of exit from the ER, the percentage of cells with the ERAC pattern was reduced, and the percentage with the ER pattern increased concomitantly (Figure 1B, bottom). The ability to form ERACs at the restrictive temperature varied from 17.4 ± 7.67% (for sec12-4) to 49.1 ± 15.8% (for sar1D32G), depending on the mutant used. The ERAC or ERAC and ER pattern of EGFP-CFTR localization was still observed in >70% of the vesicle targeting and fusion mutants sec17-1 and sec18-1 at the restrictive temperature, indicating that a block in the late step of ER-to-Golgi transport does not significantly influence EGFP-CFTR sequestration. These results confirm and extend earlier observations of the participation of COPII components in EGFP-CFTR–induced ERAC formation that is independent of their vesicular trafficking function.
FIGURE 1:
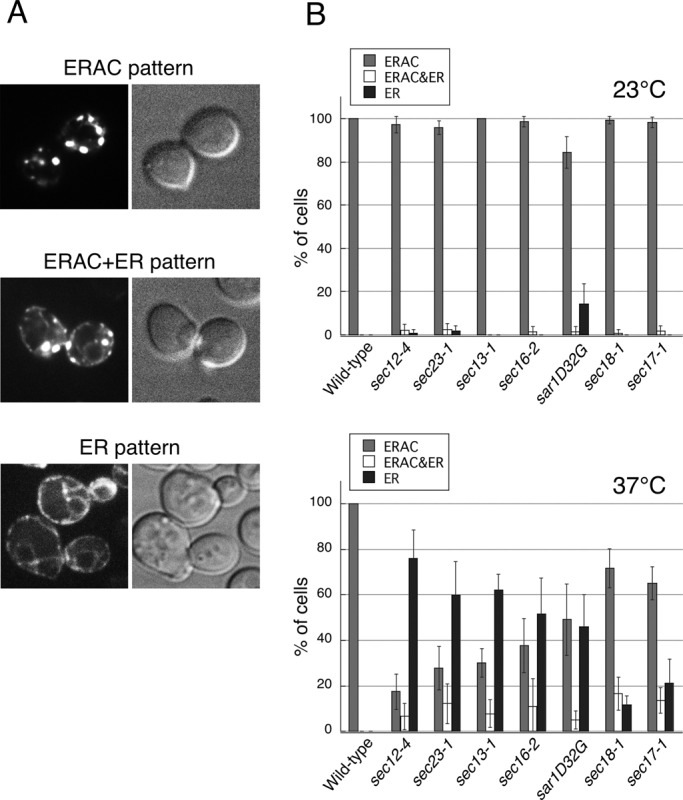
The formation of EGFP-CFTR–induced ERACs requires COPII function. (A) Wild-type (ANY21) cells containing the EGFP-CFTR expression plasmid were grown to mid–log phase at 23ºC and incubated with copper-containing medium for 2 h to induce expression, followed by observation with confocal microscopy. (B) Wild-type and sec12-4, sec23-1, sec13-1, sec16-2, sar1D32G, sec18-1, and sec17-1 strains were grown to mid–log phase and induced with copper at 23ºC (top) or 37ºC (bottom) for 2 h to induce EGFP-CFTR expression. ERAC formation is quantified using the three categories that were defined in A, and the data are presented as the percentage of cells expressing EGFP-CFTR. The experiments were repeated at least three times, and >100 cells were analyzed for each condition.
Genetic analysis of ERAC formation
To ensure that the foregoing results with the sec mutants were not a consequence of indirect effects of traffic inactivation, we next used genetic analysis to provide additional evidence for the role of COPII components in the formation of ERACs. The temperature-sensitive growth phenotype of certain ER budding mutants can be suppressed by overexpressing other genes involved in the ER-to-Golgi transport (Nakano and Muramatsu, 1989; d'Enfert et al., 1991; Oka and Nakano, 1994; Gimeno et al., 1995; Saito et al., 1999; Kurihara et al., 2000; Buchanan et al., 2010). To study whether the sequestration of EGFP-CFTR into ERACs and COPII-mediated vesicle formation function in parallel or as part of an integrated system, we tested whether the ERAC formation defect of the COPII vesicle budding mutants also could be suppressed by overexpressing other genes that are required for COPII vesicle formation. A multicopy plasmid containing the gene required for the biogenesis of COPII vesicles or an empty vector control was introduced into several temperature-sensitive mutants that were defective at the stage of ER vesicle budding, and the suppression activity was monitored by growth at 37ºC (Figure 2A). Among them, the high-copy expression of SAR1 suppressed the temperature-sensitive growth defect of the sec12-4 and sec16-2 mutants, which was also reported previously (Nakano and Muramatsu, 1989; d'Enfert et al., 1991). As expected, the ERAC formation defect of the sec12-4 mutant was almost fully corrected by SAR1 overexpression (Figure 3B). In contrast, the high gene dosage of SAR1 could not rescue the failure of EGFP-CFTR to be sequestered into ERACs in sec16-2 cells (Figure 3C). As controls, we verified that adding back each gene (SEC23, SEC12, or SEC16) on a high-copy plasmid reversed both the temperature sensitivity and the ERAC formation defect (Figures 2 and 3). Furthermore, we found that although overexpression of SEC16 did not suppress the temperature-sensitive growth defect of the sec23-1 mutant (Figure 2A), it significantly rescued the ERAC formation defect at the restrictive temperature (Figure 3A). A similar effect was observed with SEC13, although the extent of the suppression was less pronounced. To confirm that these suppressions were not due to the incomplete block of the ER-to-Golgi transport, we followed the transport of the vacuolar protease carboxypeptidase Y (CPY) as a marker for early events in the secretory pathway under the same conditions as described. As shown in Figure 2B, the ER form of CPY (p1) accumulated within the sec23-1 cells overexpressing SEC16, confirming that the foregoing conditions indeed caused a severe block in the ER-to-Golgi transport of secretory proteins. The mutant lacking ERV29, which encodes a sorting receptor for CPY export from the ER (Belden and Barlowe, 2001), was used as a control for transport inhibition. These results clearly indicate that the suppression of the temperature-sensitive growth defects of the COPII mutants is not necessarily correlated with the ability to rescue the ERAC formation defect.
FIGURE 2:
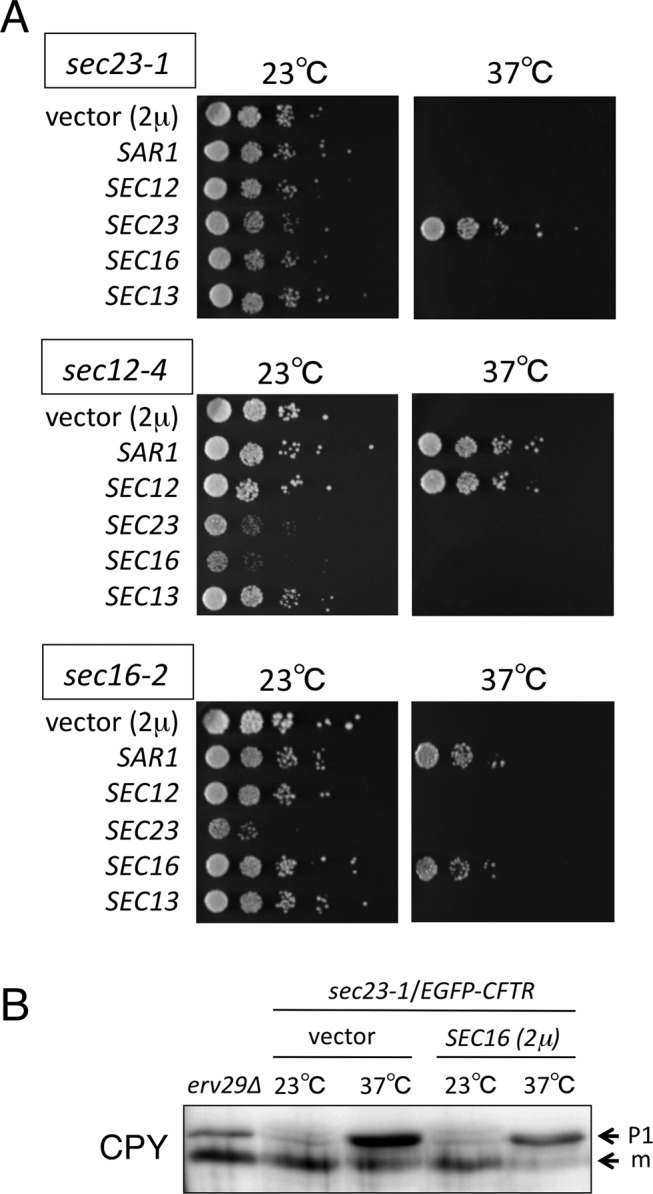
The effect of overexpressing individual COPII components on the growth of temperature-sensitive mutants defective in COPII vesicle formation. (A) sec23-1 (MBY8-20C), sec12-4 (MBY10-7A), and sec16-2 (MBY4-1A) cells harboring pEGFP-CFTR were transformed with pYO324 (2 μ vector), pSAR1 (2 μ SAR1), pSEC12 (2 μ SEC12), pSEC23 (2 μ SEC23), pSEC16 (2 μ SEC16), or pSEC13 (2 μ SEC13), and the transformants were grown to saturation at 23ºC in MCD medium lacking uracil and tryptophan (MCD-Ura-Trp). The cells were diluted to a final concentration of 0.3 at OD600, and 5 μl of 10-fold dilutions was spotted onto MCD-Ura-Trp plates. The plates were incubated at 23 or 37ºC as indicated. (B) sec23-1 (MBY8-20C) cells transformed with pYO324 (2 μ vector) or pSEC16(2 μ SEC16) were grown at 23 or 37ºC in selective media. Equal amounts of whole-cell extracts were subjected to immunoblotting with anti-CPY antibody. The erv29∆ strain was used as a control. The ER (p1) and mature (m) forms of CPY are indicated.
FIGURE 3:

The effect of overexpressing individual COPII components on ERAC formation in temperature-sensitive mutants defective in COPII vesicle formation. (A–C) The transformants used in Figure 2 were grown to mid–log phase and treated with copper at 23ºC (left) or 37ºC (right) for 2 h to induce EGFP-CFTR expression. ERAC formation was quantified as described in Figure 1B. The experiments were repeated at least three times, and >70 cells were analyzed for each condition.
Localization of COPII components relative to ERAC
We next examined the cellular localization of COPII components relative to EGFP-CFTR–induced ERACs. Previous studies reported that CFTR-induced ERACs were subdomains of ER and that they consisted of a tubular network of ER membranes (Fu and Sztul, 2003; Huyer et al., 2004). We first examined the localization of Sar1p (which normally resides throughout the entire ER) relative to the EGFP-CFTR–enriched ERACs. When coexpressed with EGFP-CFTR, we found that Sar1p-mCherry colocalized well with EGFP-CFTR ERACs in addition to its well-established ER localization (Figure 4A). However, it is not certain whether Sar1p is specifically targeted to ERACs or arrives there simply because of its association with the ER. Therefore we examined whether the inactive GDP form of mutant Sar1p (Sar1p-D32G) (Saito et al., 1998) was also present in ERACs. Similar to the wild-type Sar1p, Sar1p-D32G-mCherry was also present both in ERACs and in the ER at the restrictive temperature (Figure 4B), suggesting that Sar1p localization to ERACs was not directly regulated in response to Sar1p activation. A common feature of most ERAC-like structures is the colocalization of Kar2p/BiP (Valetti et al., 1991; Kiser et al., 2001; Zhang et al., 2001; Fu and Sztul, 2003; Sullivan et al., 2003; Huyer et al., 2004). However, we found that another ER-resident protein, a subunit of the ER translocon, Sec71p-mCherry, which had nonchaperone functions, was also clearly present in ERACs (Figure 4C). These results suggest that components of the ER are also localized within EGFP-CFTR ERACs.
FIGURE 4:
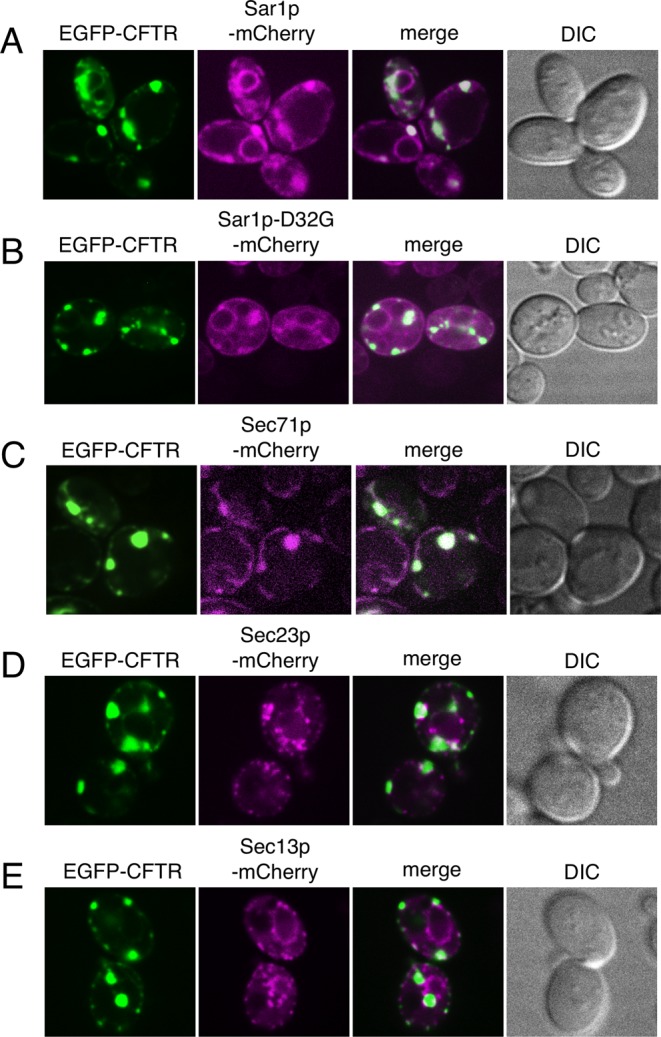
The relationship between the components of the endoplasmic reticulum and ERAC structures. Log-phase cultures of wild-type cells (ANY21) coexpressing EGFP-CFTR (induced as described in Figure 1A) and Sar1p-mCherry (A), Sar1p-D32G-mCherry (B), Sec71p-mCherry (C), Sec23p-mCherry (D), or Sec13p-mCherry (E) were visualized by confocal microscopy at 23ºC, except B, which was observed at 37ºC.
ERES are also subdomains of the ER that are detected as small punctate structures scattered throughout the ER and colocalize with COPII coat subunits Sec23/24p and Sec13/31p (Connerly et al., 2005; Castillon et al., 2009; Shindiapina and Barlowe, 2010; Yorimitsu and Sato, 2012). We used mCherry-fused Sec23p and Sec13p to explore the relationship between ERES and EGFP-CFTR–induced ERACs. In cells expressing EGFP-CFTR, several ERES-like punctate structures were often observed within large foci of the EGFP-CFTR–containing ERACs in addition to the typical ERES punctate pattern distributed throughout the ER (Figure 4, D and E). Therefore ERES appear morphologically distinct from ERACs, and punctate structures visualized by Sec23p-mCherry and Sec13p-mCherry within ERACs most likely represent ERES that have been incidentally engulfed into ERACs. Taken together, these findings led to the conclusion that there was no specific accumulation of COPII components in the already formed ERACs.
A diacidic ER exit code of CFTR is dispensable for its entry into ERACs
An earlier study showed that the COPII machinery was required for CFTR to be exported out of the ER in mammalian cells (Yoo et al., 2002). A diacidic ER exit code was identified in the CFTR molecule that directly interacted with the COPII coat subunit Sec23/24. It was demonstrated that mutation of the diacidic motif DAD (residues 565–567) to DAA disrupted Sec23/24 binding (Wang et al., 2004). Because the human homologue of the yeast COPII component, Sec23A, complements the temperature-sensitive growth of the yeast sec23-1 mutant at the restrictive temperature (37ºC; Paccaud et al., 1996), it is possible that the yeast COPII coat can bind to the diacidic sequence of EGFP-CFTR expressed in yeast cells and may have some role in coordinating the sequestration process. Therefore we examined the ability of the diacidic EGFP-CFTR-D567A mutant to be sequestered into ERACs, but this did not significantly affect its entry into the ERAC (Figure 5). Thus the diacidic ER export motif of CFTR, although necessary for ER export in mammalian cells, does not participate in the ERAC formation process in yeast.
FIGURE 5:
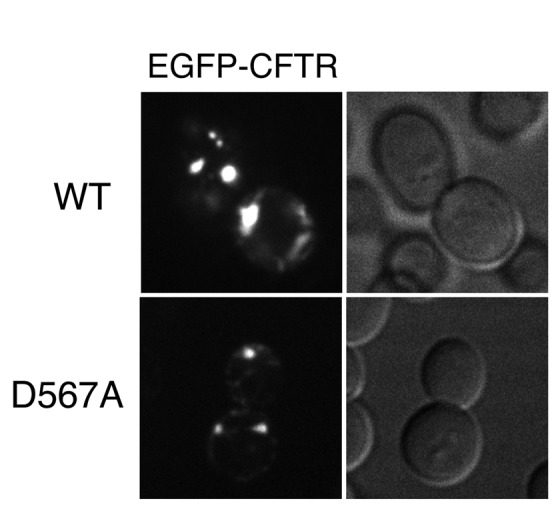
The diacidic ER export motif of CFTR is dispensable for its entry into ERACs. Wild-type (ANY21) cells containing the EGFP-CFTR plasmid (top) or EGFP-CFTR-D567A plasmid (bottom) were grown to mid–log phase at 23ºC and incubated with copper-containing medium for 2 h to induce expression. The cells were then visualized by confocal microscopy.
Involvement of Hsp40s in ERAC formation
Although much less is known about how misfolded membrane proteins are sorted to the ERAD pathway, it has been reported that the cytosolic Hsp70–Hsp40 chaperone system affects the degradation of several integral membrane ERAD substrates (Strickland et al., 1997; Meacham et al., 1999; Hill and Cooper, 2000; Zhang et al., 2001; Youker et al., 2004; Ahner et al., 2007), although precisely how these chaperones facilitate the turnover of integral membrane ERAD substrates is not completely understood. CFTR degradation was slowed in yeast mutated for either SSA1-4 (cytosolic Hsp70s) or YDJ1/HLJ1 (ER-associated Hsp40; Zhang et al., 2001; Youker et al., 2004). S. cerevisiae has four Ssa Hsp70 proteins (Ssa1–4p), and a temperature-sensitive ssa1-45 mutant strain lacking functional SSA2/3/4 was shown to retain the ability to form CFTR-induced ERACs at the restrictive temperature (Zhang et al., 2001). To test whether Hsp40 cochaperones Ydj1p and Hlj1p are involved in the sequestration of EGFP-CFTR into ERACs, we assessed ERAC formation in ydj1∆ or hlj1∆ mutants and in an isogenic wild-type strain (Figure 6). We found that ydj1∆ and hlj1∆ mutant strains were moderately defective in ERAC formation relative to the wild-type strain; nearly 60% of the mutant cells displayed the ER pattern of localization. Because Ydj1p and Hlj1p have been demonstrated to function redundantly to facilitate the degradation of CFTR in conjunction with Hsp70 Ssa proteins (Youker et al., 2004), it is possible that the loss of one protein may not completely impair ERAC formation. In contrast to ydj1∆ and hlj1∆ mutants, the cells with a deletion of the other cytosolic Hsp40 chaperone, APJ1 (Kryndushkin et al., 2002), showed no effect on EGFP-CFTR ERAC formation under the same conditions (Figure 6B), implying a direct and specific effect of Ydj1p/Hlj1p. We also examined the stability of EGFP-CFTR in ydj1∆ or hlj1∆ mutants by cycloheximide chase analysis. We observed that deletion of either HLJ1 or YDJ1 was sufficient to slow degradation of EGFP-CFTR relative to the isogenic wild-type strain (Figure 6C). Taken together, these results demonstrate that ER-associated Hsp40 cochaperones Ydj1p and Hlj1p participate in the sequestration of EGFP-CFTR into ERACs.
FIGURE 6:
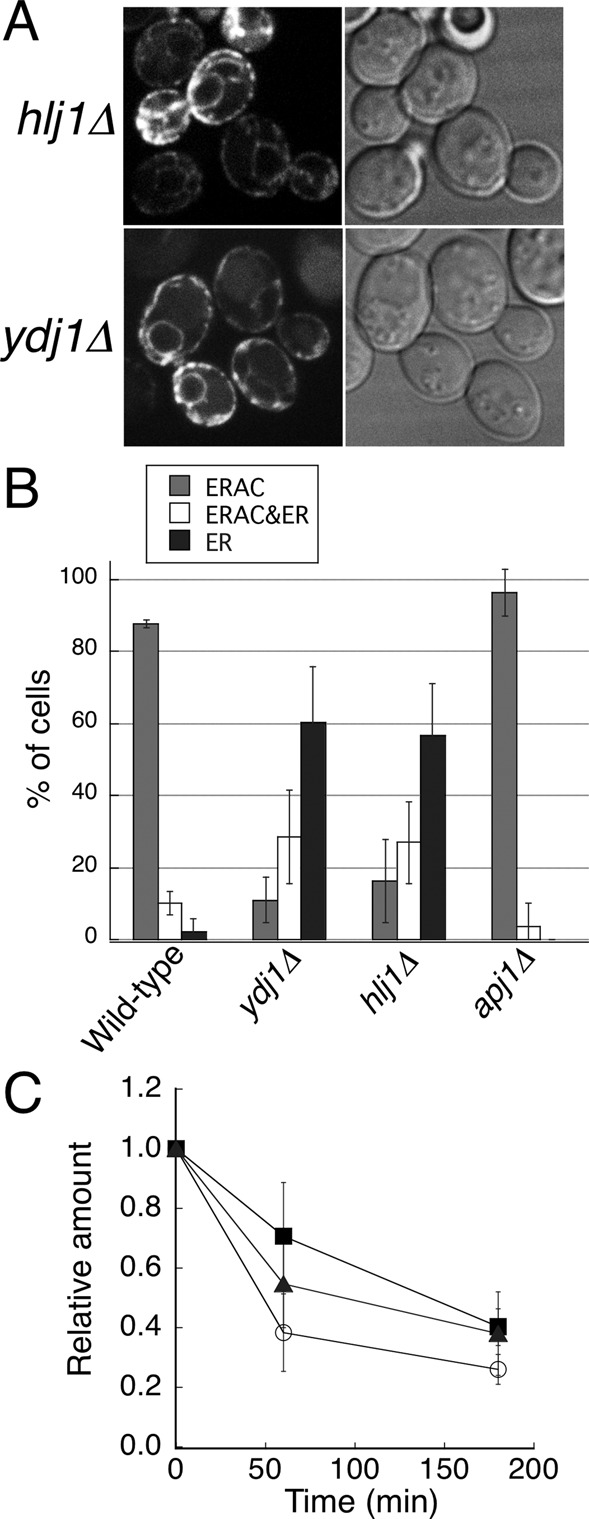
The formation of EGFP-CFTR–induced ERACs is ER associated and Hsp40 dependent. (A) ydj1∆ and hlj1∆ cells containing the EGFP-CFTR plasmid were grown to mid–log phase at 23ºC and incubated with copper-containing medium for 2 h to induce EGFP-CFTR expression, followed by observation with confocal microscopy. (B) Wild-type, hlj1∆, ydj1∆, and apj1∆ cells harboring pEGFP-CFTR were grown to mid–log phase and induced with copper at 23ºC for 2 h to induce EGFP-CFTR expression. ERAC formation was quantified as described in Figure 1B. The experiments were repeated at least three times, and >50 cells were analyzed for each condition. (C) Wild-type, hlj1∆, and ydj1∆ cells expressing EGFP-CFTR were subjected to cycloheximide chase analysis as described in Materials and Methods, and the amount of EGFP-CFTR at time point 0 was set to 1.0. The relative amounts of EGFP-CFTR remaining in wild-type (open circles), hlj1∆ (closed squares), and ydj1∆ (closed triangles) strains over time were plotted.
In general, Hsp40 proteins can bind to abnormally folded polypeptides and deliver them to specific Hsp70 partners to prevent aggregation and facilitate refolding (Craig et al., 2006; Buck et al., 2007). In addition, in some cases, Hsp40 protein itself can act as a chaperone, avoiding protein aggregation by forming stable complexes with folded and unfolded polypeptides (Szabo et al., 1994; Rudiger et al., 2001). Therefore, to determine whether a direct interaction could be observed between EGFP-CFTR and the Hsp40 chaperone in the ER, we constructed cells expressing both glutathione S-transferase–tagged Hlj1p (GST-Hlj1p) and EGFP-CFTR. GST-Hlj1p complemented the EGFP-CFTR–induced ERAC formation defect of the hlj1∆ strain (unpublished data). ER-enriched microsomes from this strain were solubilized with a detergent, and then a portion of the GST-Hlj1p was pulled down by glutathione beads. The amounts of adsorbed proteins were determined by immunoblotting. The results in Figure 7 show that GST-Hlj1p pulled down a significant amount of EGFP-CFTR but not the ER-resident proteins Sec61p and Sec12p, indicating a specific interaction between Hlj1p and EGFP-CFTR in the ER. We further examined whether COPII components were also recovered in this complex, but no appreciable amount of Sar1p or Sec23p was detected, implying that Hlj1p and COPII components do not function in the same stable complex. We also characterized the effects of EGFP-CFTR expression on mCherry-Hlj1p distribution in the ER (Figure 8A). GFP-Hlj1p has been reported to localize throughout the ER at a steady state (Youker et al., 2004). With EGFP-CFTR expression, mCherry-Hlj1p, in addition to its typical ER localization, also localized to EGFP-CFTR–induced ERACs, although the particular action of Hlj1p after entry into the ERAC is not yet defined. Taken together, our data support the notion that Hlj1p plays a role in the sequestration of EGFP-CFTR into ERACs by directly binding to EGFP-CFTR in the ER.
FIGURE 7:
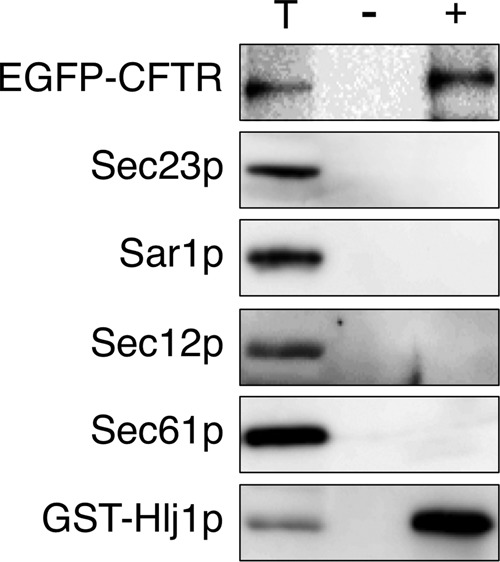
EGFP-CFTR forms a complex with Hlj1p in the ER. Microsomes (100 μg) expressing EGFP-CFTR (induced for 2 h) with (+) or without (–) GST-Hlj1p were solubilized with 1% Triton X-100. The resulting extract was cleared by centrifugation, and the supernatant was incubated with glutathione–Sepharose beads. The beads were washed, and proteins were released in the presence of 10 mM glutathione. The released proteins were analyzed by SDS–PAGE, followed by immunoblotting with antibodies to GFP, Sec23p, Sar1p, Sec12p, Sec61p, and GST. The total lane (T) represents 10% of the starting microsomal extract used in the lane labeled (+).
FIGURE 8:
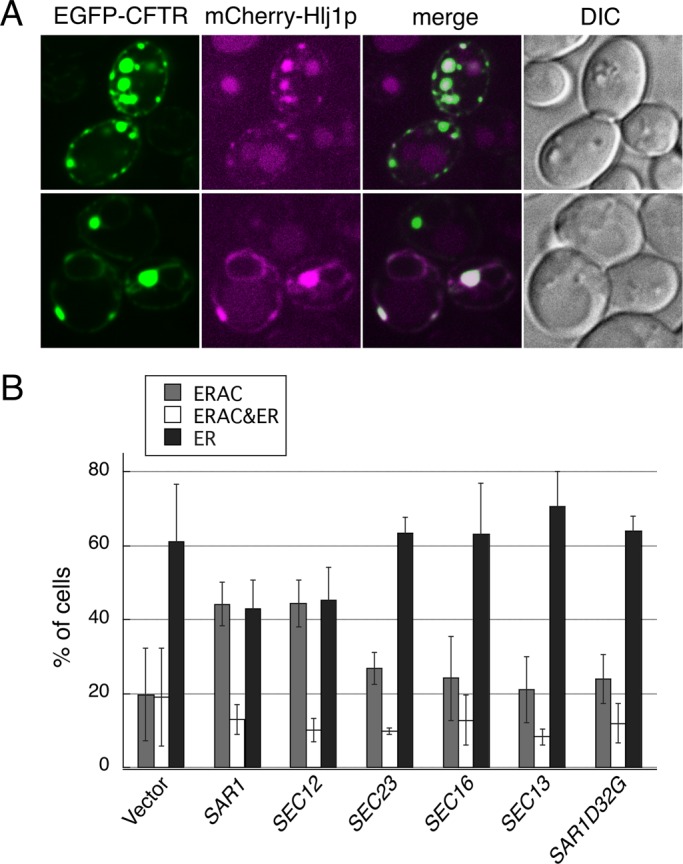
Hsp40s and COPII components act in the same pathway to drive ERAC formation. (A) hlj1∆ cells coexpressing EGFP-CFTR (induced as described in Figure 1A) and mCherry-Hlj1p were observed by confocal microscopy. (B) hlj1∆ cells harboring pEGFP-CFTR were transformed with pYO324 (2 μ vector), pSAR1 (2 μ SAR1), pSEC12 (2 μ SEC12), pSEC23 (2 μ SEC23), pSEC16 (2 μ SEC16), pSEC13 (2 μ SEC13), or pSAR1D32G (2 μ SAR1D32G), and the transformants were grown to mid–log phase and induced with copper at 23ºC for 2 h to induce EGFP-CFTR expression. ERAC formation was quantified as described in Figure 1B. The experiments were repeated at least three times, and >150 cells were analyzed for each condition.
Hsp40 and COPII components function in the same pathway to drive ERAC formation
Given the role of both Hlj1p and COPII components in ERAC formation, a crucial question is whether these components participate in the same functional pathway. To explore this, we examined whether overexpression of a protein involved in COPII vesicle formation could rescue the ERAC formation defect of hlj1∆ cells. We forced hlj1∆ cells to overexpress one of the COPII proteins from a multicopy plasmid used in Figure 2A and monitored the EGFP-CFTR–induced ERAC formation by confocal microscopy (Figure 8B). Of the five COPII components (Sar1p, Sec12p, Sec23p, Sec13p, Sec16p) tested, overexpression of Sar1p and Sec12p significantly increased the number of ERAC patterns of localization by ∼2.2-fold (44.2 ± 5.98% for Sar1p, 44.4 ± 6.40% for Sec12p) compared with the vector control (19.8 ± 12.5%). No obvious increase in ERAC pattern was detected with Sec23p, Sec16p, Sec13p, or Sar1p-D32G. These genetic relationships indicate that Hlj1p acts upstream or in conjunction with COPII components during ERAC formation. The ability of high-copy expression of Sar1p or Sec12p to suppress defects in ERAC formation is consistent with the fact that these two components act the most upstream in COPII assembly of the investigated COPII components. Collectively these findings suggest that overexpression of some of the COPII genes can, at least partially, bypass the ERAC formation defect of hlj1∆ cells, which confirms that Hlj1p and COPII components act in the same ERAC formation pathway.
DISCUSSION
A previous study demonstrated a requirement for the COPII components in the sequestration of the transmembrane ERAD substrate CFTR into ERACs in S. cerevisiae (Fu and Sztul, 2003). However, the molecular mechanism by which the COPII machinery contributes to this process is unclear. To achieve a better understanding of the involvement of COPII proteins in ERAC formation, we expanded the previous study by examining the localization of EGFP-CFTR in various temperature-sensitive mutants with a deficiency in ER-to-Golgi transport. The pattern of EGFP-CFTR localization in the sar1D32G, sec12-4, sec23-1, sec13-1, and sec16-2 strains, all of which were defective in the stage of COPII vesicle budding, showed significant changes at the restrictive temperature: EGFP-CFTR was homogeneously dispersed throughout the ER. In contrast, when the vesicle targeting and fusion mutants sec17-1 and sec18-1 were grown at the restrictive temperature, >70% of the mutant cells displayed the ERAC or ERAC and ER pattern of EGFP-CFTR localization, confirming that the sequestration of EGFP-CFTR is independent of ER-to-Golgi trafficking (Figure 1B).
These experiments require a temperature shift from the permissive temperature (23ºC) to the restrictive temperature (37ºC) to evaluate the effect of the mutant COPII component on ERAC formation. Thus one might expect that cytosolic and luminal chaperones that are induced in response to a rapid shift of yeast cells to the restrictive temperature prevent the formation of protein aggregates and/or promote protein refolding, which may act to limit ERAC formation. In fact, others have observed that an elevated temperature markedly stabilizes wild-type CFTR in mammalian cells and its temperature-sensitive folding mutant, ∆F508-CFTR, from ERAD in response to the induction of heat shock proteins (Strickland et al., 1997; Loo et al., 1998; Meacham et al., 1999; Choo-Kang and Zeitlin, 2001). However, this is certainly not the case for EGFP-CFTR in yeast, because ERAC formation was not significantly altered upon the temperature shift in either wild-type or sec17-1 and sec18-1 mutants (Figure 1B). Alternatively, the accumulation of aberrant proteins in the ER generally leads to the induction of the unfolded protein response (UPR; Walter and Ron, 2011), which also induces the expression of a variety of genes required for protein folding and ER export (Travers et al., 2000). Upregulation of COPII-pathway genes may be of significance in view of the data presented here and in previous work (Fu and Sztul, 2003). However, CFTR fails to induce the UPR (Zhang et al., 2001); other transmembrane ERAD substrates expressed in yeast also fail to induce the UPR (Ferreira et al., 2002; Huyer et al., 2004). Indeed, EGFP-CFTR was normally sequestered into ERACs in cells lacking IRE1, the master membrane kinase involved in transmitting the unfolded protein signal to activate UPR target genes (Supplemental Figure S1).
Additional evidence for the involvement of a traffic-independent function of COPII components in ERACs formation comes from multicopy suppression analyses. The temperature-sensitive growth defects in many mutants defective in COPII vesicle formation can be suppressed by high-copy expression of other genes involved in this process. However, we observed that such multicopy suppressors were not always able to rescue the ERAC formation defect. In addition, we also found that the overexpression of a number of genes involved in COPII vesicle formation that fail to suppress the ER-to-Golgi transport defect suppresses the ERAC formation defect (Figure 3). These collective genetic data provide circumstantial evidence that the COPII machinery has at least two different functions in the ER. One function involves ER-to-Golgi transport, and the other involves the sorting of integral membrane ERAD substrates into specialized ER subdomains.
ERAC-like structures that form in response to certain mutant or heterologously expressed proteins have been observed both in yeast and in mammalian cells (Valetti et al., 1991; Hobman et al., 1992, 1998; Supply et al., 1993; Nishikawa et al., 1994; Raposo et al., 1995; Kamhi-Nesher et al., 2001; Ferreira et al., 2002). Because certain misfolded proteins reside only transiently in the ERAC-like structures and eventually enter the secretory pathway, some of these structures have been proposed to represent expanded ER exit sites (Nishikawa et al., 1994; Hobman et al., 1998; Kamhi-Nesher et al., 2001; Ferreira et al., 2002). However, we could not observe the specific localization of ERES marker proteins within the ERACs induced by EGFP-CFTR. Furthermore, a constitutively inactive form of Sar1p (Sar1p-D32G), which fails to be targeted to ERES (Yorimitsu and Sato, 2012), was also found to localize to ERACs (Figure 4). Therefore it is likely that at least the EGFP-CFTR–induced ERACs are not expanded ER exit sites. Instead, some small punctate structures labeled by ERES markers, which closely resemble the structures characteristic of ERES, were often associated within ERACs. Because EGFP-CFTR–induced ERACs are accumulations of tubular membrane extensions connected to the ER (Fu and Sztul, 2003; Huyer et al., 2004) and a resident transmembrane ER protein (Sec71p) is also present in ERACs (Figure 4), we assume that these small puncta represent ERES that had been coincidentally incorporated into the ERAC during its formation. However, we do not have data to indicate that these punctate signals are functionally equivalent to ERES observed within the ER.
The recognition of ERAD substrates is believed to be mediated primarily by molecular chaperones. Integral membrane ERAD substrates with large cytoplasmic domains have been proposed to bind to cytosolic Hsp70s and Hsp40s. In fact, CFTR includes two large cytosolic nucleotide-binding domains, and cytosolically localized Hsp70 and Hsp40s have been shown to facilitate the ERAD of CFTR in both yeast (Zhang et al., 2001; Youker et al., 2004) and mammals (Meacham et al., 1999; Rubenstein and Zeitlin, 2000). The contributions of Hsp40 proteins in the process of ERAD have been observed to require their interaction with partner Hsp70 proteins. In general, Hsp40 binds directly to substrate polypeptides first and then, in nearly all cases, transfers the bound substrates to its cognate Hsp70 (Craig et al., 2006; Buck et al., 2007). We could indeed detect a direct interaction between ER-localized Hsp40, Hlj1p, and EGFP-CFTR (Figure 7). We demonstrated that the maximal formation of ERAC requires at least two ER-associated Hsp40s, Ydj1p and Hlj1p. However, the function of the Hlj1p/Ydj1p Hsp70 cognate, Ssa proteins, has been shown to be dispensable for CFTR-induced ERAC formation (Zhang et al., 2001). Although it has been shown that Hlj1p and Ydj1p redundantly function to facilitate CFTR degradation (Youker et al., 2004), our data showed that deletion of either one caused defects in both sequestration and degradation of EGFP-CFTR (Figure 6). One possible reason for this discrepancy could be due to a difference in expression level of CFTR: the previous report (Youker et al., 2004) used the constitutive PGK promoter, whereas we used the inducible CUP1 promoter. When expressed from the relatively weak PGK promoter, the cells with the loss of either HLJ1 or YDJ1 may still manage to sequester CFTR into ERACs, whereas CFTR expressed from the strong inducible CUP1 promoter requires full Hsp40 activity. Our finding is the first example in which ER-associated Hsp40s are shown to play a role in a step upstream of ERAC formation.
Several lines of genetic evidence presented here revealed partial overlaps in the functions of COPII proteins and ER-associated Hsp40s during ERAC formation. It is therefore likely that the substrate-binding activities of Hlj1p involved in ERAC formation have a close mechanistic link with COPII functions rather than simply delivering bound substrates to its partner Hsp70. However, we could not detect COPII components in the same Hlj1p-EGFP-CFTR protein complex under detergent-solubilized conditions (Figure 7). This might be due to several factors, including a breakdown of the interactions in the presence of detergent and/or an inability to maintain the complex under detergent-solubilized membrane-free conditions. Understanding this key step will be important to exploring how COPII might facilitate the sorting of EGFP-CFTR into specialized ER subdomains. Because ERACs are extensive networks of tubulovesicular structures, we speculate that the membrane-deforming ability of Sar1p and COPII coats (Bi et al., 2002; Bielli et al., 2005; Lee et al., 2005; Stagg et al., 2008; Long et al., 2010; O'Donnell et al., 2011) might contribute to drive ERAC formation. Moreover, there is a report that at least some ERACs are cleared from cells through the autophagy-dependent pathway (Fu and Sztul, 2009). Therefore, taking into account that the requirement for a subgroup of COPII proteins in autophagosome formation has been demonstrated in yeast (Ishihara et al., 2001; Hamasaki et al., 2003), it is possible to consider ERACs to be a kind of preautophagosomal-like structure accumulated by COPII components.
In summary, our results demonstrated that the inhibition of ERAC formation in the absence of COPII functions was not due to indirect effects of inactivating ER-to-Golgi trafficking; instead, COPII components played a certain role during ERAC formation. Furthermore, we found that the ER-associated Hsp40s were specifically involved in the substrate sequestration process. We propose that a continued characterization of the underlying mechanisms that govern substrate partitioning to ERACs will yield additional insights into the varied activities of COPII machinery.
MATERIALS AND METHODS
Yeast strains and plasmids
The S. cerevisiae strains used in this study are listed in Supplemental Table S1. The yeast strains were grown in MCD medium (0.67% yeast nitrogen base without amino acids [Difco Laboratories, Franklin Lakes, NJ], 2% glucose, and 0.5% casamino acids with auxotrophic supplements). The coding sequences of the SAR1, SEC12, SEC23, SEC13, and SEC16 genes, together with upstream and downstream flanking regions, were amplified by PCR from S. cerevisiae genomic DNA and inserted into the SacI and XhoI sites of pYO324 (2 μ, TRP1), yielding pSAR1, pSEC12, pSEC23, pSEC13, and pSEC16, respectively. The coding sequences of the SEC71 and SEC23 genes, together with upstream and downstream flanking regions, were amplified from genomic DNA by PCR and inserted into the BamHI and XhoI sites (for SEC71) or SacI and XhoI sites (for SEC23) of pRS314 (CEN, TRP1). For fusion of fluorescent proteins to the C-terminus, a SphI site (for SEC71) or a BamHI site (for SEC23) was created just before the stop codon of each gene, and the fragment encoding mCherry, which was PCR amplified from pmCherry (Clontech, Mountain View, CA), was inserted into these sites to yield pSEC71-mCherry or pSEC23-mCherry. The coding sequence of the HLJ1 gene, together with upstream and downstream flanking regions, was amplified from the genomic DNA by PCR and inserted into the SacI–XhoI sites of pYO324. A SphI site was created just before the start codon, and the fragment encoding mCherry or GST was inserted into the SphI site to yield pmCherry-HLJ1 or pGST-Hlj1, respectively. The generation of plasmids expressing Sar1p-mCherry, Sar1p-D32G-mCherry, and Sec13p-mCherry was described previously (Yorimitsu and Sato, 2012). The plasmid expressing EGFP-tagged human CFTR under a copper-inducible promoter (pCU426CUP1/EGFP-CFTR) was a generous gift from Elizabeth Sztul and Lianwu Fu (University of Alabama). The mutation to alanine at position 567 of CFTR was introduced into the gene of EGFP-CFTR in plasmid pEGFP-CFTR using primer-directed mutagenesis, yielding pEGFP-CFTR-D567A.
Fluorescence microscopy
Yeast cells were grown to an OD600 of 0.5 at 23ºC, and EGFP-CFTR expression was induced with 100 μM CuSO4 for 2 h. To analyze ERAC formation in temperature-sensitive mutants, yeast cells containing the EGFP-CFTR plasmid were grown to an OD600 of 0.5 at 23ºC. Then each culture was separated into two aliquots, and the aliquots were incubated at 23 or 37ºC for 10 min. Cells were then induced to express EGFP-CFTR in the presence of 100 μM CuSO4 for 2 h. Fluorescence microscopy observation was carried out using an Olympus IX71 microscope equipped with a CSU10 spinning-disk confocal scanner (Yokogawa Electric Corporation, Tokyo, Japan) and an electron-multiplying charge-coupled device camera (iXon, DU897; Andor Technology, South Windsor, CT). The acquired images were analyzed by using Andor iQ (Andor Technology). In this setting, a 473-nm solid-state laser (Showa Optronics, Tokyo, Japan J050BS) was used to excite AcGFP and mCherry at 561 nm (Jive; Cobolt, Solna, Sweden).
Cycloheximide chase assay
Cells containing CUP1 promoter regulated EGFP-CFTR were grown to an OD600 of 0.5 at 23ºC, and EGFP-CFTR was induced with 100 μM CuSO4 for 2 h. To begin the chase, protein synthesis was stopped by the addition of cycloheximide to a final concentration of 50 μg/ml with continued incubation at 23ºC. At specified time points, 3.0 OD of cells were collected and 1/10 volume of ice-cold 100% trichloroacetic acid (TCA) was added to terminate the chase. The cells were washed with cold water and disrupted by vortexing with glass beads in lysis buffer (50 mM Tris-Cl, pH 7.5, 0.5% SDS). The cleared lysate was precipitated using TCA and analyzed by immunoblotting.
Pull-down assays
Microsomes were prepared from cells expressing EGFP-CFTR (induced for 2 h) and GST-Hlj1p as described previously (Sato and Nakano, 2003). For the pull-down assays, 100 μg of microsomes was solubilized with 1% Triton X-100 in B88-8 (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid–KOH, pH 8.0, 150 mM KOAc, 5 mM MgOAc, and 250 mM sorbitol). After 30 min on ice, the insoluble material was removed by centrifugation, and then the supernatant was incubated with glutathione–Sepharose beads (GE Healthcare, Piscataway, NJ) at 4ºC for 1 h. The beads were washed with the same buffer, and bound proteins were eluted using 10 mM glutathione in buffer B88-8. The samples were subjected to TCA precipitation, followed by SDS–PAGE and immunoblotting. The anti-GFP antibody (Living Colors A.v. Monoclonal Antibody) was obtained from Clontech. The anti-Sec23p polyclonal antibody was generated against glutathione-Sepharose–purified GST-Sec23p. The anti-Sec61p polyclonal antibody was raised against a synthetic peptide corresponding to residues 12–30. Antibodies directed against Sar1p (Saito-Nakano and Nakano, 2000) and Sec12p (Nishikawa and Nakano, 1993) were described previously.
Supplementary Material
Acknowledgments
We are grateful to Elizabeth Sztul and Lianwu Fu for providing the EGFP-tagged construct of the CFTR plasmid. We also thank members of the Sato laboratory for helpful discussions. This work was supported by a Grant-in-Aid for Scientific Research of the Japan Society for the Promotion of Science (T.Y. and K.S.) and in part by the Targeted Proteins Research Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan (K.S.).
Abbreviations used:
- CFTR
cystic fibrosis transmembrane conductance regulator
- COPII
coat protein complex II
- ERAC
ER-associated compartment
- ERAD
ER-associated degradation
- GST
glutathione-S-transferase
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-08-0639) on January 9, 2013.
REFERENCES
- Ahner A, Nakatsukasa K, Zhang H, Frizzell RA, Brodsky JL. Small heat-shock proteins select deltaF508-CFTR for endoplasmic reticulum-associated degradation. Mol Biol Cell. 2007;18:806–814. doi: 10.1091/mbc.E06-05-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Balch WE. Kinase signaling initiates coat complex II (COPII) recruitment and export from the mammalian endoplasmic reticulum. J Biol Chem. 2000;275:35673–35676. doi: 10.1074/jbc.C000449200. [DOI] [PubMed] [Google Scholar]
- Bannykh SI, Rowe T, Balch WE. The organization of endoplasmic reticulum export complexes. J Cell Biol. 1996;135:19–35. doi: 10.1083/jcb.135.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Barlowe C, Schekman R. SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature. 1993;365:347–349. doi: 10.1038/365347a0. [DOI] [PubMed] [Google Scholar]
- Belden WJ, Barlowe C. Role of Erv29p in collecting soluble secretory proteins into ER-derived transport vesicles. Science. 2001;294:1528–1531. doi: 10.1126/science.1065224. [DOI] [PubMed] [Google Scholar]
- Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- Bielli A, Haney CJ, Gabreski G, Watkins SC, Bannykh SI, Aridor M. Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission. J Cell Biol. 2005;171:919–924. doi: 10.1083/jcb.200509095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumental-Perry A, Haney CJ, Weixel KM, Watkins SC, Weisz OA, Aridor M. Phosphatidylinositol 4-phosphate formation at ER exit sites regulates ER export. Dev Cell. 2006;11:671–682. doi: 10.1016/j.devcel.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan R, Kaufman A, Kung-Tran L, Miller EA. Genetic analysis of yeast Sec24p mutants suggests cargo binding is not co-operative during ER export. Traffic. 2010;11:1034–1043. doi: 10.1111/j.1600-0854.2010.01080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck TM, Wright CM, Brodsky JL. The activities and function of molecular chaperones in the endoplasmic reticulum. Semin Cell Dev Biol. 2007;18:751–761. doi: 10.1016/j.semcdb.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillon GA, Watanabe R, Taylor M, Schwabe TM, Riezman H. Concentration of GPI-anchored proteins upon ER exit in yeast. Traffic. 2009;10:186–200. doi: 10.1111/j.1600-0854.2008.00857.x. [DOI] [PubMed] [Google Scholar]
- Choo-Kang LR, Zeitlin PL. Induction of HSP70 promotes DeltaF508 CFTR trafficking. Am J Physiol Lung Cell Mol Physiol. 2001;281:L58–L68. doi: 10.1152/ajplung.2001.281.1.L58. [DOI] [PubMed] [Google Scholar]
- Connerly PL, Esaki M, Montegna EA, Strongin DE, Levi S, Soderholm J, Glick BS. Sec16 is a determinant of transitional ER organization. Curr Biol. 2005;15:1439–1447. doi: 10.1016/j.cub.2005.06.065. [DOI] [PubMed] [Google Scholar]
- Craig EA, Huang P, Aron R, Andrew A. The diverse roles of J-proteins, the obligate Hsp70 co-chaperone. Rev Physiol Biochem Pharmacol. 2006;156:1–21. doi: 10.1007/s10254-005-0001-0. [DOI] [PubMed] [Google Scholar]
- d'Enfert C, Barlowe C, Nishikawa S, Nakano A, Schekman R. Structural and functional dissection of a membrane glycoprotein required for vesicle budding from the endoplasmic reticulum. Mol Cell Biol. 1991;11:5727–5734. doi: 10.1128/mcb.11.11.5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancourt J, Barlowe C. Protein sorting receptors in the early secretory pathway. Annu Rev Biochem. 2010;79:777–802. doi: 10.1146/annurev-biochem-061608-091319. [DOI] [PubMed] [Google Scholar]
- Ferreira T, Mason AB, Pypaert M, Allen KE, Slayman CW. Quality control in the yeast secretory pathway: a misfolded PMA1 H+-ATPase reveals two checkpoints. J Biol Chem. 2002;277:21027–21040. doi: 10.1074/jbc.M112281200. [DOI] [PubMed] [Google Scholar]
- Fu L, Sztul E. Traffic-independent function of the Sar1p/COPII machinery in proteasomal sorting of the cystic fibrosis transmembrane conductance regulator. J Cell Biol. 2003;160:157–163. doi: 10.1083/jcb.200210086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Sztul E. ER-associated complexes (ERACs) containing aggregated cystic fibrosis transmembrane conductance regulator (CFTR) are degraded by autophagy. Eur J Cell Biol. 2009;88:215–226. doi: 10.1016/j.ejcb.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillon AD, Latham CF, Miller EA. Vesicle-mediated ER export of proteins and lipids. Biochim Biophys Acta. 2012;1821:1040–1049. doi: 10.1016/j.bbalip.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno RE, Espenshade P, Kaiser CA. SED4 encodes a yeast endoplasmic reticulum protein that binds Sec16p and participates in vesicle formation. J Cell Biol. 1995;131:325–338. doi: 10.1083/jcb.131.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Noda T, Ohsumi Y. The early secretory pathway contributes to autophagy in yeast. Cell Struct Funct. 2003;28:49–54. doi: 10.1247/csf.28.49. [DOI] [PubMed] [Google Scholar]
- Higashio H, Sato K, Nakano A. Smy2p participates in COPII vesicle formation through the interaction with Sec23p/Sec24p subcomplex. Traffic. 2008;9:79–93. doi: 10.1111/j.1600-0854.2007.00668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill K, Cooper AA. Degradation of unassembled Vph1p reveals novel aspects of the yeast ER quality control system. EMBO J. 2000;19:550–561. doi: 10.1093/emboj/19.4.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobman TC, Woodward L, Farquhar MG. The rubella virus E1 glycoprotein is arrested in a novel post-ER, pre-Golgi compartment. J Cell Biol. 1992;118:795–811. doi: 10.1083/jcb.118.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobman TC, Zhao B, Chan H, Farquhar MG. Immunoisolation and characterization of a subdomain of the endoplasmic reticulum that concentrates proteins involved in COPII vesicle biogenesis. Mol Biol Cell. 1998;9:1265–1278. doi: 10.1091/mbc.9.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyer G, Longsworth GL, Mason DL, Mallampalli MP, McCaffery JM, Wright RL, Michaelis S. A striking quality control subcompartment in Saccharomyces cerevisiae: the endoplasmic reticulum-associated compartment. Mol Biol Cell. 2004;15:908–921. doi: 10.1091/mbc.E03-07-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, Kihara A, Yoshimori T, Noda T, Ohsumi Y. Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell. 2001;12:3690–3702. doi: 10.1091/mbc.12.11.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamhi-Nesher S, Shenkman M, Tolchinsky S, Fromm SV, Ehrlich R, Lederkremer GZ. A novel quality control compartment derived from the endoplasmic reticulum. Mol Biol Cell. 2001;12:1711–1723. doi: 10.1091/mbc.12.6.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser GL, Gentzsch M, Kloser AK, Balzi E, Wolf DH, Goffeau A, Riordan JR. Expression and degradation of the cystic fibrosis transmembrane conductance regulator in Saccharomyces cerevisiae. Arch Biochem Biophys. 2001;390:195–205. doi: 10.1006/abbi.2001.2385. [DOI] [PubMed] [Google Scholar]
- Kodera C, Yorimitsu T, Nakano A, Sato K. Sed4p stimulates Sar1p GTP hydrolysis and promotes limited coat disassembly. Traffic. 2011;12:591–599. doi: 10.1111/j.1600-0854.2011.01173.x. [DOI] [PubMed] [Google Scholar]
- Kryndushkin DS, Smirnov VN, Ter-Avanesyan MD, Kushnirov VV. Increased expression of Hsp40 chaperones, transcriptional factors, ribosomal protein Rpp0 can cure yeast prions. J Biol Chem. 2002;277:23702–23708. doi: 10.1074/jbc.M111547200. [DOI] [PubMed] [Google Scholar]
- Kuehn MJ, Herrmann JM, Schekman R. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature. 1998;391:187–190. doi: 10.1038/34438. [DOI] [PubMed] [Google Scholar]
- Kurihara T, Hamamoto S, Gimeno RE, Kaiser CA, Schekman R, Yoshihisa T. Sec24p and Iss1p function interchangeably in transport vesicle formation from the endoplasmic reticulum in Saccharomyces cerevisiae. Mol Biol Cell. 2000;11:983–998. doi: 10.1091/mbc.11.3.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122:605–617. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Lee TH, Linstedt AD. Potential role for protein kinases in regulation of bidirectional endoplasmic reticulum-to-Golgi transport revealed by protein kinase inhibitor H89. Mol Biol Cell. 2000;11:2577–2590. doi: 10.1091/mbc.11.8.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KR, Yamamoto Y, Baker AL, Watkins SC, Coyne CB, Conway JF, Aridor M. Sar1 assembly regulates membrane constriction and ER export. J Cell Biol. 2010;190:115–128. doi: 10.1083/jcb.201004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo MA, Jensen TJ, Cui L, Hou Y, Chang XB, Riordan JR. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 1998;17:6879–6887. doi: 10.1093/emboj/17.23.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999;18:1492–1505. doi: 10.1093/emboj/18.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, Orci L, Schekman R. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell. 2003;114:497–509. doi: 10.1016/s0092-8674(03)00609-3. [DOI] [PubMed] [Google Scholar]
- Mossessova E, Bickford LC, Goldberg J. SNARE selectivity of the COPII coat. Cell. 2003;114:483–495. doi: 10.1016/s0092-8674(03)00608-1. [DOI] [PubMed] [Google Scholar]
- Nakano A, Muramatsu M. A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol. 1989;109:2677–2691. doi: 10.1083/jcb.109.6.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Brodsky JL. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic. 2008;9:861–870. doi: 10.1111/j.1600-0854.2008.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S, Nakano A. Identification of a gene required for membrane protein retention in the early secretory pathway. Proc Natl Acad Sci USA. 1993;90:8179–8183. doi: 10.1073/pnas.90.17.8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S, Hirata A, Nakano A. Inhibition of endoplasmic reticulum (ER)-to-Golgi transport induces relocalization of binding protein (BiP) within the ER to form the BiP bodies. Mol Biol Cell. 1994;5:1129–1143. doi: 10.1091/mbc.5.10.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell J, Maddox K, Stagg S. The structure of a COPII tubule. J Struct Biol. 2011;173:358–364. doi: 10.1016/j.jsb.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Oka T, Nakano A. Inhibition of GTP hydrolysis by Sar1p causes accumulation of vesicles that are a functional intermediate of the ER-to-Golgi transport in yeast. J Cell Biol. 1994;124:425–434. doi: 10.1083/jcb.124.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Ravazzola M, Meda P, Holcomb C, Moore HP, Hicke L, Schekman R. Mammalian Sec23p homologue is restricted to the endoplasmic reticulum transitional cytoplasm. Proc Natl Acad Sci USA. 1991;88:8611–8615. doi: 10.1073/pnas.88.19.8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paccaud JP, Reith W, Carpentier JL, Ravazzola M, Amherdt M, Schekman R, Orci L. Cloning and functional characterization of mammalian homologues of the COPII component Sec23. Mol Biol Cell. 1996;7:1535–1546. doi: 10.1091/mbc.7.10.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, van Santen HM, Leijendekker R, Geuze HJ, Ploegh HL. Misfolded major histocompatibility complex class I molecules accumulate in an expanded ER-Golgi intermediate compartment. J Cell Biol. 1995;131:1403–1419. doi: 10.1083/jcb.131.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismanchi N, Puertollano R, Blackstone C. STAM adaptor proteins interact with COPII complexes and function in ER-to-Golgi trafficking. Traffic. 2009;10:201–217. doi: 10.1111/j.1600-0854.2008.00856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of DeltaF508-CFTR. Am J Physiol Cell Physiol. 2000;278:C259–C267. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- Rudiger S, Schneider-Mergener J, Bukau B. Its substrate specificity characterizes the DnaJ co-chaperone as a scanning factor for the DnaK chaperone. EMBO J. 2001;20:1042–1050. doi: 10.1093/emboj/20.5.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Kimura K, Oka T, Nakano A. Activities of mutant Sar1 proteins in guanine nucleotide binding, GTP hydrolysis, and cell-free transport from the endoplasmic reticulum to the Golgi apparatus. J Biochem. 1998;124:816–823. doi: 10.1093/oxfordjournals.jbchem.a022185. [DOI] [PubMed] [Google Scholar]
- Saito Y, Yamanushi T, Oka T, Nakano A. Identification of SEC12, SED4, truncated SEC16, and EKS1/HRD3 as multicopy suppressors of ts mutants of Sar1 GTPase. J Biochem. 1999;125:130–137. doi: 10.1093/oxfordjournals.jbchem.a022249. [DOI] [PubMed] [Google Scholar]
- Saito-Nakano Y, Nakano A. Sed4p functions as a positive regulator of Sar1p probably through inhibition of the GTPase activation by Sec23p. Genes Cells. 2000;5:1039–1048. doi: 10.1046/j.1365-2443.2000.00391.x. [DOI] [PubMed] [Google Scholar]
- Sato K, Nakano A. Oligomerization of a cargo receptor directs protein sorting into COPII-coated transport vesicles. Mol Biol Cell. 2003;14:3055–3063. doi: 10.1091/mbc.E03-02-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Nakano A. Dissection of COPII subunit-cargo assembly and disassembly kinetics during Sar1p-GTP hydrolysis. Nat Struct Mol Biol. 2005;12:167–174. doi: 10.1038/nsmb893. [DOI] [PubMed] [Google Scholar]
- Sato K, Nakano A. Mechanisms of COPII vesicle formation and protein sorting. FEBS Lett. 2007;581:2076–2082. doi: 10.1016/j.febslet.2007.01.091. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Stephens DJ. Cargo loading at the ER. Mol Membr Biol. 2010;27:398–411. doi: 10.3109/09687688.2010.506203. [DOI] [PubMed] [Google Scholar]
- Shindiapina P, Barlowe C. Requirements for transitional endoplasmic reticulum site structure and function in Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:1530–1545. doi: 10.1091/mbc.E09-07-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg SM, LaPointe P, Razvi A, Gurkan C, Potter CS, Carragher B, Balch WE. Structural basis for cargo regulation of COPII coat assembly. Cell. 2008;134:474–484. doi: 10.1016/j.cell.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland E, Qu BH, Millen L, Thomas PJ. The molecular chaperone Hsc70 assists the in vitro folding of the N-terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1997;272:25421–25424. doi: 10.1074/jbc.272.41.25421. [DOI] [PubMed] [Google Scholar]
- Sullivan ML, Youker RT, Watkins SC, Brodsky JL. Localization of the BiP molecular chaperone with respect to endoplasmic reticulum foci containing the cystic fibrosis transmembrane conductance regulator in yeast. J Histochem Cytochem. 2003;51:545–548. doi: 10.1177/002215540305100417. [DOI] [PubMed] [Google Scholar]
- Supply P, Wach A, Thines-Sempoux D, Goffeau A. Proliferation of intracellular structures upon overexpression of the PMA2 ATPase in Saccharomyces cerevisiae. J Biol Chem. 1993;268:19744–19752. [PubMed] [Google Scholar]
- Szabo A, Langer T, Schroder H, Flanagan J, Bukau B, Hartl FU. The ATP hydrolysis-dependent reaction cycle of the Escherichia coli Hsp70 system DnaK, DnaJ, and GrpE. Proc Natl Acad Sci USA. 1994;91:10345–10349. doi: 10.1073/pnas.91.22.10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata KV, Sato K, Ide T, Nishizaka T, Nakano A, Noji H. Visualization of cargo concentration by COPII minimal machinery in a planar lipid membrane. EMBO J. 2009;28:3279–3289. doi: 10.1038/emboj.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Valetti C, Grossi CE, Milstein C, Sitia R. Russell bodies: a general response of secretory cells to synthesis of a mutant immunoglobulin which can neither exit from, nor be degraded in, the endoplasmic reticulum. J Cell Biol. 1991;115:983–994. doi: 10.1083/jcb.115.4.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang X, Matteson J, An Y, Moyer B, Yoo JS, Bannykh S, Wilson IA, Riordan JR, Balch WE. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J Cell Biol. 2004;167:65–74. doi: 10.1083/jcb.200401035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki A, Tani K, Yamamoto A, Kitamura N, Komada M. The Ca2+-binding protein ALG-2 is recruited to endoplasmic reticulum exit sites by Sec31A and stabilizes the localization of Sec31A. Mol Biol Cell. 2006;17:4876–4887. doi: 10.1091/mbc.E06-05-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JS, Moyer BD, Bannykh S, Yoo HM, Riordan JR, Balch WE. Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J Biol Chem. 2002;277:11401–11409. doi: 10.1074/jbc.M110263200. [DOI] [PubMed] [Google Scholar]
- Yorimitsu T, Sato K. Insights into structural and regulatory roles of Sec16 in COPII vesicle formation at ER exit sites. Mol Biol Cell. 2012;23:2930–2942. doi: 10.1091/mbc.E12-05-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshibori M, Yorimitsu T, Sato K. Involvement of the penta-EF-hand protein Pef1p in the Ca(2+)-dependent regulation of COPII subunit assembly in Saccharomyces cerevisiae. PLoS One. 2012;7:e40765. doi: 10.1371/journal.pone.0040765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youker RT, Walsh P, Beilharz T, Lithgow T, Brodsky JL. Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol Biol Cell. 2004;15:4787–4797. doi: 10.1091/mbc.E04-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, Brodsky JL. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell. 2001;12:1303–1314. doi: 10.1091/mbc.12.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.