

Abstract
DNA polymerases (Pols) ε and δ perform the bulk of yeast leading- and lagging-strand DNA synthesis. Both Pols possess intrinsic proofreading exonucleases that edit errors during polymerization. Rare errors that elude proofreading are extended into duplex DNA and excised by the mismatch repair (MMR) system. Strains that lack Pol proofreading or MMR exhibit a 10- to 100-fold increase in spontaneous mutation rate (mutator phenotype), and inactivation of both Pol δ proofreading (pol3-01) and MMR is lethal due to replication error-induced extinction (EEX). It is unclear whether a similar synthetic lethal relationship exists between defects in Pol ε proofreading (pol2-4) and MMR. Using a plasmid-shuffling strategy in haploid Saccharomyces cerevisiae, we observed synthetic lethality of pol2-4 with alleles that completely abrogate MMR (msh2Δ, mlh1Δ, msh3Δ msh6Δ, or pms1Δ mlh3Δ) but not with partial MMR loss (msh3Δ, msh6Δ, pms1Δ, or mlh3Δ), indicating that high levels of unrepaired Pol ε errors drive extinction. However, variants that escape this error-induced extinction (eex mutants) frequently emerged. Five percent of pol2-4 msh2Δ eex mutants encoded second-site changes in Pol ε that reduced the pol2-4 mutator phenotype between 3- and 23-fold. The remaining eex alleles were extragenic to pol2-4. The locations of antimutator amino-acid changes in Pol ε and their effects on mutation spectra suggest multiple mechanisms of mutator suppression. Our data indicate that unrepaired leading- and lagging-strand polymerase errors drive extinction within a few cell divisions and suggest that there are polymerase-specific pathways of mutator suppression. The prevalence of suppressors extragenic to the Pol ε gene suggests that factors in addition to proofreading and MMR influence leading-strand DNA replication fidelity.
Keywords: DNA replication fidelity, antimutator, proofreading, mismatch repair, error catastrophe
ORGANISMS must accurately duplicate their genomes to avoid loss of long-term fitness. Consequently, cells employ high-fidelity DNA polymerases (Pols) equipped with proofreading exonucleases to replicate their DNA (reviewed in McCulloch and Kunkel 2008; Reha-Krantz 2010). Mismatch repair (MMR) further ensures the integrity of genetic information by targeting mismatches for excision from newly replicated DNA (reviewed in Kolodner and Marsischky 1999; Iyer et al. 2006; Hsieh and Yamane 2008). These polymerase error-correcting mechanisms, together with DNA damage repair (Friedberg et al. 2006), maintain the genome with less than one mutation per 109 nucleotides per cell division (Drake et al. 1998). Defects in proofreading or MMR result in mutator phenotypes characterized by increased rates of spontaneous mutation (Kolodner and Marsischky 1999; Iyer et al. 2006; Hsieh and Yamane 2008; McCulloch and Kunkel 2008; Reha-Krantz 2010).
Mutator phenotypes can be an important source of genetic diversity, which facilitates adaptation to environmental change. In bacterial and yeast populations, unstable environments favor mutator strains that readily acquire adaptive mutations (Chao and Cox 1983; Mao et al. 1997; Sniegowski et al. 1997; Giraud et al. 2001a; Notley-McRobb et al. 2002; Nilsson et al. 2004; Thompson et al. 2006; Desai et al. 2007). In mammals, mutator phenotypes are proposed to accelerate the process of somatic cell evolution during tumorigenesis (Loeb et al. 1974, 2008). Deep sequencing of spontaneous tumors provides evidence for a mutator phenotype (Fox et al. 2009; Loeb 2011), and genetic defects in MMR or Pol proofreading elevate cancer susceptibility (Wei et al. 2002; Peltomäki 2005; Preston et al. 2010). However, mutator phenotypes do not persist indefinitely. Loss of fitness accompanies sustained expression of a mutator phenotype in a variety of organisms, including viruses (Smith et al. 2005), bacteria (Funchain et al. 2000; Giraud et al. 2001a), yeast (Wloch et al. 2001; Zeyl and De Visser 2001; Herr et al. 2011a), worms (Estes et al. 2004), and mammals (Albertson et al. 2009). Thus, following adaptation, selection pressure favors restoration of low mutation rates, which can occur through the elimination of mutator alleles or the acquisition of mutator suppressors (i.e., antimutators). A limited number of antimutator variants in the DNA replication machinery have been described (reviewed in Herr et al. 2011b).
In the budding yeast Saccharomyces cerevisiae, DNA polymerases epsilon (Pol ε) and delta (Pol δ) are thought to perform the bulk of leading- and lagging-strand DNA synthesis, respectively (Pursell et al. 2007; Kunkel and Burgers 2008; Nick McElhinny et al. 2008; Larrea et al. 2010; Pavlov and Shcherbakova 2010). Both polymerases are accurate and possess intrinsic proofreading exonucleases that edit mispaired primer termini during polymerization (Morrison et al. 1991; Simon et al. 1991; Shimizu et al. 2002; Shcherbakova et al. 2003; Fortune et al. 2005). Defects in Pol ε or Pol δ proofreading increase the spontaneous mutation rate in a manner consistent with major roles for these polymerases in leading- and lagging-strand synthesis (Morrison et al. 1991; Simon et al. 1991; Morrison and Sugino 1994; Shcherbakova et al. 1996; Tran et al. 1999; Karthikeyan et al. 2000; Greene and Jinks-Robertson 2001). Interestingly, the Pol δ proofreading defect generates a mutator phenotype 5- to 30-fold greater than that observed in Pol ε proofreading-deficient strains (Morrison and Sugino 1994; Shcherbakova et al. 1996; Tran et al. 1999; Datta et al. 2000; Karthikeyan et al. 2000; Greene and Jinks-Robertson 2001; Pavlov et al. 2004), and the spectra of spontaneous mutations that arise in Pol ε and Pol δ proofreading-deficient strains differ, which may reflect distinct error specificities of the polymerases as well as strand-specific effects (Morrison and Sugino 1994; Karthikeyan et al. 2000; Pavlov et al. 2002, 2003; Shcherbakova et al. 2003; Fortune et al. 2005). Mouse cells with defects in Pol ε or Pol δ proofreading also exhibit increased mutation rates (Goldsby et al. 2002; Albertson et al. 2009), and, consistent with distinct roles in DNA replication, the types of tumors that develop in Pol ε and Pol δ proofreading-deficient mice differ markedly (Albertson et al. 2009). Thus, avoidance of errors during eukaryotic DNA replication depends on both Pol ε and Pol δ proofreading (McCulloch and Kunkel 2008; Preston et al. 2010).
The extent to which Pol ε and Pol δ proofreading contribute to DNA replication fidelity is obscured by MMR. The eukaryotic MMR machinery consists of homologs of bacterial MutS and MutL proteins (reviewed in Iyer et al. 2006; Hsieh and Yamane 2008). MutS homolog 2 (Msh2) associates with Msh6 or Msh3 to form two different heterodimers with partially overlapping activities. Msh2-Msh6 recognizes and binds to base-base and small insertion/deletion mispairs, while Msh2-Msh3 recognizes small and larger insertion/deletion mispairs and a subset of base-base mispairs (Harrington and Kolodner 2007). Once bound to mismatched DNA, the Msh proteins recruit heterodimers of MutL homologs (Mlh). Mlh1 is the common subunit for two complexes. Mlh1-Pms1 (Mlh1-Pms2 in mammals) functions with both Msh2-Msh6 and Msh2-Msh3, while Mlh1-Mlh3 works primarily with Msh2-Msh3. Pms1 and Mlh3 contain latent endonucleases that cleave the nascent DNA strand, providing entry points for removal of mismatches and error-free DNA resynthesis (Kadyrov et al. 2006, 2007; Nishant et al. 2008).
Consistent with these biochemical properties, genetic studies in yeast reveal overlapping mutator phenotypes when individual MMR genes are deleted. Deletion of MSH2 eliminates both Msh6- and Msh3-dependent repair, effectively abrogating MMR and conferring a strong base-substitution and frameshift mutator phenotype. Deletion of MSH6 or MSH3 alone only partially inactivates MMR. msh6Δ mutants are strong base-substitution but weak frameshift mutators, msh3Δ strains are weak frameshift and duplication/deletion mutators (with increases in some base substitutions), and msh3Δ msh6Δ double mutants recapitulate the strong mutator phenotype of msh2Δ (Reenan and Kolodner 1992; New et al. 1993; Johnson et al. 1996; Marsischky et al. 1996; Greene and Jinks-Robertson 1997; Sia et al. 1997; Flores-Rozas and Kolodner 1998; Tran et al. 1999; Harrington and Kolodner 2007). Similar to msh2Δ, deletion of MLH1 inactivates MMR, resulting in a strong base-substitution and frameshift mutator phenotype. pms1Δ mutants are also strong base-substitution and frameshift mutators, while mlh3Δ strains are weak frameshift and duplication/deletion mutators (Williamson et al. 1985; Strand et al. 1993; Prolla et al. 1994; Greene and Jinks-Robertson 1997; Flores-Rozas and Kolodner 1998; Yang et al. 1999; Harfe et al. 2000; Harrington and Kolodner 2007). Yeast pms1Δ mlh3Δ double mutants have strong mutator phenotypes, similar to or stronger than pms1Δ and mlh1Δ single mutants (Flores-Rozas and Kolodner 1998). In mice, deletion of both Mlh3 and Pms2 (equivalent to yeast PMS1) is required to recapitulate the strong mutator and cancer phenotypes caused by deletion of Mlh1 alone (Chen et al. 2005). Collectively, these studies indicate that Msh2-Msh6 and Mlh1-Pms1 (Mlh1-Pms2 in mammals) are the primary complexes that function in eukaryotic MMR, while Msh2-Msh3 and Mlh1-Mlh3 play important secondary roles.
Elimination of MMR in Pol ε or Pol δ proofreading-deficient cells results in a multiplicative increase in mutation rate in diploid yeast, suggesting that proofreading and MMR act in series to correct polymerase errors (Morrison et al. 1993; Morrison and Sugino 1994). In haploids, combined inactivation of Pol δ proofreading (via the pol3-01 allele) and any one of several MMR components (msh6Δ, msh2Δ, or pms1Δ) is lethal, presumably due to unrestrained mutagenesis during replication (Morrison et al. 1993; Tran et al. 1999; Greene and Jinks-Robertson 2001). We recently used a collection of pol3 mutator alleles to define the maximal mutation rate compatible with haploid yeast viability (Herr et al. 2011a). Cell populations become inviable when mutation rates exceed ∼10−3 inactivating mutations/gene/cell division. This “error-induced extinction” (EEX) phenotype is readily suppressed by antimutator mutations encoding amino-acid substitutions in the catalytic subunit of Pol δ, as well as suppressor mutations in undefined genes. Thus, variants that escape error-induced extinction (eex mutants) provide a means to probe mechanisms of adaptation to mutator phenotypes (Herr et al. 2011a,b).
Whether Pol ε errors are also sufficient to trigger error-induced extinction remains unclear. Morrison and Sugino (1994) reported that the pol2-4 allele, which inactivates Pol ε proofreading, was not synthetically lethal with pms1Δ in haploid yeast, but noted that the resulting colonies were heterogeneous in size and grew slowly. Interestingly, pol2-4 pms1Δ isolates exhibited varying mutation rates, suggesting that mutator suppressors may arise in these strong mutator strains. Tran et al. (1999) also described haploid strains with Pol ε proofreading and MMR defects: pol2-4 msh2Δ and pol2-4 msh3Δ msh6Δ. Mutation rate increases relative to pol2-4, msh2Δ, and msh3Δ msh6Δ strains were consistent with a multiplicative relationship between MMR and Pol ε proofreading (Tran et al. 1999). However, Greene and Jinks-Robertson (2001) later reported that they could not obtain a viable pol2-4 msh2Δ strain using either gene disruption or plasmid-shuffling methods. Thus, it remains unresolved whether the magnitude of Pol ε errors is sufficient for error-induced extinction.
Here, we show that defective Pol ε proofreading is lethal to haploid yeast in the absence of MMR, providing evidence that, when left unrepaired, leading-strand errors exceed a mutation threshold. Moreover, we show that spontaneous mutants escape this Pol ε error-induced extinction and that eex alleles function as antimutators. We discuss possible mechanisms of escape and mutator suppression. Our studies corroborate the unstable nature of mutators and provide a tractable system to investigate adaptive antimutator mutations that influence leading-strand DNA replication fidelity.
Materials and Methods
Media and growth conditions
Standard media and growth conditions were used in the propagation of yeast strains (Sherman 2002). Cells were grown nonselectively using YPD or synthetic complete (SC) media with 2% dextrose. Selective growth was on SC media containing 2% dextrose and lacking the appropriate amino acid(s). Preformulated SC amino acid supplement was purchased from Bufferad, and supplements lacking defined amino acids were made from individual components as described (Sherman 2002). URA3-deficient cells were selected with 5-fluoroorotic acid (FOA, 1 mg/ml; Zymo Research) media (Boeke et al. 1984). can1 mutants were selected on SC lacking arginine and supplemented with 60 μg/ml of canavanine. Unless otherwise specified, reagents were purchased from Sigma-Aldrich or Fisher Scientific.
Plasmids and strain constructions
POL2 plasmids:
pRS416, a CEN6/ARS4/URA3 plasmid (Brachmann et al. 1998), was engineered to carry the wild-type POL2 gene under control of its native promoter. The genomic sequence of POL2 was amplified from BY4733 yeast using Expand Hi-Fidelity DNA polymerase (Roche) and the following primers and PCR conditions: Pol2-XhoI (5′-ACTCGGTACTCGAGGCGCTCTGCCCTAGTTGGAATG-3′; XhoI site underlined) and PolED2 (5′-GATATTCCGAGCTCGCAACTTCCGGAGTGGTCAC-3′; SacI site underlined); 94°, 1 min; 29× (94°, 15 sec; 58°, 20 sec; 68°, 8 min); 68°, 16 min. The resulting 7.5-kb fragment and pRS416 were digested with SacI and XhoI, ligated together with T4 DNA ligase (Gibco BRL), and then introduced into Escherichia coli. Transformed clones were isolated, and a correct POL2-containing plasmid was confirmed by sequencing the entire insert. This vector (pRS416POL2-5′YIF1) did not fully complement the growth deficiency of our pol2Δ mutants. pRS416POL2-5′YIF1 contains the entire POL2 coding sequence as well as 592 bp of upstream sequence, including 371 bp of noncoding sequence containing the promoter and 221 bp of the 5′ end of the YIF1 gene, transcribed in the opposite direction. We hypothesized that transcription of the truncated YIF1 gene may suppress POL2 expression in our vector. Thus, we eliminated the YIF1 sequences by replacing the XhoI-SalI 5′ fragment of pRS416POL2-5′YIF1 with DNA amplified from this plasmid using PCR primers Pol2-7386bp-XhoIF (5′-ATGACTCGAGGTATGGGCCTTTGGTTTTCGT-3′) and Pol2-8161bpR (5′-GTTACACGCAATAAAGAAGTATGG-3′). The PCR product and pRS416POL2-5′YIF1 were digested with BamHI and SalI and ligated together, and E. coli were transformed with the ligation product. The entire POL2 gene from a transformant was again sequenced to verify its integrity, and this new vector (pRS416POL2) fully complemented the growth deficiency of pol2Δ yeast. We subcloned the functional POL2 fragment from pRS416POL2 into the XhoI and SalI sites of the related plasmid pRS415 (CEN6/ARS4/LEU2) (Brachmann et al. 1998) to obtain pRS415POL2. pRS416POL2 and pRS415POL2 contain the full-length POL2 coding sequence plus 368 bp upstream of the POL2 start site (corresponding to nucleotides 147844 to 155125 of yeast chromosome XIV). The pol2-4 mutation and all eex mutations were introduced into pRS415POL2 using the primers listed in supporting information, Table S1 (see also References for Supporting Tables) the QuikChange protocol (Wang and Malcolm 1999); Phusion polymerase (New England Biolabs); and the following PCR conditions: 95°, 1 min; 16× (95°, 40 sec; 53°, 1 min; 68°, 7 min). The entire pol2 gene was sequenced in each case to verify the presence of desired mutations and the absence of other mutations. All Pol δ-expressing plasmids are previously described (Herr et al. 2011a). pRS vectors (Brachmann et al. 1998) were used as templates for chromosomal gene disruptions (see below). pUG6 served as a template for kanMX (Guldener et al. 1996) and pFv199 as a template for natMX (Stulemeijer et al. 2011).
Strains:
BY4733 and Y7092 haploid yeast strains were engineered to carry alleles of DNA polymerase and MMR genes (Table S2). BY4733 (MATa leu2Δ0 ura3Δ0 met15Δ0 trp1Δ63 his3Δ200), a S288C descendant (Brachmann et al. 1998), was obtained by sporulating a BY4733 × BY4734 diploid (kindly provided by Tim Formosa, University of Utah). All engineered BY4733 strains (Table S2) originated from the same spore. Y7092 (MATα can1Δ::STE2pr_his5 lyp1Δ ura3Δ0 leu2Δ0 his3Δ1 met15Δ0), also a S288C descendant, is a BY4742 derivative (Brachmann et al. 1998) modified by Boone and colleagues to use in synthetic genetic array analyses (Tong and Boone 2007). Chromosomal gene disruptions were made using PCR products generated with Phusion polymerase (New England Biolabs) and the primers, templates, and PCR conditions indicated in Table S3. Yeast were transformed with the resulting PCR products using lithium acetate transformation (Gietz and Woods 2002). Cells from transformant colonies were treated with Zymolyase (ICN Biomedicals; 50 units/ml in 10 mM Tris–HCl/0.1 mM EDTA, pH 7.5, at 37° for 30 min and then at 95° for 10 min) and subjected to junction-specific PCR with primers in the transgenes and flanking endogenous loci to detect correct insertion/deletion mutants (primers and PCR conditions available upon request).
Plasmid shuffling
Plasmid shuffling strains contained either pol2Δ::kanMX or pol3Δ::HIS3 chromosomal gene disruptions and the URA3 plasmids pRS416POL2 or pGL310 (POL3) (Simon et al. 1991) to provide the essential activity of Pol ε or Pol δ (Table S2). Following transformation with CEN/LEU2 plasmids encoding mutant polymerases (YCplac111pol3-01, pRS415pol2-4, pRS415pol2-4,eex, or pRS415pol2-eex), transformants were selected on SC media lacking leucine and uracil. YCplac111POL3, pRS415POL2, and the pRS415-unmodified plasmid were used as positive and negative controls. Colonies (1–2 mm) from the selection plates were picked and suspended in 200 μl sterile water, and 25 μl was plated onto FOA-containing media (Boeke et al. 1984) in serial dilutions to select for spontaneous loss of the URA3 plasmid expressing the wild-type polymerase.
eex mutant screen
eex mutants were isolated using the shuffling protocol. pol2Δ msh2Δ strains with the pRS416POL2–URA3 plasmid were transformed with pRS415pol2-4–LEU2. Forty-eight individual colonies (1–2 mm) were picked from the SC media lacking uracil and leucine transformation plates and separately suspended in 240 μl sterile water, and 25-μl aliquots from each independent transformant were spotted in separate patches on FOA-containing media in a 6 × 8 grid. pRS415POL2–LEU and unmodified pRS415–LEU plasmids were used as positive and negative controls, respectively. After incubation at 30° for 3 days, individual colonies were isolated and genotyped to identify clones carrying the pol2-4 allele and no wild-type POL2. Sequences encoding the Pol ε exonuclease domain were amplified using primers Pol2-4U (5′-ATAACACTCTCAGGGGACAAGTATAT-3′) and Pol2s5 (5′-AGAATATTCGGAAAGGTGCTG-3′) and the following PCR conditions: 98°, 1 min; 30× (98°, 10 sec; 54°, 60 sec; 72°, 90 sec); 72°, 60 sec. PCR products were then digested with Alu1 at 37° for at least 6 hr. The wild-type POL2 allele results in a prominent 824-bp product as well as a number of smaller DNA fragments (275, 114, 84, 79, 53, and 17 bp). The pol2-4 mutation introduces an additional AluI site within the 824-bp fragment, resulting in 447- and 377-bp products. Using this assay, we eliminated strains with pol2-4 to POL2 gene conversions (colonies with only the POL2 allele) or other mutations (e.g., ura3) that allow cells to retain POL2 when grown on FOA (colonies with both pol2-4 and POL2). Strains containing only the pol2-4 allele were considered bona fide eex mutants. To identify eex mutants that are intragenic to pol2-4, plasmids were recovered from each mutant, introduced into E. coli, purified, and reshuffled into a fresh pol2Δ msh2Δ yeast strain. Plasmids that retained the ability to induce pol2-4 msh2Δ synthetic lethality were considered to be from strains with an eex mutation extragenic to the pol2-4 plasmid (“chromosomal eex”). Plasmids that failed to recapitulate the pol2-4 msh2Δ synthetic lethality were hypothesized to carry eex mutations within pol2-4 (“intragenic eex”). These mutations were identified by sequencing the pol2 gene. All intragenic eex alleles were re-engineered into fresh pRS415POL2 and pRS415pol2-4 plasmids and reshuffled for final confirmation of suppression. The re-engineered eex plasmids were used for all subsequent analyses.
Mutation and suppression rates
Mutation rates:
Canavanine resistance (Canr) mutation rates were measured as described (Herr et al. 2011a). Briefly, a freshly streaked pol2Δ msh6Δ strain containing pRS416POL2 was transformed with pRS415POL2, pRS415pol2-4, pRS415pol2-eex, or pRS415pol2-4,eex, and multiple transformants were plated onto FOA media to obtain shuffled colonies. Twenty-four colonies for each allele were picked, suspended in water, and vigorously vortexed to disperse the cells. Diluted aliquots from each colony were then plated onto SC (to estimate the number of cell divisions during colony formation) and canavanine selection plates (to determine the number of Canr mutants). Colonies were counted after 3–4 days at 30°. Mutation rates were calculated by the MMS maximum-likelihood method using the web-based program Fluctuation AnaLysis CalculatOR (FALCOR; http://www.keshavsingh.org/protocols/FALCOR.html) (Hall et al. 2009).
Rates of escape from error-induced extinction:
Rates of escape were determined from the plates shown in Figure 3 by counting the number of colonies in grid positions with verified eex mutants (patches with colonies that had pol2-4 → POL2 gene conversions or other FOA-resistant mutations were not included in this calculation) and the number of colonies from wild-type controls. The shuffling efficiency was assessed by plating aliquots of cells from an individual colony onto FOA (to determine the number of viable cells that lose the ura3 plasmid) and synthetic complete media (to determine the total number of viable cells plated). The percentage of cells that lose the ura3 plasmid (i.e., the shuffling efficiency) was ∼1% in our system. Therefore, colony counts were multiplied by 100, and FALCOR was used to calculate the rate of escape from error-induced extinction (Hall et al. 2009). Totals from the grid positions with verified eex mutants were entered into FALCOR as number of mutants (r), and totals from wild-type control positions were entered as number of viable cells (Nt). The FALCOR program uses these values to calculate a maximum-likelihood estimate of mutation rate expressed per cell division (see Hall et al. 2009 and references therein).
Figure 3.

Escape from error-induced extinction. (A and B) Plasmid shuffling was used to screen for eex mutants that suppress pol2-4 msh2Δ synthetic lethality. pol2Δ msh2Δ POL2–URA3 strains derived from (A) BY4733 and (B) Y7092 were transformed with pol2-4, POL2, or vector-only LEU2 plasmids. Approximately 104–105 cells from 48 independent pol2-4 transformants of each strain were spotted separately in a 6 × 8 grid on FOA-containing media to select for loss of the POL2–URA3 plasmid and to isolate suppressor mutants. Bona fide suppressors containing pol2-4 as the sole source of Pol ε arose at a rate of 1.9 × 10−4 eex mutants/cell division in the BY4733 strain [95% confidence interval (C.I.) = 2.6 × 10−4 – 1.3 × 10−4] and 4.0 × 10−3 in the Y7092 background (95% CI = 4.4 × 10−3 – 3.6 × 10−3). (C) A similar plasmid-shuffling strategy was used to estimate the rate of escape from pol3-01 msh2Δ synthetic lethality in the Y7092 background (8.8 × 10−6 eex mutants/cell division; 95% CI = 2.0 × 10−5 – 1.6 × 10−6). In C, 10-fold fewer viable cells were plated in each grid position compared to A and B. The POL2 control patches in A are two independent transformants. The POL2 and POL3 control patches in B and C are also from replicate transformants and include 10-fold dilutions of each. Rates (eex mutants/cell division) were calculated as described in Materials and Methods. Red boxes indicate grid positions magnified below each plate.
CAN1 mutation spectra
For each strain, ∼55 Canr colonies were isolated from 55 independent shuffling experiments. Cells were treated with Zymolyase, and the can1 gene was PCR-amplified and sequenced as previously described (Herr et al. 2011a). Mutation spectra were compared statistically using iMARS (Morgan and Lewis 2006) and Fisher’s exact test.
Results
Synthetic lethal interactions of Pol ε proofreading and MMR defects
To investigate error-induced extinction due to Pol ε replication errors, we introduced pol2-4, which encodes proofreading-deficient Pol ε, into newly constructed haploid strains containing deletions of individual MMR genes (msh2Δ, msh3Δ, msh6Δ, mlh1Δ, mlh3Δ, or pms1Δ). A plasmid-shuffling strategy was used, starting with MMR mutant strains that also carry a chromosomal deletion of the Pol ε gene (pol2Δ) covered by a POL2–URA3 plasmid. Following transformation with a pol2-4–LEU2 plasmid, cells were plated on 5-FOA-containing media to select for spontaneous loss of POL2–URA3, and the viabilities of the resultant pol2-4 mmrΔ double mutants were assessed following incubation at 30° for 2–3 days.
We observed that pol2-4 was synthetically lethal with msh2Δ or mlh1Δ, but not with msh3Δ, msh6Δ, pms1Δ, or mlh3Δ (Figure 1). The pol2-4 msh6Δ and pol2-4 pms1Δ strains exhibited slow-growth phenotypes, as evidenced by the reduced size and number of colonies compared to POL2msh6Δ and POL2pms1Δ strains (Figure 1A). In contrast, when pol3-01 (which encodes proofreading-deficient Pol δ) was introduced by plasmid shuffling into analogous MMR mutant strains (pol3Δ mmrΔ), synthetic lethality occurred with all MMR gene deletions except msh3Δ and mlh3Δ (Figure 1).
Figure 1.

Genetic interactions between Pol ε and Pol δ proofreading and MMR. Using a plasmid-shuffling strategy, proofreading-deficient variants of Pol ε (encoded by pol2-4) or Pol δ (encoded by pol3-01) or wild-type controls (POL2 or POL3) were introduced via LEU2 plasmids into BY4733 pol2Δ or pol3Δ strains harboring deletion mutations that partially (A) or completely (B) abrogate MMR. Serial dilutions were plated on FOA-containing media to select for loss of complementing POL2– or POL3–URA3 plasmids and reveal the synthetic phenotype. POL and MMR alleles are indicated at left and above the corresponding panels, respectively.
The pattern of pol2-4 (in)viability with each of the single MMR gene deletions suggested that complete loss of MMR is required for synthetic lethality. To further investigate this relationship, we shuffled pol2-4 into strains lacking both Msh2 partners (msh3Δ and msh6Δ) or both Mlh1 partners (pms1Δ and mlh3Δ). pol2-4 msh3Δ msh6Δ and pol2-4 pms1Δ mlh3Δ triple mutants were inviable (Figure 1B). Considered together, these data suggest that Msh2-Msh3, Msh2-Msh6, Mlh1-Pms1, and Mlh1-Mlh3 complexes all play important roles in suppressing lethal Pol ε errors and that the Msh2-Msh6 and Mlh1-Pms1 complexes predominate.
The reduced growth of strains lacking Pol ε proofreading and either Msh2-Msh6 (msh6Δ) or Mlh1-Pms1 (pms1Δ) suggests that these strains accumulate deleterious mutations that compromise replicative fitness. pol2-4 msh6Δ cells were most compromised (Figure 1A, top left), resembling strong mutator variants previously shown to exist near the maximum tolerated mutation rate of haploid yeast (Herr et al. 2011a). msh2Δ and mlh1Δ are stronger mutator alleles than msh6Δ (Greene and Jinks-Robertson 1997; Flores-Rozas and Kolodner 1998; Tran et al. 1999; Li et al. 2005), suggesting that pol2-4 msh2Δ and pol2-4 mlh1Δ cells are inviable because they exceed the maximum tolerated rate. Although pol2-4 msh2Δ cells did not form visible colonies (Figure 1B, top left), when viewed under the microscope we found abundant microcolonies of ∼100 cells. Thus, pol2-4 msh2Δ cells initially divide but fail to continue after 6–7 mitotic cycles. This pattern of abortive growth typifies cells undergoing replication error-induced extinction (Morrison et al. 1993; Herr et al. 2011a).
Pols ζ and η do not mediate pol2-4 msh2Δ synthetic lethality
The mutator phenotypes of many DNA replication mutants are dependent on the specialized DNA polymerase Pol ζ (Shcherbakova et al. 1996; Pavlov et al. 2001; Kai and Wang 2003; Northam et al. 2006; Aksenova et al. 2010; Northam et al. 2010). This dependency may reflect a role for Pol ζ in rescuing stalled DNA replication forks (Northam et al. 2010) and may relate to Pol ζ’s ability to efficiently extend primers with 3′-terminal mismatches (Johnson et al. 2000; Guo et al. 2001; Haracska et al. 2001; Simhadri et al. 2002). To determine whether Pol ζ is required for pol2-4 msh2Δ lethal mutagenesis, we used our plasmid-shuffling strategy to introduce pol2-4 into msh2Δ cells that also lack REV3 (which encodes the catalytic subunit of Pol ζ). rev3Δ did not rescue pol2-4 msh2Δ synthetic lethality (Figure 2). Similarly, deletion of RAD30, which encodes the translesion DNA polymerase η (Waters et al. 2009), also failed to rescue pol2-4 msh2Δ synthetic lethality (Figure 2). These data show that Pols ζ and η do not mediate pol2-4 msh2Δ synthetic lethality.
Figure 2.
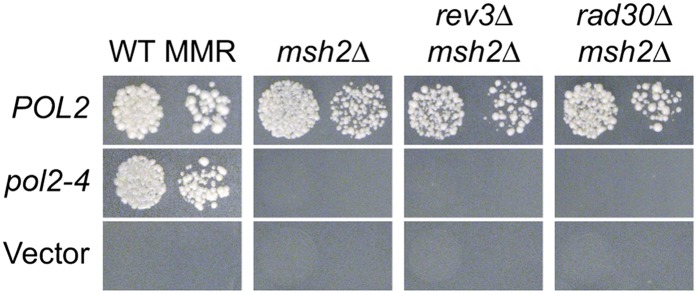
Effects of Pols ζ and η on pol2-4 msh2Δ synthetic lethality. pol2Δ msh2Δ POL2–URA3 cells defective for Pol ζ (rev3Δ) or Pol η (rad30Δ) were transformed with POL2– or pol2-4–LEU2 plasmids, and 10-fold serial dilutions of isolated transformants were plated onto FOA-containing media as in Figure 1. For comparison, the POL2– and pol2-4–LEU2 plasmids were similarly shuffled into strains that were wild type (WT) for Pols ζ and η (REV3 RAD30) and either msh2Δ or MSH2 (WT MMR). The LEU2 plasmid with no POL2 or pol2-4 gene served as the vector-only control. Colony formation was assessed after incubation at 30° for 3 days. Neither rev3Δ nor rad30Δ rescued pol2-4 msh2Δ synthetic lethality.
Mutants escape pol2-4 msh2Δ synthetic lethality
We occasionally observed macroscopic colonies that survived when pol2-4 was shuffled into the msh2Δ, mlh1Δ, msh3Δ msh6Δ, or pms1Δ mlh3Δ strains. In light of our recent discovery of Pol δ antimutators (Herr et al. 2011a), we hypothesized that these surviving colonies may have acquired suppressor mutations that improve Pol ε fidelity. Thus, we systematically screened for error-induced extinction (eex) mutants that survived pol2-4 msh2Δ synthetic lethality. Using the plasmid-shuffling strategy, multiple independent pol2-4 msh2Δ transformants were separately plated on FOA-containing medium so that rare survivors could be detected and counted (Figure 3, A and B). There was wide fluctuation in the number and size of surviving colonies, suggesting that escape variants arise randomly prior to selection on FOA.
After eliminating clones with pol2-4 → POL2 gene conversions or other mutations conferring FOA resistance (e.g., ura3), 81 independent eex mutants were isolated (Table 1). To distinguish between eex mutations in the plasmid-borne pol2-4 gene (intragenic eex) and eex mutations located elsewhere in the yeast genome (chromosomal eex), pol2-4 plasmids were rescued from the eex mutants and used to transform a fresh msh2Δ strain. Among the 81 eex mutants characterized, 76 (94%) carried plasmids that were lethal when reshuffled into msh2Δ cells. Thus, the majority of eex mutants contained suppressor mutations extragenic to pol2-4. Five mutants carried pol2-4 plasmids that did not induce pol2-4 msh2Δ synthetic lethality. DNA sequencing revealed that, in addition to the pol2-4 allele, each of these plasmids encoded a single amino-acid substitution in Pol ε: G435C, V522A, T850M, K966Q, or A1153D. The amino-acid changes were located in regions that are moderately or highly conserved in DNA polymerases ε and δ (Figure 4). When the corresponding mutations were re-engineered into fresh pol2-4 plasmid backbones and shuffled into msh2Δ cells, they conferred an eex phenotype (Figure 5A). pol2-4,eex msh2Δ strains were viable, forming small clearly visible colonies (Figure 5A, right). Moreover, the eex alleles completely rescued the slow-growth phenotype of pol2-4 msh6Δ cells (Figure 5A, center). These data show that secondary amino-acid substitutions within proofreading-deficient Pol ε can rescue pol2-4 msh2Δ synthetic lethality and restore normal growth to pol2-4 msh6Δ cells.
Table 1. Genotypes of candidate eex mutants.
| Mutants isolated from: |
|||
|---|---|---|---|
| Class of FOAr mutant | BY4733 | Y7092 | Total no. of mutants |
| eex | |||
| Intragenic to pol2-4 | 4 | 1 | 5 |
| Extragenic to pol2-4 | 50 | 26 | 76 |
| Total | 54 | 27 | 81 |
| pol2-4 → POL2 conversion | 26 | 3 | 29 |
| Other (e.g., ura3 mutations) | 23 | 0 | 23 |
| Total | 103 | 30 | 133 |
FOA-resistant (FOAr) colonies from 133 different pol2-4–LEU2 POL2–URA3 msh2Δ parent clones were isolated, and POL2 was genotyped to distinguish genuine eex mutants from pol2-4 → POL2 gene conversions and other mutations that confer FOA resistance (e.g., ura3). Clones harboring only pol2-4 were considered eex mutants. Plasmids from eex mutants were recovered and retested in a fresh msh2Δ strain to distinguish second-site suppressors within pol2-4 (“intragenic eex”) from eex mutations located extragenic to pol2-4 (“chromosomal eex”). eex mutants were isolated from two different strains: BY4733 and Y7092 (Figure 3, A and B).
Figure 4.

Amino acid changes in Pol ε eex mutants. Aligned amino-acid sequences of the catalytic subunits of Pols ε and δ (S.c. pol e, S. cerevisiae Pol ε; H.s. pol e, Homo sapiens Pol ε; S.c. pol d, S. cerevisiae Pol δ; H.s. pol d, H. sapiens Pol δ). Secondary structural elements of yeast Pol δ (Swan et al. 2009) are indicated below the alignment and color-coded to depict their domain location (as in Figure 7): rectangles, α-helices; arrows, β-strands; solid lines, loops; dotted line, structure unknown. Amino-acid substitutions encoded by Pol ε eex mutations are shown in black type above the sequence, with the corresponding yeast Pol δ residues in parentheses. Conserved polymerase and exonuclease motifs are framed in green and blue, respectively (Bernad et al. 1989; Wang et al. 1989). Three additional regions of homology (C-1, C-2, and C-3) are framed in black (Huang et al. 1999). Amino-acid conservation is indicated using the following color scheme: red, residues conserved in all four sequences; yellow, residues conserved in three sequences; gray, similar amino acids in at least three sequences.
Figure 5.

Growth and antimutator phenotypes conferred by pol2-4 intragenic eex. (A) eex mutations reverse synthetic growth defects associated with pol2-4. The pol2-4,eex mutations were re-engineered into fresh pol2-4–LEU2 plasmids and introduced into wild-type (WT) MMR, msh6Δ, or msh2Δ strains for plasmid shuffling. Strains harboring POL2– or pol2-4–LEU2 plasmids served as controls. Transformants were serially diluted and spotted onto FOA-containing media to assess colony-forming capacity after incubation at 30° for 3 days (WT MMR and msh6Δ) or 4 days (msh2Δ). (B) eex mutations confer antimutator phenotypes. Rates of spontaneous mutation, expressed as Canr mutants/cell division, were determined from multiple independent fluctuation analyses of each strain. Confidence intervals (95%) for each mutation rate are shown as error bars. The downward red arrow in the gray box indicates the antimutator effect of eex alleles on the pol2-4 mutator phenotype. Symbol patterns indicate POL2 and MSH6 allele status: black left half, POL2; black right half, MSH6; solid black, POL2 MSH6; unfilled left half, pol2-4; unfilled right half, msh6Δ; completely unfilled, pol2-4 msh6Δ. Symbol shapes indicate eex allele status (see key insert): star and hexagon, no eex; triangle, G435C; inverted triangle, V522A; circle, T850M; square, K966Q; diamond, A1153D.
Strain-dependent differences in pol2-4 msh2Δ suppression
In our screen for eex mutants, we used two related yeast strains derived from S288C: BY4733 (Brachmann et al. 1998) and Y7092 (Tong and Boone 2007). eex alleles intragenic and extragenic to pol2-4 were recovered from both strains (Table 1). However, BY4733 displayed a reproducibly lower frequency of pol2-4 msh2Δ suppression than Y7092 (compare Figures 3, A and B). Independent pol2-4 msh2Δ clones from either genetic background yielded variable numbers of escape mutants (compare different grid positions on the same plate), further indicating that suppressors originate during clonal expansion prior to plating on FOA. Accordingly, intragenic escape mutants isolated from a common parent clone (i.e., from the same grid position) harbored identical eex mutations. Based on these observations, we used fluctuation analyses to calculate the rates of escape from error-induced extinction in each strain. Transformation of POL2–URA3msh2Δ cells with the wild-type POL2–LEU2 plasmid served as a control to assess shuffling efficiencies and estimate the number of cell divisions during colony outgrowth (see Materials and Methods). Escape mutants arose at a rate of 2 × 10−4 per cell division in the BY4733 strain and at a 20-fold higher rate in the Y7092 strain (4 × 10−3 escape mutants per cell division). These results show that genetic background influences the propensity of strains to escape the synthetic lethal interaction of pol2-4 and msh2Δ.
To compare rates of escape from Pol ε and Pol δ proofreading deficiency, we quantified eex mutants in pol3-01 msh2Δ Y7092 cells using an analogous plasmid shuffling strategy (Herr et al. 2011a) (Figure 3C). Suppressors arose at a rate 450 times lower in the pol3-01 msh2Δ strain (9 × 10−6 escape mutants per cell division) compared to pol2-4 msh2Δ (4 × 10−3) in the same genetic background. Thus, escape from error-induced extinction occurs more readily in Pol ε than Pol δ proofreading-deficient msh2Δ cells.
Intragenic eex mutants suppress mutation rates
If pol2-4 msh2Δ synthetic lethality is due to error-induced extinction, then Pol ε eex substitutions may promote escape by increasing the fidelity of Pol ε. To determine whether the eex substitutions suppressed the pol2-4 mutator phenotype, we compared mutation rates conferred by pol2-4 and pol2-4,eex alleles in an msh6Δ strain derived from BY4733. All of the eex mutations suppressed the pol2-4 msh6Δ mutator phenotype between 3- and 23-fold (Figure 5B, shaded box). Thus, the eex mutations likely promote escape from pol2-4 msh2Δ synthetic lethality by lowering mutation rates below an error threshold. We also determined whether the eex alleles influence mutation rates in the presence of Pol ε proofreading by introducing each eex mutation into the otherwise wild-type POL2 gene. The resultant pol2-eex alleles had modest effects on background msh6Δ mutation rates (Figure 5B, right). pol2-G435C msh6Δ and pol2-A1153D msh6Δ were indistinguishable from POL2msh6Δ. pol2-K966Q and pol2-T850M increased the msh6Δ mutation rate two- to threefold, while pol2-V522A lowered the rate threefold.
Distinct mutation spectra of eex mutants
To gain insight into how the eex substitutions suppress mutation rates, we determined the types of mutations that spontaneously arise in pol2-4 msh6Δ and pol2-4,eex msh6Δ strains (Figure 6 and Table 2). Multiple independent Canr colonies were isolated (∼50 per strain), and the CAN1 genes were sequenced to determine the spectrum of spontaneous mutations in each strain. Mutations were distributed throughout the CAN1 sequence (Figure 6), with recurrent mutations observed at several nucleotide positions (i.e., “hotspots”). In all strains, the majority of mutations were base substitutions (Table 2), consistent with the expected synergy of a proofreading-deficient polymerase with msh6Δ (Tran et al. 1999).
Figure 6.

Spontaneous CAN1 mutations from pol2-4 and pol2-4,eex msh6Δ cells. The can1 genes from ∼50 independent Canr mutants of each strain were PCR-amplified and sequenced. Mutations identified in different strains are color-coded according to the key at the bottom. Each base letter above the wild-type CAN1 sequence indicates an independent base substitution or frameshift (+ or −) mutation. Horizontal lines indicate CAN1 sequences involved in complex mutations, duplications, and deletions. The wild-type POL2 and POL2 msh6Δ spectra are from Herr et al. (2011a).
Table 2. Types of spontaneous CAN1 mutations in pol2-4,eex msh6Δ strains.
|
pol2-4,eex msh6Δ |
||||||||
|---|---|---|---|---|---|---|---|---|
| WTa | msh6Δa | pol2-4 msh6Δ | G435C | V522A | T850Mb | K966Qb | A1153D | |
| Transitions | ||||||||
| G→A | 8 (24) | 16 (34) | 20 (36) | 19 (41) | 10 (19) | 21 (41) | 26 (48) | 21 (41) |
| C→T | 3 (9) | 2 (4) | 5 (9) | 4 (9) | 8 (15) | 4 (8) | 6 (11) | 4 (8) |
| A→G | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (2) | 0 (0) | 0 (0) |
| T→C | 0 (0) | 3 (6) | 0 (0) | 0 (0) | 0 (0) | 1 (2) | 4 (7)c | 1 (2) |
| Total | 11 (33) | 21 (45) | 25 (45) | 23 (50) | 18 (35) | 27 (51) | 36 (67)c | 26 (51) |
| Transversions | ||||||||
| G→T | 2 (6) | 5 (11) | 21 (38) | 23 (50) | 29 (56) | 6 (12)c | 12 (22) | 19 (37) |
| C→A | 3 (9) | 10 (21) | 1 (2) | 0 (0) | 0 (0) | 8 (16)c | 4 (7) | 1 (2) |
| G→C | 1 (3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (2) |
| C→G | 2 (6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| A→C | 1 (3) | 1 (2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| T→G | 1 (3) | 2 (4) | 0 (0) | 0 (0) | 0 (0) | 1 (2) | 0 (0) | 0 (0) |
| A→T | 1 (3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (2) | 0 (0) | 0 (0) |
| T→A | 1 (3) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (2) | 0 (0) | 0 (0) |
| Total | 12 (36) | 18 (38) | 22 (39) | 23 (50) | 29 (56) | 17 (33) | 16 (30) | 21 (41) |
| Frameshifts | ||||||||
| +1 | 2 (6) | 3 (6) | 9 (16) | 0 (0)c | 3 (6) | 3 (6) | 2 (4)c | 3 (6) |
| −1 | 2 (6) | 1 (2) | 0 (0) | 0 (0) | 2 (4) | 5 (10)c | 0 (0) | 1 (2) |
| 4 (12) | 4 (9) | 9 (16) | 0 (0)c | 5 (10) | 8 (16) | 2 (4)c | 4 (8) | |
| Other | 6 (18) | 4 (9) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Multiple | 0 (0) | 0 (0) | 1 (2) | 1 (2) | 1 (2) | 0 (0) | 1 (2) | 0 (0) |
| Total | 33 | 47 | 56 | 46 | 52 | 51 | 54 | 51 |
The CAN1 genes were sequenced from independent Canr mutants of each strain. The numbers of mutations of each subtype are shown with percentages in parentheses. Transitions and transversions indicate base changes observed in the coding strand of CAN1 (as in Figure 6). “Other” includes duplications, deletions, and complex mutations. Some mutants had two mutations separated by >10 bp (61–1150 bp); these are reported under ‘”Multiple”; each mutation in this category was scored as an independent event and added to the relevant subclass tally, although they may be mechanistically linked. See Figure 6 for locations of mutations in the CAN1 gene.
WT and msh6Δ data from Herr et al. (2011a).
Distribution of base substitutions significantly different from pol2-4 msh6Δ (P ≤ 0.05, Monte Carlo hypergeometric test).
Significantly different from pol2-4 msh6Δ (P ≤ 0.05, Fisher’s exact test).
We analyzed the mutation spectra using iMARS (Morgan and Lewis 2006) to compare each pol2-4,eex msh6Δ spectrum to that produced by pol2-4 msh6Δ in a pair-wise fashion. This statistical analysis considers both the type of mutation and its position in the CAN1 sequence (Figure 6). All but one eex mutant, pol2-4,A1153D msh6Δ (P = 0.10), displayed significantly different mutation spectra from the pol2-4 msh6Δ strain (P ≤ 0.05; Monte Carlo hypergeometric test). When only base substitutions are considered with no regard to sequence position (Table 2), the distributions of mutation types in pol2-4,T850M msh6Δ and pol2-4,K966Q msh6Δ were significantly different from the distribution in pol2-4 msh6Δ. Specifically, K966Q increased the percentage of T→C substitutions in the CAN1-coding sequence, and T850M increased the C→A percentage while decreasing the proportion of G→T substitutions. T850M also increased the percentage of −1 frameshifts, and G435C and K966Q decreased the proportion of +1 frameshifts.
To quantify the effects of eex alleles on individual mutation types, we converted mutation frequencies (Table 2) to mutation rates (Table 3). Only the most frequent mutation types were included in this analysis. When type-specific rates in pol2-4 msh6Δ and pol2-4,eex msh6Δ strains were compared (Table 3), several patterns of mutator suppression were evident. The two strongest eex alleles (T850M and A1153D) suppressed all mutation types to a similar degree (94–99% suppression relative to pol2-4 msh6Δ). K966Q also exhibited a nearly uniform pattern of suppression (70–94%) with some preference for +1 frameshifts. In contrast, G435C only weakly suppressed base substitutions (50–60%) while strongly suppressing +1 frameshifts (>95%), and V522A preferentially suppressed both G→A substitutions and +1 frameshifts (94–96%) with a somewhat weaker effect on C→T and G→T substitutions (80%). Collectively, the data in Tables 2 and 3 indicate that there are multiple mechanisms of mutator suppression.
Table 3. Rates of spontaneous CAN1 mutations in pol2-4,eex msh6Δ strains.
|
pol2-4,eex msh6Δ |
||||||
|---|---|---|---|---|---|---|
| pol2-4 msh6Δ | G435C | V522A | T850M | K966Q | A1153D | |
| Transitionsa | ||||||
| G→A (C•dATP) | 360 (1.0) | 150 (0.4) | 20 (0.06) | 18 (0.05) | 120 (0.3) | 25 (0.07) |
| C→T (G•dTTP) | 90 (1.0) | ∼32 (∼0.4) | 16 (0.2) | ∼3 (∼0.04) | 27 (0.3) | ∼5 (∼0.05) |
| Transversionsa | ||||||
| G→T (C•dTTP) | 380 (1.0) | 180 (0.5) | 58 (0.2) | 5 (0.01) | 55 (0.1) | 22 (0.06) |
| C→A (G•dATP) | ∼18 (1.0) | <8 (ND) | <2 (ND) | 7 (ND) | ∼18 (ND) | ∼1 (ND) |
| Frameshifts | ||||||
| +1 | 160 (1.0) | <8 (<0.05) | ∼6 (∼0.04) | ∼3 (∼0.02) | ∼9 (∼0.06) | ∼4 (∼0.02) |
| −1 | <18 (1.0) | <8 (ND) | ∼4 (ND) | 4 (ND) | <5 (ND) | ∼1 (ND) |
| Overall | 1001 (1.0) | 365 (0.36) | 104 (0.10) | 43 (0.04) | 247 (0.25) | 60 (0.06) |
Expressed as the number of Canr mutants per cell division (×10−7). Rates for individual mutation types were calculated by multiplying the overall Canr mutation rate of a strain (last row of table) by the percentage of the corresponding mutation in the CAN1 mutation spectrum from that strain (Table 2). Rates relative to those in pol2-4 msh6Δ are in parentheses. Values were calculated to three decimal places and then rounded to one or two significant figures. < or ∼: incidence values (Table 2) were zero or not significantly different from zero, respectively (P > 0.05, Fisher’s exact test). ND: relative rates could not be reliably determined because mutations of this type were not detected at significant levels in the pol2-4 msh6Δ strain (Table 2).
Base changes in the CAN1 coding sequence are shown with presumed causal mispairs in parentheses (template base•incoming dNTP), if Pol ε catalyzes leading-strand DNA synthesis (Pursell et al. 2007; Kunkel and Burgers 2008; Pavlov and Shcherbakova 2010) initiated from the ARS507 origin of replication (Raghuraman et al. 2001; Yabuki et al. 2002; Kumar et al. 2011; Siow et al. 2012).
Discussion
Adaptation necessitates genetic diversity. Large cell populations with low DNA replication error rates may contain sufficient variation to surmount a single selective barrier. However, rapid environmental changes impose additional selection pressures that may exceed the ability of wild-type cells to adapt. Natural populations of bacteria and yeast often harbor low levels of mutator cells that emerge under such conditions (Giraud et al. 2001b; Elena and Lenski 2003). However, sustained expression of mutator phenotypes compromises long-term fitness through the accumulation of deleterious mutations. Thus, following adaptation to external pressures, selection would favor cells that have either evolved mutator suppressors or eliminated the mutator allele. In this manner, mutator phenotypes rise and fall as cells shift between periods of relative stability and environmental change (Taddei et al. 1997; Giraud et al. 2001b).
Here we investigated pathways of mutator suppression that restore fidelity to an error-prone variant of Pol ε, the primary leading-strand DNA polymerase in yeast (Pursell et al. 2007; Nick McElhinny et al. 2008). We show that cells defective for both Pol ε proofreading and MMR are inviable (Figure 1). However, spontaneous mutants readily escape this synthetic lethality (Figure 3). Five percent of these escape mutants encode secondary amino-acid substitutions in Pol ε (Figure 4) that suppress the Pol ε proofreading-deficient phenotype (Figure 5). This study complements our recent investigation of Pol δ (Herr et al. 2011a), where we defined the maximal mutation rate sustainable by haploid yeast cells and described variants that suppress the mutator phenotype of cells deficient for Pol δ proofreading. Our findings highlight the unstable nature of mutator phenotypes and suggest multiple mechanisms for their suppression.
Pol ε errors and lethal mutagenesis
It is well established that combined defects in Pol δ proofreading and MMR are synthetically lethal in haploid yeast (Morrison et al. 1993; Tran et al. 1999; Greene and Jinks-Robertson 2001). Loss of Msh6 or Pms1 (msh6Δ or pms1Δ) is sufficient to extinguish Pol δ proofreading-deficient (pol3-01) cells (Figure 1), the apparent consequence of mutation accumulation during DNA replication (Morrison et al. 1993; Herr et al. 2011a). Consistent with a mechanism of error-induced lethality, yeast cells with Pol δ proofreading and MMR defects form microcolonies of ∼100 cells arrested at various stages of the cell cycle (Morrison et al. 1993) and with diverse cell morphologies resembling inactivation of different essential genes (Yu et al. 2006). The maximum tolerated mutation rate in haploid yeast (∼10−3 inactivating mutations/gene/cell division ≈ 3 × 10−6 mutations/base pair/cell division) is consistent with random inactivation of essential genes as the cause of extinction (Herr et al. 2011a).
In contrast to Pol δ, the combined loss of Pol ε proofreading (pol2-4) and Msh6 (msh6Δ) is not synthetically lethal, although it does compromise growth (Figure 1). The different fates of msh6Δ cells with defects in Pol δ proofreading (inviable) or Pol ε proofreading (slow-growing, strong mutators) are likely due to quantitative differences in cellular mutation burden. While pol3-01 msh6Δ cells exceed the maximum tolerated mutation rate (Herr et al. 2011a), pol2-4 msh6Δ cells mutate at a rate 10 times lower than the maximum (10−4 Canr mutants/cell division ≈ 10−4 inactivating mutations/gene/cell division) (Figure 5B). We observe that pol2-4 cells become inviable when both Msh6- and Msh3-dependent MMR are disrupted (Figure 1B). Thus, the additional burden of unrepaired Msh2-Msh3 substrates is sufficient to push pol2-4 msh6Δ cells over the lethal threshold. Similarly, pol2-4 pms1Δ cells become inviable when MLH3 is deleted. Our finding that unrepaired Pol ε errors are lethal provides additional evidence that Pol ε plays a substantial role in genome replication (Pursell and Kunkel 2008). Our data also indicate that loss of genetic information on either DNA strand [Pol ε errors on leading-strands and Pol δ errors on lagging-strands (Pursell et al. 2007; Nick McElhinny et al. 2008)] is sufficient to drive extinction of a population of cells.
MMR pathways suppressing lethal Pol ε errors
The MMR pathways that suppress deleterious Pol ε errors are sharply delineated in proofreading-MMR double mutants (Figure 1). Pol ε’s bias for generating more base substitutions than frameshifts (Shcherbakova et al. 2003) is consistent with our observation that pol2-4 cells defective for base-base mismatch repair (msh6Δ or pms1Δ) exhibit slow-growth phenotypes, while pol2-4 cells defective primarily for frameshift repair (msh3Δ or mlh3Δ) grow normally (Figure 1). Morrison and Sugino (1994) also observed compromised growth of pol2-4 pms1Δ cells. Surprisingly, we found that deletion of MSH6 impacted cell growth more severely than deletion of PMS1 (Figure 1A), even though Msh2-Msh6 mediates repair through its interaction with Mlh1-Pms1 (Habraken et al. 1998; Iyer et al. 2006; Hsieh and Yamane 2008). This indicates that some Msh6-dependent repair can occur in the absence of Mlh1-Pms1, perhaps by using Mlh1-Mlh3 in place of Mlh1-Pms1 (Cannavo et al. 2005). Our observation that pol2-4 pms1Δ cells become inviable when MLH3 is deleted (Figure 1) further implicates Mlh1-Mlh3 in the repair of deleterious Pol ε errors. Thus, both Pms1 and Mlh3 play important protective roles. Synthetic lethality with pol2-4 occurs only when both Mlh1-Pms1 and Mlh1-Mlh3 are eliminated by deleting either MLH1 alone or PMS1 and MLH3 together (Figure 1). Mouse Mlh1, Mlh3, and Pms2 (equivalent to yeast PMS1) have a similar relationship in preventing spontaneous mutations and cancer (Chen et al. 2005). Considered together, our data show that multiple MMR components protect cells from deleterious Pol ε errors, which include base substitutions, frameshifts, and possibly other mutation types (Harrington and Kolodner 2007).
Contribution of Pol ε errors to cellular mutation burden
What accounts for the differences in synthetic phenotypes of pol2-4 and pol3-01 with MMR defects? Biochemical studies indicate that exonuclease-deficient Pols ε and δ have similar overall fidelities in vitro (Shimizu et al. 2002; Shcherbakova et al. 2003; Fortune et al. 2005). They generate distinct mutational spectra, but both primarily produce base-base mispairs and frameshift errors, the preferred substrates for MMR (Kolodner and Marsischky 1999; Iyer et al. 2006; Hsieh and Yamane 2008). Nevertheless, in MMR-proficient cells, the pol3-01 mutation rate is 5–30 times higher than that of pol2-4 (Morrison and Sugino 1994; Shcherbakova et al. 1996; Tran et al. 1999; Datta et al. 2000; Karthikeyan et al. 2000; Greene and Jinks-Robertson 2001; Pavlov et al. 2004). MMR preferentially repairs lagging-strand errors (Pavlov et al. 2003; Hombauer et al. 2011). Consistent with this bias, we observe that deletion of MSH6 increases the mutation rate of pol3-01 variants ∼160-fold (Herr et al. 2011a) and the pol2-4 mutation rate ∼50-fold (Figure 5B). Shcherbakova et al. (2003) see a similar impact of msh6Δ on pol2-4 mutation rates (40- to 50-fold). However, this preference of MMR for Pol δ errors contradicts the observation that loss of Pol δ proofreading results in higher mutation rates than loss of Pol ε proofreading.
There are several possible explanations for this apparent discrepancy. As previously suggested by others (Morrison and Sugino 1994; Pavlov and Shcherbakova 2010), Pol ε may replicate less of the genome than Pol δ. Replacement of Pol ε with Pol δ near the end of a replicon may be necessary to enable ligation of the leading strand to the downstream Okazaki fragment from an adjacent replicon (Garg et al. 2004). If replacement occurs randomly or in response to Pol ε errors or pausing at DNA lesions (Pavlov and Shcherbakova 2010), then Pol δ would synthesize variable amounts of each leading-strand fragment and overall more of the genome than Pol ε. Gap-filling synthesis is also catalyzed predominantly by Pol δ during DNA repair, recombination, and telomere replication (Pavlov et al. 2006b).
Another possibility is that Pol ε creates fewer mutations in vivo than predicted by in vitro measurements of fidelity. The overall error rate of Pol ε in vivo should approximately correspond to the mutation rate of cells that lack both Pol ε proofreading and MMR. pol2-4 msh6Δ cells generate 10−4 Canr mutants/cell division or ∼3 × 10−7 mutations/base pair/cell division (calculated as described in Herr et al. 2011a). However, this likely underestimates the Pol ε error rate. Other enzymes may excise Pol ε errors [e.g., Pol δ may proofread for Pol ε (Pavlov and Shcherbakova 2010)], and Msh2-Msh3-mediated MMR, which is still active in these cells, also repairs some Pol ε errors. Tran et al. (1999) show that loss of Msh2-Msh3 (msh3Δ) further increases the mutation rate of pol2-4 msh6Δ cells about threefold, while we find that msh3Δ is lethal in pol2-4 msh6Δ cells and therefore must increase mutations ≥10-fold. A 10-fold increase corresponds to ∼3 × 10−6 mutations/base pair, which is substantially lower than the error rate of proofreading-deficient Pol ε in vitro [∼3 × 10−4 errors/base pair (Shcherbakova et al. 2003)]. Furthermore, the spectra of mutations observed in vivo and in vitro are quite different (Figure 6 and Table 2) (Shcherbakova et al. 2003). These differences are not likely due to differential extension of mispairs in vitro because Pol ε is present in great excess to maximize mispair extension in the M13 fidelity assay (Bebenek and Kunkel 1995; Shcherbakova et al. 2003). Thus, in vitro fidelity measurements do not recapitulate the mutational events caused by Pol ε in vivo. As previously suggested (Shcherbakova et al. 2003), Pols ε and δ may replicate similar amounts of the yeast genome, but may do so with different accuracies, perhaps by utilizing strand- or polymerase-specific accessory factors or repair pathways that decrease mutations from Pol ε or by triggering the mutagenic Dun1 pathway that increases mutations from Pol δ (Datta et al. 2000; Reha-Krantz et al. 2011). Interestingly, the error-prone DNA polymerases ζ and η do not significantly impact mutagenesis by proofreading-deficient Pol ε or Pol δ (Figure 2) (Shcherbakova et al. 1996; Datta et al. 2000). Additional studies are required to examine in greater detail the roles of Pol ε and Pol δ in vivo and the pathways that mediate mutagenesis when these polymerases err.
Escape from Pol ε error-induced extinction
In our plasmid-shuffling experiments, colonies frequently emerged that escape pol2-4 msh2Δ lethality (Figure 3). This escape from Pol ε error-induced extinction is similar to that recently described for Pol δ (Herr et al. 2011a). Morrison and Sugino (1994) also observed mutator suppression in a pol2-4 pms1 clone, but this clone was not further characterized. In our studies, escape results from genetic suppressors that are either second-site mutations within pol2-4 (intragenic suppressors) or alleles affecting unknown genes elsewhere in the genome and extragenic to pol2-4 (chromosomal suppressors) (Table 1). Five intragenic suppressors were identified (Figure 4). Each of these individually conferred escape from pol2-4 msh2Δ lethality (Figure 5A) and suppressed the mutator phenotype of Pol ε proofreading deficiency 3- to 23-fold (Figure 5B). These data strongly suggest that pol2-4 msh2Δ inviability results from lethal mutagenesis and that intragenic suppressors lower the spontaneous mutation rate below the maximum tolerated threshold. The existence of a relatively narrow lethal threshold was previously shown using a collection of pol3 mutator and antimutator alleles (Herr et al. 2011a). Our new data with pol2 alleles validate this threshold and are consistent with a maximal mutation rate in haploid yeast of ∼10−3 inactivating mutations/gene/cell division (Herr et al. 2011a).
Both pol3-01 msh6Δ and pol2-4 msh2Δ strains exceed the lethal error threshold, and antimutator variants that lower mutation rates below the threshold were readily obtained in both strains (data herein and Herr et al. 2011a). Yet chromosomal suppressors of pol2-4 msh2Δ lethality represented a greater proportion of eex mutants (94%) (Table 1) than those that suppressed pol3-01 msh6Δ lethality (65%) (Herr et al. 2011a). We also observed that eex mutants arise more readily in Pol ε than in Pol δ proofreading-deficient msh2Δ cells (Figure 3, B and C). This may reflect differences in how close the mutation rates of the strains are to the lethal threshold. Both weak and strong antimutators would rescue pol2-4 msh2Δ and pol3-01 msh6Δ cells, which reside relatively close to the threshold (see above and Herr et al. 2011a). In contrast, the combination of pol3-01 and msh2Δ imparts a much higher mutation rate, which would be overcome by only the strongest antimutators. It is also possible that a wider array of genes influences Pol ε replication fidelity than Pol δ fidelity, and thus the probability of isolating chromosomal suppressor alleles is greater for Pol ε. pol3-01 msh6Δ cells still have Msh2-Msh3 MMR activity that protects against frameshift mutations (Marsischky et al. 1996; Palombo et al. 1996), while Pol ε error-induced extinction depends on abrogation of the Msh2-Msh3 pathway (Figure 1). Thus, the higher proportion of chromosomal suppressors in pol2-4 msh2Δ cells may also be due to mutations in genes encoding replication components that affect frameshift mutagenesis.
An unexpected finding from our study is that different strains escape from error-induced extinction at different rates (Figure 3). The facile emergence of pol2-4 msh2Δ eex mutants in Y7092 compared to BY4733 cells suggests that the former strain acquired a weak antimutator prior to our screening experiments. A pre-existing weak antimutator allele may be insufficient to trigger escape but could cooperate with a second nascent antimutator to shift mutation rates below the lethal threshold and into the viable range. Alternatively, a pre-existing weak mutator allele in Y7092 cells may generate suppressors at a higher rate. However, the infrequency of FOA resistance due to ura3 and other mutations is inconsistent with a higher overall mutation rate in Y7092 (Table 1). The Y7092 strain has undergone a number of genetic manipulations (Tong and Boone 2007), thus providing opportunity for variants to arise. These data suggest that strain differences or the presence of unknown suppressor alleles may explain inconsistencies in the literature regarding Pol ε proofreading and MMR synthetic lethal interactions (Morrison and Sugino 1994; Tran et al. 1999; Greene and Jinks-Robertson 2001).
Possible mechanisms of mutator suppression by Pol ε variants
Our screen for suppressors uncovered five novel Pol ε antimutator variants: G435C, V522A, T850M, K966Q, and A1153D (Figure 4). The Pol ε structure is unknown, but the positions of antimutator residues in Pol ε can be deduced from structures of related B-family DNA polymerases (Wang et al. 1997; Swan et al. 2009). Mapping the Pol ε eex substitutions onto the S. cerevisiae Pol δ structure indicates that the Pol ε substitutions are scattered throughout the domains of the protein (Figure 7A), similar to the wide distribution of eex substitutions found in Pol δ (Figure 7B) (Herr et al. 2011a). No eex mapped to the C-terminal half of Pol ε, which performs an essential, noncatalytic role in yeast cells (Dua et al. 1999; Kesti et al. 1999; Feng and D’Urso 2001) and is not present in Pol δ or other B-family DNA polymerases.
Figure 7.

Locations of Pol ε eex amino-acid substitutions mapped onto the Pol δ structure. (A) Overall distribution of Pol ε eex amino-acid substitutions. The catalytic subunit of yeast Pol δ is depicted as a ribbon diagram with the following color-coded elements: exonuclease domain, red; thumb domain, green; fingers domain, blue; palm domain, purple; amino domain, gray; DNA template strand, brown sticks; primer strand, tan sticks; catalytic carboxylate residues in the polymerase and exonuclease active sites, gray CPK sticks; metal ions, small black spheres; incoming dCTP, green CPK sticks; template G nucleotide, orange CPK sticks. Locations of eex-encoded changes are shown as yellow spheres and labeled by the Pol ε amino-acid substitution with the corresponding Pol δ residue in parentheses. The A1153D substitution is not pictured because it falls in a region where the Pol δ structure is unknown. (B) Locations of Pol ε eex substitutions (yellow spheres) relative to Pol δ eex substitutions (aqua spheres; see (Herr et al. 2011a). The purple sphere at the Exo-Thumb interface is Pol ε G435C, which aligns with Pol δ R475I/G. (C, D, E, and F) Close-up depictions of Pol ε eex substitutions. Important residues are highlighted as space-filling spheres, with yellow indicating positions corresponding to Pol ε eex substitutions. Structure of S. cerevisiae Pol δ is from Swan et al. (2009) (Protein Data Bank accession code 3IAY).
The locations of the eex-encoded amino-acid substitutions (Figure 7) and the mutation spectra resulting from each (Figure 6 and Tables 2 and 3) suggest several possible antimutator mechanisms for the Pol ε variants. Two amino-acid substitutions, T850M and K966Q, are within the predicted palm domain of Pol ε (Figure 7A). T850 is in conserved motif B (Figure 4), proximal to the polymerase active site (Figure 7, A and C) and adjacent to Y831, a highly conserved residue, which, when mutated, also suppresses the pol2-4 mutator phenotype (Pavlov et al. 2004). This suggests that T850M increases Pol ε fidelity through its interaction with Y831, thereby changing the active-site geometry and reducing the formation or extension of mispairs and strand slippage intermediates (Table 3). The K966Q eex substitution, also located in the palm domain, maps to the first lysine in an absolutely conserved KKRYA sequence (Figure 4) that interacts with the minor groove of the primer•template (Figure 7, A and D). K966Q significantly decreased the rate of +1 frameshift mutations (Table 3), suggesting that K966Q decreases Pol ε pausing through homonucleotide runs, thereby reducing the opportunity for slippage (Viguera et al. 2001; Johnson et al. 2003).
Interestingly, the T850M and K966Q antimutator variants confer a mild mutator phenotype (two- to threefold) in msh6Δ cells when the catalytic residues of Pol ε’s exonuclease are restored to wild type (Figure 5B). Studies of T4 antimutators show that enhanced discrimination against some errors is commonly accompanied by weakened discrimination against others (Drake 1993). Polymerase variants with increased fidelity often have reduced overall activity (Loh et al. 2007), which can improve accuracy (Clayton et al. 1979; Kunkel et al. 1994; Joyce and Benkovic 2004; Johnson 2010; Herr et al. 2011b), but will also increase error rates for some mutation types (Kunkel et al. 1994) and may trigger mutagenic replication processes in the cell (Northam et al. 2006; Aksenova et al. 2010; Northam et al. 2010). Additionally, T850M and K966Q would increase mutagenesis if they hamper partitioning of nascent errors to the exonuclease active site (Donlin et al. 1991; Reha-Krantz 2010).
The remaining Pol ε antimutator substitutions affect other domains in the polymerase structure (Figures 4 and 7). V522A maps to the amino domain in an α-helix previously implicated in pol3-01 mutator suppression (Figure 7, A, B, and E) (Herr et al. 2011a). The same types of spontaneous mutations arise in pol2-4 msh6Δ cells with or without the V522A allele (Table 2). Thus, V522A suppresses the most prevalent mutations generated by proofreading-deficient Pol ε to a similar degree. This pattern of suppression is consistent with a mutant polymerase that exhibits either increased overall nucleotide selectivity or increased facility to undergo extrinsic proofreading (Albertson and Preston 2006; Nick McElhinny et al. 2006; Pavlov et al. 2006a). The G435C antimutator maps to Pol δ R475 in a loop of the exonuclease domain that extends into and interacts with the thumb domain (Figure 7, A and F). We previously identified R475 substitutions in Pol δ that suppress pol3-01 msh6Δ synthetic lethality (Herr et al. 2011a). The recurrence of eex mutants with substitutions at this position is striking and suggests that Pols δ and ε share structural features that govern fidelity. Finally, the Pol ε A1153D antimutator likely resides in the thumb domain of the polymerase (Figure 4), although the structure of this region in Pol δ is unknown (Swan et al. 2009). A1153D suppressed the overall mutation rate 17-fold (Figure 5B), but did not significantly alter the mutation spectrum (Tables 2 and 3). Thus, A1153D may uniformly increase dNTP selectivity by Pol ε or may promote uniform extrinsic proofreading of Pol ε errors.
Perspectives and conclusions
Previous studies identified antimutator variants of bacteriophage T4 and E. coli DNA polymerases (reviewed in Reha-Krantz 1995; Schaaper 1998; Herr et al. 2011b). Similar to the yeast variants that we describe (data herein and Herr et al. 2011a), E. coli antimutators were isolated in the absence of native proofreading and are due to individual amino-acid substitutions throughout the polymerase structures (Schaaper 1998; Loh et al. 2007; Herr et al. 2011b). Accordingly, Schaaper and colleagues proposed two general mechanisms for polymerase antimutators: (1) increased dNTP discrimination and (2) increased dissociation from DNA to allow editing by alternate pathways (Fijalkowska and Schaaper 1995). Our data are consistent with these mechanisms. Biochemical analyses will be required to assess the fidelities and processivities of our Pol ε variants and the potential contributions of these parameters to antimutagenesis.
The idea of dissociation and alternative editing raises the question: What other enzymes might remove Pol ε errors in the antimutator strains? One candidate is proofreading by Pol δ. Yeast defective for both Pol δ and Pol ε proofreading exhibit a synergistic increase in mutation rate, suggesting that one or both polymerases may proofread for the other (Morrison and Sugino 1994). Pavlov and Shcherbakova (2010) present evidence that mistakes made by Pol ε are corrected by the proofreading activity of Pol δ, but not vice versa. Other candidates for extrinsic proofreading include the 3′→5′ exonuclease activities of Mre11 (Trujillo and Sung 2001) and Apn2 (Unk et al. 2001) or endonucleases such as Rad1/Rad10 or Mus81/Mms4 that cleave 3′ flap structures during replication fork restart (Bardwell et al. 1994; Boddy et al. 2001; Chen et al. 2001; Kaliraman et al. 2001; Bastin-Shanower et al. 2003).
The majority of eex mutants isolated in our screen were extragenic to pol2-4 (Table 1). Which genes mediate antimutagenesis in these eex mutants? What are the functions of the proteins affected by these eex alleles? And what are the roles of the proteins in eukaryotic DNA replication? Antimutators could result from upregulation of DNA repair or recombination proteins that, when overexpressed, are able to remove nascent 3′ mispairs or mismatches in duplex DNA. MMR proteins, other 3′ nucleases (see above), and recombination proteins and resolvases are potential candidates for this mechanism. Antimutators could also result from changes in proteins that function at the DNA replication fork. Proteins directly affecting stalled replication forks are candidates as are checkpoint sensors and indirect effectors of these processes. The error-prone Pols ζ and η are unlikely eex candidates because neither polymerase is required for pol2-4 msh2Δ synthetic lethality (Figure 2). Previous studies show that the mutator phenotype caused by defective Pol δ proofreading (pol3-01) is partially dependent on Dun1 (Datta et al. 2000), a checkpoint kinase that up-regulates dNTP synthesis (Zhou and Elledge 1993; Zhao and Rothstein 2002; Tsaponina et al. 2011). However, the pol2-4 mutator phenotype does not appear dependent on Dun1 (Datta et al. 2000). Thus, mutations in DUN1 are unlikely eex candidates. There are a number of other possible antimutagenesis mechanisms involving multiple pathways and candidate genes (Herr et al. 2011b). Whole-genome sequencing should greatly facilitate identification of the chromosomal eex alleles (Birkeland et al. 2010; Wenger et al. 2010; Dunham 2012). These eex alleles may reveal novel pathways of mutation suppression and help clarify the division of labor between Pols ε and δ at the replication fork.
Our studies illustrate the inherent instability of eukaryotic mutators. Antimutators readily emerge from yeast harboring defects in either Pol ε or Pol δ proofreading (Figure 3) (Herr et al. 2011a). Suppressors of diverse mutator phenotypes (proofreading, MMR, and DNA damage repair) also frequently arise in E. coli (Tröbner and Piechocki 1984; Schaaper and Cornacchio 1992; Fijalkowska et al. 1993; Fijalkowska and Schaaper 1995; Schaaper 1996; Giraud et al. 2001b; Notley-McRobb et al. 2002). Thus, it appears that mutator phenotypes in general are prone to suppression. We speculate that antimutators moderate high mutation rates and minimize deleterious mutations during microbial adaptation and mammalian oncogenesis.
Supplementary Material
Acknowledgments
We thank Tim Formosa and Brian Piening for yeast strains; Masanori Ogawa, Michel Simon, and Gerard Faye for plasmid constructions; and Scott Kennedy for critical reading of the manuscript. This work was supported by the National Institutes of Health (R01 ES09927, R01 CA098243, R01 CA111582, P20 CA103728, and P01 AG01751) and the National Institute of Environmental Health Sciences-sponsored Center for Ecogenetics and Environmental Health at the University of Washington (P30 ES07033). L.N.W. was supported by a Public Health Service National Research Service Award (T32 GM07270). A.J.H. held a Hitchings-Elion Fellowship from the Burroughs Wellcome Fund.
Footnotes
Communicating editor: J. A. Nickoloff
Literature Cited
- Aksenova A., Volkov K., Maceluch J., Pursell Z. F., Rogozin I. B., et al. , 2010. Mismatch repair-independent increase in spontaneous mutagenesis in yeast lacking non-essential subunits of DNA polymerase epsilon. PLoS Genet. 6: e1001209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertson T. M., Preston B. D., 2006. DNA replication fidelity: proofreading in trans. Curr. Biol. 16: R209–R211. [DOI] [PubMed] [Google Scholar]
- Albertson T. M., Ogawa M., Bugni J. M., Hays L. E., Chen Y., et al. , 2009. DNA polymerase ε and δ proofreading suppress discrete mutator and cancer phenotypes in mice. Proc. Natl. Acad. Sci. USA 106: 17101–17104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell A. J., Bardwell L., Tomkinson A. E., Friedberg E. C., 1994. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 265: 2082–2085. [DOI] [PubMed] [Google Scholar]
- Bastin-Shanower S. A., Fricke W. M., Mullen J. R., Brill S. J., 2003. The mechanism of Mus81-Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1-Rad10. Mol. Cell. Biol. 23: 3487–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebenek K., Kunkel T. A., 1995. Analyzing fidelity of DNA polymerases. Methods Enzymol. 262: 217–232. [DOI] [PubMed] [Google Scholar]
- Bernad A., Blanco L., Lazaro J. M., Martin G., Salas M., 1989. A conserved 3′→5′ exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell 59: 219–228. [DOI] [PubMed] [Google Scholar]
- Birkeland S. R., Jin N., Ozdemir A. C., Lyons R. H., Jr, Weisman L. S., et al. , 2010. Discovery of mutations in Saccharomyces cerevisiae by pooled linkage analysis and whole-genome sequencing. Genetics 186: 1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy M. N., Gaillard P. H., McDonald W. H., Shanahan P., Yates J. R., III, et al. , 2001. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107: 537–548. [DOI] [PubMed] [Google Scholar]
- Boeke J. D., LaCroute F., Fink G. R., 1984. A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol. Gen. Genet. 197: 345–346. [DOI] [PubMed] [Google Scholar]
- Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., et al. , 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14: 115–132. [DOI] [PubMed] [Google Scholar]
- Cannavo E., Marra G., Sabates-Bellver J., Menigatti M., Lipkin S. M., et al. , 2005. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Res. 65: 10759–10766. [DOI] [PubMed] [Google Scholar]
- Chao L., Cox E. C., 1983. Competition between high and low mutating strains of Escherichia coli. Evolution 37: 125–134. [DOI] [PubMed] [Google Scholar]
- Chen P. C., Dudley S., Hagen W., Dizon D., Paxton L., et al. , 2005. Contributions by MutL homologues Mlh3 and Pms2 to DNA mismatch repair and tumor suppression in the mouse. Cancer Res. 65: 8662–8670. [DOI] [PubMed] [Google Scholar]
- Chen X. B., Melchionna R., Denis C. M., Gaillard P. H., Blasina A., et al. , 2001. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol. Cell 8: 1117–1127. [DOI] [PubMed] [Google Scholar]
- Clayton L. K., Goodman M. F., Branscomb E. W., Galas D. J., 1979. Error induction and correction by mutant and wild type T4 DNA polymerases. Kinetic error discrimination mechanisms. J. Biol. Chem. 254: 1902–1912. [PubMed] [Google Scholar]
- Datta A., Schmeits J. L., Amin N. S., Lau P. J., Myung K., et al. , 2000. Checkpoint-dependent activation of mutagenic repair in Saccharomyces cerevisiae pol3–01 mutants. Mol. Cell 6: 593–603. [DOI] [PubMed] [Google Scholar]
- Desai M. M., Fisher D. S., Murray A. W., 2007. The speed of evolution and maintenance of variation in asexual populations. Curr. Biol. 17: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlin M. J., Patel S. S., Johnson K. A., 1991. Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry 30: 538–546. [DOI] [PubMed] [Google Scholar]
- Drake J. W., 1993. General antimutators are improbable. J. Mol. Biol. 229: 8–13. [DOI] [PubMed] [Google Scholar]
- Drake J. W., Charlesworth B., Charlesworth D., Crow J. F., 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dua R., Levy D. L., Campbell J. L., 1999. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae Pol ε and its unexpected ability to support growth in the absence of the DNA polymerase domain. J. Biol. Chem. 274: 22283–22288. [DOI] [PubMed] [Google Scholar]
- Dunham M. J., 2012. Two flavors of bulk segregant analysis in yeast. Methods Mol. Biol. 871: 41–54. [DOI] [PubMed] [Google Scholar]
- Elena S. F., Lenski R. E., 2003. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat. Rev. Genet. 4: 457–469. [DOI] [PubMed] [Google Scholar]
- Estes S., Phillips P. C., Denver D. R., Thomas W. K., Lynch M., 2004. Mutation accumulation in populations of varying size: the distribution of mutational effects for fitness correlates in Caenorhabditis elegans. Genetics 166: 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W., D’Urso G., 2001. Schizosaccharomyces pombe cells lacking the amino-terminal catalytic domains of DNA polymerase epsilon are viable but require the DNA damage checkpoint control. Mol. Cell. Biol. 21: 4495–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowska I. J., Schaaper R. M., 1995. Effects of Escherichia coli dnaE antimutator alleles in a proofreading-deficient mutD5 strain. J. Bacteriol. 177: 5979–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowska I. J., Dunn R. L., Schaaper R. M., 1993. Mutants of Escherichia coli with increased fidelity of DNA replication. Genetics 134: 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Rozas H., Kolodner R. D., 1998. The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc. Natl. Acad. Sci. USA 95: 12404–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune J. M., Pavlov Y. I., Welch C. M., Johansson E., Burgers P. M., et al. , 2005. Saccharomyces cerevisiae DNA polymerase δ: high fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J. Biol. Chem. 280: 29980–29987. [DOI] [PubMed] [Google Scholar]
- Fox E. J., Salk J. J., Loeb L. A., 2009. Cancer genome sequencing: an interim analysis. Cancer Res. 69: 4948–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., et al. , 2006. DNA Repair and Mutagenesis. ASM Press, Washington, DC. [Google Scholar]
- Funchain P., Yeung A., Stewart J. L., Lin R., Slupska M. M., et al. , 2000. The consequences of growth of a mutator strain of Escherichia coli as measured by loss of function among multiple gene targets and loss of fitness. Genetics 154: 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg P., Stith C. M., Sabouri N., Johansson E., Burgers P. M., 2004. Idling by DNA polymerase δ maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 18: 2764–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R. D., Woods R. A., 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method, pp. 87–96 in Part B: Guide to Yeast Genetics and Molecular and Cell Biology, edited by Guthrie C., Fink G. R. Academic Press, New York. [DOI] [PubMed] [Google Scholar]
- Giraud A., Matic I., Tenaillon O., Clara A., Radman M., et al. , 2001a Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291: 2606–2608. [DOI] [PubMed] [Google Scholar]
- Giraud A., Radman M., Matic I., Taddei F., 2001b The rise and fall of mutator bacteria. Curr. Opin. Microbiol. 4: 582–585. [DOI] [PubMed] [Google Scholar]
- Goldsby R. E., Hays L. E., Chen X., Olmsted E. A., Slayton W. B., et al. , 2002. High incidence of epithelial cancers in mice deficient for DNA polymerase δ proofreading. Proc. Natl. Acad. Sci. USA 99: 15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene C. N., Jinks-Robertson S., 1997. Frameshift intermediates in homopolymer runs are removed efficiently by yeast mismatch repair proteins. Mol. Cell. Biol. 17: 2844–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene C. N., Jinks-Robertson S., 2001. Spontaneous frameshift mutations in Saccharomyces cerevisiae: accumulation during DNA replication and removal by proofreading and mismatch repair activities. Genetics 159: 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldener U., Heck S., Fielder T., Beinhauer J., Hegemann J. H., 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 24: 2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D., Wu X., Rajpal D. K., Taylor J. S., Wang Z., 2001. Translesion synthesis by yeast DNA polymerase ζ from templates containing lesions of ultraviolet radiation and acetylaminofluorene. Nucleic Acids Res. 29: 2875–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habraken Y., Sung P., Prakash L., Prakash S., 1998. ATP-dependent assembly of a ternary complex consisting of a DNA mismatch and the yeast MSH2–MSH6 and MLH1–PMS1 protein complexes. J. Biol. Chem. 273: 9837–9841. [DOI] [PubMed] [Google Scholar]
- Hall B. M., Ma C. X., Liang P., Singh K. K., 2009. Fluctuation AnaLysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbrück fluctuation analysis. Bioinformatics 25: 1564–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haracska L., Unk I., Johnson R. E., Johansson E., Burgers P. M., et al. , 2001. Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes Dev. 15: 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe B. D., Minesinger B. K., Jinks-Robertson S., 2000. Discrete in vivo roles for the MutL homologs Mlh2p and Mlh3p in the removal of frameshift intermediates in budding yeast. Curr. Biol. 10: 145–148. [DOI] [PubMed] [Google Scholar]
- Harrington J. M., Kolodner R. D., 2007. Saccharomyces cerevisiae Msh2-Msh3 acts in repair of base-base mispairs. Mol. Cell. Biol. 27: 6546–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr A. J., Ogawa M., Lawrence N. A., Williams L. N., Eggington J. M., et al. , 2011a Mutator suppression and escape from replication error–induced extinction in yeast. PLoS Genet. 7: e1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr A. J., Williams L. N., Preston B. D., 2011b Antimutator variants of DNA polymerases. Crit. Rev. Biochem. Mol. Biol. 46: 548–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombauer H., Campbell C. S., Smith C. E., Desai A., Kolodner R. D., 2011. Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell 147: 1040–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh P., Yamane K., 2008. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 129: 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D., Knuuti R., Palosaari H., Pospiech H., Syväoja J. E., 1999. cDNA and structural organization of the gene Pole1 for the mouse DNA polymerase ε catalytic subunit. Biochim. Biophys. Acta 1445: 363–371. [DOI] [PubMed] [Google Scholar]
- Iyer R. R., Pluciennik A., Burdett V., Modrich P. L., 2006. DNA mismatch repair: functions and mechanisms. Chem. Rev. 106: 302–323. [DOI] [PubMed] [Google Scholar]
- Johnson K. A., 2010. The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta 1804: 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. E., Kovvali G. K., Prakash L., Prakash S., 1996. Requirement of the yeast MSH3 and MSH6 genes for MSH2-dependent genomic stability. J. Biol. Chem. 271: 7285–7288. [DOI] [PubMed] [Google Scholar]
- Johnson R. E., Washington M. T., Haracska L., Prakash S., Prakash L., 2000. Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 406: 1015–1019. [DOI] [PubMed] [Google Scholar]
- Johnson S. J., Taylor J. S., Beese L. S., 2003. Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc. Natl. Acad. Sci. USA 100: 3895–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce C. M., Benkovic S. J., 2004. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry 43: 14317–14324. [DOI] [PubMed] [Google Scholar]
- Kadyrov F. A., Dzantiev L., Constantin N., Modrich P., 2006. Endonucleolytic function of MutLα in human mismatch repair. Cell 126: 297–308. [DOI] [PubMed] [Google Scholar]
- Kadyrov F. A., Holmes S. F., Arana M. E., Lukianova O. A., O’Donnell M., et al. , 2007. Saccharomyces cerevisiae MutLα is a mismatch repair endonuclease. J. Biol. Chem. 282: 37181–37190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M., Wang T. S., 2003. Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev. 17: 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliraman V., Mullen J. R., Fricke W. M., Bastin-Shanower S. A., Brill S. J., 2001. Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev. 15: 2730–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthikeyan R., Vonarx E. J., Straffon A. F., Simon M., Faye G., et al. , 2000. Evidence from mutational specificity studies that yeast DNA polymerases δ and ε replicate different DNA strands at an intracellular replication fork. J. Mol. Biol. 299: 405–419. [DOI] [PubMed] [Google Scholar]
- Kesti T., Flick K., Keranen S., Syväoja J. E., Wittenberg C., 1999. DNA polymerase ε catalytic domains are dispensable for DNA replication, DNA repair, and cell viability. Mol. Cell 3: 679–685. [DOI] [PubMed] [Google Scholar]
- Kolodner R. D., Marsischky G. T., 1999. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 9: 89–96. [DOI] [PubMed] [Google Scholar]
- Kumar D., Abdulovic A. L., Viberg J., Nilsson A. K., Kunkel T. A., et al. , 2011. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 39: 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel T. A., Burgers P. M., 2008. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 18: 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel T. A., Patel S. S., Johnson K. A., 1994. Error-prone replication of repeated DNA sequences by T7 DNA polymerase in the absence of its processivity subunit. Proc. Natl. Acad. Sci. USA 91: 6830–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrea A. A., Lujan S. A., Nick McElhinny S. A., Mieczkowski P. A., Resnick M. A., et al. , 2010. Genome-wide model for the normal eukaryotic DNA replication fork. Proc. Natl. Acad. Sci. USA 107: 17674–17679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Murphy K. M., Kanevets U., Reha-Krantz L. J., 2005. Sensitivity to phosphonoacetic acid: a new phenotype to probe DNA polymerase δ in Saccharomyces cerevisiae. Genetics 170: 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb L. A., 2011. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat. Rev. Cancer 11: 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb L. A., Springgate C. F., Battula N., 1974. Errors in DNA replication as a basis of malignant changes. Cancer Res. 34: 2311–2321. [PubMed] [Google Scholar]
- Loeb L. A., Bielas J. H., Beckman R. A., 2008. Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 68: 3551–3557. [DOI] [PubMed] [Google Scholar]
- Loh E., Choe J., Loeb L. A., 2007. Highly tolerated amino acid substitutions increase the fidelity of Escherichia coli DNA polymerase I. J. Biol. Chem. 282: 12201–12209. [DOI] [PubMed] [Google Scholar]
- Mao E. F., Lane L., Lee J., Miller J. H., 1997. Proliferation of mutators in a cell population. J. Bacteriol. 179: 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsischky G. T., Filosi N., Kane M. F., Kolodner R., 1996. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 10: 407–420. [DOI] [PubMed] [Google Scholar]
- McCulloch S. D., Kunkel T. A., 2008. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 18: 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan C., Lewis P. D., 2006. iMARS—mutation analysis reporting software: an analysis of spontaneous cII mutation spectra. Mutat. Res. 603: 15–26. [DOI] [PubMed] [Google Scholar]
- Morrison A., Sugino A., 1994. The 3′→5′ exonucleases of both DNA polymerases δ and ε participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol. Gen. Genet. 242: 289–296. [DOI] [PubMed] [Google Scholar]
- Morrison A., Bell J. B., Kunkel T. A., Sugino A., 1991. Eukaryotic DNA polymerase amino acid sequence required for 3′ → 5′ exonuclease activity. Proc. Natl. Acad. Sci. USA 88: 9473–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison A., Johnson A. L., Johnston L. H., Sugino A., 1993. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 12: 1467–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New L., Liu K., Crouse G. F., 1993. The yeast gene MSH3 defines a new class of eukaryotic MutS homologues. Mol. Gen. Genet. 239: 97–108. [DOI] [PubMed] [Google Scholar]
- Nick McElhinny S. A., Pavlov Y. I., Kunkel T. A., 2006. Evidence for extrinsic exonucleolytic proofreading. Cell Cycle 5: 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny S. A., Gordenin D. A., Stith C. M., Burgers P. M. J., Kunkel T. A., 2008. Division of labor at the eukaryotic replication fork. Mol. Cell 30: 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson A. I., Kugelberg E., Berg O. G., Andersson D. I., 2004. Experimental adaptation of Salmonella typhimurium to mice. Genetics 168: 1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishant K. T., Plys A. J., Alani E., 2008. A mutation in the putative MLH3 endonuclease domain confers a defect in both mismatch repair and meiosis in Saccharomyces cerevisiae. Genetics 179: 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northam M. R., Garg P., Baitin D. M., Burgers P. M., Shcherbakova P. V., 2006. A novel function of DNA polymerase ζ regulated by PCNA. EMBO J. 25: 4316–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northam M. R., Robinson H. A., Kochenova O. V., Shcherbakova P. V., 2010. Participation of DNA polymerase ζ in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics 184: 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notley-McRobb L., Seeto S., Ferenci T., 2002. Enrichment and elimination of mutY mutators in Escherichia coli populations. Genetics 162: 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palombo F., Iaccarino I., Nakajima E., Ikejima M., Shimada T., et al. , 1996. hMutSβ, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Curr. Biol. 6: 1181–1184. [DOI] [PubMed] [Google Scholar]
- Pavlov Y. I., Shcherbakova P. V., 2010. DNA polymerases at the eukaryotic fork—20 years later. Mutat. Res. 685: 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov Y. I., Shcherbakova P. V., Kunkel T. A., 2001. In vivo consequences of putative active site mutations in yeast DNA polymerases α, ε, δ, and ζ. Genetics 159: 47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov Y. I., Newlon C. S., Kunkel T. A., 2002. Yeast origins establish a strand bias for replicational mutagenesis. Mol. Cell 10: 207–213. [DOI] [PubMed] [Google Scholar]
- Pavlov Y. I., Mian I. M., Kunkel T. A., 2003. Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Curr. Biol. 13: 744–748. [DOI] [PubMed] [Google Scholar]
- Pavlov Y. I., Maki S., Maki H., Kunkel T. A., 2004. Evidence for interplay among yeast replicative DNA polymerases alpha, delta and epsilon from studies of exonuclease and polymerase active site mutations. BMC Biol. 2: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov Y. I., Frahm C., McElhinny S. A., Niimi A., Suzuki M., et al. , 2006a Evidence that errors made by DNA polymerase α are corrected by DNA polymerase δ. Curr. Biol. 16: 202–207. [DOI] [PubMed] [Google Scholar]
- Pavlov Y. I., Shcherbakova P. V., Rogozin I. B., 2006b Roles of DNA polymerases in replication, repair, and recombination in eukaryotes. Int. Rev. Cytol. 255: 41–132. [DOI] [PubMed] [Google Scholar]
- Peltomäki P., 2005. Lynch syndrome genes. Fam. Cancer 4: 227–232. [DOI] [PubMed] [Google Scholar]
- Preston B. D., Albertson T. M., Herr A. J., 2010. DNA replication fidelity and cancer. Semin. Cancer Biol. 20: 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prolla T. A., Christie D. M., Liskay R. M., 1994. Dual requirement in yeast DNA mismatch repair for MLH1 and PMS1, two homologs of the bacterial mutL gene. Mol. Cell. Biol. 14: 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursell Z. F., Kunkel T. A., 2008. DNA polymerase ε: a polymerase of unusual size (and complexity). Prog. Nucleic Acid Res. Mol. Biol. 82: 101–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursell Z. F., Isoz I., Lundstrom E. B., Johansson E., Kunkel T. A., 2007. Yeast DNA polymerase ε participates in leading-strand DNA replication. Science 317: 127–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman M. K., Winzeler E. A., Collingwood D., Hunt S., Wodicka L., et al. , 2001. Replication dynamics of the yeast genome. Science 294: 115–121. [DOI] [PubMed] [Google Scholar]
- Reenan R. A., Kolodner R. D., 1992. Characterization of insertion mutations in the Saccharomyces cerevisiae MSH1 and MSH2 genes: evidence for separate mitochondrial and nuclear functions. Genetics 132: 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reha-Krantz L. J., 1995. Learning about DNA polymerase function by studying antimutator DNA polymerases. Trends Biochem. Sci. 20: 136–140. [DOI] [PubMed] [Google Scholar]
- Reha-Krantz L. J., 2010. DNA polymerase proofreading: multiple roles maintain genome stability. Biochim. Biophys. Acta 1804: 1049–1063. [DOI] [PubMed] [Google Scholar]
- Reha-Krantz L. J., Siddique M. S., Murphy K., Tam A., O’Carroll M., et al. , 2011. Drug-sensitive DNA polymerase δ reveals a role for mismatch repair in checkpoint activation in yeast. Genetics 189: 1211–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaper R. M., 1996. Suppressors of Escherichia coli mutT: antimutators for DNA replication errors. Mutat. Res. 350: 17–23. [DOI] [PubMed] [Google Scholar]
- Schaaper R. M., 1998. Antimutator mutants in bacteriophage T4 and Escherichia coli. Genetics 148: 1579–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaper R. M., Cornacchio R., 1992. An Escherichia coli dnaE mutation with suppressor activity toward mutator mutD5. J. Bacteriol. 174: 1974–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova P. V., Noskov V. N., Pshenichnov M. R., Pavlov Y. I., 1996. Base analog 6-N-hydroxylaminopurine mutagenesis in the yeast Saccharomyces cerevisiae is controlled by replicative DNA polymerases. Mutat. Res. 369: 33–44. [DOI] [PubMed] [Google Scholar]
- Shcherbakova P. V., Pavlov Y. I., Chilkova O., Rogozin I. B., Johansson E., et al. , 2003. Unique error signature of the four-subunit yeast DNA polymerase ε. J. Biol. Chem. 278: 43770–43780. [DOI] [PubMed] [Google Scholar]
- Sherman F., 2002. Getting started with yeast, pp. 3–41 in Part B: Guide to Yeast Genetics and Molecular and Cell Biology, edited by Guthrie C., Fink G. R. Academic Press, San Diego. [Google Scholar]
- Shimizu K., Hashimoto K., Kirchner J. M., Nakai W., Nishikawa H., et al. , 2002. Fidelity of DNA polymerase ε holoenzyme from budding yeast Saccharomyces cerevisiae. J. Biol. Chem. 277: 37422–37429. [DOI] [PubMed] [Google Scholar]
- Sia E. A., Kokoska R. J., Dominska M., Greenwell P., Petes T. D., 1997. Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol. Cell. Biol. 17: 2851–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simhadri S., Kramata P., Zajc B., Sayer J. M., Jerina D. M., et al. , 2002. Benzo[a]pyrene diol epoxide-deoxyguanosine adducts are accurately bypassed by yeast DNA polymerase ζ in vitro. Mutat. Res. 508: 137–145. [DOI] [PubMed] [Google Scholar]
- Simon M., Giot L., Faye G., 1991. The 3′ to 5′ exonuclease activity located in the DNA polymerase δ subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J. 10: 2165–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siow C. C., Nieduszynska S. R., Müller C. A., Nieduszynski C. A., 2012. OriDB, the DNA replication origin database updated and extended. Nucleic Acids Res. 40: D682–D686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. A., Loeb L. A., Preston B. D., 2005. Lethal mutagenesis of HIV. Virus Res. 107: 215–228. [DOI] [PubMed] [Google Scholar]
- Sniegowski P. D., Gerrish P. J., Lenski R. E., 1997. Evolution of high mutation rates in experimental populations of E. coli. Nature 387: 703–705. [DOI] [PubMed] [Google Scholar]
- Strand M., Prolla T. A., Liskay R. M., Petes T. D., 1993. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 365: 274–276. [DOI] [PubMed] [Google Scholar]
- Stulemeijer I. J., Pike B. L., Faber A. W., Verzijlbergen K. F., van Welsem T., et al. , 2011. Dot1 binding induces chromatin rearrangements by histone methylation-dependent and -independent mechanisms. Epigenetics Chromatin 4: 2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan M. K., Johnson R. E., Prakash L., Prakash S., Aggarwal A. K., 2009. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase δ. Nat. Struct. Mol. Biol. 16: 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei F., Radman M., Maynard-Smith J., Toupance B., Gouyon P. H., et al. , 1997. Role of mutator alleles in adaptive evolution. Nature 387: 700–702. [DOI] [PubMed] [Google Scholar]
- Thompson D. A., Desai M. M., Murray A. W., 2006. Ploidy controls the success of mutators and nature of mutations during budding yeast evolution. Curr. Biol. 16: 1581–1590. [DOI] [PubMed] [Google Scholar]
- Tong A. H. Y., Boone C., 2007. High-throughput strain construction and systematic synthetic lethal screening in Saccaromyces cerevisiae, pp. 369–386 and 706–707 in Methods in Microbiology, edited by Stansfield I., Stark M. J. R. Academic Press, New York. [Google Scholar]
- Tran H. T., Gordenin D. A., Resnick M. A., 1999. The 3′→5′ exonucleases of DNA polymerases δ and ε and the 5′→3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol. Cell. Biol. 19: 2000–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tröbner W., Piechocki R., 1984. Selection against hypermutability in Escherichia coli during long term evolution. Mol. Gen. Genet. 198: 177–178. [DOI] [PubMed] [Google Scholar]
- Trujillo K. M., Sung P., 2001. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50·Mre11 complex. J. Biol. Chem. 276: 35458–35464. [DOI] [PubMed] [Google Scholar]
- Tsaponina O., Barsoum E., Astrom S. U., Chabes A., 2011. Ixr1 is required for the expression of the ribonucleotide reductase Rnr1 and maintenance of dNTP pools. PLoS Genet. 7: e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk I., Haracska L., Prakash S., Prakash L., 2001. 3′-Phosphodiesterase and 3 → 5 exonuclease activities of yeast Apn2 protein and requirement of these activities for repair of oxidative DNA damage. Mol. Cell. Biol. 21: 1656–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera E., Canceill D., Ehrlich S. D., 2001. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 20: 2587–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Sattar A. K., Wang C. C., Karam J. D., Konigsberg W. H., et al. , 1997. Crystal structure of a pol α family replication DNA polymerase from bacteriophage RB69. Cell 89: 1087–1099. [DOI] [PubMed] [Google Scholar]
- Wang T. S.-F., Wong S. W., Korn D., 1989. Human DNA polymerase α: predicted functional domains and relationships with viral DNA polymerases. FASEB J. 3: 14–21. [DOI] [PubMed] [Google Scholar]
- Wang W., Malcolm B. A., 1999. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques 26: 680–682. [DOI] [PubMed] [Google Scholar]
- Waters L. S., Minesinger B. K., Wiltrout M. E., D’Souza S., Woodruff R. V., et al. , 2009. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 73: 134–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei K., Kucherlapati R., Edelmann W., 2002. Mouse models for human DNA mismatch-repair gene defects. Trends Mol. Med. 8: 346–353. [DOI] [PubMed] [Google Scholar]
- Wenger J. W., Schwartz K., Sherlock G., 2010. Bulk segregant analysis by high-throughput sequencing reveals a novel xylose utilization gene from Saccharomyces cerevisiae. PLoS Genet. 6: e1000942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson M. S., Game J. C., Fogel S., 1985. Meiotic gene conversion mutants in Saccharomyces cerevisiae. I. Isolation and characterization of pms1–1 and pms1–2. Genetics 110: 609–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wloch D. M., Szafraniec K., Borts R. H., Korona R., 2001. Direct estimate of the mutation rate and the distribution of fitness effects in the yeast Saccharomyces cerevisiae. Genetics 159: 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuki N., Terashima H., Kitada K., 2002. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells 7: 781–789. [DOI] [PubMed] [Google Scholar]
- Yang Y., Karthikeyan R., Mack S. E., Vonarx E. J., Kunz B. A., 1999. Analysis of yeast pms1, msh2, and mlh1 mutators points to differences in mismatch correction efficiencies between prokaryotic and eukaryotic cells. Mol. Gen. Genet. 261: 777–787. [DOI] [PubMed] [Google Scholar]
- Yu L., Pena Castillo L., Mnaimneh S., Hughes T. R., Brown G. W., 2006. A survey of essential gene function in the yeast cell division cycle. Mol. Biol. Cell 17: 4736–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeyl C., de Visser J. A. G. M., 2001. Estimates of the rate and distribution of fitness effects of spontaneous mutation in Saccharomyces cerevisiae. Genetics 157: 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Rothstein R., 2002. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc. Natl. Acad. Sci. USA 99: 3746–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Elledge S. J., 1993. DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell 75: 1119–1127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.