

Abstract
Haemophilus influenzae protein E (PE) is a multifunctional adhesin involved in direct interactions with lung epithelial cells and host proteins, including plasminogen and the extracellular matrix proteins vitronectin and laminin. We recently crystallized PE and successfully collected X-ray diffraction data at 1.8 Å. Here, we solved the structure of a recombinant version of PE and analyzed different functional regions. It is a dimer in solution and in the asymmetric unit of the crystals. The dimer has a structure that resembles a flattened β-barrel. It is, however, not a true β-barrel, as there are differences in both the hydrogen-bonding pattern and the shape. Each monomer consisted of a 6-stranded antiparallel β-sheet with a rigid α-helix at the C terminus tethered to the concave side of the sheet by a disulfide bridge. The laminin/plasminogen binding region (residues 41 to 68) is exposed, while the vitronectin binding region (residues 84 to 108) is partially accessible in the dimer. The dimerized PE explains the simultaneous interaction with laminin and vitronectin. In addition, we found this unique adhesin to be present in many bacterial genera of the family Pasteurellaceae and also orthologues in other, unrelated species (Enterobacter cloacae and Listeria monocytogenes). Peptides corresponding to the surface-exposed regions PE 24 to 37, PE 74 to 89, and PE 134 to 156 were immunogenic in the mouse. Importantly, these peptide-based antibodies also recognized PE at the bacterial surface. Taken together, our detailed structure of PE explains how this important virulence factor of H. influenzae simultaneously interacts with host vitronectin, laminin, or plasminogen, promoting bacterial pathogenesis.
INTRODUCTION
Haemophilus influenzae is an important Gram-negative respiratory pathogen that causes, for example, acute otitis media in children and exacerbations in patients with chronic obstructive pulmonary disease (COPD), but also invasive diseases, such as meningitis and sepsis (1). Encapsulated H. influenzae is categorized into six different serotypes, a to f, whereas the remaining noncapsulated H. influenzae is designated nontypeable H. influenzae (NTHI) (2). H. influenzae resides in the mucosa, and NTHI is mainly associated with infections in the respiratory tract, whereas encapsulated H. influenzae, including H. influenzae type b (Hib), causes invasive disease. Until the 1990s, Hib was the most common serotype, but a dramatic reduction in Hib cases was observed after the introduction of a conjugate vaccine against Hib. However, an increasing incidence of invasive disease caused by non-type b H. influenzae has recently been reported from several countries (2–4).
In contrast to the very efficient vaccine against Hib, no suitable vaccine has been implemented for NTHI. Lipopolysaccharide (LPS) and surface-exposed antigenic proteins of Gram-negative pathogens are generally predicted to be putative starting points for screening of suitable vaccine candidates (5). However, not all surface proteins or LPS are suitable for eliciting protection in the host against a particular pathogen. In recent years, several surface adhesin proteins, including HMW-1 and -2 (6), PilA (7), P6 (8), and protein D (9), have been analyzed for their vaccine potential. Some of these NTHI surface proteins showed initial protection in experimental models, and protein D is now included in the vaccine Synflorix, giving partial protection against H. influenzae in humans (10, 11). Structural data are, however, available for only a few of these vaccine candidates.
We have described the role of a hitherto unknown H. influenzae protein E (PE) in interactions with host epithelial cells and in subversion of the host innate immune response (12). Protein E is a 16-kDa surface lipoprotein of H. influenzae that functions as an adhesin and induces a proinflammatory response during infection, leading to interleukin 8 (IL-8) secretion and upregulation of ICAM-1 (CD54) in both cell lines and primary epithelial cells originating from patients with COPD. An isogenic pe mutant showed defective adhesion and internalization of host epithelial cells. Furthermore, by using a peptide-mapping approach, we suggested that the amino acid region 84 to 108 is involved in binding to epithelial cells. Importantly, immunization with the PE amino acid (aa) 84 to 108 peptide showed significantly better pulmonary clearance in a mouse model than immunization with an unrelated control peptide (12). When the pe gene was sequenced in all Haemophilus spp., including Hib and NTHI clinical isolates, we found that PE is a ubiquitous Haemophilus outer membrane protein (13). The active vitronectin-binding region PE aa 84 to 106 was found to be 100% conserved. PE homologues were also present in other members of the family Pasteurellaceae, including Aggregatibacter spp., Actinobacillus spp., Mannheimia succiniciproducens, and Pasteurella multocida (13).
Vitronectin (Vn) and laminin (Ln) are among other proteins found in the extracellular matrix (ECM) (14, 15). In addition, Vn plays a crucial role in maintaining homeostasis in the regulation of the complement system, i.e., the innate immunity. We recently observed that NTHI binds Vn via surface-exposed PE and that this interaction leads to increased serum resistance. The peptide region covered by amino acids 84 to 108 is the Vn interaction domain (14–16), and further analysis revealed that in particular, the residues K85 and R86 are involved in Vn binding (17). In addition to the PE-dependent Vn binding, we observed that the region comprising PE amino acids 41 to 68 interacts with Ln, an abundant ECM protein in the basement membrane, and that this interaction leads to better adhesion of NTHI to host tissues (18). Interestingly, the Ln and Vn binding sites on the PE molecule are completely separate and do not interfere with each other during binding. The outer membrane PE thus both functions as an adhesin and concurrently protects bacteria from complement-mediated killing (18). More recently, we observed that PE also binds plasminogen, which is ultimately converted into plasmin, and this consequently leads to degradation of complement 3 (C3) and dampening of the host innate response (19).
We recently published a technical report on crystallization data for a recombinant variant PE and an Se-methionine-labeled PE (SeMet PE) (20). In the present study, the X-ray diffraction data at 1.8-Å resolution were analyzed in detail to obtain the structure of PE. Based upon the crystal, we show that the PE molecule is a dimer in the asymmetric unit. The structure of the monomer contained six antiparallel β-sheets connected with loops. At the C-terminal end, a rigid α-helix was found that was fixed in its position by a disulfide bond to the top of the β-sheet. The laminin and plasminogen binding regions of PE were exposed at the surface of the molecule, while the Vn binding region, previously defined by a peptide-mapping approach, was partially exposed. Furthermore, we used structure-based selection of the exposed regions to verify their localizations and respective immunogenicities by raising antibodies in mice. These structural findings provide insight into the regions that are involved in interactions with host proteins and a possible mode of PE-mediated host interaction by H. influenzae.
MATERIALS AND METHODS
Protein expression, purification, and crystallization.
PE and SeMet PE were recombinantly expressed in Escherichia coli as inclusion bodies, and purification was performed as described previously (20). In brief, E. coli BL21(DE3) harboring plasmids encoding PE and SeMet PE were grown in 1 liter LB with 50 μg/ml kanamycin, and protein expression was induced by addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). The cells were lysed by sonication, and inclusion bodies were collected by centrifugation. The inclusion bodies were washed with 5 M urea and dissolved in 10 ml of 8 M urea. Refolding of PE was performed by the dilution method in refolding buffer containing 50 mM Tris-HCl, pH 7.8, 500 mM NaCl, 5 mM dithiothreitol (DTT), 0.005% Tween 20, and 2 M urea. The refolded proteins were dialyzed against 50 mM Tris-HCl, pH 7.8, buffer containing 135 mM NaCl and loaded into a Q-Sepharose Fast Flow anion-exchange column (GE Healthcare Biosciences, Uppsala, Sweden) equilibrated with 50 mM Tris-HCl, pH 7.8, buffer containing 135 mM NaCl and 2 mM DTT. The flowthrough of the column that contained PE was collected and further purified with a Superdex 200 gel filtration column (GE Healthcare Biosciences) equilibrated with 50 mM Tris-HCl, pH 7.5, buffer containing 500 mM NaCl and 2 mM DTT (20). The protein purity was estimated by SDS-PAGE, and concentrations were measured with a Nano-Drop spectrophotometer.
The recombinant variant of PE and SeMet PE were concentrated up to 5 mg/ml. The crystallization conditions were screened by using commercial kits as described elsewhere (20). Crystals of the recombinant variant PE were obtained in 100 mM sucrose-phosphate-glutamic acid (SPG) buffer, pH 6.0, 25% (wt/vol) polyethylene glycol (PEG) 1500. The SeMet PE produced crystals in 100 mM MES (morpholineethanesulfonic acid), pH 6, 200 mM NaCl, and 20% PEG 6000. The size of the SeMet PE crystals was further improved by using the microseeding technique. Both PE proteins produced crystals within 4 to 5 days of incubation at 25°C. Details of the crystallization strategies were published previously (20).
Data collection and refinement.
Crystals were quickly cooled in the presence of a cryoprotectant solution in a MiTeGen loop (Ithaca, NY) using a stream of nitrogen gas at 100 K before exposing them to the X-ray beam. The universal cryosolution, containing 32% (wt/vol) glycerol, 32% (wt/vol) ethylene, 36% (wt/vol) sucrose, and 2% glucose, was used for the recombinant variant PE crystals. Suitable crystals were incubated in drops of mother liquor to which an equal volume of the cryosolution was added. After about 30 s of incubation, they were mounted in loops and placed in a cryogenic N2 gas stream at 100 K. In contrast to the cryoconditions for the recombinant variant PE crystals, 15% (wt/vol) PEG 400 in reservoir solution was used for SeMet PE crystals. All data collections were performed at station I911-3 at the MAX IV Laboratory (Lund University, Sweden). The station was equipped with an MD2 goniostat (Maatel, France) and a MAR225 charge-coupled-device (CCD) detector (Marresearch, Norderstedt, Germany).
To obtain reliable phase information, three data sets, peak (PK), point of inflection (PI), and remote (RM), were collected at the K edge of selenium (around 0.9795 Å) from the same SeMet-containing crystal (Table 1 shows data collection details and statistics). Data for PK and PI sets were collected first, with reduced exposure times, so that the influence of radiation damage on the phasing could be minimized. The data for RM, however, were collected with longer exposure times in order to obtain data to the diffraction limit of the crystals (Table 1). All data were integrated and scaled using the program XDS (21). The phasing power was monitored by using HKL2MAP (22), a graphical interface to a set of programs from the SHELX suite. Six of the 8 possible Se positions could be identified using Patterson methods in SHELXC (23). Subsequent refinement and phasing of the structure was achieved by using autoSHARP (24), using data from all 3 data sets.
Table 1.
Data processing, phasing, and refinement statistics
| Parametera | Value |
||||
|---|---|---|---|---|---|
| PE 1 | PE 2 | SeMet peak | SeMet inf | SeMet rem | |
| Space group | P21 | P21 | P21 | P21 | P21 |
| a (Å) | 44.2 | 54.7 | 44.1 | 44.1 | 44.1 |
| b (Å) | 57.3 | 42.5 | 56.9 | 56.9 | 56.9 |
| c (Å) | 61.4 | 56.8 | 61.4 | 61.4 | 61.4 |
| β (°) | 96.05 | 116.3 | 97.1 | 97.1 | 97.1 |
| Wavelength (Å) | 1.000 | 1.000 | 0.97918 | 0.97942 | 0.9700 |
| Maximum resolution (Å) | 1.8 | 2.1 | 2.6 | 2.7 | 2.3 |
| VMatthews(Da/Å3) dimer in asymmetric unit | 2.28 | 1.75 | 2.26 | 2.26 | 2.26 |
| Solvent content (%) | 46.0 | 29.7 | 45.7 | 45.7 | 45.7 |
| Total no. of observations | 115,628 | 32,032 | 62,016 | 60,725 | 101,090 |
| No. of unique reflections | 28,219 | 12,867 | 17,365 | 16,137 | 26,407 |
| Rmerge (%) | 6.2 (45.7) | 5.6 (45.9) | 5.5 (20.2) | 4.7 (15.7) | 9.4 (50.4) |
| Completeness (%) | 99.5 (98.6) | 91.4 (79.8) | 94.4 (68.2) | 98.8 (94.3) | 99.0 (95.1) |
| Avg (I)/(ΣI) | 12.34 (2.64) | 11.94 (2.03) | 16.37 (4.19) | 20.45 (6.76) | 11.40 (2.85) |
| Multiplicity | 4.09 | 2.49 | 3.57 | 3.76 | 3.83 |
| Phasing | |||||
| Figure of merit | 0.32 | ||||
| Phasing power | |||||
| Isomorphous | 0 | 0.713 (0.538) | 0.331 (0.253) | ||
| Anomalous | 1.079 | 1.050 | 0.416 | ||
| Cullis R factor | |||||
| Isomorphous | 0 | 0.617 (0.642) | 0.899 (0.976) | ||
| Anomalous | 0.843 | 0.826 | 0.958 | ||
| Refinement | |||||
| Refinement range (Å) | 29–1.8 | 28–2.1 | 29–2.3 | ||
| Rcrys (%) | 20.07 | 18.17 | 18.68 | ||
| Rfree (%) | 23.10 | 24.46 | 25.49 | ||
| No. of water molecules | 127 | 50 | 119 | ||
| Other | 3 GOL, 2 EDO | 1 GOL, 2 EDO | |||
| RMSD bond length (Å) | 0.013 | 0.07 | 0.009 | ||
| RMSD angle (°) | 1.498 | 1.055 | 1.208 | ||
| Favorable region | 99.2 | 98.4 | 98.1 | ||
| Additional allowed region | 0.8 | 1.6 | 1.9 | ||
| Disallowed | 0 | 0 | 0 | ||
RMSD, root mean square difference.
autoSHARP built 246 residues out of 282 possible using ARP/wARP (25). The model was inspected using Coot (26), and subsequent rounds of refinement and model building were performed by using the PHENIX package (27) and Coot. In total, 130 residues for each chain could be traced, with 10 residues missing at the N terminus and 1 residue at the C-terminal end of each of the protein chains. During refinement, an additional 127 water molecules were added to the model. The final model had good stereochemistry (the root mean square deviation [RMSD] from ideal geometry in bonds was 0.017 Å and in angle was 1.65°), and 97.3% of the residues were in the most favored part of the Ramachandran plot.
Data for the two different crystal forms of the recombinant variant PE were also collected. XDS was used for data processing and scaling (Table 1). The solvent content of the 2nd type of crystals was relatively low (around 30%), which probably was the reason for the diffracting power of these crystals. Both crystal forms were monoclinic, and details of the different data sets are described in Table 1. The structure of the SeMet mutant form of PE was used as a starting model in molecular replacement using Phaser (28) for the first crystal form. For the 2nd crystal form of the recombinant variant PE protein, the first form was used as a search model and Phaser was used for the molecular replacement. Both structures were refined using TLS refinement within the Phenix suite, and model inspection was performed using Coot. During refinement, difference density indicated the presence of ethylene and glycerol molecules from the cryosolutions, and they were placed accordingly.
Immunization of mice.
Based upon the crystal structure of the PE, peptides were designed for immunization and conjugated with keyhole limpet hemocyanin (KLH) (Innovagen AB, Lund, Sweden). Mice (BALB/c; 6 in each group) were immunized with KLH-conjugated peptides according to a standard immunization protocol (12). In brief, 50 μg peptide with complete Freund's adjuvant was subcutaneously administered. After 4 weeks, booster doses of 50 μg peptide with aluminum hydroxide were injected over the following 3 weeks. Blood was drawn 1 week after the last booster dose, and specific antibodies (Abs) were immunopurified using the same peptide, followed by testing in sandwich enzyme-linked immunosorbent assays (ELISAs) against the various peptides or recombinant PE. The animal experiments were done according to the general rules established by the Swedish Government (Svensk författnings-samling 1988:534 and Ändringsförfattning 2012:256; Riksdagen, Stockholm, Sweden). An ethics permit (no. M193-11) was obtained from Malmö/Lund District Court (Djurförsöksetiska nämnden, Tingsrätten, Lund, Sweden). This body approved the protocol used in the study.
ELISA.
Ninety-six-well PolySorb microtiter plates (Nunc-Immuno, Roskilde, Denmark) were coated with PE peptides or recombinant PE 22 to 160 (100 ng) in 100 μl of 100 mM Tris-HCl, pH 9.0, for 15 h at 4°C. The coated plates were washed 3 times with phosphate-buffered saline (PBS) and blocked with 2.5% bovine serum albumin (BSA) in PBS containing 0.05% Tween 20 (PBST). Serum or purified antibodies in 2.5% BSA plus PBST were added to the wells and allowed to bind for 1 h at room temperature. After washes with PBST, horseradish peroxidase (HRP)-conjugated rabbit anti-mouse polyclonal Ab (PAb) (Dako, Denmark) were added in 2.5% BSA plus PBST. Finally, the plates were washed 4 times with PBST and developed with HRP substrate containing 20 mM tetramethylbenzidine and 0.1 M potassium citrate. After color development, the reactions were terminated with 1 M H2SO4, and finally, the plates were read at 450 nm in a microplate reader.
Flow cytometry.
NTHI 3655 (12) from overnight cultures was grown in broth to an optical density at 600 nm (OD600) of 0.8. Thereafter, the bacteria were washed twice with PBS containing 1% BSA and incubated with purified mouse anti-PE peptide antiserum according to a standard protocol. After washing, the bacteria were incubated with fluorescein isothiocyanate (FITC)-conjugated secondary rabbit-anti-mouse PAbs (Dako Sweden, Stockholm, Sweden) followed by two washes and then flow cytometry analysis (Epics XL-MCL; Coulter, Hialeah, FL).
Dynamic light scattering and electron microscopy.
Dynamic-light-scattering (DLS) experiments were performed by using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, United Kingdom). Different concentrations of PE (1 mg/ml, 3 mg/ml, and 5 mg/ml) were used to collect data in triplicate, and mean values were plotted. We used negative staining and transmission electron microscopy (TEM) to visualize the PE molecules, as described elsewhere (29).
Protein structure accession numbers.
All coordinates and structure factors for the different models were submitted to the Protein Data Bank (PDB) with codes 3zh6 for the SeMet mutant, 3zh5 for the recombinant PE variant PE 1, and 3zh7 for the recombinant PE variant PE 2.
RESULTS
Protein E is a dimer.
The gel filtration profile showed that the majority (>85%) of recombinant PE molecules existed as dimers in solution, whereas only a minor fraction consisted of an oligomeric form under these conditions (Fig. 1A and B). The pooled fractions 13 to 18 in Fig. 1A were also analyzed by dynamic light scattering. These experiments confirmed that approximately 85% of PE that eluted as a single peak represented dimers, whereas the minor peak containing the remaining 10 to 15% represented tetramers (Fig. 1C). In addition, TEM (negative staining) of recombinant PE also verified that >85% of PE existed in dimers, with a minor fraction as tetramers and a few monomers (Fig. 1D). Taken together, these results clearly indicated that PE is a dimer.
Fig 1.

Purification of recombinant PE. (A) The gel filtration profile of the recombinant variant PE (approximately 2 mg protein purified by anion exchange as described in Materials and Methods) was injected into an equilibrated Superdex 200 column. The separation chromatogram of PE is shown, along with a standard molecular weight filtration marker. mAu, milliabsorbance unit. (B) Fractions collected from panel A were separated using 12% SDS-PAGE, followed by staining with Coomassie blue R250. A similar gel filtration profile and purity pattern were also observed with SeMet PE (data not shown). (C) DLS pattern of the recombinant variant PE purified by gel filtration. (D) Transmission electron microscopy showing PE molecules (arrowheads). Bar, 100 nM.
When the crystal structure was determined, we found that PE is also present as a dimer in the asymmetric unit. Each PE monomer consists of a β-sheet formed by 6 antiparallel β-strands (β1, residues 39 to 43; β2, 46 to 59; β3, 64 to 78; β4, 88 to 99; β5, 102 to 114; β6a, 118 to 122; and β6b, 130 to 134) (Fig. 2A). In addition, a longer α-helix (residues 138 to 151) packs on the concave face of the sheet (Fig. 2B), and strands β1, β2, and β3 are curved around it. Strand β6 consists of two shorter strands, β6a and β6b, interrupted by a short loop from residues 123 to 129. After the loop, β2b lines up to strand β3, and proper hydrogen bond interactions for a sheet are formed. The edge of the sheet is formed by strand β1 and part of strand β2 at one side and strands β6a and β6b at the other side. The α-helix is tethered to the β-sheet through a conserved disulfide bond between cysteine residues 99 and 148. This disulfide bond connects the top of the central strand of the β-sheet and one end of the α-helix. The α-helix shields the upper half of the concave β-sheet, and in particular, the part formed by strands β1, β2, β3, and partly β4 is protected from solvent by the helix while the remaining part of the face is exposed.
Fig 2.

Secondary-structure elements and the PE monomer. (A) PE 28 to 159 amino acids showing the secondary structure. In total, 6 β-strands, 8 loops, and one C-terminal helix exist. (B) In the monomer, 6 antiparallel β-strands form the β-sheet. A longer α-helix packs on the concave face of the sheet, where strands β1, β2, and β3 are curved around it. It is tethered to the β-sheet through a conserved disulfide bond between cysteines 99 and 148.
The convex side of the sheet is not protected by other structural elements in the PE monomer. However, the two monomers in the asymmetric unit pack together through the convex side of each of their β-sheets to form a nearly continuous antiparallel β-sheet that resembles a flattened barrel-like dimer structure. The dimer formations are nearly identical in the different crystal forms presented. The resulting dimer is 55 Å by 44 Å by 25 Å in size (Fig. 3A and B). The barrel has a wedge-like shape, with the top narrower than the bottom. Loops connecting the β-strands at the top of the barrel (Fig. 2, loops 3, 5, and 7) are smaller than the loops found at the bottom of the barrel (loops 1, 2, 4, and 6). No electron density for amino acids 17 to 27 (before loop 1) of PE could be observed in the crystal structure, and hence, they are not modeled. At each side of the barrel, strand β6b from one monomer and the end of strand β2 and loop 3 from another monomer lined up to close the barrel (Fig. 4A), with similar interactions between the two monomers at each side of the barrel. The interactions between these two segments are not through main-chain atom hydrogen bonding interactions, as seen in true β-barrels, but rather through interactions of atoms from side chains of residues in loop 52 to 61 from one monomer (named PEA) with main-chain atoms in strand β6b in the other monomer (named PEB). The central 2 residues are Asn60 and TrpA57. The Nδ2 of Asn60 forms a hydrogen bond with the backbone oxygen of Thr131, whereas the Oδ1 makes a hydrogen bond with the backbone nitrogen from the same Thr131. The side chain of Trp57 occupies a hydrophobic pocket formed by the aliphatic part of the side chain of Lys129, the methyl moiety of Thr105, and the side chain of His67. The Nε1 of Trp57 makes a hydrogen bond with the backbone oxygen of Lys129 (Fig. 4A). Other interactions are formed between Asp59 and Thr131, Gln61, and Ser133. Tyr94 from strand β4 completes the main interactions through a hydrogen bond to the Nz of Lys129 (Fig. 4A). The dimer interactions are not extensive, and only 625 Å2 is buried in the interface, according to an analysis using Pisa (30).
Fig 3.

The PE dimer. (A) Cartoon representation of the PE dimer. (B) Surface of the PE dimer shown from the top and bottom cavities, in addition to the charge distribution of the molecule. The top surface of the molecule is neutral in charge, whereas the bottom side of the dimer is basic. The positive and negative charges are shown in blue and red, respectively.
Fig 4.

Dimer interface and PE internal cavities. (A) Stereoview showing side chains in the loop from residues 52 to 61 from one monomer (green A) to main-chain atoms in the strand β6b in the other monomer (red B). Two central residues are AsnA60 and TrpA57. The Nδ2 of AsnA60 forms a hydrogen bond with the backbone oxygen of ThrB131, and Oδ1 makes a hydrogen bond with the backbone nitrogen from the same ThrB131. The side chain of TrpA57 occupies a hydrophobic pocket formed by the aliphatic part of the side chain of Lys B129, the methyl moiety of Thr B105, and the side chain of His A67. The Nε1 of TrpA57 makes a hydrogen bond with the backbone oxygen of LysB129. Other interactions are formed between AspA59 and ThrB131, Gln A61, and Ser133. Tyr A94 from strand β4 completes the main interactions through a hydrogen bond to the Nz of Lys B129. (B) Top and bottom pockets of the dimer (red and blue, respectively). The side chains of Arg108 and Arg121 in each monomer are shown. (C) Schematic drawing of the residues lining the walls of the top pocket of the dimer. The colored halves represent the two monomers, and the colored residues inside the pocket are located at the bottom wall of the pocket.
Both the top and bottom sides of the barrel-like feature are hollow, and deep pockets are formed (Fig. 4B). The pocket at the wider bottom is larger and not well defined or well protected from solvent. The volume of the pocket is 857 Å3 as calculated by Pocketfinder (http://www.modelling.leeds.ac.uk/pocketfinder/). This pocket is lined with a number of charged residues, most notably 4 arginines (Arg108 and Arg121 from each subunit), that come together in the center of the pocket to form a ring-like arrangement (Fig. 4B). Two Asp residues (Asp110 from each monomer) are positioned between two of the Arg residues and provide some charge compensation (see Fig. S1 in the supplemental material). Further charged residues that line the inner pocket are Lys125 and Lys126 (see Fig. S1).
The second pocket at the top of the barrel is much more confined in space and better defined. It has a flattened shape, with dimensions of 14 Å by 8 Å and is about 10 Å deep, with a volume of 363 Å3 (Fig. 4B). The central part of the pocket is lined with four Ile residues (Ile65 and Ile96 from each monomer), whereas at the inside walls of the pocket, more polar residues (His67, Asp59, and Thr105 from each monomer) are found (Fig. 4B and C). The rim of the pocket is also polar in character, and residues His103, Asn98, and Asn101 are located there. Since this pocket is very well defined in shape and protected from the solvent, it seems likely that it has a ligand binding feature. The residues that line the walls of both pockets, however, are not particularly conserved within a family of homologous proteins from different pathogens, as described below.
Functional host factor-binding regions of PE are exposed at the surface of the molecule.
We previously used a peptide-mapping approach to identify the PE amino acid regions that were involved in binding to epithelial cells (12). A peptide consisting of the PE 84 to 108 amino acid region directly interacted with lung epithelial cells. More recently, a similar region was verified that bound Vn (16, 17). In the present crystal structure, the 84 to 108 amino acid region is located partly in loops 4 and 5 and in beta strands 4 and 5 (Fig. 5A). The structure shows that loop 4 plays a role at the base of the protein, where amino acids Asn73-Arg89 are well exposed (Fig. 5B). In contrast, the Ser90-Arg108 part is located in beta strands 4 and 5, which are positioned in the core of the PE dimer. Thus, the loop 4 region up to amino acid 84 seems more likely to be the real epithelial-cell-interacting domain (Fig. 5B). Recently, we showed that amino acids Lys85 and Arg86 are important for the PE-dependent interaction with vitronectin (17). Importantly, in loop 4, the side chains for Lys85 and Arg86 are well exposed (Fig. 5A and B). The linear sequence of loop 4 in all Haemophilus spp. is highly conserved, which suggests similar ligand specificities, while in other pathogens this loop is only partially conserved (Fig. 6A).
Fig 5.

Binding sites of the various host factors on the PE dimer. (A) Ribbon diagram of a PE dimer that shows the Vn binding region. The two separate monomers are shown in green and light blue, whereas the amino acid region 84 to 108 (based on peptide mapping) is shown in dark magenta and dark blue in the two monomers. (B) Surface of the PE dimer with exposed K85 and R86 side chains, along with other residues. (C) The Ln/plasminogen binding region of PE. The ribbon model shows the Ln/plasminogen binding region in red. The amino acids of the binding region are shown as stick models. For ease of following the chain, several residues along this fragment are numbered. (D) Surface of PE with the Ln and plasminogen binding region indicated in red.
Fig 6.

PE homologues also exist in other pathogens. (A) BLAST results revealed that several other pathogens have PE homologues. Retrieved sequences were aligned using the MultAlin online tool (42). Highly conserved amino acids are shown in red, and partially conserved amino acids are in blue. Secondary structures are shown at the top, with beta strands as arrows and helices as cylinders. (B) Identity and similarity matrix of protein sequences analyzed by using the pairwise alignment tool Needle (http://www.ebi.ac.uk). (C) Rooted phylogenetic tree showing 5 different clusters. Haemophilus spp. formed a single cluster, and distantly related sequences of Pasteurella spp. and E. cloacae and L. monocytogenes formed two separate clusters. The sequences were analyzed for phylogenetic relations by using MEGA5 software (43).
Since the multifunctional PE also interacts with laminin, we previously applied a peptide-mapping approach to determine the specific laminin binding amino acid region in PE. Interestingly, the region consisting of PE aa 41 to 68 was directly interacting with Ln (18). The same peptide also has affinity for plasminogen (19). This particular region is located in loop 1 between strands β1 and β2 and beta strand β2 and loop 2. The whole region is partly exposed at the surface and presents diagonally, along with the whole length of the dimer (Fig. 5C and D). In parallel with loop 4, the laminin/plasminogen binding region (loop 3) is also conserved among Haemophilus spp., but it is diverse in other pathogens. However, loop 3 has acidic amino acids that are highly conserved in most of the PE homologues for which sequence information is available (Fig. 6A).
PE is a unique adhesin of the family Pasteurellaceae.
All pathogenic bacterial species are equipped with specific adhesins that very precisely recognize host tissues in particular anatomical niches. This group of proteins has numerous structurally diverse members. In general, adhesins may be fimbriae, pili, or other, nonpilus adhesins (31). There are only a limited number of adhesins for which structures have been determined, and that limits the categorization of this group of proteins (http://supfam.cs.bris.ac.uk). The structural classification of adhesins on the basis of domains involved in recognition of host proteins was performed in the database available at http://supfam.cs.bris.ac.uk/SUPERFAMILY/cgi-bin/scop.cgi?sunid=49401. The database includes a limited number of proteins, such as collagen-binding proteins, fibrinogen-binding proteins, pilus subunits, PapG adhesion receptor-binding proteins, F17-C type adhesin, and Dr family adhesins. However, a large number of other known bacterial adhesins are not yet categorized due to lack of structural information on the domain recognition.
Pasteurellaceae is a large and diverse family of Gram-negative proteobacteria (32). Recently, the family was classified into 17 different genera (http://www.pasteurellaceae.life.ku.dk/). The latest BLAST update in the ExPASy (NCBI BLAST2) database showed that there are 11 different bacterial pathogens that have a pe gene homologue (Fig. 6A). The similarity matrix showed that H. influenzae has 34.1 to 98.8% identity and 55.3 to 99.4% similarity with other members of Pasteurellaceae (Fig. 6B), and a primary amino acid sequence-based phylogenetic tree showed 5 separate clusters (Fig. 6C). Haemophilus spp. grouped into a single cluster with 65.3 to 98.8% identity and 77.0 to 99.4% similarity. Actinobacillus actinomycetemcomitans showed 70.9 to 78.4% sequence identity and 80.8 to 88.6% similarity with Actinobacillus segnis and Actinobacillus aphrophilus, respectively. In the third cluster, Actinobacillus succinogenes showed 43.6% identity and 65.4% similarity to M. succiniciproducens (Fig. 6B). Pasteurella spp. also have PE analogues that are approximately 34% similar to H. influenzae PE, whereas PE derived from P. multocida is only 45.9% identical and 64.8% similar to Pasteurella dagmatis PE. In addition to these Pasteurellaceae family members, Enterobacter cloacae and Listeria monocytogenes have PE orthologues (E3G4R8 and E1UDL7, respectively), albeit with lower homology (Fig. 6B and C). We suggest that PE belongs to a unique group of bacterial outer membrane proteins in the family Pasteurellaceae.
PE belongs to the cystatin family of proteins.
To verify the folding pattern, the PE structure was matched with a fold database (33). The arrangement of a 6-stranded antiparallel β-sheet and α-helix packing at one face, as seen within the monomer, was observed in a number of different proteins. In these proteins, however, the other side of the sheet is normally shielded by other secondary-structure elements, and thus, the sheet and helix are part of larger fold arrangements that are not related to each other. The closest structures, which have the same minimal arrangement, can be seen in the monellin/cystatin family fold (34), which is also composed of a single antiparallel β-sheet with an α-helix protecting the concave face of the sheet. Proteins in this family are either cysteine protease inhibitors (cystatin-like) or sweet proteins from plants (monellin-like). The basic folds of, e.g., monellin and PE superimpose well (Fig. 7). However, the position of the helix within the secondary-structure topology of the monellin/cystatin family fold is different than it is within the PE structure. In the monellin/cystatin family, it is placed after the first strand of the sheet, whereas it is positioned at the C-terminal end of the PE molecule. The helix for the monellin/cystatin family is positioned in the center of the beta-sheet face and protects the whole face of the protein. The helix in PE, however, is tethered to the top of the sheet by the disulfide bridge, and therefore, part of the sheet is more exposed. Thus, it seems that PE and monellin/cystatin probably are analogues of each other and probably are products of convergent evolution.
Fig 7.
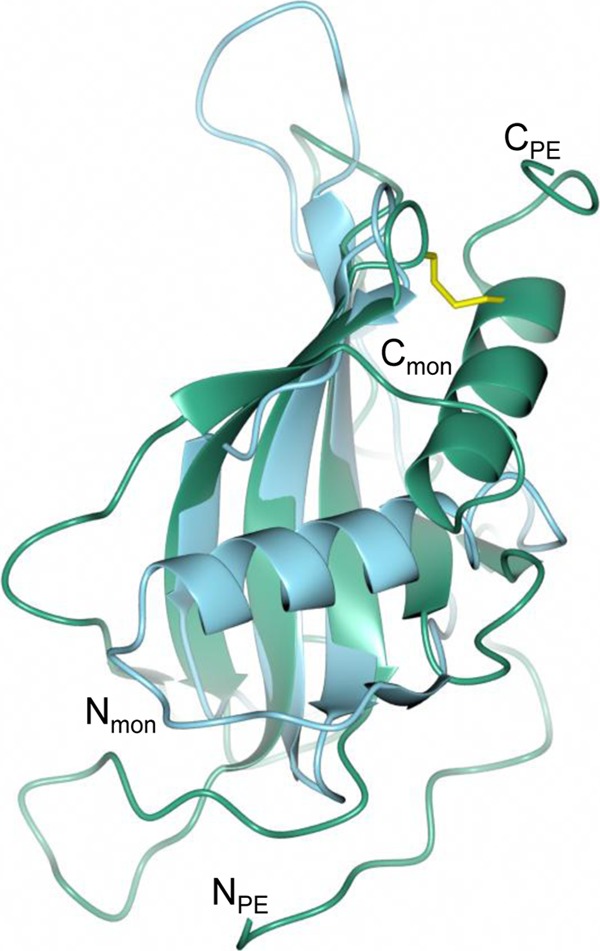
PE has a monellin-like fold. Shown are the superimposed structures of monellin (mon) (PDB code 2O9U) chain A (blue) and PE monomer (green). The beta sheet arrangements in the protein backbone are very similar. However, helices are present at the C terminus in PE and at the N terminus in monellin.
The most homologous regions for all the species are located in the secondary-structure elements, especially within the central-strand β3. Interestingly, one region with very high homology is not associated with a secondary-structure element, but instead, it is found in loop 6 and consists of the fingerprint (FY)111-(YH)-X-(DE)-F-W-G-X-G119. This loop between β5 and β6 folds back over the convex inner side of the sheet and forms a small hydrophobic core on the inside bottom of the β-barrel-like dimer. The aromatic residues in the fingerprint interact with residues from the different strands, as well as with the N-terminal loop 1. In particular, the residues Tyr/His112 and Trp116 are involved in multiple interactions with residues from different secondary-structure elements (Fig. 6A). Using the Prosite Web server (35) to scan the database for sequences with similar fingerprints, it seems that this fingerprint is unique, and thus, it might be an indicator of this particular fold family.
Structural approach for mapping immunogenic and surface-exposed regions of PE by raising peptide antibodies.
Previously, we reported that immunization with PE 84 to 108 mediated protection in a mouse pulmonary clearance model (12). The ubiquitous presence of PE among NTHI clinical isolates further signified the importance of the protein as a vaccine candidate (13). To verify our crystal model, as well as to analyze the immunogenic capacities of various surface-exposed regions of PE, we designed a series of peptides, as outlined in Fig. 8A and B (PE 24 to 37, PE 74 to 89, PE 84 to 108, and PE 134 to 156). In addition, a peptide based upon the cryptic region PE 104 to 128 was used as a negative control. KLH-conjugated peptides were used for subcutaneous immunization of BALB/c mice. Sera from immunized mice (n = 6 in each group) were pooled, and peptide Abs were purified using affinity chromatography and finally tested in ELISA. All peptides produced specific Abs directed against their corresponding peptides (Fig. 8C). In contrast, the resulting antibodies showed different recognition patterns against recombinant full-length PE 22 to 160 in ELISA. Antibodies directed against PE 24 to 37, PE 74 to 89, and PE 134 to 156 recognized the PE 22 to 160 molecule, whereas the anti-PE 84 to 108 Ab recognized PE 22 to 160 with less efficiency (Fig. 8D). However, purified peptide antibodies directed against PE 24 to 37 and PE 84 to 108 displayed cross-reactivity against both peptides in ELISA. As expected, the negative-control Ab against PE 104 to 128 did not detect full-length PE.
Fig 8.

Mapping of surface-exposed immunogenic regions using mouse anti-PE peptide Abs. (A) Ribbon diagram showing PE amino acid regions (indicated with different colors) that were selected for immunization of a series of mice. (B) Surface structure of selected regions as shown in panel A. (C) Results from ELISA that demonstrate Ab recognition of peptides with which microtiter plates were coated. (D) Recognition of full-length recombinant PE 22 to 160 by peptide Abs shown in ELISA. (E) Flow cytometry profiles of NTHI showing surface recognition of PE at the surfaces of bacteria using anti-PE peptide Abs.
We also wanted to test whether our peptide Abs recognized PE when the molecule was surface exposed in its native PE form on the clinical isolate NTHI 3655. Bacteria were incubated with Abs, followed by flow cytometry analysis. In parallel with the results obtained by ELISA, anti-PE 24 to 37, anti-PE 74 to 89, and anti-PE 134 to 156 Abs all detected PE at the bacterial surface, whereas the anti-84 to 108 Ab showed a minor shift compared to the negative control consisting of the secondary antibody only (Fig. 8E). Taken together, Abs directed against surface-exposed epitopes of the PE molecule that were defined by the designed peptides fit very well with the crystal structure.
DISCUSSION
Protein E is an outer membrane protein found in both encapsulated H. influenzae and NTHI. We have shown that NTHI equipped with the multifunctional adhesin PE binds to several different host proteins, including Vn, Ln, and plasminogen (13, 16, 17, 19). The majority of NTHI infections are in general noninvasive, although an increasing trend of invasive NTHI disease has been observed the last 5 years (3). Active degradation of host tissues and the ECM, however, is a property of typeable H. influenzae, such as Hib. Adherence mediated by the PE-Ln interaction thus could provide a firm anchorage for host basal lamina and tissues (Fig. 9). In contrast to Hib, NTHI may also use Ln as a ligand, particularly when the epithelial mucosa is destroyed by a prior viral infection or mixed bacterial infections. In parallel, plasminogen bound to PE degrades C3b and inhibits the complement-mediated innate immunity. The protease activity of plasmin also degrades the ECM (Fig. 9), which eventually increases the host damage. Experimental evidence suggests that Ln and plasminogen share similar binding domains in PE (16, 19). This might, of course, be conditional according to the abundance and affinity of ligands. An interesting finding is that recombinant PE simultaneously binds Ln and Vn (18). We have also observed that laminin LG1 to -5 domains of the alpha chain interact with PE and that this interaction can be inhibited by heparin (18). The PE loop 3 region has a few acidic residues (residues D59 and E62) that might be involved in binding to the LG4 and -5 domains of laminin. The present data on a dimerized PE molecule (Fig. 3A) explain these in vitro observations. However, it is at present unclear whether the dual-ligand-binding mode of the PE molecule also exists in vivo.
Fig 9.

Schematic representation of the multiple functions of PE. (Left) H. influenzae PE interacts with the host epithelium. The PE molecule mediates binding to the epithelial surface utilizing a hitherto unknown receptor. This interaction contributes to bacterial adhesion and induction of a proinflammatory response by the epithelial cells. PE binds to Ln, which contributes to adhesion of H. influenzae to the basement membrane and the host ECM. In addition to Ln, H. influenzae binds plasminogen by using PE. When plasminogen is bound to PE, it is converted into active plasmin by host urokinase plasminogen activator (uPA) or tissue plasminogen activator (tPA). Active plasmin may help in bacterial invasion and degradation of the ECM. (Right) PE inhibits the MAC at the surface of H. influenzae, which is accomplished by the binding of Vn. Vitronectin is a well-known complement regulator that inhibits the terminal complement pathway by interacting with the C5b-7 complex assembly and also inhibits C9 polymerization during formation of a lytic pore. PE is able to bind both Ln and Vn at the same time, and the two ligands do not interfere with each other; thus, adhesion and MAC inhibition can be carried out simultaneously.
Protein E is a lipoprotein that has a signal peptide at the N terminus, followed by Cys16 (12). According to the general described mechanisms of lipoprotein transport and lipidation mechanisms, the protein is transported to the outer membrane, followed by addition of lipid chains and removal of the signal peptide (36, 37). The Cys16 residue is thus predicted to be involved in lipidation and functions as an anchor of PE on the outer membrane in bacteria. The N terminus of the dimer is supposed to face toward the membrane side of the bacteria, whereas the C terminus faces the outside.
The presence of a well-formed pocket on the top side of the dimer (Fig. 3B) is intriguing. The shape and accessibility of the pocket is very reminiscent of binding pockets in smaller ligand binding proteins. However, a clear function for this pocket cannot be deduced, and due to the variability of the residues lining the pocket, it does not seem to be associated with a conserved function within the family of PE-like proteins. It has been suggested, however, that a high level of diversity within interacting surfaces of adhesins when in contact with their host proteins stems from the need of the pathogen to change these recognition sites in order to avoid an immune response while retaining a common biological function (38). This possibility needs to be further investigated. Identification of a putative ligand to the smaller and well-defined pocket of PE would be an asset in these investigations.
Protein E is known as one of the H. influenzae Vn binding proteins with the highest affinity (dissociation constant [Kd] = 4.0 × 10−7 M), and most importantly, Vn bound to PE protects NTHI from the membrane attack complex (MAC). The vitronectin-binding region PE 84 to 108 that is embedded in the structure is mostly located in the dimer interface. Recently, we further suggested that Lys85 and Arg86 of PE are involved in binding to Vn (17). Lys85 and Arg86 are located in the end of the exposed loop 4. Thus, a probable region for Vn interactions may be mostly located between amino acids Leu78 and Ala88 of loop 4. Vn is also known to function as a bridge molecule that interacts with bacterial surface proteins and the host integrins via an RGD motif (14, 39). Similarly, the PE-mediated recruitment of Vn by H. influenzae might also be involved in adhesion to and internalization into epithelial cells.
Protein E meets the criteria to be suitable as a vaccine candidate, i.e., its ubiquitous presence, highly conserved nature, favored immunogenicity, and protective role in model animals (12). In our preliminary experiments, the structure-based approach for designing antibodies against the surface-exposed part of the PE molecule recognized recombinant PE, in addition to PE at the surfaces of bacteria (Fig. 8E). This indicated that these peptides may be appropriate for immunization against NTHI.
PE has a unique three-dimensional structure that is involved in adhesion of H. influenzae to epithelial cells and binds and/or recruits multiple host proteins. The PE structure is formed by β-sheets, with an α-helix covering one side. Within the crystal structure, the protein is present as a dimer, with the two monomers forming a β-barrel-like structure. The dimer of PE resembles a flattened β-barrel-like structure, as it seems to form a continuous up and down antiparallel β-sheet, resulting in the formation of large pockets inside the molecule. However, in a number of aspects, it differs from true β-barrel structure. The first is that at the sides of the monomers interacting with each other, the contact surfaces are not made through β-strands, but instead, loops are used in an extended conformation. The hydrogen-bonding system is not like that observed in a β-sheet; here, instead, hydrogen bonds are built up through side chain interactions. Although the interaction area is limited, the presence of dimers is also seen in gel filtration, DLS, and TEM experiments, indicating that this is not likely to be an artifact of crystallization. The other unusual aspect of this “barrel-like” structure is the way the barrel fans out at the bottom. This is mainly achieved by the high twist in the separate sheets of each monomer. Only the top part of the barrel is intact and continuous, while the bottom part is sheared and open at the sides. This is an unusual arrangement and is not observed in other proteins, as judged by fold recognition searches using Dali (33).
The protein-folding pattern has a partial match with that of the monellin/cystatin family. However, cystatins are in general monomeric, whereas monellin is a heterodimer formed by an A chain of 42 aa and a B chain of 50 aa. A higher oligomeric variant of monellin has been observed in crystal structures (40). The dimer arrangement of PE, however, is different from that of monellin, and in PE, a more side-to-side barrel-like dimer is found. Proteins in the monellin/cystatin family have been studied extensively for their capacity to aggregate (41). However, the aggregation of PE has not yet been fully investigated. Besides this similarity to monellin/cystatin, there are no other bacterial adhesins that have this kind of structural appearance. However, orthologues belonging to the PE adhesin subfamily are also present in E. cloacae and L. monocytogenes. We conclude from sequence comparisons within this set of proteins that the residues that are important for the specific features of the fold are very well conserved. These conserved patches at the surface of PE that might be important for the structure of the common fold are shown in Fig. S2 in the supplemental material. The specific roles of these proteins in the various bacterial species are not yet elucidated. In fact, the best-characterized member of this family is PE, which is described in this paper. We would therefore propose that PE and its homologues belong to a new group of adhesins that in particular present in the family of Pasteurellaceae.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Alfred Österlund, Åke-Wiberg, Anna and Edwin Berger, Greta and Johan Kock, Lars Hierta, FLÄK, the Gyllenstiernska Krapperup, the Marianne and Marcus Wallenberg Foundation, the Physiographical Society, the Swedish Medical Research Council (grant number 521-2010-4221; http://www.vr.se), the Cancer Foundation at the University Hospital in Malmö, the Skåne County Council’s research and development foundation, and GlaxoSmithKline-Rixensart, Belgium.
We thank the staff at the MAX IV laboratories and the crystallization facility for their valuable help.
Footnotes
Published ahead of print 28 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01111-12.
REFERENCES
- 1.Murphy TF, Faden H, Bakaletz LO, Kyd JM, Forsgren A, Campos J, Virji M, Pelton SI. 2009. Nontypeable Haemophilus influenzae as a pathogen in children. Pediatr. Infect. Dis. J. 28:43–48 [DOI] [PubMed] [Google Scholar]
- 2.van Wessel K, Rodenburg GD, Veenhoven RH, Spanjaard L, van der Ende A, Sanders EA. 2011. Nontypeable Haemophilus influenzae invasive disease in The Netherlands: a retrospective surveillance study 2001–2008. Clin. Infect. Dis. 53:e1–e7 doi:10.1093/cid/cir268. [DOI] [PubMed] [Google Scholar]
- 3.Resman F, Ristovski M, Ahl J, Forsgren A, Gilsdorf JR, Jasir A, Kaijser B, Kronvall G, Riesbeck K. 2011. Invasive disease caused by Haemophilus influenzae in Sweden 1997–2009; evidence of increasing incidence and clinical burden of non-type b strains. Clin. Microbiol. Infect. 17:1638–1645 [DOI] [PubMed] [Google Scholar]
- 4.Rubach MP, Bender JM, Mottice S, Hanson K, Weng HY, Korgenski K, Daly JA, Pavia AT. 2011. Increasing incidence of invasive Haemophilus influenzae disease in adults, Utah, USA. Emerg. Infect. Dis. 17:1645–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grandi G. 2001. Antibacterial vaccine design using genomics and proteomics. Trends Biotechnol. 19:181–188 [DOI] [PubMed] [Google Scholar]
- 6.Giufre M, Carattoli A, Cardines R, Mastrantonio P, Cerquetti M. 2008. Variation in expression of HMW1 and HMW2 adhesins in invasive nontypeable Haemophilus influenzae isolates. BMC Microbiol. 8:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jurcisek JA, Bookwalter JE, Baker BD, Fernandez S, Novotny LA, Munson RS, Jr, Bakaletz LO. 2007. The PilA protein of non-typeable Haemophilus influenzae plays a role in biofilm formation, adherence to epithelial cells and colonization of the mammalian upper respiratory tract. Mol. Microbiol. 65:1288–1299 [DOI] [PubMed] [Google Scholar]
- 8.Chang A, Kaur R, Michel LV, Casey JR, Pichichero M. 2011. Haemophilus influenzae vaccine candidate outer membrane protein P6 is not conserved in all strains. Hum. Vaccin. 7:102–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dicko A, Odusanya OO, Diallo AI, Santara G, Barry A, Dolo A, Diallo A, Kuyinu YA, Kehinde OA, Francois N, Borys D, Yarzabal JP, Moreira M, Schuerman L. 2011. Primary vaccination with the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV) in infants in Mali and Nigeria: a randomized controlled trial. BMC Public Health 11:882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prymula R, Kriz P, Kaliskova E, Pascal T, Poolman J, Schuerman L. 2009. Effect of vaccination with pneumococcal capsular polysaccharides conjugated to Haemophilus influenzae-derived protein D on nasopharyngeal carriage of Streptococcus pneumoniae and H. influenzae in children under 2 years of age. Vaccine 28:71–78 [DOI] [PubMed] [Google Scholar]
- 11.Murphy TF. 2009. Current and future prospects for a vaccine for nontypeable Haemophilus influenzae. Curr. Infect. Dis. Rep. 11:177–182 [DOI] [PubMed] [Google Scholar]
- 12.Ronander E, Brant M, Eriksson E, Morgelin M, Hallgren O, Westergren-Thorsson G, Forsgren A, Riesbeck K. 2009. Nontypeable Haemophilus influenzae adhesin protein E: characterization and biological activity. J. Infect. Dis. 199:522–531 [DOI] [PubMed] [Google Scholar]
- 13.Singh B, Brant M, Kilian M, Hallstrom B, Riesbeck K. 2010. Protein E of Haemophilus influenzae is a ubiquitous highly conserved adhesin. J. Infect. Dis. 201:414–419 [DOI] [PubMed] [Google Scholar]
- 14.Singh B, Su YC, Riesbeck K. 2010. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Mol. Microbiol. 78:545–560 [DOI] [PubMed] [Google Scholar]
- 15.Singh B, Fleury C, Jalalvand F, Riesbeck K. 2012. Human pathogens utilize host extracellular matrix proteins laminin and collagen for adhesion and invasion of the host. FEMS Microbiol. Rev. 36:1122–1180 [DOI] [PubMed] [Google Scholar]
- 16.Hallstrom T, Blom AM, Zipfel PF, Riesbeck K. 2009. Nontypeable Haemophilus influenzae protein E binds vitronectin and is important for serum resistance. J. Immunol. 183:2593–2601 [DOI] [PubMed] [Google Scholar]
- 17.Singh B, Jalalvand F, Morgelin M, Zipfel P, Blom AM, Riesbeck K. 2011. Haemophilus influenzae protein E recognizes the C-terminal domain of vitronectin and modulates the membrane attack complex. Mol. Microbiol. 81:80–98 [DOI] [PubMed] [Google Scholar]
- 18.Hallstrom T, Singh B, Resman F, Blom AM, Morgelin M, Riesbeck K. 2011. Haemophilus influenzae protein E binds to the extracellular matrix by concurrently interacting with laminin and vitronectin. J. Infect. Dis. 204:1065–1074 [DOI] [PubMed] [Google Scholar]
- 19.Barthel D, Singh B, Riesbeck K, Zipfel PF. 2012. Haemophilus influenzae uses the surface protein E to acquire human plasminogen and to evade innate immunity. J. Immunol. 188:379–385 [DOI] [PubMed] [Google Scholar]
- 20.Singh B, Al Jubair T, Fornvik K, Thunnissen MM, Riesbeck K. 2012. Crystallization and X-ray diffraction analysis of a novel surface-adhesin protein: protein E from Haemophilus influenzae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68:222–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kabsch W. 2010. XDS. Acta Crystallogr. D Biol. Crystallogr. 66:125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pape T, Schneider TR. 2004. HKL2MAP: a graphical user interface for phasing with SHELX programs. J. Appl. Crystallogr. 37:843–844 [Google Scholar]
- 23.Sheldrick GM. 2008. A short history of SHELX. Acta Crystallogr. A 64:112–122 [DOI] [PubMed] [Google Scholar]
- 24.Vonrhein C, Blanc E, Roversi P, Bricogne G. 2007. Automated structure solution with autoSHARP. Methods Mol. Biol. 364:215–230 [DOI] [PubMed] [Google Scholar]
- 25.Langer G, Cohen SX, Lamzin VS, Perrakis A. 2008. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3:1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engel J, Furthmayr H. 1987. Electron microscopy and other physical methods for the characterization of extracellular matrix components: laminin, fibronectin, collagen IV, collagen VI, and proteoglycans. Methods Enzymol. 145:3–78 [DOI] [PubMed] [Google Scholar]
- 30.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797 [DOI] [PubMed] [Google Scholar]
- 31.Soto GE, Hultgren SJ. 1999. Bacterial adhesins: common themes and variations in architecture and assembly. J. Bacteriol. 181:1059–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuhnert P, Christensen H. (ed). 2008. Pasteurellaceae: biology, genomics and molecular aspects. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 33.Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38:W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esposito V, Temussi PA. 2011. Cystatins: a versatile family. BioMol. Concepts 2:95–102 [DOI] [PubMed] [Google Scholar]
- 35.de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. 2006. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34:W362–W365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayashi S, Wu HC. 1990. Lipoproteins in bacteria. J. Bioenerg. Biomembr. 22:451–471 [DOI] [PubMed] [Google Scholar]
- 37.Nakayama H, Kurokawa K, Lee BL. 2012. Lipoproteins in bacteria: structures and biosynthetic pathways. FEBS. J. 279:4247–4268 [DOI] [PubMed] [Google Scholar]
- 38.Johnson S, Tan L, van der Veen S, Caesar J, Goicoechea De Jorge E, Harding RJ, Bai X, Exley RM, Ward PN, Ruivo N, Trivedi K, Cumber E, Jones R, Newham L, Staunton D, Ufret-Vincenty R, Borrow R, Pickering MC, Lea SM, Tang CM. 2012. Design and evaluation of meningococcal vaccines through structure-based modification of host and pathogen molecules. PLoS Pathog. 8:e1002981 doi:10.1371/journal.ppat.1002981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bergmann S, Lang A, Rohde M, Agarwal V, Rennemeier C, Grashoff C, Preissner KT, Hammerschmidt S. 2009. Integrin-linked kinase is required for vitronectin-mediated internalization of Streptococcus pneumoniae by host cells. J. Cell Sci. 122:256–267 [DOI] [PubMed] [Google Scholar]
- 40.Bujacz G, Miller M, Harrison R, Thanki N, Gilliland GL, Ogata CM, Kim SH, Wlodawer A. 1997. Structure of monellin refined to 2.3 A resolution in the orthorhombic crystal form. Acta Crystallogr. D Biol. Crystallogr. 53:713–719 [DOI] [PubMed] [Google Scholar]
- 41.Esposito V, Guglielmi F, Martin SR, Pauwels K, Pastore A, Piccoli R, Temussi PA. 2010. Aggregation mechanisms of cystatins: a comparative study of monellin and oryzacystatin. Biochemistry 49:2805–2810 [DOI] [PubMed] [Google Scholar]
- 42.Corpet F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16:10881–10890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.