

Background: Noncanonical functions of matrix metalloproteases are poorly characterized.
Results: The prodomain of MMP23 co-localizes with and traps KV1.3 channels intracellularly, thereby suppressing KV1.3 currents.
Conclusion: A novel metalloprotease-independent channel-modulating function of the MMP23 prodomain has been identified.
Significance: The topological similarity of the prodomain of MMP23 and KCNE proteins suggests a shared mechanism of channel modulation.
Keywords: Endoplasmic Reticulum (ER), Matrix Metalloproteinase (MMP), Membrane Trafficking, NMR, Potassium Channels, Kv1.2, Kv1.3, MMMP23B, MMP23, Prodomain
Abstract
Matrix metalloproteases (MMPs) are endopeptidases that regulate diverse biological processes. Synthesized as zymogens, MMPs become active after removal of their prodomains. Much is known about the metalloprotease activity of these enzymes, but noncanonical functions are poorly defined, and functions of the prodomains have been largely ignored. Here we report a novel metalloprotease-independent, channel-modulating function for the prodomain of MMP23 (MMP23-PD). Whole-cell patch clamping and confocal microscopy, coupled with deletion analysis, demonstrate that MMP23-PD suppresses the voltage-gated potassium channel KV1.3, but not the closely related KV1.2 channel, by trapping the channel intracellularly. Studies with KV1.2-1.3 chimeras suggest that MMP23-PD requires the presence of the KV1.3 region from the S5 trans-membrane segment to the C terminus to modulate KV1.3 channel function. NMR studies of MMP23-PD reveal a single, kinked trans-membrane α-helix, joined by a short linker to a juxtamembrane α-helix, which is associated with the surface of the membrane and protected from exchange with the solvent. The topological similarity of MMP23-PD to KCNE1, KCNE2, and KCNE4 proteins that trap KV1.3, KV1.4, KV3.3, and KV3.4 channels early in the secretory pathway suggests a shared mechanism of channel regulation. MMP23 and KV1.3 expression is enhanced and overlapping in colorectal cancers where the interaction of the two proteins could affect cell function.
Introduction
Matrix metalloproteases (MMPs)4 are a family of zinc-dependent endopeptidases that degrade extracellular matrix proteins, cleave cell surface receptors, release apoptotic ligands, and activate chemokines and cytokines (1). Their metalloprotease activities are involved in tissue remodeling, cell proliferation, cell migration, differentiation, angiogenesis, apoptosis, and the immune response (2, 3). MMPs are synthesized as zymogens, from which the active enzymes are released after proteolytic cleavage and removal of their prodomain. Current knowledge about MMPs is derived almost exclusively from studies of the active enzymes and their metalloprotease activity. The only described functions of the prodomains also involve metalloprotease-dependent regulation (4, 5). Noncanonical functions of MMPs are poorly defined.
MMP23, a 391-residue type-II trans-membrane protein, is synthesized as a zymogen, which is anchored to ER/nuclear membranes via a trans-membrane segment located within an N-terminal 80-residue prodomain (see Fig. 1A) (6–10). The MMP23 prodomain lacks the cysteine switch (PRCGXPD motif) present in other MMPs, which maintains these enzymes in an inactive state (9). However, MMP23 contains an RRRR motif that can be cleaved by the serine protease furin (7), separating the membrane-tethered prodomain from the secreted active enzyme. The secreted protein contains three domains (see Fig. 1A) (2, 3, 10). At the N terminus is a Zn2+-dependent catalytic domain (Cat domain) like those in other metalloproteases. This is followed by a cysteine-rich region (TxD) with sequence and structural similarity to BgK and ShK, 35-residue peptide inhibitors of KV1 channels from the sea anemones Bunodosoma granulifera and Stichodactyla helianthus, respectively. At the C-terminal end is an immunoglobulin-like cell adhesion molecule (IgCAM) domain with sequence similarity to the ROBO1, ROBO3, and ROBO4 proteins (supplemental Fig. S1).
FIGURE 1.

MMP23-PD suppresses KV1.3 but not KV1.2 currents. A, schematic of full-length MMP23 (MMP23-FL) showing its functional domains. B, schematic of DsRed-tagged MMP23-FL and C-terminal deletion constructs. CatDom, catalytic domain. C, KV1.3 and KV1.2 currents in the presence or absence of MMP23-FL (red) and MMP23-PD (green). Scale bars represent 1 nA and 50 ms, respectively. D, scattergrams showing channel numbers/cell of KV1.3 and KV1.2 channels in the presence or absence of MMP23-FL or deletion constructs. Means are determined from n samples of 8–38 for KV1.3 and 6–17 for KV1.2, respectively. Error bars indicate S.E. The membrane capacitances of the patched cells were 15.1 ± 1.0 pF (cells co-expressing the DsRed vector), 15.4 ± 1.1 pF (cells co-expressing DsRed-MMP23-FL), and 18.36 ± 1.6 pF (cells co-expressing DsRed-MMP23-PD). Statistical significance is determined by Student's t test and indicated by p values.
We reported previously a novel channel-modulating, metalloprotease-independent, function for MMP23 (10). Full-length MMP23 zymogen suppresses voltage-gated KV1.3 and KV1.6 potassium channels by intracellular retention, but has no effect on related KV1 channels (e.g. KV1.2, KV1.7) (10). We hypothesized that this trapping function resided in the KV1.3- and KV1.6-blocking TxD in MMP23.
In the present study, we used deletion analysis to test this hypothesis. To our surprise, MMP23 retained its trapping function even after deletion of the TxD. Here we report that the prodomain of MMP23 (MMP23-PD) co-localizes with and suppresses KV1.3 channels, but not closely related KV1.2 channels, by intracellular trapping. This trapping is likely to take place in the ER because MMP23 co-localizes with the ER protein Stim-1. Purified MMP23-PD in lipid micelles is shown by NMR to consist of a single bent trans-membrane α-helix joined by a short linker to a juxtamembrane α-helix, which is associated with the surface of the membrane. Experiments with KV1 chimeras suggest that MMP23-PD requires the presence of the region of KV1.3 from the S5 segment to the C terminus in order for it to co-localize and trap the channel intracellularly. MMP23 and KV1.3 proteins are both present in normal human colon epithelium, and both are up-regulated in colonic cancers where channel modulation may occur.
EXPERIMENTAL PROCEDURES
Cell Lines, Transfection, and Cell Culture
Enhanced GFP-tagged human KV1.2 and human KV1.3 channels and their chimeric channels were transiently co-transfected with either DsRed monomer or one of the DsRed-tagged MMP23 constructs in COS-7 cells for 24–48 h. COS-7 cells were maintained in Dulbecco's modified Eagle's medium containing 10% heat-inactivated FCS (Summit Biotechnology), 4 mm glutamine, 1 mm sodium pyruvate, and 500 μg/ml G418 (Calbiochem) to 60–80% confluence in culture and were transfected with 0.4–1 μg of DNA using Lipofectamine 2000 (Invitrogen) in Opti-MEM I medium as per the manufacturer's protocol. After 48 h, transfection efficiency was assessed by fluorescence microscopy (Olympus).
The human KV1.2 channel was GFP-tagged on the C terminus in the pCMV6-AC-GFP vector (OriGene). The human KV1.3 cDNA coding region was amplified from a pCDNA3.1 vector using primers containing SgfI (AsiSI) and MluI restriction sites. This KV1.3 PCR fragment was then directionally cloned into the pCMV6-AC-GFP plasmid to create a GFP-tagged KV1.3 channel similar to the KV1.2 channel.
We exploited the unique BstXI restriction site located after Phe-305 (KV1.2 numbering) to construct KV1.3-1.2-GFP and KV1.2-1.3-GFP-tagged chimeric channels. This domain swap created a KV1.2-1.3 chimera containing the KV1.2 region from its N terminus up to its trans-membrane segment S4 (KV1.2 Phe-305) and KV1.3 trans-membrane segment S4-S5 linker up to the KV1.3 C terminus (see Fig. 3A). The KV1.3-1.2 chimera was constructed similarly utilizing the complementary domain exchange.
FIGURE 3.

MMP23-PD suppresses activity of the KV1.2-KV1.3 chimera but not the KV1.3-KV1.3 chimera. A, diagram showing the construction of the KV1.2-1.3 and the KV1.3-1.2 chimeras from domains of their respective KV1.2 and KV1.3 parental channels. B, the KV1.2-KV1.3 chimera containing the KV1.3 pore domain exhibits cumulative inactivation, a unique property of the Kv1.3 pore domain. C, the KV1.3-KV1.2 chimera containing the KV1.2 pore domain exhibits use-dependent potentiation, a unique property of the KV1.2 channel pore domain. Red traces represent the first traces of the currents elicited by pulses to +40 mV in 1-s intervals, and black traces represent subsequent current thereafter. Scale bars indicate 1 nA and 100 ms, respectively. D and E, scattergrams showing channel numbers/cell in cells expressing the KV1.2-1.3 and KV1.3-1.2 chimeras in the presence of DsRed vector or DsRed-MMP23-PD. Means are determined from n samples of 9–13. Error bars indicate S.E. Membrane capacitances of the patched cells were 17.1 ± 0.9 pF (cells co-expressing KV1.2-1.3 + the DsRed vector), 15.3 ± 0.5 pF (cells co-expressing KV1.2-1.3 + DsRed-MMP23-PD), 18.0 ± 1.1 pF (cells co-expressing KV1.3-1.2 + the DsRed vector), and 15.6 ± 0.8 pF (cells co-expressing KV1.3-1.3 + DsRed-MMP23-PD). Statistical significance is determined by Student's t test and indicated by p values.
The rat MMP23 cDNA was cloned into the pDsRed vector, and subsequent deletion constructs of the MMP23 protein were created by PCR-mediated site-directed mutagenesis using this vector as the template. Point mutations were inserted to create AUU stop codons, and changes were confirmed by sequencing. The YFP-Stim-1 plasmid was a kind gift from Dr. Michael D. Cahalan (University of California Irvine).
Electrophysiology
The whole-cell patch clamp technique (11) was used to characterize the current amplitude of COS-7 cells transiently expressing KV channels as described previously (12, 13), in the presence of different MMP23 constructs. Currents were recorded in normal Ringer solution with a Ca2+-free pipette solution containing (in mm): 145 KF, 10 HEPES, 10 EGTA, and 2 MgCl2, pH 7.2, 300 mosm. Transfected cells were visualized by epifluorescence microscopy of co-expressed green and red fluorescent proteins. Data acquisition and analysis were performed using pClamp software.
Confocal Microscopy
COS-7 cells were transiently transfected with either DsRed monomer or DsRed-MMP23 with hKV1.3, KV1.2, KV1.2-1.3, or KV1.3-1.2. Co-transfected cells were allowed to adhere to poly-l-lysine (Sigma)-coated coverslips overnight prior to fixing (4% paraformaldehyde, Sigma). Cells were imaged by confocal microscopy (LSM Zeiss 700). Images were analyzed for co-localization using LSM Zeiss Zen 2011 software (n = 3 independent experiments; 15–30 cells were imaged for quantification of co-localization).
Immunohistochemistry
Human colon sections (6 μm; formalin-fixed, paraffin-embedded) were obtained from the University of California Irvine (UCI) Experimental Tissue Resource. The slides were de-identified so that no private health information was discoverable. Sections were dewaxed with xylene (3 times, 5 min each) and rehydrated through graded alcohols (100–30%). Before staining with the primary antibody, citrate buffer-based (Vector H3300) antigen retrieval was performed for 20 min to unmask antigens masked by paraffin embedding. Endogenous peroxidase activity was blocked by 1.5% hydrogen peroxide solution in PBS for 20 min. After blocking with 5% corresponding serum, 5% BSA, and 0.1% sodium azide in PBS, sections were incubated with primary antibody overnight at 4 °C. We used a rabbit anti-KV1.3 polyclonal antibody (a gift from Dr. Hans Gunther-Knaus, University of Innsbruck, 1:50 dilution) and rabbit anti-MMP23 polyclonal antibody (Millipore, 1:100 dilution) described in the Human Protein Atlas. As the isotype control, we used rabbit polyclonal IgG in place of primary antibodies. Bound primary Abs and the isotype control were detected with a goat anti-rabbit biotinylated secondary antibody followed by an HRP-conjugated avidin complex using VECTASTAIN Elite ABC kit (Vector Laboratories). Peroxidase activity was visualized with 3,3′-diaminobenzidine using the diaminobenzidine substrate kit for peroxidase (Vector Laboratories). Sections were counterstained with hematoxylin (Fisher Scientific) and bluing solution, dehydrated through graded alcohol and xylene washes, and mounted with Permount (Fisher Scientific). Two independent investigators scored the intensity of KV1.3 and MMP23 expression on a scale of 1–3 in normal epithelium, colon adenomas, and colorectal cancers.
Cloning, Expression, and Purification of MMP23-PD
The cDNA for the prodomain of human MMP23 (residues 1–78) was amplified by PCR from the vector pCMV6-XL5 (OriGene Technologies Inc., Rockville MD) using the forward primer 5′-TAA ACC ATG GCA CTG GAA GTT CTG TTT CAG GGC CCG CCC TTC CCC ACG GTG GCC ACC ACC CCA CCG-3′ and the reverse primer 5′-TTT TAC AAG CTT CTA TTA TCA GCG GCG TCT GCG GGG GGC CAG TGG-3′. The amplified product was cloned into the expression vector pET32a(+) (Novagen Inc., La Jolla, CA) via the NcoI and HindIII restriction sites. The final constructs contained cDNA for the N-terminal thioredoxin fusion protein followed by a hexahistidine tag, a HRV 3C protease cleavage site, and the MMP23-PD protein.
The expression construct containing the MMP23-PD gene was transformed into Escherichia coli BL21(DE3) cells (Novagen/Merck). Transformants were grown in M9 minimal medium containing 100 μg/μl ampicillin with [15N]H4Cl and d-[13C6]glucose as the sole sources of nitrogen and carbon, respectively. Cells were grown at 37 °C to an A600 of 0.5–1.0 and then overnight at 28 °C following the addition of 1 mm isopropyl-1-thio-β-d-galactopyranoside to induce expression of the thioredoxin-MMP23 prodomain fusion protein (Trx-MMP23-PD). Cells were harvested by centrifugation, and the pellet was resuspended in lysis buffer composed of BugBuster protein extraction reagent (Merck) supplemented with 10 mm CHAPS (Sigma-Aldrich), 1 mm PMSF, EDTA-free Complete® protease inhibitor mixture (Roche Applied Science), 1.0 mg/ml lysozyme, and 5 μg/ml DNase I. The cell suspension was probe-sonicated (XL-2000 ultrasonic liquid processor; Misonix, Farmingdale, NY; power level of 8.5, 30-s pulses separated by 30 s) for 15 min on ice. The lysate was centrifuged at 4 °C and 35,000 × g for 20 min to remove insoluble cell debris. The supernatant containing the Trx-MMP23-PD fusion protein was loaded at room temperature onto a 5-ml HiTrap (Amersham Biosciences) chelating column charged with nickel. The column was extensively washed with buffer A (20 mm Tris buffer, pH 8.0, containing 150 mm NaCl and 10 mm CHAPS) containing 5 mm imidazole followed by buffer A containing 50 mm imidazole. The protein was eluted with buffer A containing 350 mm imidazole. Cleavage of the hexahistidine-tagged fusion protein was carried out overnight at 4 °C at a protein concentration of 1.0 mg/ml using 1.0 unit of HRV 3C protease (Novagen) per mg of protein. The Trx tag was removed by reverse-phase HPLC on a Luna C8 column equilibrated in solvent A (99.9% water and 0.01% TFA). Proteins were eluted with an acetonitrile gradient in 0.01% TFA (0–40% over 10 min followed by 40–80% over 30 min). Eluted fractions containing MMP23-PD were lyophilized, dissolved in 20 mm sodium citrate, pH 5.0, containing 20 mm TCEP, and 10 mm CHAPS, and then purified on the same column using an acetonitrile gradient in 0.01% TFA of 0–50% over 5 min followed by 50–70% over 40 min. Fractions containing MMP23-PD were lyophilized and stored at −20 °C. The concentration of the protein was determined from the A280 using an extinction coefficient of 0.962 mg/ml protein per A280 unit (14)in a 1-cm path length cell.
Circular Dichroism Spectroscopy
CD experiments were performed on a model 410SF CD spectropolarimeter (Aviv Biomedical). Far UV CD spectra were recorded in a 0.01-cm path length quartz cuvette over the wavelength range 190–260 nm, with a resolution of 0.5 nm, a bandwidth of 1 nm, and a response time of 1 s. Spectra were averaged over eight separate scans and corrected for background. The sample contained 14 μm MMP23-PD in 20 mm sodium citrate buffer, pH 5.0, containing 100 mm deuterated n-dodecylphosphocholine (d38-DPC; Sigma-Aldrich).
NMR Spectroscopy
Lyophilized uniformly 15N- and 13C-15N-labeled MMP23-PD was dissolved in a buffer containing 20 mm sodium citrate (pH 5.0), 100 mm deuterated DPC, 20 mm TCEP, 0.02% (w/v) sodium azide (NaN3), 90% H2O, and 10% 2H2O, at a final protein concentration of 0.7 mm. Two-dimensional 1H-15N heteronuclear single quantum coherence (HSQC) spectra of MMP23-PD in DPC micelles were recorded on a Varian Inova 600-MHz spectrometer equipped with a cryogenically cooled triple-resonance probe at various temperatures to determine the optimal conditions for subsequent experiments. 256 and 1024 complex points were acquired in the t1 (15N dimension) and t2 (1H dimension) time domains, respectively. The data were zero-filled to 512 × 2048 and apodized using a sine-bell squared window function prior to Fourier transformation using NMRPipe (version 3.0).
NMR data were also acquired on Bruker Avance spectrometers operating at 1H frequencies of 600 and 800 MHz and equipped with cryogenically cooled triple-resonance probes. Backbone resonance assignments were obtained at 45 °C using the following three-dimensional experiments, employing conventional or nonuniform sampling in the 15N and 13C dimensions: HN(CA)CO, HNCO, HN(CO)CA, HNCA, CBCA(CO)NH, and HNCACB (15). Nonuniform sampling datasets were processed using the multidimensional decomposition algorithm (16). Uniformly sampled datasets were processed using TOPSPIN (version 3.0) from BrukerBioSpin or NMRPipe (17). Data were analyzed using NMRView (18), and initial automated assignments were obtained with the program PINE (19).
15N relaxation experiments were performed at 30 °C on a Varian 600-MHz spectrometer. R1 values were obtained from a series of 1H-15N correlation spectra with 0-, 10-, 50-, 100-, 300-, 500-, 800-, 1000-, 1500-, 2000-, and 2500-ms relaxation delays. R2 values were acquired with 10-, 30-, 50-, 70-, 90-, 110-, 130-, 150-, 170-, 190-, and 210-ms delays. The relaxation rates were calculated in NMRView by least square fitting of peak intensities versus relaxation delay times to an equation for single-order exponential decay. The reported errors were standard deviations derived from the fit of the data. Steady-state 1H-15N NOE values were determined from the ratio of peak intensities for spectra collected with and without 3-s proton presaturation. The experimental uncertainty was estimated from the signal-to-noise ratio for each spectrum.
The rotational correlation time (τc) was determined using the following equation where, in the limit of slow molecular motion (τc ≫ 0.5 ns), the correlation time of a protein is related to the ratio of the longitudinal (t1) and transverse (t2) relaxation times and the nuclear frequency (νN)
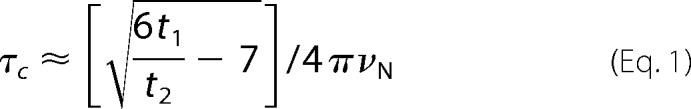 |
where τc is the effective rotational correlation coefficient and νN is the 15N resonance frequency. Equation 1 was derived from Equation 8 in Kay et al. (20) by considering only J(0) and J(ω) spectral densities and neglecting higher frequency terms. The effective hydrodynamic radius a was calculated from Stokes law
where η is the viscosity of the solvent, κ is the Boltzmann constant, and T is the temperature.
The Clean SEA-HSQC experiment (21) was performed on a 0.7 mm uniformly 15N-labeled sample of MMP23-PD in 20 mm sodium citrate buffer containing 20 mm TCEP, 100 mm DPC, 90% H2O, 10% 2H2O, and 0.02% NaN3 at pH 5.0. Spectra were acquired on a Varian Inova 600-MHz spectrometer at 30 °C. The mixing time was 10 ms, and the prescan delay was 3.0 s.
Paramagnetic relaxation enhancement experiments were carried out by the addition of 10 μl of 0.5 m Gd(DTPA-BMA) (22) to 500 μl of 0.5 mm 15N-labeled MMP23-PD in 20 mm sodium citrate buffer, pH 5.0, containing 20 mm TCEP, 100 mm DPC, 90% H2O, 10% 2H2O, and 0.02% NaN3 at 30 °C. 1H-15N HSQC spectra were collected using eight scans and 1024 × 128 complex points for a total acquisition time of 30 min.
Molecular Modeling
All molecular dynamics calculations were performed using the GROMACS (version 4.5.5) package of programs (23) employing the Martini force field (24, 25). The time step used was 25 fs. Peptide, lipid, and solvent (including ions) were coupled separately to a thermal bath at 320 K employing velocity rescaling (26), applied with a coupling time of 1.0 ps. The pressure was maintained at 1 bar by applying a semi-isotropic coupling to a Berendsen barostat (27) with a coupling constant of 5.0 ps and compressibility of 4.5 × 10−5 bar. A shifted nonbonded potential was used, with a distance cutoff of 12 Å and a shift distance of 9 Å, applying a neighbor list update frequency of 10 steps (250 fs). Coulombic interactions were screened using a relative dielectric constant of 15.
An initial model of the prodomain (PD) peptide was generated employing only the secondary structure restraints determined from the NMR experiments; residues Gly-15 to Ala-27, Leu-28 to Leu-31, Pro-32 to Leu-40, and Ala-47 to Leu-58 were restrained to adopt an α-helical conformation. Generation of this atomistic model used the MODELLER program (version 9.10 (28)). From this atomistic model, a coarse-grain (CG) model was created, and the trans-membrane helix aligned along the Z-coordinate axis. The PD was inserted into a CG representation of a preformed dioleoylphosphatidylcholine lipid bilayer, with water (and antifreeze particles, 10%) and sufficient Na+ and Cl− ions to provide a solvent ionic concentration of 0.15 m. The insertion employed a simple transmutation in which ghost atoms (those in which the Lennard-Jones interaction parameters were set to zero) were transformed into the desired CG representation; this adiabatic transformation was conducted over 1.25 ns with the PD restrained at its original position. Following PD insertion, the simulation was continued with no restraints for a further 500 ns. An atomistic model of the final geometry from the molecular dynamics simulation was created using the CG representation of the PD as a template in comparative modeling using the MODELLER program; secondary structure restraints (described above) were applied, along with distance restraints between Cα carbons restrained to the separation between the equivalent backbone CG particles.
RESULTS
The Prodomain of MMP23 (MMP23-PD) Selectively Suppresses KV1.3 Channels without Affecting Closely Related KV1.2 Channels
Full-length rat MMP23 (MMP23-FL), tagged at the N terminus with DsRed-monomer (Fig. 1B), was co-expressed in COS-7 cells with C-terminally GFP-tagged KV1.3 or KV1.2 channels. Whole-cell patch clamp experiments performed 48 h later showed that MMP23-FL suppressed KV1.3 currents but not KV1.2 (Fig. 1, C and D). Cells co-transfected with KV1.3 and vector expressed 2967 ± 434 channels/cell (mean ± S.E.), whereas cells co-transfected with KV1.3 MMP23-FL expressed 1256 ± 216 channels/cell (p < 5 × 10−4). KV1.3 channels in cells co-transfected with rat MMP23-FL were indistinguishable in terms of voltage dependence of activation or inactivation from cells expressing KV1.3 alone (supplemental Fig. S2). This result suggests that MMP23-FL decreases the number of functional KV1.3 channels on the cell surface but does not alter the properties of the channel. Because MMP23-FL has not been detected in the plasma membrane (6, 7, 10), it is possible that the KV1.3 channels on the surface of MMP23-FL-expressing cells are not associated, directly or indirectly, with MMP23-FL and may represent channels that have escaped from an intracellular pool.
To define the region of rat MMP23-FL responsible for KV1.3 suppression, we generated three C-terminal deletion constructs of MMP23, tagged at the N terminus with DsRed monomer, by progressively removing the immunoglobulin-like cell adhesion molecule domain, TxD, and Cat domain (Fig. 1B). All three MMP23 deletion constructs suppressed KV1.3 but not KV1.2 currents (Fig. 1, C and D), highlighting the specificity of this suppression. MMP23-PD, the shortest construct, caused equivalent suppression of KV1.3 to that of MMP23-FL (Fig. 1, C and D; 1181 ± 269 KV1.3 channels/cell, p < 5 × 10−5). MMP23-PD did not change the properties of KV1.3 channels (supplemental Fig. S2). These data suggest that MMP23-FL and MMP23-PD have the same ability to serve as functional modulators of KV1.3 channels, they both deplete the number of functional channels on the cell surface, and MMP23-PD is likely the region of MMP23 responsible for KV1.3 suppression.
MMP23-PD corresponds exactly to the naturally occurring prodomain that is liberated by furin cleavage in human and mouse MMP23 (supplemental Fig. S3) (6, 7). In an earlier study (7), protein sequencing of the secreted processed mouse MMP23 enzyme showed an N-terminal sequence of YTLTPARL (residues 79–86), which indicates that the prodomain remaining after furin cleavage ends with the C-terminal sequence RRRR (residues 75–78) and is the same Arg-78 residue at the C-terminal end of rat MMP23-PD (supplemental Fig. S3). Although rat MMP23-FL contains the furin cleavage site (supplemental Fig. S3), it is not processed in mammalian cells for reasons that remain unclear (6). Because we used COS-7 cells for our experiments, rat MMP23-FL would not have been processed, suggesting that the full-length MMP23 protein has the potential to act as a KV1.3 channel suppressor without being cleaved.
MMP23-PD Co-localizes Intracellularly with KV1.3, but Not KV1.2 Channels
We used confocal microscopy to determine whether KV1.3 channel suppression was associated with intracellular trapping of the channel. First, we studied the localization of MMP23-FL and MMP23-PD. C-terminally tagged mouse, rat, and human MMP23 all localize to perinuclear regions and the ER in mammalian cells (6, 7). N-terminally GFP-tagged rat MMP23-FL also co-localizes with the ER marker sarcoplasmic reticulum Ca2+-ATPase (SERCA) (10). Corroborating the published data, N-terminally DsRed-tagged MMP23-FL and MMP23-PD both co-localized with the ER marker YFP-Stim-1 in COS-7 cells (Fig. 2, A and B). These results together with the published data demonstrate that MMP23 and MMP23-PD, either N-terminally or C-terminally tagged, localize to ER/Golgi and perinuclear membranes.
FIGURE 2.

Co-localization of MMP23 with KV1.3 but not KV1.2. A and B, confocal microscopy demonstrating that MMP23-FL and MMP23-PD co-localize with YFP-Stim-1, an ER marker. YFP-Stim-1: pseudocolor green was used for the presentation. C, quantification of co-localization of MMP23-FL and MMP23-PD with KV1.3 and KV1.2. Means are determined from n samples of 10–35 for KV1.3 and 6–14 for KV1.2, respectively. Error bars indicate S.E. Statistical significance is determined by Student's t test and indicated by p values. D and E, confocal images showing that DsRed-MMP23-FL and DsRed-MMP23-PD co-localize with KV1.3-GFP but not with KV1.2-GFP, whereas the DsRed vector does not co-localize with either channel.
We then examined whether DsRed-tagged MMP23-FL and MMP23-PD co-localized with GFP-tagged KV1.3 expressed in COS-7 cells. Both MMP23 proteins exhibited strong co-localization with KV1.3 but not with the vector control or with KV1.2 (Fig. 2, C–E). The MMP23-TxD and MMP23-Cat domain proteins also co-localized with KV1.3 (supplemental Fig. S4). Quantification of the degree of co-localization (co-localization percentage) indicated that MMP23-FL and MMP23-PD co-localized significantly and to equivalent extents with KV1.3 but not with KV1.2 (Fig. 2C). This co-localization is likely to be in an intracellular compartment because fewer KV1.3 channels/cell are detected on the cell surface by patch clamp (Fig. 1D), and MMP23-FL and MMP-23-PD are both present in ER/Golgi and perinuclear membranes (Fig. 2, A and B). In support of this, less KV1.3 protein was detected on the cell surface in cells co-expressing MMP23-FL and MMP23-PD than cells co-expressing the DsRed vector control (supplemental Fig. S5).For each cell, we plotted cell surface KV1.3 expression versus intracellular KV1.3 co-localization with DsRed vector, DsRed-MMP23-FL, or DsRed-MMP23-PD. Intracellular co-localization was inversely related to KV1.3 surface expression; as intracellular co-localization increased, cell surface KV1.3 decreased. These results suggest that MMP23-FL and MMP23-PD deplete KV1.3 channels on the cell surface by a mechanism consistent with trapping KV1.3 in intracellular compartments, most likely the ER. This co-localization and trapping may be via a direct or an indirect interaction.
The KV1.3 Region from the S5 Segment to the C Terminus Is Required for MMP23-PD-mediated Suppression and Co-localization
We constructed C-terminal GFP-tagged chimeras of MMP23-modulated KV1.3 and MMP23-resistant KV1.2 to identify the KV1.3 region required for modulation by MMP23-PD.The KV1.2-1.3 chimera contains the N terminus through to the end of the S4 segment of KV1.2 and S5 through to the C-terminal end of KV1.3. The KV1.3-1.2 chimera contains the N terminus through to the end of the S4 segment of KV1.3 and S5 through to the C-terminal end of KV1.2 (Fig. 3A). Notably, KV1.2 and KV1.3 share identical sequences across the S4-S5 chimera ligation region. Both chimeras produced robust currents when expressed in COS-7 cells (Fig. 3, B and C) and behaved as expected. Cumulative inactivation, a unique property of the KV1.3 pore domain (29), was exhibited by the KV1.2-1.3 chimera (containing the KV1.3 pore domain) but not by the KV1.3-1.2 chimera (containing the KV1.2 pore domain) (Fig. 3, B and C). In contrast, the KV1.3-KV1.2 chimera but not the KV1.2-1.3 chimera exhibited use-dependent activation, a unique property of the pore domain of KV1.2 (Fig. 3, B and C). Next, we expressed MMP23-PD in COS-7 cells together with the KV1.2-1.3 or KV1.3-1.2 chimeras. MMP23-PD decreased the number of functional KV1.2-1.3 channels on the cell surface from 2573 ± 714 channels/cell in cells co-expressing the DsRed vector to 764 ± 313 channels/cell in cells co-expressing DsRed-MMP23-PD (Fig. 3D; p < 0.05). In contrast, MMP23-PD had no effect on the KV1.3-1.2 chimera (Fig. 3E). Cells co-transfected with the KV1.3-1.2 chimera and the DsRed vector expressed 1028 ± 203 channels/cell, and cells co-transfected with the KV1.3-1.2 chimera and DsRed-MMP23-PD expressed 1104 ± 299 channels/cell (Fig. 3E). In confocal experiments, MMP23-FL and MMP23-PD both co-localized significantly with the KV1.2-1.3 chimera, but not with the KV1.3-1.2 chimera (Fig. 4, A–C). Together, these results demonstrate that the region of KV1.3 extending from the S5 segment through to the C-terminal end is required for MMP23-PD-mediated depletion of functional channels on the cell surface and for intracellular co-localization with MMP23-PD.
FIGURE 4.

MMP23-PD co-localizes with the KV1.2-1.3 but not the KV1.3-1.2 chimera. A and B, confocal microscopic images showing that DsRed-MMP23-FL and DsRed-MMP23-PD co-localize with the GFP-KV1.2-1.3 chimera but not the GFP-KV1.3-1.2 chimera. C, quantification of co-localization of DsRed-MMP23-FL and MMP23-PD with GFP-KV1.2-1.3 versus GFP-KV1.3-1.2. Means are determined from n samples of 9–17. Error bars indicate S.E. Statistical significance is determined by Student's t test and indicated by p values.
KV1.3 and MMP23 Share Overlapping Expression in the Human Colon
The Human Protein Atlas reports strong expression of MMP23 in human colonic epithelium, and an earlier study described KV1.3 expression in human colonic epithelium, especially in colorectal cancers (30, 31). We therefore performed immunohistochemistry studies to determine whether the two proteins exhibited a similar expression pattern in colonic epithelium. In normal colon, we observed equivalent intensity of staining of MMP23 and KV1.3 in colonic epithelium (Fig. 5, A and E). In tissues from patients with colon cancer, we scored for staining intensity in normal-looking epithelium, in adenomas, and in malignant epithelium. Expression of both KV1.3 and MMP23 was lower in the normal-looking epithelium and in adenomas when compared with malignant epithelium (Fig. 5, B and E). Higher magnification images were consistent with KV1.3 and MMP23 being mainly intracellular, both in primary colorectal tumors and in metastatic tumors in the lymph node (Fig. 5C) and liver (not shown). The overlapping intracellular pattern of MMP23 and KV1.3, particularly in colorectal cancers, suggests that the two proteins may interact, directly or indirectly, in colonic epithelium.
FIGURE 5.

Expression of KV1.3 and MMP23 in human colon. A, immunohistochemistry of normal human colon (at 20×) showing KV1.3 (left) and MMP23 (right) in epithelial cells. Inset, the same isotype control was used for both KV1.3 and MMP23; polyclonal rabbit IgG was used in place of the primary rabbit anti-KV1.3 or rabbit anti-MMP23 antibodies. B, KV1.3 (left, 20×) and MMP23 (right, 20×) expression in colorectal cancer. Arrows indicate adenoma (yellow) and KV1.3- and MMP23-expressing malignant colorectal epithelium (red). C, higher magnification images of colorectal cancer showing KV1.3 (left, 100×) and MMP23 (right, 100×). Note intracellular staining of both proteins. D, KV1.3 (left, 20×) and MMP23 (right, 20×) in metastatic colorectal epithelium in lymph nodes. The red arrows highlight the KV1.3- and MMP23-expressing metastatic colorectal epithelium. The surrounding lymph node is not stained. E, scoring of staining intensity on a scale of 1–3 of KV1.3 and MMP23 in epithelium from normal colon and from three regions of colonic tissue from patients with colorectal cancer: normal-looking crypts, adenoma, and malignant epithelium.
Expression and Purification of MMP23-PD
Purified recombinant MMP23-PD eluted as two separate peaks on a reverse-phase HPLC column, corresponding to monomeric and dimeric forms of the protein (Fig. 6, B and C). SDS-PAGE analysis under reducing and nonreducing conditions confirmed that the dimer resulted from intermolecular disulfide formation via the single Cys residue in MMP23-PD. Monomeric 15N-labeled MMP23-PD ran as a single band on SDS-PAGE, and electrospray mass spectrometry analysis gave a mass of 8161.06 ± 0.75 Da, consistent with the theoretical value of 8161.5 Da. MMP23-PD contains a stretch of predominately hydrophobic amino acid residues (Trp-20–Leu-40) identified as a potential trans-membrane domain (TMD) (supplemental Fig. S6) that overlaps a region predicted to adopt a helical conformation (supplemental Fig. S7A). The N-terminal region of this helix (17ERRW20) contains residues that are often found at the membrane interface of TMDs (32). The prediction of secondary structure is variable in the region just C-terminal to the TMD, although there is a propensity for helical content in this region. In support of these predictions, the CD spectrum of MMP23-PD in DPC micelles exhibited two prominent minima at 208 and 222 nm, indicative of significant α-helical content (supplemental Fig. S7B).
FIGURE 6.

Purification and backbone resonance assignments for MMP23-PD. A, MMP23-PD construct used for these studies. Trx, thioredoxin; 6-His, hexahistidine tag; 3C, 3C protease cleavage site; Pro, MMP23 prodomain. B, reverse-phase HPLC purification of MMP23-PD. The monomeric and dimeric forms of MMP23-PD eluted in fractions 4 and 5, respectively. C, SDS-PAGE analysis of purified MMP23-PD. Reverse-phase HPLC fraction numbers are shown in lanes 1 and 2, respectively. D, amino acid sequence of MMP23-PD with the predicted TMD highlighted in red. Spectral overlap prevented assignment of Val-26 (underlined). The sequence includes the N-terminal residual tag residues Gly-1 and Pro-0. E, 1H-15N HSQC spectrum of uniformly 13C-15N-labeled MMP23-PD in 20 mm sodium citrate buffer, pH 5.0, containing 100 mm DPC, 20 mm TCEP, 10% 2H2O, 90% H2O, and 0.02% w/v NaN3 at a final protein concentration of 0.7 mm. Inset: Trp indole NH cross-peaks (bottom right) and one of the Gln-52 side chain resonances (center right). Aliased resonances from the nine Arg side chains are highlighted with red boxes.
NMR Assignments of MMP23-PD in DPC Micelles
1H-15N HSQC spectra of MMP23-PD were recorded at 30, 40, and 45 °C (supplemental Fig. S8). Several backbone amide resonance peaks that appeared weak at 30 °C were significantly more intense at higher temperatures. The 600-MHz two-dimensional 1H-15N HSQC spectrum acquired at 45 °C for 0.7 mm 13C-15N-labeled MMP23-PD is shown in Fig. 6E. The spectrum revealed relatively narrow peak dispersion in the 1H dimension, typical for a single α-helical trans-membrane protein. The spectrum contained the correct number of resonances for a protein of this size, and the majority were well resolved. Several weaker peaks were observed in the 1H-15N HSQC spectrum, suggesting a slow equilibrium of two conformations, presumably due to Pro cis/trans-isomerization. However, only the cross-peaks from the major conformer were analyzed here.
Backbone resonance assignments for MMP23-PD in DPC micelles at 45 °C were determined using three-dimensional heteronuclear NMR experiments (Fig. 7). The majority (97%) of the 68 nonproline backbone 1H and 15N assignments for MMP23-PD were completed (excluding the two N-terminal residues arising from the vector). 13Cα and 13Cβ backbone assignments were both 97% complete. Backbone amide resonances corresponding to Val-26 could not be assigned due to resonance overlap. The 13Cβ chemical shift for Cys-29 at 27.5 ppm clearly indicated that the thiol group for this residue existed in the reduced state and did not engage in any intermolecular disulfide bonding. Secondary 1Hα, 1HN, 13Cα, and 13Cβ chemical shifts (Fig. 7, B–F) and predicted N- and C-helical capping regions (Fig. 7, G and H) indicated that MMP23-PD contains a relatively long helix (α1) (residues Glu-17–Leu-40) incorporating the TMD, which is joined by a short linker to a juxtamembrane helical region (α2) (residues Ala-47–Leu-58). These results are similar to predictions obtained using TALOS+ (33) (data not shown) and Motif Identification from Chemical Shifts (MICS) (34) (Fig. 7I). Identification of helical capping motifs using the MICS program was used to aid in assigning the N- and C-terminal boundaries for each helix (Fig. 7, G and H). Three potential N-terminal helical capping motifs were identified at residues Glu-17, Ala-47, and Asp-54, whereas residues Leu-40 and Leu-58 were predicted as C-terminal helical capping motif. N-terminal helical capping motif residues Glu-17 and Ala-47 are located at the N termini of helices α1 and α2, respectively, whereas Asp-54 lies in the middle of α2. Secondary chemical shift data also indicate a possible disruption of helix α2 at residue 53, suggesting the possibility that helix α2 may in fact consist of two short α-helices. The data also suggest a disruption in the TMD α1 helix near residues Ala-27–Leu-28, roughly one helical turn N-terminal to Pro-32. Consistent with this observation, the resonances for Cys-29 and Ala-33 were shifted well up-field of other resonances, possibly due to distortion of the α1 helix within this region.
FIGURE 7.

Secondary structure of MMP23-PD. A, amino acid sequence of MMP23-PD where α-helical regions are boxed in cyan and residues in the predicted trans-membrane domain are colored red. B–F, deviations of 13Cα-13Cβ (B), 13Cα(C), 13Cβ (D), 1Hα (E), and 1HN (F) chemical shifts from random coil values (56). Sites for which no values were determined are denoted by a P for proline residues or an asterisk for unassigned. Secondary structure elements derived from CSI values are illustrated at the top and boxed by a dotted line in each figure. Helix α1 extends from residue Glu-17 to Leu-40 and helix α2 from residue Ala-47 to Leu-58. G and H, prediction of N-terminal (G) (Ncap) and C-terminal (H) helical capping (Ccap) motifs using the MICS algorithm. I, helical regions for MMP23-PD determined by MICS (34). MICS is similar to TALOS+ (33), which uses an artificial neural network to predict protein ϕ and ψ backbone torsion angles and protein secondary structure for a given residue along with its two flanking residues using a combination of six kinds of chemical shifts (HN, HA, CA, CB, CO, N) and the amino acid sequence. On the other hand, MICS uses an artificial neural network trained to recognize additional structural features such as N- and C-terminal helical capping motifs and the most common types of β-turns for a given residue (i) within a hexapeptide spanning residues i - 1 to 1 + 4. J, model of MMP23-PD in a lipid membrane. Shown is a model of TMD (α1 helix) and juxtamembrane (α2) helix of MMP23-PD in a lipid membrane. Atoms of the side chain groups of residues in the two helices are highlighted. Phosphate groups of the lipid are represented as blue spheres; lipid choline, glycerol and acyl chains, and water are omitted for clarity.
Employing only the secondary structure information obtained above, we constructed a model of MMP23-PD in a lipid membrane (Fig. 7J). The model illustrates how the TMD α1 helix can span the membrane with a slight kink associated with Pro-32, and the α2 helix remains associated with the lipid head groups and, along with the linker region, is consequently largely shielded from solvent.
Backbone Dynamics of MMP23-PD in DPC Micelles
Backbone 1H-15N steady-state NOEs and 15N longitudinal (R1) and transverse (R2) rate constants for MMP23-PD are shown in Fig. 8. The average 1H-15N NOE, R1, and R2 values for the full-length protein and α-helical regions of MMP23-PD are given in supplemental Table S1. The observation of mostly positive NOE values for residues 14–63 showed that the conformation of α1 (containing the TMD), α2, and the intervening linker was restrained, whereas the N and C termini (residues 1–13 and 64–78, respectively) were disordered (Fig. 8A). Residues in helices α1 and α2 displayed large transverse relaxation rates (R2) (Fig. 8C), giving rise to broader line widths and reduced peak intensities when compared with other regions of the protein (supplemental Fig. S9). Peak intensities over this region of MMP23-PD increased at higher temperatures, primarily due to a decrease in the correlation time for the MMP23-PD/micelle complex. R1, R2 and R1/R2 values indicated that the degree of rigidity within this region varied, with the backbone of α2 being slightly more mobile than α1 (Fig. 8, B and D). This was expected because the TMD of α1 is buried and dynamically constrained by lipid within the DPC micelles. However, R2 values indicated some degree of flexibility in the TM α1 helix around residues Ala-27 to Leu-28. Based on the R1/R2 values for residues within the TMD (residues Glu-17–Leu-40), it was possible to calculate an overall isotropic global correlation time (τc) of 14 ns for MMP23-PD in DPC micelles at 30 °C. This corresponds to the rotation of a globular particle with an effective hydrodynamic radius of 26.4 Å and an apparent volume of ∼77.1 nm3 (assuming that the MMP23-PD-DPC micelles are spherical), comparable with values measured for other peptides embedded in DPC micelles (35).
FIGURE 8.

NMR relaxation parameters and membrane topology of MMP23-PD in DPC micelles at 30 °C and 600 MHz. A–D, 1H-15N steady-state NOE values (A), longitudinal (R1) (B), transverse (R2) 15N relaxation rates (C), and ratio of R1 to R2 for MMP23-PD (D). E, ratio of peak intensities before and after the addition of the water-soluble paramagnetic spin label Gd(DTPA-BMA) (19). F, peak intensities for the Clean SEA-HSQC spectrum (Cleanex) (18) of MMP23-PD in DPC micelles, measuring the exchange of backbone amide protons with solvent. Sites for which no values were determined are denoted by a P for proline residues or an asterisk for unassigned residues. Secondary structure elements derived from CSI values are illustrated at the top and boxed by a dotted line in each figure.
Paramagnetic Relaxation Enhancement and Solvent Exchange Studies of MMP23-PD in DPC Micelles
Protein localization within the DPC micelles was assessed by measuring the decrease in peak intensities in the 1H-15N HSQC spectrum of MMP23-PD in the presence of the water-soluble paramagnetic relaxation agent Gd(DTPA-BMA) (22). Comparison of 1H-15N HSQC peak intensities before and after adding the relaxation agent to MMP23-PD in 100 mm DPC (Fig. 8E) showed that residues in the MMP23-PD TMD were not accessible to Gd(DTPA-BMA), presumably because they were buried within the DPC micelles. Peak intensities for residues at the N-terminal end of the α1 helix (residues Glu-17–Arg-19) adjacent to the predicted TMD (residues Trp-20–Leu-40) were significantly attenuated, suggesting that these residues protruded from the micelle and were exposed to the solvent. Backbone amides for residues in helix α2 and the linker region between α1 and α2 experienced only a moderate paramagnetic relaxation enhancement effect and were presumably located close to the surface of the DPC micelle with only limited access to Gd(DTPA-BMA).
The Clean SEA-HSQC experiment detects the exchange of amide protons with solvent and provides a convenient method for identifying solvent-exposed residues (21). Backbone amide protons for residues Arg-19 to Ser-61, which incorporate the TMD, helix α2, and the linker region, exhibited negligible solvent exchange, indicating that these residues were associated with the DPC micelles and protected from solvent exchange (Fig. 8F). In contrast, residues at the N terminus of helix α1 (Glu-17–Arg-18) adjacent to the TMD exhibited high levels of exchange, implying that this region of the helix extends from the DPC micelles and is exposed to the solvent.
DISCUSSION
In this study, we have identified and characterized a noncanonical, metalloprotease-independent, voltage-gated K+ channel-modulating role for the 81-residue single helical TMD-containing prodomain of MMP23, MMP23-PD. MMP23-FL and MMP23-PD caused an equivalent ∼2-fold decrease in the number of functional KV1.3 channels/cell, whereas not suppressing KV1.2. The residual KV1.3 currents in MMP23-FL- and MMP23-PD-expressing cells exhibited the same biophysical properties as wild-type KV1.3. Because MMP23 is not present in the surface membrane (6, 7, 10), it is likely that these channels are not associated with MMP23-FL or MMP23-PD. MMP23-FL and MMP23-PD both co-localized with the ER marker YFP-Stim-1, confirming earlier studies reporting that mouse, rat, and human MMP23 are ER proteins (6, 7, 10). Both MMP23-FL and MMP23-PD co-localized equivalently with KV1.3, but not with KV1.2 or the DsRed vector. Furthermore, KV1.3 expression on the cell membrane was reduced as more of the channel co-localized intracellularly with MMP23-FL and MMP23-PD. KV1.3 channel suppression may be mediated by a mechanism consistent with intracellular trapping of the channel in the ER, although we have no evidence as to whether this is through a direct or an indirect interaction. Studies with chimeras of KV1.2 and KV1.3 show that the KV1.3 region from S5 to the C terminus is required for MMP23-PD-dependent channel modulation.
Structural analysis by NMR reveals that MMP23-PD contains a 24-residue α-helical region encompassing residues Glu-17 to Leu-40 (α1), incorporating the TMD (Fig. 7). Helix α1 is slightly longer than expected for typical trans-membrane proteins found within the ER or Golgi (20 residues) (36). However, the N-terminal region of helix α1 (residues Glu-17–Trp-20, comprising the N-terminal capping motif) contains residues that are known to prefer the polar/nonpolar interface of lipid bilayers (32). These residues are partially flexible and solvent-exposed, suggesting that the N-terminal portion of α1 may protrude from the surface of the membrane. Thus, the TMD of MMP23-PD likely extends from Trp-20 to Leu-40 (supplemental Fig. S6) and is the appropriate length for an ER trans-membrane protein. The data indicate that a disruption in the TMD α1 helix several residues upstream of Pro-32 around residues Ala-27–Leu-28 introduces some flexibility into the helix in this region. Proline residues are known to be helix-destabilizing in water-soluble proteins but are surprisingly common in trans-membrane proteins (32, 37). They often introduce a region of flexibility and a kink in trans-membrane helices because of steric conflicts with the preceding residue and loss of a backbone hydrogen bond (38). However, although Pro residues are often found associated with kinks in single-helical trans-membrane proteins, they usually occur several residues C-terminal to the bend (37), as observed here. We note also that the micelle environment may influence the nature of the kink, which might not be as pronounced in a bilayer membrane.
Binding of MMP23-PD to KV1.3 channels would be likely to leave one face of the TMD lipid-exposed while the other face interacts with the channel. Comparison of MMP23-PDs from various species shows that conserved residues are localized to two separate regions on the surface of the TMD (supplemental Fig. S10); either or both of these may be required for MMP23-TM-mediated modulation of KV1.3 trafficking. These residues may be optimally oriented to interact with KV1.3 as a consequence of the likely kink in the TMD around residues 26–28.
The α-helical TMD of MMP23-PD is linked to a shorter membrane-interacting α-helical region (Ala-47–Leu-58) (Fig. 7J). Intra- or extracellular helical segments adjacent to TMDs are not uncommon (39, 40) and often play a functional role (41). KCNE1, another K+ channel-modulating protein, also contains an α-helical trans-membrane domain linked to two membrane-interacting juxtamembrane helical domains (40). Like MMP23-PD, KCNE1 and the related KCNE2 protein both suppress currents generated by homomeric KV1.4, KV3.3, and KV3.4 channels by trapping them early in the secretory pathway (42–44). Furthermore, KCNE4, a related protein, suppresses currents through KV1.3-containing homotetramers and heterotetramers by trapping the channels within the ER (45). KCNE1 has additional channel-modulating effects on KV7.1 channels mediated by the C-terminal juxtamembrane helical domain, (46) which was not found with MMP23-PD.
We have shown expression of MMP23 and KV1.3 in normal colonic epithelium and overlapping staining of both proteins in human colorectal cancers. It is tempting to speculate that MMP23-PD may regulate the surface expression of KV1.3 channels in primary and metastatic colorectal cancer cells, and thereby regulate cellular function. Colon cancer cells may secrete processed active MMP23 enzyme containing the KV1.3 channel-blocking toxin domain, which would block KV1.3 channels (IC50 = 2.8 μm) on infiltrating anti-tumor T cells and suppress them as a means of immune evasion. The tumor cell would protect itself from the channel-blocking toxin domain because its KV1.3 channels are trapped intracellularly by MMP23-PD. In support of this idea, human malignant melanoma tumors with higher MMP23 expression contain significantly fewer tumor-infiltrating lymphocytes and are associated with a greater risk of recurrence among patients treated with immune biologics (47). We cannot exclude a role for KV1.3 in the endoplasmic reticulum of tumors. MMP23 and KV1.3 also exhibit overlapping tissue expression in many other tissues where they may interact: lung, heart, uterus, placenta, ovary, testis seminiferous tubules, prostate, intestine, pancreatic islets, cingulate cortex, adrenal cortex, osteoblasts, chondroblasts, cartilage, synovium, natural killer cells, dendritic cells, and tendons (6, 8, 10, 48–55) (see BioGPS web site).
In conclusion, our studies reveal a novel noncanonical, channel-modulating role for a MMP. The prodomain of MMP23, an integral membrane domain, co-localizes and traps KV1.3 channels intracellularly, resulting in suppression of the KV1.3 current. MMP23 is present in many tissues that express KV1.3 channels. In these tissues, trapping of KV1.3 channels by MMP23-PD could deplete channels from the cell surface and thereby alter cellular function. The similarity in overall architecture and trapping function between MMP23-PD and the KV-trapping KCNE proteins suggests a common evolutionary mechanism of channel modulation and interaction by these trans-membrane proteins.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant NS48252 (to K. G. C.). This work was also supported by the Australian Research Council (Grant DP1093909 to R. S. N., B. J. S., and K. G. C.). Colorectal cancer tissue procurement was supported by Award Number P30CA062203 from the National Institutes of Health through the NCI.

This article contains supplemental Table S1 and Figs. S1–S10.
The 1H, 13C, and 15N chemical shifts of the backbone resonances from this publication have been submitted to the BioMagResBank database (http://www.bmrb.wisc.edu) and assigned the accession number 18676.
- MMP
- matrix metalloprotease
- ER
- endoplasmic reticulum
- PD
- prodomain
- FL
- full-length
- TCEP
- tris(2-carboxyethyl)phosphine
- DPC
- n-dodecylphosphocholine
- HSQC
- heteronuclear single quantum coherence
- Gd(DTPA-BMA)
- 2-[bis[2-[[2-(methylamino)-2-oxoethyl]-(2-oxido-2-oxoethyl)amino]ethyl]amino] acetate gadolinium(3+)
- CG
- coarse-grain
- Cat domain
- catalytic domain
- TMD
- trans-membrane domain
- Trx
- thioredoxin
- pF
- picofarads
- CatDom
- catalytic domain
- TxD
- toxin domain.
REFERENCES
- 1. Mannello F., Medda V. (2012) Nuclear localization of matrix metalloproteinases. Prog. Histochem. Cytochem. 47, 27–58 [DOI] [PubMed] [Google Scholar]
- 2. Nagase H., Visse R., Murphy G. (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 69, 562–573 [DOI] [PubMed] [Google Scholar]
- 3. Page-McCaw A., Ewald A. J., Werb Z. (2007) Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8, 221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Golubkov V. S., Chernov A. V., Strongin A. Y. (2011) Intradomain cleavage of inhibitory prodomain is essential to protumorigenic function of membrane type-1 matrix metalloproteinase (MT1-MMP) in vivo. J. Biol. Chem. 286, 34215–34223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jozic D., Bourenkov G., Lim N. H., Visse R., Nagase H., Bode W., Maskos K. (2005) X-ray structure of human proMMP-1: new insights into procollagenase activation and collagen binding. J. Biol. Chem. 280, 9578–9585 [DOI] [PubMed] [Google Scholar]
- 6. Ohnishi J., Ohnishi E., Jin M., Hirano W., Nakane D., Matsui H., Kimura A., Sawa H., Nakayama K., Shibuya H., Nagashima K., Takahashi T. (2001) Cloning and characterization of a rat ortholog of MMP-23 (matrix metalloproteinase-23), a unique type of membrane-anchored matrix metalloproteinase and conditioned switching of its expression during the ovarian follicular development. Mol. Endocrinol. 15, 747–764 [DOI] [PubMed] [Google Scholar]
- 7. Pei D., Kang T., Qi H. (2000) Cysteine array matrix metalloproteinase (CA-MMP)/MMP-23 is a type II transmembrane matrix metalloproteinase regulated by a single cleavage for both secretion and activation. J. Biol. Chem. 275, 33988–33997 [DOI] [PubMed] [Google Scholar]
- 8. Velasco G., Pendás A. M., Fueyo A., Knäuper V., Murphy G., López-Otín C. (1999) Cloning and characterization of human MMP-23, a new matrix metalloproteinase predominantly expressed in reproductive tissues and lacking conserved domains in other family members. J. Biol. Chem. 274, 4570–4576 [DOI] [PubMed] [Google Scholar]
- 9. Pei D. (1999) CA-MMP: a matrix metalloproteinase with a novel cysteine array, but without the classic cysteine switch. FEBS Lett. 457, 262–270 [DOI] [PubMed] [Google Scholar]
- 10. Rangaraju S., Khoo K. K., Feng Z. P., Crossley G., Nugent D., Khaytin I., Chi V., Pham C., Calabresi P., Pennington M. W., Norton R. S., Chandy K. G. (2010) Potassium channel modulation by a toxin domain in matrix metalloprotease 23. J. Biol. Chem. 285, 9124–9136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391, 85–100 [DOI] [PubMed] [Google Scholar]
- 12. Beeton C., Smith B. J., Sabo J. K., Crossley G., Nugent D., Khaytin I., Chi V., Chandy K. G., Pennington M. W., Norton R. S. (2008) The d-diastereomer of ShK toxin selectively blocks voltage-gated K+ channels and inhibits T lymphocyte proliferation. J. Biol. Chem. 283, 988–997 [DOI] [PubMed] [Google Scholar]
- 13. Beeton C., Wulff H., Singh S., Botsko S., Crossley G., Gutman G. A., Cahalan M. D., Pennington M., Chandy K. G. (2003) A novel fluorescent toxin to detect and investigate KV1.3 channel up-regulation in chronically activated T lymphocytes. J. Biol. Chem. 278, 9928–9937 [DOI] [PubMed] [Google Scholar]
- 14. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 15. Rovnyak D., Frueh D. P., Sastry M., Sun Z. Y., Stern A. S., Hoch J. C., Wagner G. (2004) Accelerated acquisition of high resolution triple-resonance spectra using non-uniform sampling and maximum entropy reconstruction. J. Magn Reson. 170, 15–21 [DOI] [PubMed] [Google Scholar]
- 16. Orekhov V. Y., Jaravine V. A. (2011) Analysis of non-uniformly sampled spectra with multi-dimensional decomposition. Prog. Nucl. Magn Reson. Spectrosc. 59, 271–292 [DOI] [PubMed] [Google Scholar]
- 17. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 18. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 19. Bahrami A., Assadi A. H., Markley J. L., Eghbalnia H. R. (2009) Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS. Comput. Biol. 5, e1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kay L. E., Torchia D. A., Bax A. (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28, 8972–8979 [DOI] [PubMed] [Google Scholar]
- 21. Lin D., Sze K. H., Cui Y., Zhu G. (2002) Clean SEA-HSQC: a method to map solvent exposed amides in large non-deuterated proteins with gradient-enhanced HSQC. J. Biomol. NMR 23, 317–322 [DOI] [PubMed] [Google Scholar]
- 22. Pintacuda G., Otting G. (2002) Identification of protein surfaces by NMR measurements with a pramagnetic Gd(III) chelate. J. Am. Chem. Soc. 124, 372–373 [DOI] [PubMed] [Google Scholar]
- 23. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 24. Monticelli L., Kandasamy S. K., Periole X., Larson R. G., Tieleman D. P., Marrink S.-J. (2008) The MARTINI coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 4, 819–834 [DOI] [PubMed] [Google Scholar]
- 25. Marrink S. J., Risselada H. J., Yefimov S., Tieleman D. P., de Vries A. H. (2007) The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 111, 7812–7824 [DOI] [PubMed] [Google Scholar]
- 26. Bussi G., Donadio D., Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 [DOI] [PubMed] [Google Scholar]
- 27. Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 28. Fiser A., Sali A. (2003) Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491 [DOI] [PubMed] [Google Scholar]
- 29. Grissmer S., Nguyen A. N., Aiyar J., Hanson D. C., Mather R. J., Gutman G. A., Karmilowicz M. J., Auperin D. D., Chandy K. G. (1994) Pharmacological characterization of five cloned voltage-gated K+ channels, types KV1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 45, 1227–1234 [PubMed] [Google Scholar]
- 30. Grunnet M., Rasmussen H. B., Hay-Schmidt A., Klaerke D. A. (2003) The voltage-gated potassium channel subunit, KV1.3, is expressed in epithelia. Biochim. Biophys. Acta 1616, 85–94 [DOI] [PubMed] [Google Scholar]
- 31. Abdul M., Hoosein N. (2002) Voltage-gated potassium ion channels in colon cancer. Oncol. Rep. 9, 961–964 [PubMed] [Google Scholar]
- 32. Baeza-Delgado C., Marti-Renom M. A., Mingarro I. (April 14, 2012) Structure-based statistical analysis of transmembrane helices. Eur. Biophys. J. 10.1007/s00249-012-0813-9 [DOI] [PubMed] [Google Scholar]
- 33. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shen Y., Bax A. (2012) Identification of helix capping and β-turn motifs from NMR chemical shifts. J. Biomol. NMR 52, 211–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beswick V., Roux M., Navarre C., Coïc Y. M., Huynh-Dinh T., Goffeau A., Sanson A., Neumann J. M. (1998) 1H- and 2H-NMR studies of a fragment of PMP1, a regulatory subunit associated with the yeast plasma membrane H+-ATPase. Conformational properties and lipid-peptide interactions. Biochimie 80, 451–459 [DOI] [PubMed] [Google Scholar]
- 36. Sharpe H. J., Stevens T. J., Munro S. (2010) A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langelaan D. N., Wieczorek M., Blouin C., Rainey J. K. (2010) Improved helix and kink characterization in membrane proteins allows evaluation of kink sequence predictors. J. Chem. Inf. Model. 50, 2213–2220 [DOI] [PubMed] [Google Scholar]
- 38. Cordes F. S., Bright J. N., Sansom M. S. (2002) Proline-induced distortions of transmembrane helices. J. Mol. Biol. 323, 951–960 [DOI] [PubMed] [Google Scholar]
- 39. Klammt C., Maslennikov I., Bayrhuber M., Eichmann C., Vajpai N., Chiu E. J., Blain K. Y., Esquivies L., Kwon J. H., Balana B., Pieper U., Sali A., Slesinger P. A., Kwiatkowski W., Riek R., Choe S. (2012) Facile backbone structure determination of human membrane proteins by NMR spectroscopy. Nat. Methods 9, 834–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Horn W. D., Vanoye C. G., Sanders C. R. (2011) Working model for the structural basis for KCNE1 modulation of the KCNQ1 potassium channel. Curr. Opin. Struct. Biol. 21, 283–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barrett P. J., Song Y., Van Horn W. D., Hustedt E. J., Schafer J. M., Hadziselimovic A., Beel A. J., Sanders C. R. (2012) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kanda V. A., Lewis A., Xu X., Abbott G. W. (2011) KCNE1 and KCNE2 provide a checkpoint governing voltage-gated potassium channel α-subunit composition. Biophys. J. 101, 1364–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kanda V. A., Lewis A., Xu X., Abbott G. W. (2011) KCNE1 and KCNE2 inhibit forward trafficking of homomeric N-type voltage-gated potassium channels. Biophys. J. 101, 1354–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanda V. A., Abbott G. W. (2012) KCNE Regulation of K+ channel trafficking – a Sisyphean task? Front Physiol. 3, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Solé L., Roura-Ferrer M., Pérez-Verdaguer M., Oliveras A., Calvo M., Fernández-Fernández J. M., Felipe A. (2009) KCNE4 suppresses KV1.3 currents by modulating trafficking, surface expression and channel gating. J. Cell Sci. 122, 3738–3748 [DOI] [PubMed] [Google Scholar]
- 46. Tapper A. R., George A. L., Jr. (2000) MinK subdomains that mediate modulation of and association with KVLQT1. J. Gen. Physiol. 116, 379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krogsgaard M., Ma M. W., Friedman E. B., Vega-Saenz de Miera E., Darvishian F., Perez-Garcia A., Berman R. S., Shapiro R. L., Christos P. J., Osman I., Pavlick A. C. (2011) An analysis of altered melanoma matrix metalloproteinase-23 (MMP-23) expression and response to immune biologic therapy. J. Clin. Oncol. 29, (suppl.) (Abstr. 8541) [Google Scholar]
- 48. Clancy B. M., Johnson J. D., Lambert A. J., Rezvankhah S., Wong A., Resmini C., Feldman J. L., Leppanen S., Pittman D. D. (2003) A gene expression profile for endochondral bone formation: oligonucleotide microarrays establish novel connections between known genes and BMP-2-induced bone formation in mouse quadriceps. Bone 33, 46–63 [DOI] [PubMed] [Google Scholar]
- 49. Fortunato S. J., Menon R. (2002) Screening of novel matrix metalloproteinases (MMPs) in human fetal membranes. J. Assist. Reprod. Genet. 19, 483–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jones G. C., Corps A. N., Pennington C. J., Clark I. M., Edwards D. R., Bradley M. M., Hazleman B. L., Riley G. P. (2006) Expression profiling of metalloproteinases and tissue inhibitors of metalloproteinases in normal and degenerate human achilles tendon. Arthritis Rheum. 54, 832–842 [DOI] [PubMed] [Google Scholar]
- 51. Okada A., Okada Y. (2009) Progress of research in osteoarthritis. Metalloproteinases in osteoarthritis. Clin. Calcium 19, 1593–1601 [PubMed] [Google Scholar]
- 52. Riddick A. C., Shukla C. J., Pennington C. J., Bass R., Nuttall R. K., Hogan A., Sethia K. K., Ellis V., Collins A. T., Maitland N. J., Ball R. Y., Edwards D. R. (2005) Identification of degradome components associated with prostate cancer progression by expression analysis of human prostatic tissues. Br. J. Cancer 92, 2171–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hegedüs L., Cho H., Xie X., Eliceiri G. L. (2008) Additional MDA-MB-231 breast cancer cell matrix metalloproteinases promote invasiveness. J. Cell. Physiol. 216, 480–485 [DOI] [PubMed] [Google Scholar]
- 54. Scrideli C. A., Carlotti C. G., Jr., Okamoto O. K., Andrade V. S., Cortez M. A., Motta F. J., Lucio-Eterovic A. K., Neder L., Rosemberg S., Oba-Shinjo S. M., Marie S. K., Tone L. G. (2008) Gene expression profile analysis of primary glioblastomas and non-neoplastic brain tissue: identification of potential target genes by oligonucleotide microarray and real-time quantitative PCR. J. Neurooncol. 88, 281–291 [DOI] [PubMed] [Google Scholar]
- 55. Davidson R. K., Waters J. G., Kevorkian L., Darrah C., Cooper A., Donell S. T., Clark I. M. (2006) Expression profiling of metalloproteinases and their inhibitors in synovium and cartilage. Arthritis Res. Ther. 8, R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wishart D. S., Bigam C. G., Holm A., Hodges R. S., Sykes B. D. (1995) 1H, 13C, and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR. 5, 67–81 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.