

Abstract
Biofilms are often associated with human bacterial infections, and the natural tolerance of biofilms to antibiotics challenges treatment. Compounds with antibiofilm activity could become useful adjuncts to antibiotic therapy. We used norspermidine, a natural trigger for biofilm disassembly in the developmental cycle of Bacillus subtilis, to develop guanidine and biguanide compounds with up to 20-fold increased potency in preventing biofilm formation and breaking down existing biofilms. These compounds also were active against pathogenic Staphylococcus aureus. An integrated approach involving structure–activity relationships, protonation constants, and crystal structure data on a focused synthetic library revealed that precise spacing of positively charged groups and the total charge at physiological pH distinguish potent biofilm inhibitors.
Most bacteria form biofilms, which are multicellular microbial communities embedded in a self-produced exopolymeric substance (EPS) largely composed of a protein anchor and different extracellular polymers. Bacteria within a mature biofilm community exist in an altered metabolic state and different physical environment than their free-floating, or planktonic, relatives. Biofilm bacteria generally tolerate antibiotic treatment,1,2 and antibiotics can induce biofilm formation.3,4 Consequently, biofilm inhibitors can be applied to decrease antibiotic tolerance of bacteria.5 Biofilms play a major role in many bacterial infections.2 In humans, the antibiotic tolerance of biofilm communities frustrates the treatment of persistent bacterial infections such as those associated with cystic fibrosis, endocarditis, joint prostheses, heart catheters, and replacement heart valves.6,7
In response to this challenge, high-throughput assays have been developed to identify small molecules with the ability to prevent biofilm formation or disrupt existing biofilms.8 We recently explored an alternative strategy that exploits the normal developmental cycle of bacteria. Biofilms form when planktonic bacteria in the aqueous phase aggregate on a solid surface or at an air–liquid interface. The biofilm colony grows both by recruitment and cell division to form a mature colony. Mature colonies eventually disintegrate, and the dispersed bacteria resume a planktonic lifestyle (Figure 1). Bacterially produced small molecules orchestrate the creation and disintegration of biofilms, and identifying these molecular signals could lead to therapeutically useful templates.
Figure 1.
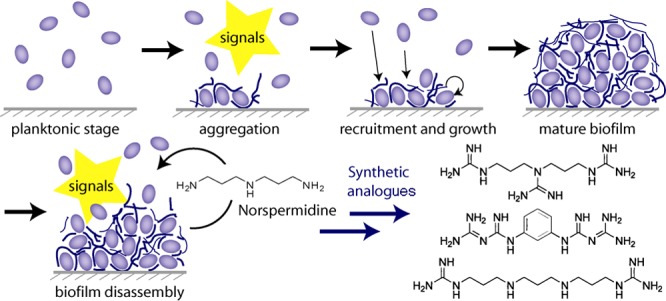
Stages in the developmental cycle of biofilm formation and disruption. Norspermidine both prevents the formation of new biofilms and collapses the structure of existing biofilms.
We previously identified d-amino acids as potent biofilm disruptors because of their ability to release the protein component of EPS from the bacterial cell wall.9 Recently we identified norspermidine as a key disruptor of the polymeric component of EPS.10 Mixtures of norspermidine with d-amino acids were found to be highly synergistic (single-digit nanomolar) in disrupting biofilms (Figure 1).10 Here we report synthetic mimics of norspermidine with increased potency and a structure-based rationale for their activity.
Norspermidine appears to disrupt biofilms by targeting the extracellular component of EPS in Bacillus subtilis, and it seemed likely that it does so by binding to negatively charged or possibly neutral groups using Coulombic attraction and hydrogen bonding as important features.10 We tested a set of commercially available polyamines. Norspermidine was most active in inhibiting biofilms for B. subtilis and Staphylococcus aureus, followed by norspermine, which has an additional aminopropyl unit in its structure [Figure S1 and Table S2 in the Supporting Information (SI)]. Perhaps surprisingly, spermidine, with one longer aminobutyl residue in place of an aminopropyl unit, and diethylenetriamine, with two shorter aminoethyl groups, were inactive in both species. This sharp length dependence indicated that matching the NH-to-NH distance of the (poly)propyleneamine motif of norspermidine and norspermine (4.9 Å) to the pitch of various helical EPS structures determined or modeled for potential exopolymers (4.6–5.3 Å; Table S1) is a key feature. Binding of these polyamines to negatively charged secondary structures would neutralize the charge and collapse the aqueous meshwork characteristic of mature biofilms.10 This simple model involving three or four positively charged groups separated by propyl units could be tested against biofilm formation of B. subtilis with synthetic mimics, and guanidines and biguanides emerged as particularly appealing substitutes for polyamines because of their potentially increased overall charge at physiological pH values.
We used three different synthetic strategies to generate a small library of compounds with guanidinyl or biguanidyl groups as chloride or sulfate salts (Figure 2). Guanidines can be conveniently prepared from amines with S-methylisothiourea (Scheme 1),11 and that reagent afforded terminal guanidines (1, 2, 4, 5, 7–9, and 12) from commercially available primary amines. Alternatively, cyanamide (or the alkylated form of its carbodiimide tautomer), which reacts with secondary amines (Scheme 1),12 was used to prepare triguanidinylated compounds (3 and 6) and alkylated guanidines (3 and 10). Finally, a biguanide (11) was synthesized from m-phenylenediamine and dicyandiamide (Scheme 1) according to Cohn.13 While aromatic amines are known to react readily with dicyandiamide,13 our attempts to extend the reaction to primary aliphatic amines were unsuccessful.
Figure 2.

Library of synthetic guanidinylated or biguanidylated polyamine analogues. For free bases, “base” appears in the “form” column; for salts, the counterion is given (for stoichiometry, see the SI).
Scheme 1.

Compounds were tested for inhibition of biofilm formation in B. subtilis, the model organism that led to norspermidine,10 and S. aureus as a related pathogenic species with high clinical relevance. The minimum biofilm inhibitory concentrations (MBICs) for all of the compounds are given in Table S2. Some of the compounds exhibited remarkable activity for the inhibition of biofilms, with 5–20-fold increased activity toward B. subtilis and >8-fold increased activity toward S. aureus relative to norspermidine (Figure 3A and Tables 1 and S2). In addition to preventing biofilm formation, the most potent compound was also able to disrupt existing biofilms (Figure 3B). Early on, it became clear that the counterion of the amine or guanidine had a significant effect on the activity. For instance, the free base of norspermidine was 3 times more active than the chloride salt, which in turn was 3 times more active than the sulfate salt in the B. subtilis assay (Table S2). Therefore, we generated the free bases of selected compounds and compared them with the corresponding salts. For B. subtilis, a free base’s activity for biofilm inhibition was greater than or equal to that of the salt, while for S. aureus there was no clear trend. Solubility products (Ks) showed no correlation with the activity of the compounds (Table S3), and bioavailability and delivery into the biofilm matrix are probably critical parameters. None of the compounds significantly inhibited bacterial growth at or close to its corresponding MBIC value, ruling out the possibility that biofilm inhibition was an artifact of reduced viability (see the SI). Only compound 7 started to affect growth in B. subtilis at concentrations above 200 μM, which is 40 times its MBIC.
Figure 3.

Enhanced activity of synthetic compounds 6a, 7a, and 11a against B. subtilis. (A) Inhibition of biofilm formation relative to the corresponding polyamines. (B) Breakdown of pre-existing biofilms within 12 h. Norspd, norspermidine; Norspn, norspermine; PBS, phosphate-buffered saline.
Table 1. Activities of Selected Compounds.
| MBIC
(μM)
at pH 7.4 |
||
|---|---|---|
| compound | B. subtilis | S. aureus |
| 4 | >1000 | 50 |
| 5a | 500 | 75 |
| 5b | 375 ± 125 | 400 |
| 6a | 10a | >1000 (500a) |
| 6b | 10 | 50 |
| 7a | 5 | 55 ± 15 |
| 7b | 2 | 250 |
| 9 | 100 | 500 |
| 10 | 30 | 20 ± 10 |
| 11a | 30 | 300 |
| 11b | 7 ± 3 | 750 ± 250 |
Incomplete inhibition.
In B. subtilis, the most active compounds were 6, 7, 8, 10, and 11 (as salts and bases; Tables 1 and S2), with MBICs between 2 μM (7b) and 30 μM (10 and 11a). Active compounds exhibited the proposed binding motif10 of three to four amino or guanidine groups formally separated by propyl chains (5–7, 10, and 11; Tables 1 and S2). Additionally, the shorter compound 9 having only a single propyl chain displayed activity only at 100 μM. The activity pattern for S. aureus was slightly different, as the minimal motif required for activity was two guanidine groups or one amino group and one guanidine group separated by a propyl chain (4–11). Compounds with ethyl instead of propyl chains (1–3 and 12) were inactive (≥1 mM) for B. subtilis and only weakly active (750 μM) or inactive for S. aureus. Biguanide itself was inactive for both species (Tables 1 and S2).
The most active inhibitors of biofilm formation by S. aureus were 4, 5a, 6b, 7a, and 10, with MBICs in the range 10–75 μM. The activities of the best biofilm inhibitors in this initial library are comparable to the lower range of what has been reported in the literature for biofilm inhibitory compounds that do not adversely affect bacterial growth.8
Our results support a model in which the binding of polyamine-based inhibitors to the exopolymer depends on the correct spacing of multiple amino or guanidine groups. The structure–activity relationship in this library further indicates that although there is a common motif in the two species, the composition and structure of biofilms of S. aureus and B. subtilis are different and allow customized inhibition of biofilm formation.
In addition to the structural properties described above, the charge of the compounds could be an important contributor to their inhibitory activity.10 To investigate this possibility, we determined the pKa values of selected compounds at 25 °C and 25 mM. Cumulative association constants were calculated by HypNMR14,15 (Figures S2–S11), and values for pKa(D2O) were finally converted to pKa(H2O).16 The pKa values for the compounds are given in Table 2. For comparison, similar pKa values have been reported previously for spermidine (pKa1 = 10.90, pKa2 = 9.71, and pKa3 = 8.25).17
Table 2. pKa values of selected compounds.
| compound | pKa1 | pKa2 | pKa3 | pKa4 |
|---|---|---|---|---|
| norspermidine | 11.1 ± 0.1 | 9.4 ± 0.2 | 7.1 ± 0.0 | – |
| norspermine | 10.6 ± 0.3 | 10.5 ± 0.3 | 8.7 ± 0.2 | 6.7 ± 0.1 |
| spermidine | 11.1 ± 0.1 | 9.8 ± 0.1 | 7.8 ± 0.1 | – |
| DETa | 11.6 ± 0.1 | 9.0 ± 0.3 | 3.9 ± 0.1 | – |
| 1 | 13.6 ± 0.1 | 12.6 ± 0.2 | – | – |
| 2 | 13.5 ± 0.0 | 12.8 ± 0.2 | 6.3 ± 0.0 | – |
| 4 | 13.7 ± 0.1 | 13.0 ± 0.3 | – | – |
| 5 | 13.4 ± 0.1 | 12.0 ± 0.3 | 8.4 ± 0.1 | – |
| 6 | 13.8 ± 0.2 | 13.2 ± 0.3 | 12.7 ± 0.5 | – |
| 7 | 13.5 ± 0.1 | 12.0 ± 0.3 | 9.8 ± 0.1 | 7.6 ± 0.1 |
| 8 | 13.6 ± 0.0 | 12.2 ± 0.1 | 9.2 ± 0.1 | – |
| 9 | 13.6 ± 0.1 | 9.4 ± 0.1 | – | – |
| 11 | 12.1 ± 0.1 | 10.6 ± 0.1 | – | – |
Diethylenetriamine.
Speciation data derived from the pKa values were generated using the program HySS (Figures S12–S14), and the average charge was plotted against pH for each molecule (Figure 4). For convenience, protonation states will be given by a string of digits, with 1 for a protonated site and 0 for a nonprotonated site. In this notation, the fully protonated state of norspermidine is denoted as (111). Diethylenetriamine (DET) has one extremely low protonation constant of 3.9 that results in one amino group being uncharged [protonation state (101)] in the wide pH range of 5–8 (Figure 4A). Although guanidine groups in the related structure 2 significantly increase the third pKa value to 6.3, the central amino group remains unprotonated at physiological pH (101), as confirmed by X-ray structure analysis (Figure S17).
Figure 4.

Protonation and charge states. (A) Average degree of protonation of polyamines and corresponding di- or triguanidines as a function of pH. (B) Classical, incorrect representation of protonated biguanides. (C) Correct protonation state for 11. (D) Biguanide moiety cropped from the crystal structure of 11.
The lack of activity of 2 at pH 7 reflects both structural and charge liabilities. Not surprisingly, guanidine groups on the scaffold of norspermidine or norspermine increased all of the individual pKa values relative to those of the corresponding polyamines (Figure 4A) causing the average degree of protonation to rise, which is in line with the increased activity of 6 over norspermidine and 7 over norspermine. However, while 5 was active, it did not display increased activity over norspermidine, despite its higher degree of protonation (Figure S16). On average, norspermine carries 3.3 positive charges at pH 7, corresponding to ∼30% fully charged (1111) molecules and the rest triply charged [(1110) or (1101)], with the protonation microstate (1101) having one noncharged secondary amine as the likely predominant species.15,17,18 The (1101) species does not comply with the triply charged (111) motif and may contribute to the higher activity of norspermidine over norspermine.
The active compound 11 exists at maximum in a doubly protonated form because of its high pKa values, and this form is virtually the only relevant species until pH 8.5 (Figure S16). In the literature, the structure and protonation of biguanides is frequently misrepresented, as reported by Bharatam et al.,19 whose computational studies indicated that the central nitrogen of a biguanide is not bonded to a hydrogen in either neutral or charged states (Figure 4B,C). This N atom is partially negatively charged, while the positive charge is delocalized between the terminal nitrogen atoms of the biguanide. The crystal structure of protonated 11 confirmed these results (Figures 4D and S19), making 11 analogous to the (1111) motif of fully charged compound 7, which explains its activity.
Finally, to confirm the importance of charge for biological activity, we determined the biofilm inhibition in a pH-dependent assay that should directly affect the average charge state of the assayed compounds. We plotted MBIC values in B. subtilis and S. aureus assays against the calculated degree of protonation (Figure 5). Although biofilm morphology and physiology as well as the bioavailability of the compounds are expected to change with pH, the potency of active compounds correlated well with the degree of protonation. For all of the active compounds, the activity generally increased (lower MBIC) for both species at higher protonation states, while the inactive compound 2 did not respond to changes in protonation. The absolute activities of different compounds, however, did not coincide with the degree of protonation, suggesting that a combination of structure and charge determine the biological activity.
Figure 5.

Inhibition of biofilm formation is dependent on the average degree of protonation. MBICs of polyamines and guanidine compounds for (A) B. subtilis and (B) S. aureus show a clear trend with degree of protonation. Error bars are standard deviations of the MBIC values.
In conclusion, chemical synthesis generated a focused library of guanidine and biguanide compounds that mimic norspermidine structurally and in some cases functionally with an ability to inhibit biofilm formation in B. subtilis and S. aureus. The best compound also mimicked norspermidine’s ability to disrupt a mature biofilm. A detailed investigation of structure–activity relationships involving protonation constants and crystal structure data provided insights into the ways that charge and spacing between positively charged groups affect biological activity.
Acknowledgments
This research was supported by a Leopoldina Research Fellowship (LPDS 2009-45) from the German Academy of Sciences Leopoldina (T.B.), a Human Frontier Science Program LTF Fellowship (I.K.-G.), NIH Grant GM086258 (J.C.), NERCE-BEID through 5U54 AI057159 (J.C.), NIH Grant GM18568 (R.L.), the NIAID-sponsored program on antibiotic resistance (P01AI083214 to R.L. and R.K.), a grant from BASF (R.L. and R.K.), and NIH Grants GM82137 and GM58213 (R.K.). We thank Dr. Shao-Liang Zheng for his help with X-ray data collection and structure determination.
Supporting Information Available
Syntheses and characterization, biofilm inhibition data, pKa measurements, speciation data, and crystal structures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fux C. A.; Costerton J. W.; Stewart P. S.; Stoodley P. Trends Microbiol. 2005, 13, 34. [DOI] [PubMed] [Google Scholar]
- Davies D. Nat. Rev. Drug Discovery 2003, 2, 114. [DOI] [PubMed] [Google Scholar]
- Hoffman L. R.; D’Argenio D. A.; MacCoss M. J.; Zhang Z.; Jones R. A.; Miller S. I. Nature 2005, 436, 1171. [DOI] [PubMed] [Google Scholar]
- Linares J. F.; Gustafsson I.; Baquero F.; Martinez J. L. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 19484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris T. L.; Worthington R. J.; Melander C. Angew. Chem., Int. Ed. 2012, 51, 11254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsek M. R.; Singh P. K. Annu. Rev. Microbiol. 2003, 57, 677. [DOI] [PubMed] [Google Scholar]
- Stewart P. S.; Costerton J. W. Lancet 2001, 358, 135. [DOI] [PubMed] [Google Scholar]
- Worthington R. J.; Richards J. J.; Melander C. Org. Biomol. Chem. 2012, 10, 7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin-Gal I.; Romero D.; Cao S.; Clardy J.; Kolter R.; Losick R. Science 2010, 328, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin-Gal I.; Cao S.; Chai L.; Böttcher T.; Kolter R.; Clardy J.; Losick R. Cell 2012, 149, 684. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Castagnolo D.; Schenone S.; Botta M. Chem. Rev. 2011, 111, 5247. [DOI] [PubMed] [Google Scholar]
- Bischoff F. J. Biol. Chem. 1928, 80, 345. [Google Scholar]
- Cohn G. J. Prakt. Chem. 1911, 84, 394. [Google Scholar]
- Frassineti C.; Ghelli S.; Gans P.; Sabatini A.; Moruzzi M. S.; Vacca A. Anal. Biochem. 1995, 231, 374. [DOI] [PubMed] [Google Scholar]
- Frassineti C.; Alderighi L.; Gans P.; Sabatini A.; Vacca A.; Ghelli S. Anal. Bioanal. Chem. 2003, 376, 1041. [DOI] [PubMed] [Google Scholar]
- Popov K.; Rönkkömäki H.; Lajunen L. H. J. Pure Appl. Chem. 2006, 78, 663. [Google Scholar]
- Kimberly M. M.; Goldstein J. H. Anal. Chem. 1981, 53, 789. [Google Scholar]
- Salehzadeh S.; Yaghoobi F.; Bayat M. Chem. Phys. 2009, 361, 18. [Google Scholar]
- Bharatam P. V.; Patel D. S.; Iqbal P. J. Med. Chem. 2005, 48, 7615. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.