

Abstract
Cleft palate is one of the most common human birth defects and is associated with multiple genetic and environmental risk factors. Although mutations in the genes encoding transforming growth factor beta (TGFβ) signaling molecules and interferon regulatory factor 6 (Irf6) have been identified as genetic risk factors for cleft palate, little is known about the relationship between TGFβ signaling and IRF6 activity during palate formation. Here, we show that TGFβ signaling regulates expression of Irf6 and the fate of the medial edge epithelium (MEE) during palatal fusion in mice. Haploinsufficiency of Irf6 in mice with basal epithelial-specific deletion of the TGFβ signaling mediator Smad4 (Smad4fl/fl;K14-Cre;Irf6+/R84C) results in compromised p21 expression and MEE persistence, similar to observations in Tgfbr2fl/fl;K14-Cre mice, although the secondary palate of Irf6+/R84C and Smad4fl/fl;K14-Cre mice form normally. Furthermore, Smad4fl/fl;K14-Cre;Irf6+/R84C mice show extra digits that are consistent with abnormal toe and nail phenotypes in individuals with Van der Woude and popliteal pterygium syndromes, suggesting that the TGFβ/SMAD4/IRF6 signaling cascade might be a well-conserved mechanism in regulating multiple organogenesis. Strikingly, overexpression of Irf6 rescued p21 expression and MEE degeneration in Tgfbr2fl/fl;K14-Cre mice. Thus, IRF6 and SMAD4 synergistically regulate the fate of the MEE, and TGFβ-mediated Irf6 activity is responsible for MEE degeneration during palatal fusion in mice.
Keywords: TGFβ, IRF6, Palatal fusion, Mouse
INTRODUCTION
Cleft palate is a serious and common craniofacial birth defect affecting millions of people worldwide (Mossey et al., 2009; Wong and Hagg, 2004). Cleft palate has ethnic and geographic variations in prevalence, and it affects feeding, swallowing, speech, hearing, middle-ear ventilation, respiration and appearance (Iwata et al., 2011). Studies in mouse models and genetic screening in humans have implicated several factors in syndromic cleft palate, such as IRF6 mutation in Van der Woude syndrome (VWS) and popliteal pterygium syndrome (PPS), SMAD4 mutation in juvenile polyposis syndrome, and TGFBR1 or TGFBR2 mutation in Loeys-Dietz syndrome (previously called Marfan syndrome type II). Mutations in TGFB3, IRF6, CYP (cytochrome P450), MSX1 and TBX10 have also been associated with non-syndromic cleft lip with or without cleft palate (NSCL/P) (Iwata et al., 2011). In addition, polymorphic variants associated with NSCL/P within human chromosomes 1q32 (IRF6), 1p22 (ABCA4), 8q24.21, 10q25 (VAX1), 17q22 and 20q12 (MAFB) have been identified by genome-wide association studies (Dixon et al., 2011). Thus, mutations in IRF6, SMAD4 and TGFBR2 confer a significant attributable risk for cleft palate.
TGFβ signaling is one of the major signaling cascades crucial for craniofacial development (Iwata et al., 2011). Epithelial-specific deletion of Tgfbr2 (Tgfbr2fl/fl;K14-Cre) in mice results in persistence of the medial edge epithelium (MEE) and submucous cleft palate (Xu et al., 2006). TGFβ transmits signals through a membrane receptor serine/threonine kinase complex that phosphorylates SMAD2 and SMAD3, followed by the formation of transcriptional complexes with SMAD4 and translocation into the nucleus (Massagué, 2012; Ross and Hill, 2008; Schmierer and Hill, 2007; Shi and Massagué, 2003). TGFβ also activates SMAD-independent signaling cascades, including mitogen-activated protein kinase (MAPK) pathways, such as p38 MAPK (MAPK14 - Mouse Genome Informatics), under certain physiological and pathological conditions (Kang et al., 2009; Xu et al., 2008; Zhang, 2009). Studies using SMAD4-deficient cells, or dominant-negative SMADs, support the possibility that MAPK activation is independent of SMADs (Chen et al., 1998; Giehl et al., 2000; Hocevar et al., 1999; Hu et al., 1999). p38 MAPK activation by TGFβ is accompanied by SMAD-independent, TRAF6 and TAK1 (MAP3K7 - Mouse Genome Informatics) phosphorylation (Iwata et al., 2012; Sorrentino et al., 2008; Yamashita et al., 2008). The balance between direct activation of SMADs and MAPK pathways often defines cellular responses to TGFβ.
IRF6 belongs to a family of transcription factors that share a highly conserved, helix-turn-helix, DNA-binding domain and a less conserved, protein-binding domain. Among the genes that have been associated with NSCL/P, IRF6 has been implicated in the largest percentage of cases (Koillinen et al., 2005; Srichomthong et al., 2005). Mutation of IRF6 can lead to the autosomal-dominant conditions VWS and PPS, which are characterized by oral clefting and lower lip pits (Kondo et al., 2002; Moretti et al., 2010). VWS and PPS are allelic variants of the same condition caused by different mutations of the same gene. PPS includes all the features of VWS, plus popliteal pterygia, syngnathia, distinct toe and nail abnormality, syndactyly and genito-urinary malformations. An arginine 84 to cysteine (R84C) mutation in IRF6 is the most common mutation found in patients with PPS (Richardson et al., 2006). Although the function of the R84C mutation is still largely unknown, recent studies have demonstrated that it results in loss of DNA binding (Kondo et al., 2002; Little et al., 2009). The primary defect in Irf6-deficient mice is in keratinocyte differentiation and proliferation. Homozygous Irf6gt1/gt1 (null) embryos exhibit abnormal skin, limb and craniofacial morphogenesis, including cleft palate (Ingraham et al., 2006). Mice homozygous for Irf6R84C, which is an R84C knock-in resulting in expression of mutant IRF6 protein, exhibit a severe intraoral epithelial adhesion caused by a failure of terminal differentiation similar to that in homozygous Irf6gt1/gt1 (null) embryos (Ingraham et al., 2006; Richardson et al., 2006).
Despite the established roles of TGFβ signaling and IRF6 activity during palate formation, the interaction between TGFβ signaling and IRF6 activity is poorly understood. In this study, we investigate the interaction between TGFβ signaling and IRF6 activity. We demonstrate that Irf6 and Smad4 interact genetically, and that TGFβ-mediated Irf6 expression is crucial for p21 (CDKN1A - Mouse Genome Informatics) expression and fate determination of the MEE cells during palatal fusion.
MATERIALS AND METHODS
Animals
To generate Smad4fl/fl;K14-Cre;Irf6+/R84C mice, we mated Smad4fl/+;K14-Cre;Irf6+/R84C with Smad4fl/fl mice. To generate Tgfbr2fl/fl;K14-Cre mice, we mated Tgfbr2fl/+;K14-Cre with Tgfbr2fl/fl mice. Genotyping was performed using PCR primers as previously described (Ito et al., 2003; Richardson et al., 2009; Xu et al., 2008; Xu et al., 2006). Human keratin 14 (K14; KRT14 - Human Gene Nomenclature Database) promoter-driven Irf6-encoding transgene was prepared as follows: an EcoRI-HindIII blunted fragment (7.3 kb) encoding mouse Irf6 was subcloned into the BamHI blunted sites of the pGEM 3Z-K14 vector to produce pG3ZK14-Irf6, resulting in a construct containing the K14 promoter (2.1 kb), the β-globin intron (736 bp), the coding sequence for Irf6 (4.1 kb) and the K14 polyadenylation signal (500 bp). The EcoRI-HindIII fragment was isolated free of vector sequence by preparative gel electrophoresis. DNA was further purified using an Elutip column (Schleicher and Schuell, Dessell, Germany) and microinjected in the pronuclei of fertilized oocytes (Jackson/B6D2F F1) following standard procedures. Transgenic founder mice were identified by PCR analysis. PCR amplification of tail genomic DNAs (0.5-1 μg) was performed on a thermal cycler, with 35 cycles consisting of 94°C, 62°C and 72°C for 1 minute each. An aliquot (15 μl) of each reaction was resolved in a 1% agarose gel, and amplified fragments were visualized by ethidium bromide staining. The PCR primers used were 5′-TCAGGAGCAGGTGCACAAGAGTT-3′ and 5′-ACTCGCATCCCTTTCCAATTTAC-3′. To generate Tgfbr2fl/fl;K14-Cre;Irf6Tg mice, we mated Tgfbr2fl/+;K14-Cre;Irf6Tg with Tgfbr2fl/fl mice. Genotyping was performed using PCR primers as previously described (Ito et al., 2003; Xu et al., 2008; Xu et al., 2006). All mouse experiments were conducted in accordance with protocols approved by the Department of Animal Resources and the Institutional Animal Care and Use Committee of the University of Southern California.
Comparative analysis of transcription factor-binding sites
Genomic sequences of the entire human and murine IRF6 (RefSeq accession NM_006147.3/hg19 and NM_016851.2/mm10), p63 (RefSeq accession NM_001114980/hg19 and NM_011641.2/mm10) and CDKN1A/p21 (RefSeq accession NM_000389.4/hg19 and NM_007669.4/mm10) genes were obtained from the University of California, Santa Cruz (UCSC) genome browser, including 2.5 kb upstream and 2.5 kb downstream of the respective transcription start sites (TSSs), based on mouse genome Build 38. These sequences were mapped to seven additional mammalian genomes [chimpanzee (Build 2.1.4), orangutan (Build 2.0.2), rhesus macaque (Build 1.0), human (Build 19), rat (Build 5), dog (Build 3.1) and horse (Build equCab2)] using the BLAT tool. Multiple alignments for these sequences were obtained using the ClustalW2 tool with default parameters and settings (Larkin et al., 2007). To account for uncertainty in the quality of the horse and dog draft genome sequences, sequence alignments with and without information from these species were performed. Transcription factor-binding motifs relevant to SMAD, p63 (TRP63 - Mouse Genome Informatics) and p38 MAPK pathway elements were searched within and proximal to these genes (i.e. 2.5 kb upstream and 2.5 kb downstream of the mouse TSS for each gene). The recognition sequences searched for SMAD binding were 5′-GTCT-3′, 5′-AGAC-3′ and 5′-GTCTAGAC-3′ (Denissova et al., 2000; Zawel et al., 1998) and for MEF2 family member binding was 5′-YTAWWWWTAR-3′ (Black and Olson, 1998). In addition, we evaluated the following potential p63 recognition sites: the canonical p53 family 20-base recognition sequence formed from duplicates of the half-site recognition sequence 5′-RRRCWWGYYY-3′ separated by any combination of 0 to 13 bases (Cai et al., 2012; el-Deiry et al., 1992) and the p63-preferential recognition sequence 5′-RRRCRWGYYYRRRC WYGYYY-3′ (Perez et al., 2007). To account for the possibility of degenerate p53 family member-binding sites, we used the p53scan and p63scan algorithms with default options (Kouwenhoven et al., 2010), which allow the consensus half-site to vary slightly in composition. Finally, we also searched for the eight-base recognition sequence 5′-AANNGAAA-3′ for the DNA-binding domains of IRF family members (Fujii et al., 1999) and the more specific ten-base recognition sequence 5′-AACCGAAACY-3′ for the IRF6 DNA-binding domain (Little et al., 2009).
Promoter-proximal search of p53-family responsive elements
The aforementioned consensus recognition sequences of p53 family members (see previous section) were used to search the murine and human p21 genomic sequences for putative p53 and p63 recognition sites proximal to the promoter. Genomic sequences were obtained from the UCSC genome browser, spanning a 5-kb window centered around the transcription start site of each p21 RefSeq transcript variant in the murine genome (build mm10, 2 transcript variants) and human genome (build hg19, 4 transcript variants).
Chromatin immunoprecipitation (ChIP) assay
Back skin was dissected from embryonic day (E) 13.5 C57B6/J mouse embryos in PBS. For the separation of skin epithelium from mesenchyme, the explants were incubated in 0.25% dispase (Invitrogen) for 30 minutes at 37°C. The epithelium was further dissociated in a 0.25% trypsin-EDTA solution for 10 minutes at 37°C, fixed with 1% formaldehyde for 15 minutes at room temperature and lysed in cell lysing buffer (Cell Signaling Technology) with a cocktail of proteinase inhibitors (Ultra Complete Mini, Roche). After pre-clearing treatment, cell extracts were incubated with anti-SMAD4, anti-MEF2 (Cell Signaling Technology), anti-p63 or IgG control (Santa Cruz Biotechnology) antibodies for two hours according to the manufacturer’s protocol (Cell Signaling Technology). We assayed for the presence of putative target sites in the immune complexes by PCR using primers amplifying the following genomic regions: mouse Irf6 promoter SMAD-binding site (BS) 1, 5′-GCAGGTCCTCGTGCTAGTTC-3′and 5′-CTGCCTCTTCGTCACCCTAC-3′; mouse Irf6 promoter SMAD BS 2, 5′-GGAAGCTATTCTGGGCCTCT-3′ and 5′-GCAGCTTTATTTGGGTGCTT-3′; mouse Irf6 promoter SMAD BS 3, 5′-TGTGCTATTAGCCCCAACCT-3′ and 5′-TGTAGGGGGTTGAGTGTGGT-3′; mouse Irf6 promoter MEF2 BS, 5′-CGATCACAACACCAATCTGC-3′ and 5′-GACAGGCTGTGCACTCTTGA-3′, mouse p21 promoter p63 BS 1, 5′-CTTAGCGCAGAGCGGTTC-3′ and 5′-ACCTCCTCGCCTGTCCTCTA-3′; and mouse p21 promoter p63 BS 2, 5′-TGGTCTCCATCGGAATAGGT-3′ and 5′-TGTTTGCCTAACTTGCTGGA-3′. Positions of PCR fragments correspond to National Center for Biotechnology Information (NCBI) mouse genome Build 33.1.
Histological examination
Hematoxylin and Eosin (H&E), immunohistochemical and 5-bromo-2′-deoxyuridine (BrdU) staining were performed as described previously (Iwata et al., 2012; Iwata et al., 2010). Antibodies used for immunohistochemistry were rabbit polyclonal antibodies against p21 (Santa Cruz Biotechnology), IRF6 (Aviva Systems Biology), phosphorylated histone H3 (Millipore) and Lex/SSEA1 (FUT4 - Mouse Genome Informatics) (Cell Signaling Technology), and mouse monoclonal antibody against p63 (Santa Cruz Biotechnology). Fluorescence images were obtained using a fluorescence microscope (Model IX71, Olympus).
Immunoblot analysis
Immunoblots were performed as described previously (Iwata et al., 2006; Iwata et al., 2010). Antibodies used for immunoblotting were rabbit polyclonal antibodies against IRF6 (Aviva Systems Biology) and p21 (Abcam), and mouse monoclonal antibodies against p63 (Santa Cruz Biotechnology) and GAPDH (Chemicon).
Palatal shelf organ culture
Timed pregnant mice were sacrificed at E13.5. Genotyping was carried out as described above. The palatal shelves were microdissected and cultured in serum-free chemically defined medium as previously described (Ito et al., 2003; Xu et al., 2008). After 48 or 72 hours in culture, palates were harvested, fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4), and processed. For p63 experiments, palatal shelves were transfected with small interfering RNA (siRNA) duplexes (500 nM) for p63 or control (Santa Cruz Biotechnology). The 500 nM siRNA solutions were prepared by diluting a siRNA stock (10 μM) in BGJb medium containing Oligofectamine (0.3%) (Invitrogen). The siRNA mixture in transfection medium was changed every 24 hours and incubated with palatal shelves up to 72 hours after siRNA treatment at 37°C in a CO2 incubator. For experiments with p21, palatal shelves were transfected with a GFP-tagged p21 overexpression or control vector (Origene). The 2-μg p21 transfection solutions were prepared in BGJb medium containing Lipofectamine LTX and PLUS reagents (Invitrogen) according to the manufacturer’s protocol. The p21 transfection mixture was changed every 24 hours and incubated with palatal shelves up to 72 hours after transfection at 37°C in a CO2 incubator. All experiments were performed with at least five samples.
Cell culture
Primary mouse keratinocytes were isolated from newborn mice and cultured in dermal cell basal medium (ATCC) supplemented with the Keratinocyte Growth Kit (ATCC). Primary mouse keratinocytes (2×106 cells) were plated in a 60-mm cell culture dish until the cells reached 60-80% confluence. Tgfbr2 and Irf6 siRNA duplexes were purchased from Santa Cruz Biotechnology. siRNA mixture in transfection medium was incubated with cells for 7 hours at 37°C in a CO2 incubator, as described previously (Iwata et al., 2010).
Quantitative reverse transcription PCR (RT-PCR)
Total RNA was isolated from dissected mouse MEE at E14.5 with the QIAshredder and RNeasy Micro Extraction Kit (QIAGEN), as described previously (Iwata et al., 2010). The following PCR primers were used: Irf6, 5′-AGGGCTCTGTCATTAATCCAG-3′ and 5′-TGATTCGGGGCTGCAGTTTC-3′; p21 (Cdkn1a), 5′-AGCCTGAAGACTGTGATGGG-3′ and 5′-AAAGTTCCACCGTTCTCGG-3′; ΔNp63, 5′-CAAAACCCTGGAAGCAGAAA-3′ and 5′-GAGGAGCCGTTCTGAATCTG-3′; and Gapdh, 5′-AACTTTGGCATTGTGGAAGG-3′ and 5′-ACACATTGGGGGTAGGAACA-3′.
Scanning electron microscopic (SEM) analysis
Samples were fixed with a modified Karnovsky fixative solution [2% paraformaldehyde and 2.5% glutaraldehyde in 0.067 M cacodylate buffer (pH 7.4)] for two days. After dehydration through a graded ethanol series, samples were critical-point dried in a Balzer Union apparatus (FL-9496), ion-sputtered with platinum-palladium (10-15 nm), and observed in JEOL JSM-6390 low vacuum scanning electron microscope (JEOL USA, MA, USA) at a low accelerating voltage of 10 kV.
Whole-mount skeletal staining and micro-CT analysis
The three-dimensional architecture of the skeleton was examined using a modified whole-mount Alcian Blue-Alizarin Red S staining protocol as previously described (Ito et al., 2003; Iwata et al., 2012). Micro-CT analysis was performed using SCANCO μCT 50 (nanoCT) at the University of Southern California Molecular Imaging Center. The data were collected at a resolution of 10 μm. The reconstruction was performed using AVIZO 7.0 (Visualization Sciences Group).
Statistical analysis
A two-tailed Student’s t-test was applied for statistical analysis. For all graphs, data are represented as mean±s.d. A P-value of less than 0.05 was considered statistically significant.
RESULTS
TGFβ signaling in the MEE regulates Irf6 expression during palatal fusion
Loss of TGFβ signaling in the basal epithelium in mice (Tgfbr2fl/fl;K14-Cre) results in diminished Irf6 expression and failure of apoptosis in MEE cells, followed by MEE persistence, suggesting that TGFβ-mediated Irf6 expression might play a role in degeneration of the MEE (Xu et al., 2006). The MEE is composed of a basal columnar cell layer covered by flat cells that constitute the periderm (Cuervo and Covarrubias, 2004). Irf6 is expressed in the periderm at E13.5, and Irf6 expression shows an obvious and consistent transition from the periderm to regions of the basal epithelial layer at E14.5 (Fakhouri et al., 2012). Expression of Irf6 mRNA (Fig. 1A) and protein (Fig. 1B,C) was decreased in Tgfbr2fl/fl;K14-Cre mice at E14.5. We found no defect in the periderm of Tgfbr2fl/fl;K14-Cre mice during palatal fusion (supplementary material Fig. S1). To test whether Irf6 expression is regulated by a SMAD-dependent pathway, we analyzed gene expression of Irf6 in Smad4fl/fl;K14-Cre mice. Irf6 expression was not significantly changed in Smad4fl/fl;K14-Cre mice (Fig. 1D), suggesting that SMAD4-independent TGFβ signaling might be involved in the induction of Irf6 in MEE cells during palatogenesis. Because SMAD4 and p38 MAPK are functionally redundant in regulating the MEE disappearance (Xu et al., 2008), we performed ex vivo organ culture of E13.5 palatal shelf explants from Smad4fl/fl;K14-Cre and Smad4fl/fl control mice with p38 MAPK inhibitor or with vehicle as control. We found that p38 MAPK inhibitors blocked both Irf6 expression (Fig. 1E) and MEE disappearance (Fig. 1F) in Smad4fl/fl;K14-Cre palates but did not affect Irf6 expression (Fig. 1E) or MEE disappearance (Fig. 1H) in control samples, indicating that SMAD and p38 MAPK activation are functionally redundant in regulating Irf6 expression and MEE disappearance.
Fig. 1.

Decreased IRF6 expression in the MEE of Tgfbr2fl/fl;K14-Cre mice. (A) Quantitative RT-PCR analyses of Irf6 expression in the palates of Tgfbr2fl/fl (control, n=6) and Tgfbr2fl/fl;K14-Cre (n=6) mice at E14.5. (B,C) Immunohistochemical analyses of IRF6 expression (red) in the palates of Tgfbr2fl/fl control (B) and Tgfbr2fl/fl;K14-Cre (C) mice at E14.5. Nuclei were counterstained with DAPI (blue). (D) Quantitative RT-PCR analyses of Irf6 expression in the palates of Smad4fl/fl (control, n=6) and Smad4fl/fl;K14-Cre (n=6) mice at E14.5. (E) Quantitative RT-PCR analyses of Irf6 expression in palate explants from Smad4fl/fl (control) and Smad4fl/fl;K14-Cre mice treated with p38 MAPK inhibitor (+) or vehicle (-) for 48 hours. n=3 per group. (F-I) H&E staining of palate explants from Smad4fl/fl;K14-Cre and Smad4fl/fl (control) mice treated with p38 MAPK inhibitor (F,H) or vehicle control (G,I) for 72 hours. n=5 per group. (J) Schematic of the upstream region of the mouse Irf6 gene (not to scale), showing locations of putative SMAD-binding (red) or MEF2-binding (green) sites tested in ChIP assays. Putative SMAD- and MEF2-binding sequences are shown below. Arrowheads indicate the position of primers used in ChIP analysis. (K) ChIP analysis of DNA fragments immunoprecipitated with a SMAD4-specific or MEF2-specific antibody or with an isotype-specific control antibody. Immunoprecipitates were PCR amplified with primers flanking the putative SMAD-binding or MEF2-binding region. Input lane shows PCR amplification of the sonicated chromatin before immunoprecipitation. Ab, antibody; BS, binding site. Error bars represent s.d. *P<0.05. Scale bars: 50 μm.
Next, we analyzed the sequence of the mouse Irf6 gene (including 2.5 kb upstream and 2.5 kb downstream of the transcription start site). The mouse Irf6 genomic region has 43 potential SMAD recognition sites (three of which are conserved in at least six mammals) (Fig. 1J; supplementary material Table S1). The MEF2 transcription factor is regulated by the p38 MAPK pathway (Han and Molkentin, 2000; Toro et al., 2004). We found a potential MEF2 recognition site in the Irf6 genomic region (Fig. 1J). We performed chromatin immunoprecipitation (ChIP) assays to test whether SMAD and MEF2 could bind to the promoter region of Irf6. Binding sites for SMAD (SMAD binding site 1: -274 bp to -271 bp; site 2: -1695 bp to -1692 bp; site 3: -2375 bp to -2372 bp) and MEF2 (MEF2 binding site: -1387 bp to -1375 bp) immunoprecipitated with SMAD4 or MEF2 antibodies, but not with control antibody (Fig. 1K). Taken together, our results suggest that Irf6 gene expression is regulated by both SMAD and p38 MAPK pathways.
Next, we investigated the functional significance of Irf6 in regulating the disappearance of MEE cells during palatal fusion. Because homozygous mutation of Irf6 results in severe intraoral epithelial adhesion, it is not known whether Irf6 is crucial for palatal fusion (Ingraham et al., 2006; Richardson et al., 2006). To investigate the fate of the MEE in Irf6R84C/R84C mice, we used the ex vivo organ culture system. E13.5 wild-type control (n=17) and Irf6+/R84C (n=39) palate explants fused and the MEE disappeared following two days of culture (Fig. 2A,B). By contrast, Irf6R84C/R84C mutant (n=19) palatal shelves failed to fuse (Fig. 2C) and the MEE persisted with continuous cell proliferation (Fig. 2D-F) and compromised cell death (Fig. 2G-I) following two days of culture with complete phenotype penetrance. We quantified the increased cell proliferation as well as decreased apoptosis in Irf6R84C/R84C MEE cells compared with wild-type littermate control and Irf6+/R84C MEE cells during palatal fusion (Fig. 2J,K). The MEE in control and Irf6+/R84C explants disappeared completely following three days in culture; however, the MEE persisted in Irf6R84C/R84C explants (supplementary material Fig. S2). These data indicate that IRF6 is crucial for MEE disappearance during palatal fusion.
Fig. 2.
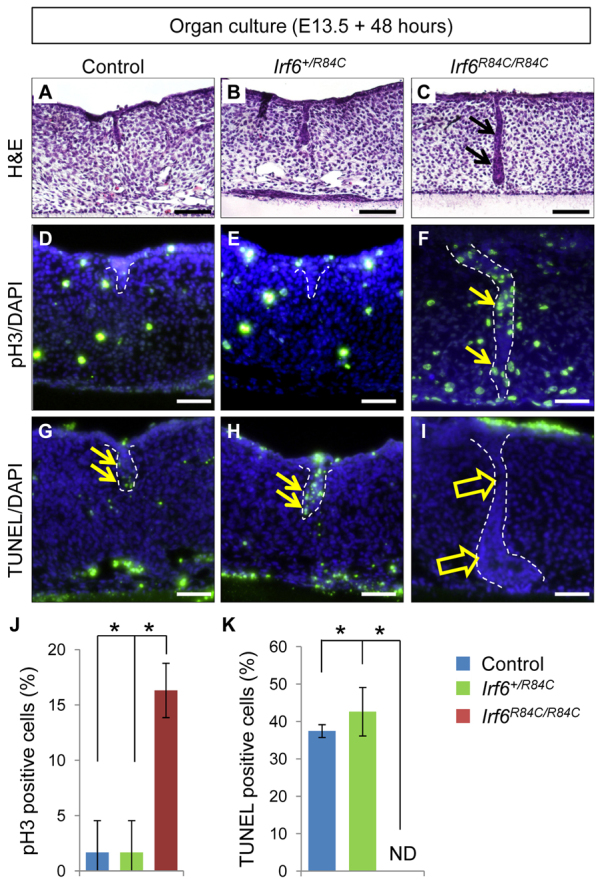
Irf6 is crucial for MEE disappearance during palate formation. (A-C) H&E staining of sections of control (A), Irf6+/R84C (B) and Irf6R84C/R84C (C) palates after culture for 48 hours. Arrows indicate MEE persistence. (D-F) Phosphorylated histone H3 (pH3) staining of sections of control (D), Irf6+/R84C (E) and Irf6R84C/R84C (F) palates after culture for 48 hours. Arrows indicate pH3-positive cells (green) in the MEE. Dashed lines outline MEE cells. Nuclei were counterstained with DAPI (blue). (G-I) TUNEL staining of sections of control (G), Irf6+/R84C (H) and Irf6R84C/R84C (I) palates after culture for 48 hours. Arrows indicate TUNEL-positive cells (green) in the MEE. Dashed lines outline MEE cells. Nuclei were counterstained with DAPI (blue). (J) Quantification of the number of phosphorylated histone H3 (pH3)-positive nuclei in the MEE in control (n=17), Irf6+/R84C (n=39) and Irf6R84C/R84C (n=19) palates. (K) Quantification of the number of TUNEL-labeled nuclei in the MEE in control (n=17), Irf6+/R84C (n=39) and Irf6R84C/R84C (n=19) palates. Error bars represent s.d. *P<0.01. ND, not detected. Scale bars: 50 μm.
Genetic interaction between Smad4 and Irf6 during palatal fusion
We hypothesized that haploinsufficiency of Irf6 in a Smad4 mutant background would cause an additional reduction of IRF6 activity and result in cleft palate. To investigate a possible genetic interaction between Irf6 and Smad4, we generated Smad4fl/fl;K14-Cre;Irf6+/R84C mice. Smad4fl/fl;K14-Cre mice have normal palates although they have open eyes, short fingers, and nail abnormalities (n=17/20) (Fig. 3; supplementary material Fig. S3). Interestingly, Smad4fl/fl;K14-Cre;Irf6+/R84C mice exhibited extra digits with complete phenotype penetrance (n=19/19) (supplementary material Fig. S3). Thus, Smad4fl/fl;K14-Cre;Irf6+/R84C mice have a more severe phenotype than do Smad4fl/fl;K14-Cre mice, suggesting that IRF6 and SMAD4 function synergistically in regulating embryogenesis. In addition, the extra digits in Smad4fl/fl;K14-Cre;Irf6+/R84C mice are consistent with the abnormal toe and nail phenotype in individuals with VWS and PPS. Because some individuals with VWS and PPS have cleft palate, we investigated the palate in Smad4fl/fl;K14-Cre;Irf6+/R84C mice in detail by scanning electron microscopy (SEM), and detected compromised rugae formation (Fig. 3G,H). By histological analysis, we found that all Smad4fl/fl;K14-Cre;Irf6+/R84C mice exhibited submucous cleft palate at birth with persistence of the MEE (n=19/19) and in some cases a stretched epithelial bridge (n=6/19) (Fig. 3I-O). Taken together, our findings suggest that a genetic interaction between Smad4 and Irf6 is responsible for MEE disappearance during palatal fusion.
Fig. 3.
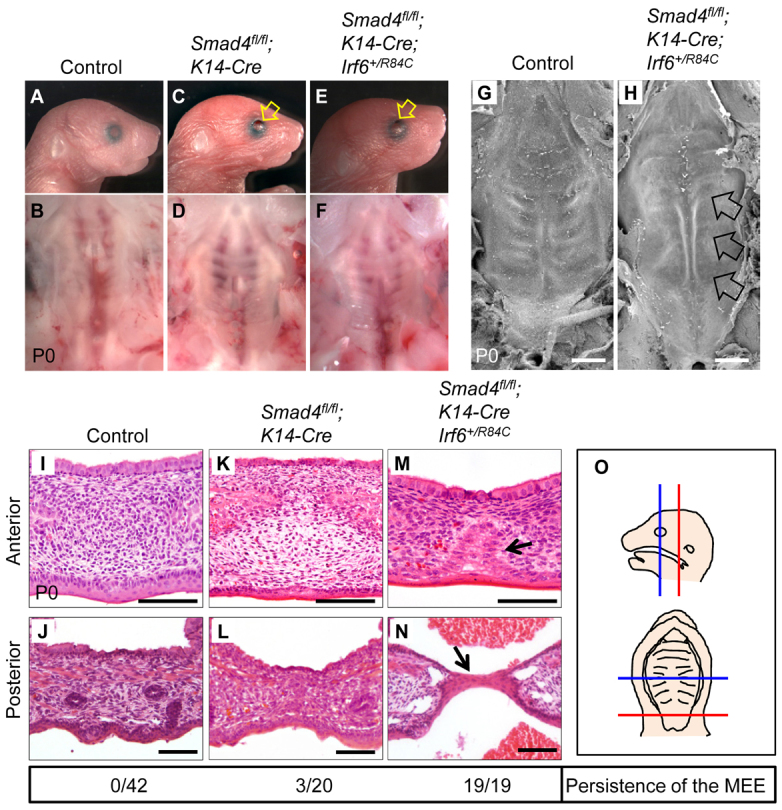
A haploinsufficiency of Irf6 in Smad4fl/fl;K14-Cre mice causes submucous cleft palate. (A-F) Morphologies of newborn control (A,B), Smad4fl/fl;K14-Cre (C,D) and Smad4fl/fl;K14-Cre;Irf6+/R84C (E,F) mice. Lower panels (B,D,F) show macroscopic appearance of palates at newborn stage. Arrows indicate open eyes. (G,H) SEM analysis of newborn control (G) and Smad4fl/fl;K14-Cre;Irf6+/R84C (H) mice. Arrows indicate lack of rugae formation. (I-N) H&E staining of sections of newborn Smad4fl/fl control (I,J; n=42), Smad4fl/fl;K14-Cre (K,L; n=20) and Smad4fl/fl;K14-Cre;Irf6+/R84C (M,N; n=19) mice. Arrows indicate MEE persistence. (O) Schematics showing the position from which the sections shown in I-N were taken. Blue lines indicate the anterior region of the palate (hard palate; I,K,M), and red lines indicate the posterior region of the palate (soft palate; J,L,N). The MEE was scored for persistence in I-N (shown below). Scale bars: 500 μm in G,H; 100 μm in I-N.
Rescue of palatal fusion in Tgfbr2fl/fl;K14-Cre mice by overexpression of Irf6
To test whether Irf6 expression level controls MEE cell fate, we generated K14-driven Irf6 transgenic (K14-Irf6Tg) mice to overexpress Irf6 in MEE cells (Fig. 4A). Most K14-Irf6Tg mice were born healthy and fertile without any noticeable pathological phenotype up to the age of one year (n=114/146) (supplementary material Table S2). We analyzed Irf6 gene expression level by quantitative RT-PCR and found that there is a significant increase in Irf6 expression (2.2-fold) in the MEE of K14-Irf6Tg mice compared with control (Fig. 4B). Moreover, we confirmed the overexpression of IRF6 protein in the MEE by immunoblotting (Fig. 4C,D) and immunostaining (Fig. 4E) at E14.5. Approximately 22% of Irf6 transgenic mice (n=32/146) exhibited absence of the calvaria and an open eye phenotype and died within one day of birth (supplementary material Table S2 and Fig. S4). Although these Irf6 transgenic mice exhibited severe calvarial defects and hypoplasia of the palatine bones (Fig. 4J,M), their palatal shelves fused normally without obvious cleft palate or persistence of the MEE (Fig. 4J). Palatal fusion appeared to be unaffected in all K14-Irf6Tg mice (n=146) (Fig. 4F-M; supplementary material Table S2).
Fig. 4.

Generation of K14-Irf6 transgenic mice. (A) Construct used to generate K14-Irf6 transgenic mice. (B) Quantitative RT-PCR analyses of Irf6 in the palates of control and K14-Irf6Tg mice at E14.5. n=6 per genotype. *P<0.05. (C) Immunoblotting analysis of IRF6 in the MEE of control and K14-Irf6Tg mice. GAPDH was used as a loading control. (D) Bar graph shows the ratio of IRF6 per GAPDH following quantitative densitometry analysis of immunoblotting data. Three samples were analyzed. (E) Immunohistochemical analysis of IRF6 in the palate of E14.5 K14-Irf6Tg mice. Arrows indicate IRF6 expression in the MEE. (F,G) H&E staining of littermate control (F) and K14-Irf6 transgenic (Irf6Tg; G) mice at E15.5. Scale bars: 50 μm. (H-J) MicroCT analysis of the soft tissues of E18.5 control (H) and K14-Irf6Tg (I,J) mice. Seventy-eight percent of K14-Irf6Tg mice exhibit apparently normal development (I), and 22% exhibit severe craniofacial deformities (J). Arrowhead indicates palatal fusion in the K14-Irf6Tg mice with severe craniofacial deformities. Arrow indicates calvarial defect in the K14-Irf6Tg mice. (K-M) MicroCT analysis of the hard tissue of E18.5 control mice (K) and K14-Irf6Tg mice with normal development (L) or with severe craniofacial deformities (M). Arrow indicates hypoplasia of the palatine bones in K14-Irf6Tg mice with severe craniofacial deformities. Error bars represent s.d.
Next, we generated Tgfbr2fl/fl;K14-Cre;Irf6Tg mice to restore Irf6 gene expression in Tgfbr2fl/fl;K14-Cre mice. By SEM analysis, rugae formation was detectable in control mice, but not in their Tgfbr2fl/fl;K14-Cre and Tgfbr2fl/fl;K14-Cre;Irf6Tg littermates (Fig. 5A-C). We also found that Tgfbr2fl/fl;K14-Cre mice exhibit cleft soft palate, which was partially rescued (in the anterior of the soft palate) in Tgfbr2fl/fl;K14-Cre;Irf6Tg mice (Fig. 5B,C). In addition, Tgfbr2fl/fl;K14-Cre mice have submucous cleft palate throughout the anterior to posterior regions of the hard palate, based on histology (Fig. 5K). At E15.5 and E16.5, the MEE disappeared in control mice, whereas MEE persistence was detectable in Tgfbr2fl/fl;K14-Cre mice (Fig. 5D,E,G,H). Tgfbr2fl/fl;K14-Cre;Irf6Tg mice show a one-day delay in the disappearance of the MEE (Fig. 5F,I). In histological sections, we detected MEE persistence (n=14/14) corresponding with the groove in the midline of the palate in Tgfbr2fl/fl;K14-Cre mice (Fig. 5K). Proper apoptotic degradation of the MEE was restored in Tgfbr2fl/fl;K14-Cre;Irf6Tg mice (n=7/7), based on histological analysis (Fig. 5L), indicating that TGFβ-mediated Irf6 signaling is functionally important and sufficient for MEE disappearance.
Fig. 5.
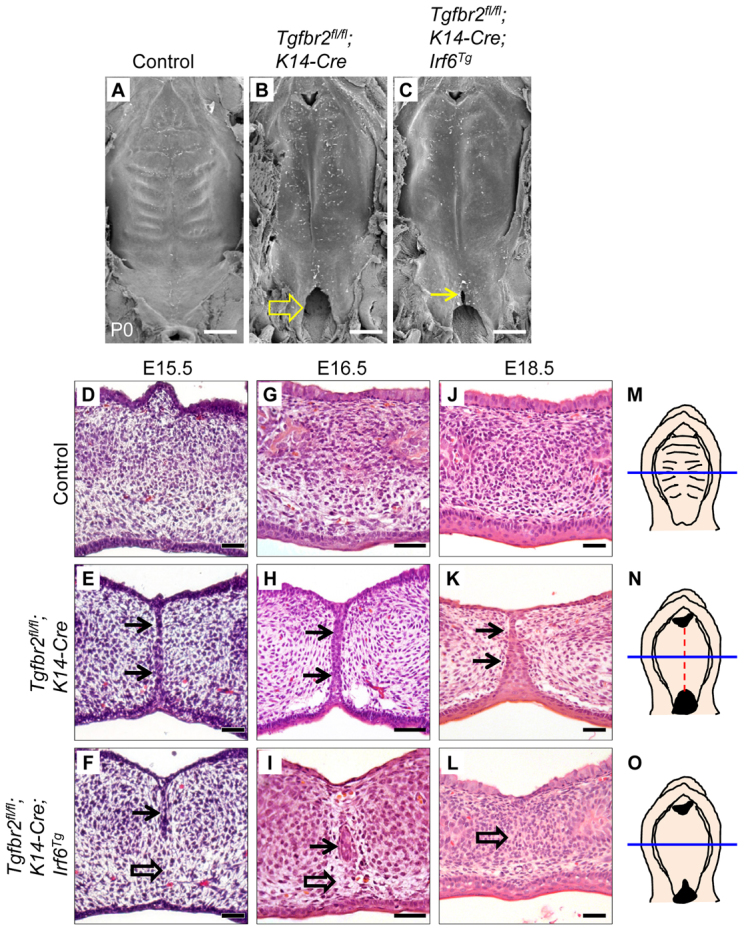
Restored MEE degeneration in Tgfbr2fl/fl;K14-Cre mice via overexpression of Irf6. (A-C) SEM images of the palates of newborn control (A), Tgfbr2fl/fl;K14-Cre (B) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (C) mice. Arrow in B indicates cleft soft palate. Arrow in C indicates a partial rescue of cleft soft palate in the anterior region of the soft palate. (D-L) H&E staining of E15.5 (D-F), E16.5 (G-I) and E18.5 (J-L) Tgfbr2fl/fl control, Tgfbr2fl/fl;K14-Cre and Tgfbr2fl/fl;K14-Cre;Irf6Tg mice. Arrows indicate MEE persistence. Open arrows indicate MEE degeneration. The MEE was scored for persistence at E18.5: control mice, n=0/28; Tgfbr2fl/fl;K14-Cre mice, n=14/14; Tgfbr2fl/fl;K14-Cre;Irf6Tg mice, n=0/7. (M-O) Schematics of E18.5 Tgfbr2fl/fl control (M), Tgfbr2fl/fl;K14-Cre (N) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (O) palates showing the position (blue lines, hard palate) from which the sections shown in D-L were taken. A dashed line (red, in N) indicates persistence of the MEE. Scale bars: 500 μm in A-C; 50 μm in D-L.
IRF6 regulates MEE disappearance via p21 expression
Apoptosis is one of the ultimate fates of the MEE during palatal fusion (Cuervo and Covarrubias, 2004). Loss of Tgfbr2 in the epithelium results in absence of apoptosis and maintenance of proliferation in MEE cells (Xu et al., 2006). We analyzed the cellular defect in persistent MEE cells by assaying both cell proliferation and apoptosis activities. We found that altered cell proliferation in the MEE was restored in Tgfbr2fl/fl;K14-Cre;Irf6Tg mice to a level comparable to the control (Fig. 6A-D). Similarly, we found that altered apoptotic activity in the MEE was also restored in Tgfbr2fl/fl;K14-Cre;Irf6Tg mice compared with the control (Fig. 6E-H). Thus, our data demonstrate that loss of Tgfbr2 results in compromised Irf6 expression, continuous cell proliferation, and failure of apoptosis in MEE cells, indicating that TGFβ-mediated Irf6 expression plays a role in the disappearance of the MEE. We previously reported that p21 (also known as Cdkn1a) was decreased in the MEE of Tgfbr2fl/fl;K14-Cre mice (Xu et al., 2008). We hypothesized that p21 expression might be regulated by the TGFβ-mediated IRF6 pathway. To investigate p21 expression, we performed immunohistochemical (Fig. 6I-K), immunoblotting (Fig. 6L) and quantitative RT-PCR (Fig. 6M,N) analyses. We found that p21 expression was restored in E14.5 Tgfbr2fl/fl;K14-Cre;Irf6Tg mice, correlating with Irf6 expression. Therefore, we propose that TGFβ-mediated IRF6 activity regulates MEE disappearance by regulating p21 expression.
Fig. 6.

p21 expression is restored in Tgfbr2fl/fl;K14-Cre;Irf6Tg mice. (A-C) Immunostaining of phosphorylated histone H3 (pH3) in Tgfbr2fl/fl control (A), Tgfbr2fl/fl;K14-Cre (B) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (C) mice at E14.5. Dashed lines indicate the MEE. Arrows indicate positive signal (green). (D) Percentage of pH3-positive nuclei in the palates of Tgfbr2fl/fl (blue bar, n=13), Tgfbr2fl/fl;K14-Cre (red bar, n=7) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (green bar, n=4) mice at E14.5. (E-G) TUNEL staining of Tgfbr2fl/fl control (E), Tgfbr2fl/fl;K14-Cre (F) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (G) mice at E14.5. Dashed lines indicate the MEE. Arrows indicate positive signal (green). (H) Percentage of TUNEL-labeled nuclei in the palates of Tgfbr2fl/fl (blue bar, n=13), Tgfbr2fl/fl;K14-Cre (red bar, n=7) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (green bar, n=4) mice at E14.5. (I-K) Immunohistochemical analyses of p21 expression in the palates of Tgfbr2fl/fl control (I), Tgfbr2fl/fl;K14-Cre (J) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (K) mice at E14.5. Dashed lines indicate the MEE. Arrows indicate positive signal (green). (L) Immunoblotting analysis of IRF6 and p21 in the MEE of Tgfbr2fl/fl control (lane 1), Tgfbr2fl/fl;K14-Cre (lane 2) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (lane 3) mice. GAPDH was used as a loading control. Bar graphs (below) show the ratio of IRF6 or p21 to GAPDH following quantitative densitometry analysis of immunoblotting data. Tgfbr2fl/fl control (blue bar), Tgfbr2fl/fl;K14-Cre (red bar), Tgfbr2fl/fl;K14-Cre;Irf6Tg (green bar). Three samples were analyzed. (M,N) Quantitative RT-PCR analyses of Irf6 (M) and p21 (N) in the MEE of Tgfbr2fl/fl (blue, n=3), Tgfbr2fl/fl;K14-Cre (red, n=3) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (green, n=3) mice at E14.5. Error bars represent s.d. *P<0.05. Scale bars: 50 μm.
Previous studies have demonstrated that IRF6 induces degradation of the p63 isoform ΔNp63 and that this is linked with the pathogenesis of VWS and PPS (Moretti et al., 2010; Thomason et al., 2010). In addition, ΔNp63 represses transcription of p21 in vitro and in vivo (Laurikkala et al., 2006; Welsh and O’Brien, 2009; Westfall et al., 2003). Because there is no conserved IRF6-binding site in the promoter-proximal genomic region of p21 (in at least six mammals, 5 kb upstream and 5 kb downstream of the TSS), we hypothesized that IRF6 might regulate p21 expression via ΔNp63. ΔNp63 is only expressed in the basal epithelial layer during palate formation (Fakhouri et al., 2012). Indeed, increased ΔNp63 expression was detectable in basal epithelial cells in Tgfbr2fl/fl;K14-Cre mice, consistent with a TGFβ-mediated IRF6/ΔNp63/p21 signaling cascade at E14.5 and E15.5 (Fig. 7A-F). Moreover, ΔNp63 expression was reversed by overexpression of Irf6 (Fig. 7C,F). We also evaluated ΔNp63 expression levels in control, Tgfbr2fl/fl;K14-Cre and Tgfbr2fl/fl;K14-Cre;Irf6Tg mice by immunoblotting (Fig. 7G). To confirm that p21 expression is regulated by p63, we analyzed the mouse p21 promoter region (2.5 kb upstream and 2.5 kb downstream of the TSS), and found two potential p63-binding sites in the mouse genomic region of p21 (Fig. 7H). We performed ChIP analysis to test whether p63 could bind to the genomic region of p21, and found that the p21 genomic region immunoprecipitated with the p63 antibody at p63-binding site 1 (+727 bp to +746 bp), but not at p63-binding site 2 (-436 bp to -455 bp) (Fig. 7I). These results indicate that p21 expression is likely to be regulated through the TGFβ/IRF6/ΔNp63 signaling cascade.
Fig. 7.
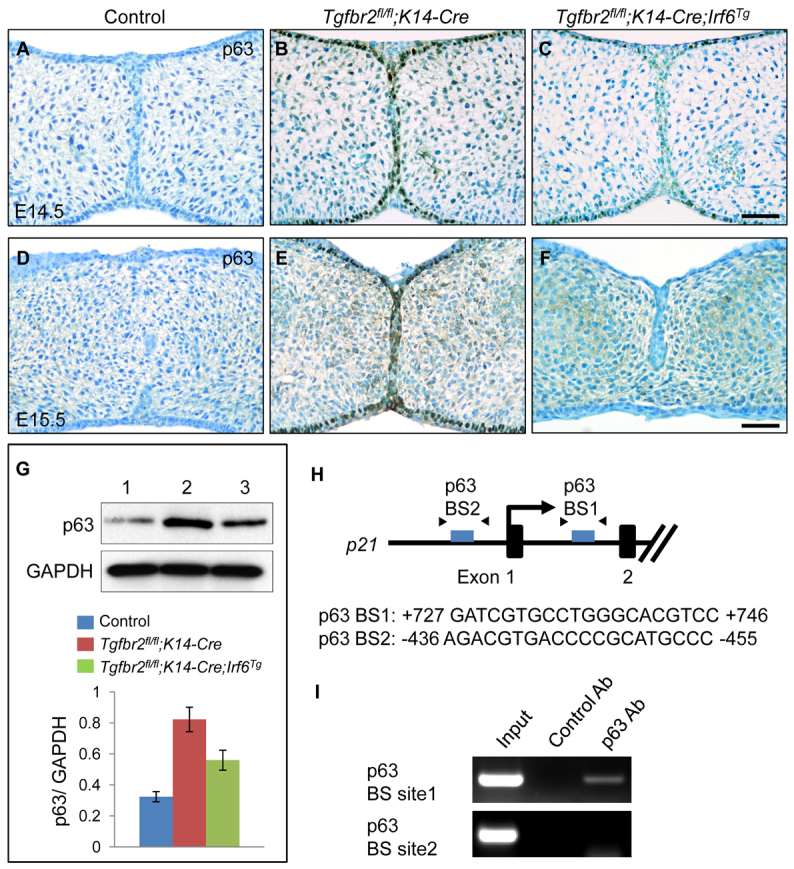
Increased ΔNp63 expression in Tgfbr2fl/fl;K14-Cre mice. (A-F) Immunohistochemical analyses of ΔNp63 expression in the palates of Tgfbr2fl/fl control (A,D), Tgfbr2fl/fl;K14-Cre (B,E) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (C,F) mice at E14.5 (A-C) and E15.5 (D-F). Brown, positive signal. Nuclei were counterstained with 0.03% Methylene Blue. Scale bars: 50 μm. (G) Immunoblotting analysis of p63 in the MEE of E14.5 Tgfbr2fl/fl control (lane 1), Tgfbr2fl/fl;K14-Cre (lane 2) and Tgfbr2fl/fl;K14-Cre;Irf6Tg (lane 3) mice. GAPDH was used as a loading control. Bar graph (below) shows the ratio of p63 to GAPDH following quantitative densitometry analysis of immunoblotting data. Three samples were analyzed. (H) Schematic of the upstream region of the mouse p21 gene (not to scale), showing locations of putative p63-binding sites tested in ChIP assays. Putative p63-binding sequences are shown below. Arrowheads indicate the position of primers used in ChIP analysis. (I) ChIP analysis of DNA fragments immunoprecipitated with a p63-specific antibody or with an isotype-specific control antibody. Immunoprecipitates were PCR amplified with primers flanking the putative p63-binding region. Input lane shows PCR amplification of the sonicated chromatin before immunoprecipitation. No amplification of target sites was detected when an isotype-specific control antibody was used. Ab, antibody; BS, binding site.
Finally, we investigated the functional significance of the IRF6/p63/p21 signaling cascade using an ex vivo organ culture system. We found that either reduction of p63 or overexpression of p21 could restore the degeneration of the MEE (Fig. 8A-F). In addition, primary mouse keratinocytes were treated with Tgfbr2, Irf6 or control siRNA (Fig. 8G). After siRNA knockdown for Tgfbr2, gene expression of Irf6 and p21 was significantly decreased and p63 gene expression was significantly (P<0.05) upregulated. Similarly, Irf6 siRNA knockdown resulted in downregulation of Irf6 and p21 gene expression and upregulation of p63 gene expression. Previous studies indicate that p63 expression is upregulated in E14.5 Irf6R84C/R84C palates (Thomason et al., 2010) and siRNA knockdown for Irf6 results in upregulated p63 expression in human keratinocytes (Moretti et al., 2010). Therefore, we investigated whether p63 expression was altered in Irf6R84C/R84C palates. The expression of p63 was increased in Irf6R84C/R84C palates, consistent with previous findings (Fig. 8H,I). Collectively, these data suggest that p21 expression is regulated through a TGFβ/IRF6/ΔNp63 signaling cascade (Fig. 8J).
Fig. 8.
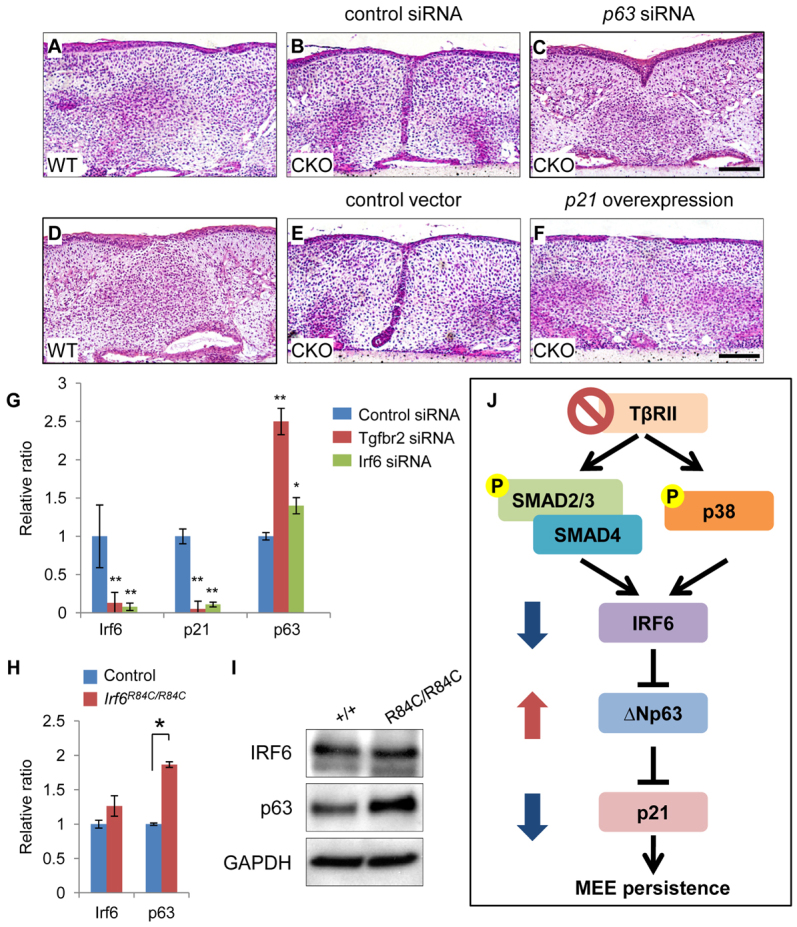
Altered p63-p21 cascade results in persistence of the MEE in Tgfbr2fl/fl;K14-Cre mice. (A-C) H&E staining of palate explants from Tgfbr2fl/fl (WT) mice and Tgfbr2fl/fl;K14-Cre (CKO) mice treated with p63 (C) or control (B) siRNA for 72 hours. n=5 per group. (D-F) H&E staining of palate explants from Tgfbr2fl/fl (WT) mice and Tgfbr2fl/fl;K14-Cre (CKO) mice treated with p21 overexpression vector (F) or control (E) for 72 hours. n=5 per group. (G) Quantitative RT-PCR analyses of Irf6, p21 and ΔNp63 expression in primary mouse keratinocytes isolated from back skin of newborn wild-type mice after treatment with control (blue bars), Tgfbr2 (red bars) or Irf6 (green bars) siRNA. Antisense siRNA was used as control. (H) Quantitative RT-PCR analyses of Irf6 and ΔNp63 expression in primary mouse keratinocytes isolated from newborn Irf6R84C/R84C (red bars) and littermate control (blue bars) back skin. (I) Immunoblotting analysis of IRF6 and p63 in the MEE of control (+/+) and Irf6R84C/R84C (R84C/R84C) mice. GAPDH was used as a loading control. (J) Schematic depicting our model of the mechanism of TGFβ-mediated Irf6-ΔNp63-p21 gene regulation in Tgfbr2fl/fl;K14-Cre palates. IRF6 expression is regulated through both SMAD and p38 MAPK pathways. In the absence of TGFβ receptor type II (TβRII), IRF6 expression decreases. ΔNp63 expression increases in the absence of TβRII resulting in reduced p21 expression in the palatal epithelium. Altered TGFβ-IRF6-ΔNp63-p21 activity results in the persistence of the MEE. P, phosphorylated. Error bars represent s.d. *P<0.05; **P<0.01. Scale bars: 50 μm.
DISCUSSION
Submucous cleft palate, which can result from MEE defects during palatal fusion, is one of the most common forms of cleft palate in humans, but the molecular and developmental mechanism of submucous cleft palate are not well studied, in part because of the paucity of animal models that exhibit this phenotype (Funato et al., 2012; Pauws et al., 2009). MEE persistence affects palatal bone formation in the hard palate and muscle development in the soft palate; consequently, patients with submucous cleft palate need surgical and other procedures to develop precise physiological functions such as speech and swallowing.
We have generated an animal model in which loss of TGFβ signaling in MEE cells results in submucous cleft palate (Xu et al., 2006). In this study, we show that TGFβ-mediated Irf6 expression is crucial for the fate of MEE cells. MEE degeneration in Tgfbr2fl/fl;K14-Cre mice was restored by overexpression of Irf6, indicating that Irf6 is functionally important for the MEE disappearance mediated by TGFβ signaling. Previous studies indicate that mice with loss of Irf6 function exhibit cleft palate owing to failure of palatal shelf elevation resulting from adhesion between the palatal shelves and the tongue, following a defect in epithelial differentiation (Ingraham et al., 2006; Richardson et al., 2006). Because of the failure of palatal elevation in Irf6R84C/R84C mice, we were previously not able to investigate the role and function of Irf6 during MEE disappearance fully. In this study, using an ex vivo organ culture system, we show for the first time that Irf6R84C/R84C mutation results in the absence of apoptosis and the maintenance of proliferation in the MEE, indicating that Irf6 is crucial for MEE cell fate during palatal fusion. Although heterozygous Irf6 mutant mice have normal palate formation, a combination of loss of Smad4 and inhibition of p38 MAPK only led to ∼50% reduction of Irf6 expression and persistence of the MEE in ex vivo organ culture. This result suggests that there are other factors or signaling pathways that might regulate Irf6 expression in addition to SMAD4/p38 MAPK signaling pathways (Ferretti et al., 2011; Letra et al., 2012). A recent study has shown that integration of IRF6 and the Notch ligand jagged 2 signaling is essential for controlling palatal adhesion and fusion during palatogenesis (Richardson et al., 2009). Interestingly, overexpression of Irf6 in Tgfbr2fl/fl;K14-Cre mice did not rescue the developmental defects in the palatal mesenchyme (supplementary material Fig. S5), suggesting that the TGFβ-mediated IRF6 signaling cascade plays a cell-autonomous role in regulating the fate of MEE cells during palatal fusion and that TGFβ regulates other downstream target genes that control the development of muscles in the soft palate through tissue-tissue interactions. Furthermore, using mutant mouse models, we found that TGFβ signaling regulates Irf6 and p21 expression and MEE disappearance via both SMAD-dependent and -independent pathways during palatal fusion.
SMAD4 mutations have been found in patients with unselected hereditary hemorrhagic telangiectasia (HHT), which is an autosomal dominant disease of vascular dysplasia (Gallione et al., 2006). The symptoms of HHT include epistaxis, telangiectases, and arteriovenous malformations, which are most often found in the lungs, brain, liver and gastrointestinal tract. In addition, SMAD4 mutations have been identified in families with juvenile polyposis, aortophathy, and mitral valve dysfunction (Andrabi et al., 2011). Approximately 15% of people with juvenile polyposis syndrome have other abnormalities, such as cleft palate, polydactyly, intestinal malrotation, heart or brain abnormalities, and abnormalities of the genitalia or urinary tract. Although most Smad4fl/fl;K14-Cre mice have normal palates, one copy of the R84C IRF6 mutation in a Smad4 mutant background (Smad4fl/fl;K14-Cre;Irf6+/R84C mice) resulted in fully penetrant submucous cleft palate and polydactyly. It is important to note that Smad4fl/fl;K14-Cre;Irf6+/R84C mice closely phenocopy individuals affected by PPS as described in humans with IRF6 mutation, whereas Irf6+/R84C mice do not show digit developmental defects. These results suggest that a compromised SMAD-dependent, TGFβ-mediated IRF6 signaling cascade might be responsible for developmental defects associated with PPS. Furthermore, an impaired TGFβ/IRF6 signaling cascade may cause submucosal cleft palate.
The secondary palate is divided into two parts: the anterior bony hard palate, which is about two-thirds of the secondary palate and is composed of bone, and the posterior fleshy soft palate, which is about one-third of the secondary palate and is composed of muscles. Our previous study suggests that loss of Tgfbr2 in the epithelium causes cleft soft palate owing to failure of muscle development and misorientation of muscle fibers (Xu et al., 2006). Overexpression of Irf6 in the MEE of Tgfbr2fl/fl;K14-Cre mice failed to restore epithelial-mesenchymal interaction and to support soft palate muscle development. We are currently investigating the molecular and cellular mechanisms of soft palate muscle defects in Tgfbr2fl/fl;K14-Cre mice.
Cleft soft palate in Tgfbr2fl/fl;K14-Cre mice was not rescued in Tgfbr2fl/fl;K14-Cre;Irf6Tg mice. One possible explanation is that there are differential gene expression patterns in the anterior versus the posterior in the MEE. Expression of Irf6, p63 and Tgfb3 is detectable throughout the oral epithelia in wild-type mice with no variation along the antero-posterior axis of the secondary palate (Fakhouri et al., 2012; Richardson et al., 2009; Yang et al., 2008). However, gene expression of Tgfb3 was compromised specifically in the posterior palate in Irf6R84C/R84C mice, indicating that Tgfb3 expression is differentially regulated between the anterior and posterior MEE (Richardson et al., 2009). It also suggests that IRF6 provides a feedback in regulating TGFβ signaling in the posterior region of developing palate.
WNT/β-catenin signaling acts upstream of Irf6 in the lip epithelium during lip fusion (Ferretti et al., 2011). A previous study has shown that loss of WNT/β-catenin signaling results in downregulation of TGFβ, whereas expression of stabilized β-catenin in the palatal epithelium can cause ectopic Tgfb3 expression and fusion of the palatal shelf and mandible (He et al., 2011). This study clearly demonstrates that a precisely controlled TGFβ signaling level is crucial for regulating the fate of MEE cells during palatal fusion. Ablation of p63 results in diminished Irf6 expression in the palate (Moretti et al., 2010; Thomason et al., 2010), and IRF6 can regulate ΔNp63 degradation in human keratinocytes (Moretti et al., 2010). However, we found that upregulated p63 expression in wild-type mice did not affect the expression of Irf6 in MEE cells (data not shown), indicating that there is no apparent feedback loop from upregulated p63 to Irf6 expression.
In summary, our data indicate that a combination of genetic mutations in Irf6 and Smad4 cause submucous cleft palate and that TGFβ-mediated IRF6 activity is crucial for MEE disappearance. This is a significant advancement in our understanding of the mechanism of TGFβ signaling-associated cleft palate. Our findings that TGFβ-mediated Irf6 expression is responsible for MEE disappearance and that overexpression of Irf6 normalizes MEE cell fate determination in Tgfbr2fl/fl;K14-Cre mice might provide potential therapeutic approaches for individuals with altered TGFβ signaling and submucous cleft palate. In addition, we propose that combined mutations in IRF6 and SMAD4 in humans might be useful diagnostic biomarkers for patients with cleft palate.
Supplementary Material
Acknowledgments
We are grateful to Dr Julie Mayo for critical reading of the manuscript, and to Dr Harold Slavkin for discussion. We thank Dr Harold Moses for Tgfbr2fl/fl mice. We thank Xuemei Deng, Toshiaki Yokota, Pablo Bringas Jr. and the Molecular Imaging Center and Center for Electron Microscopy and Microanalysis of USC for technical assistance.
Footnotes
Funding
This study was supported by grants from the National Institutes of Health National Institute of Dental and Craniofacial Research [DE020065 and DE012711 to Y.C,]; and from the UK Medical Research Council [G0901539 to M.J.D.]. Deposited in PMC for release after 6 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.089615/-/DC1
References
- Andrabi S., Bekheirnia M. R., Robbins-Furman P., Lewis R. A., Prior T. W., Potocki L. (2011). SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am. J. Med. Genet. A. 155, 1165–1169 [DOI] [PubMed] [Google Scholar]
- Black B. L., Olson E. N. (1998). Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 14, 167–196 [DOI] [PubMed] [Google Scholar]
- Cai B. H., Chao C. F., Lu M. H., Lin H. C., Chen J. Y. (2012). A half-site of the p53-binding site on the keratin 14 promoter is specifically activated by p63. J. Biochem. 152, 99–110 [DOI] [PubMed] [Google Scholar]
- Chen R. H., Su Y. H., Chuang R. L., Chang T. Y. (1998). Suppression of transforming growth factor-beta-induced apoptosis through a phosphatidylinositol 3-kinase/Akt-dependent pathway. Oncogene 17, 1959–1968 [DOI] [PubMed] [Google Scholar]
- Cuervo R., Covarrubias L. (2004). Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development 131, 15–24 [DOI] [PubMed] [Google Scholar]
- Denissova N. G., Pouponnot C., Long J., He D., Liu F. (2000). Transforming growth factor beta -inducible independent binding of SMAD to the Smad7 promoter. Proc. Natl. Acad. Sci. USA 97, 6397–6402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon M. J., Marazita M. L., Beaty T. H., Murray J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry W. S., Kern S. E., Pietenpol J. A., Kinzler K. W., Vogelstein B. (1992). Definition of a consensus binding site for p53. Nat. Genet. 1, 45–49 [DOI] [PubMed] [Google Scholar]
- Fakhouri W. D., Rhea L., Du T., Sweezer E., Morrison H., Fitzpatrick D., Yang B., Dunnwald M., Schutte B. C. (2012). MCS9.7 enhancer activity is highly, but not completely, associated with expression of Irf6 and p63. Dev. Dyn. 241, 340–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti E., Li B., Zewdu R., Wells V., Hebert J. M., Karner C., Anderson M. J., Williams T., Dixon J., Dixon M. J., et al. (2011). A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev. Cell 21, 627–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Y., Shimizu T., Kusumoto M., Kyogoku Y., Taniguchi T., Hakoshima T. (1999). Crystal structure of an IRF-DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J. 18, 5028–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato N., Nakamura M., Richardson J. A., Srivastava D., Yanagisawa H. (2012). Tbx1 regulates oral epithelial adhesion and palatal development. Hum. Mol. Genet. 21, 2524–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallione C. J., Richards J. A., Letteboer T. G., Rushlow D., Prigoda N. L., Leedom T. P., Ganguly A., Castells A., Ploos van Amstel J. K., Westermann C. J., et al. (2006). SMAD4 mutations found in unselected HHT patients. J. Med. Genet. 43, 793–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giehl K., Seidel B., Gierschik P., Adler G., Menke A. (2000). TGFbeta1 represses proliferation of pancreatic carcinoma cells which correlates with Smad4-independent inhibition of ERK activation. Oncogene 19, 4531–4541 [DOI] [PubMed] [Google Scholar]
- Han J., Molkentin J. D. (2000). Regulation of MEF2 by p38 MAPK and its implication in cardiomyocyte biology. Trends Cardiovasc. Med. 10, 19–22 [DOI] [PubMed] [Google Scholar]
- He F., Xiong W., Wang Y., Li L., Liu C., Yamagami T., Taketo M. M., Zhou C., Chen Y. (2011). Epithelial Wnt/β-catenin signaling regulates palatal shelf fusion through regulation of Tgfβ3 expression. Dev. Biol. 350, 511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocevar B. A., Brown T. L., Howe P. H. (1999). TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 18, 1345–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P. P., Shen X., Huang D., Liu Y., Counter C., Wang X. F. (1999). The MEK pathway is required for stimulation of p21(WAF1/CIP1) by transforming growth factor-beta. J. Biol. Chem. 274, 35381–35387 [DOI] [PubMed] [Google Scholar]
- Ingraham C. R., Kinoshita A., Kondo S., Yang B., Sajan S., Trout K. J., Malik M. I., Dunnwald M., Goudy S. L., Lovett M., et al. (2006). Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 38, 1335–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y., Yeo J. Y., Chytil A., Han J., Bringas P., Jr, Nakajima A., Shuler C. F., Moses H. L., Chai Y. (2003). Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development 130, 5269–5280 [DOI] [PubMed] [Google Scholar]
- Iwata J., Ezaki J., Komatsu M., Yokota S., Ueno T., Tanida I., Chiba T., Tanaka K., Kominami E. (2006). Excess peroxisomes are degraded by autophagic machinery in mammals. J. Biol. Chem. 281, 4035–4041 [DOI] [PubMed] [Google Scholar]
- Iwata J., Hosokawa R., Sanchez-Lara P. A., Urata M., Slavkin H., Chai Y. (2010). Transforming growth factor-beta regulates basal transcriptional regulatory machinery to control cell proliferation and differentiation in cranial neural crest-derived osteoprogenitor cells. J. Biol. Chem. 285, 4975–4982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J., Parada C., Chai Y. (2011). The mechanism of TGF-β signaling during palate development. Oral Dis. 17, 733–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J., Hacia J. G., Suzuki A., Sanchez-Lara P. A., Urata M., Chai Y. (2012). Modulation of noncanonical TGF-β signaling prevents cleft palate in Tgfbr2 mutant mice. J. Clin. Invest. 122, 873–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J. S., Liu C., Derynck R. (2009). New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol. 19, 385–394 [DOI] [PubMed] [Google Scholar]
- Koillinen H., Lahermo P., Rautio J., Hukki J., Peyrard-Janvid M., Kere J. (2005). A genome-wide scan of non-syndromic cleft palate only (CPO) in Finnish multiplex families. J. Med. Genet. 42, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S., Schutte B. C., Richardson R. J., Bjork B. C., Knight A. S., Watanabe Y., Howard E., de Lima R. L., Daack-Hirsch S., Sander A., et al. (2002). Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 32, 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouwenhoven E. N., van Heeringen S. J., Tena J. J., Oti M., Dutilh B. E., Alonso M. E., de la Calle-Mustienes E., Smeenk L., Rinne T., Parsaulian L., et al. (2010). Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet. 6, e1001065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- Laurikkala J., Mikkola M. L., James M., Tummers M., Mills A. A., Thesleff I. (2006). p63 regulates multiple signalling pathways required for ectodermal organogenesis and differentiation. Development 133, 1553–1563 [DOI] [PubMed] [Google Scholar]
- Letra A., Fakhouri W., Fonseca R. F., Menezes R., Kempa I., Prasad J. L., McHenry T. G., Lidral A. C., Moreno L., Murray J. C., et al. (2012). Interaction between IRF6 and TGFA genes contribute to the risk of nonsyndromic cleft lip/palate. PLoS ONE 7, e45441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little H. J., Rorick N. K., Su L. I., Baldock C., Malhotra S., Jowitt T., Gakhar L., Subramanian R., Schutte B. C., Dixon M. J., et al. (2009). Missense mutations that cause Van der Woude syndrome and popliteal pterygium syndrome affect the DNA-binding and transcriptional activation functions of IRF6. Hum. Mol. Genet. 18, 535–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. (2012). TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti F., Marinari B., Lo Iacono N., Botti E., Giunta A., Spallone G., Garaffo G., Vernersson-Lindahl E., Merlo G., Mills A. A., et al. (2010). A regulatory feedback loop involving p63 and IRF6 links the pathogenesis of 2 genetically different human ectodermal dysplasias. J. Clin. Invest. 120, 1570–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossey P. A., Little J., Munger R. G., Dixon M. J., Shaw W. C. (2009). Cleft lip and palate. Lancet 374, 1773–1785 [DOI] [PubMed] [Google Scholar]
- Pauws E., Hoshino A., Bentley L., Prajapati S., Keller C., Hammond P., Martinez-Barbera J. P., Moore G. E., Stanier P. (2009). Tbx22null mice have a submucous cleft palate due to reduced palatal bone formation and also display ankyloglossia and choanal atresia phenotypes. Hum. Mol. Genet. 18, 4171–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C. A., Ott J., Mays D. J., Pietenpol J. A. (2007). p63 consensus DNA-binding site: identification, analysis and application into a p63MH algorithm. Oncogene 26, 7363–7370 [DOI] [PubMed] [Google Scholar]
- Richardson R. J., Dixon J., Malhotra S., Hardman M. J., Knowles L., Boot-Handford R. P., Shore P., Whitmarsh A., Dixon M. J. (2006). Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat. Genet. 38, 1329–1334 [DOI] [PubMed] [Google Scholar]
- Richardson R. J., Dixon J., Jiang R., Dixon M. J. (2009). Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum. Mol. Genet. 18, 2632–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S., Hill C. S. (2008). How the Smads regulate transcription. Int. J. Biochem. Cell Biol. 40, 383–408 [DOI] [PubMed] [Google Scholar]
- Schmierer B., Hill C. S. (2007). TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982 [DOI] [PubMed] [Google Scholar]
- Shi Y., Massagué J. (2003). Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- Sorrentino A., Thakur N., Grimsby S., Marcusson A., von Bulow V., Schuster N., Zhang S., Heldin C. H., Landström M. (2008). The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 10, 1199–1207 [DOI] [PubMed] [Google Scholar]
- Srichomthong C., Siriwan P., Shotelersuk V. (2005). Significant association between IRF6 820G->A and non-syndromic cleft lip with or without cleft palate in the Thai population. J. Med. Genet. 42, e46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason H. A., Zhou H., Kouwenhoven E. N., Dotto G. P., Restivo G., Nguyen B. C., Little H., Dixon M. J., van Bokhoven H., Dixon J. (2010). Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J. Clin. Invest. 120, 1561–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro R., Saadi I., Kuburas A., Nemer M., Russo A. F. (2004). Cell-specific activation of the atrial natriuretic factor promoter by PITX2 and MEF2A. J. Biol. Chem. 279, 52087–52094 [DOI] [PubMed] [Google Scholar]
- Welsh I. C., O’Brien T. P. (2009). Signaling integration in the rugae growth zone directs sequential SHH signaling center formation during the rostral outgrowth of the palate. Dev. Biol. 336, 53–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall M. D., Mays D. J., Sniezek J. C., Pietenpol J. A. (2003). The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol. Cell. Biol. 23, 2264–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F. K., Hagg U. (2004). An update on the aetiology of orofacial clefts. Hong Kong Med. J. 10, 331–336 [PubMed] [Google Scholar]
- Xu X., Han J., Ito Y., Bringas P., Jr, Urata M. M., Chai Y. (2006). Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev. Biol. 297, 238–248 [DOI] [PubMed] [Google Scholar]
- Xu X., Han J., Ito Y., Bringas P., Jr, Deng C., Chai Y. (2008). Ectodermal Smad4 and p38 MAPK are functionally redundant in mediating TGF-beta/BMP signaling during tooth and palate development. Dev. Cell 15, 322–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M., Fatyol K., Jin C., Wang X., Liu Z., Zhang Y. E. (2008). TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 31, 918–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. T., Li W. Y., Kaartinen V. (2008). Tissue-specific expression of Cre recombinase from the Tgfb3 locus. Genesis 46, 112–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L., Dai J. L., Buckhaults P., Zhou S., Kinzler K. W., Vogelstein B., Kern S. E. (1998). Human Smad3 and Smad4 are sequence-specific transcription activators. Mol. Cell 1, 611–617 [DOI] [PubMed] [Google Scholar]
- Zhang Y. E. (2009). Non-Smad pathways in TGF-beta signaling. Cell Res. 19, 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.