

Abstract
Neurodegenerative disorders show an increasing prevalence in a number of highly developed countries. Often, these diseases require life-long treatment mostly with drugs which are costly and mostly accompanied by more or less serious side-effects. Their heterogeneous manifestation, severity and outcome pose the need for individualised treatment options. There is an intensive search for new strategies not only for treating but also for preventing these diseases. Green tea and green tea extracts seem to be such a promising and safe alternative. However, data regarding the beneficial effects and possible underlying mechanism, specifically in clinical trials, are rare and rather controversial or non-conclusive. This review outlines the existing evidence from preclinical studies (cell and tissue cultures and animal models) and clinical trials regarding preventive and therapeutic effects of epigallcatechin-3-gallate in neurodegenerative diseases and considers antioxidative vs. pro-oxidative properties of the tea catechin important for dosage recommendations.
Keywords: Neurological diseases, Predictive and personalised medicine, Targeted prevention, Green tea, Epigallocatechin-3-gallate, Tailored therapy
Review
Introduction
Neurodegenerative disorders show an increasing prevalence in a number of highly developed countries partly attributable to the still increasing life expectancy in these countries but also other still not clearly identified and characterised causal factors. Depending on the disease, sensory, motor, cognitive and autonomic functions might be affected differently. Once diagnosed, neurodegenerative diseases often require life-long treatment, mostly with drugs which are costly and mostly accompanied by more or less serious side-effects. Governments and healthcare providers have to face the emerging health burden of neurodegenerative diseases. Therefore, there is an intensive search for new strategies not only for treating but also for preventing these diseases. At the time of diagnosis, when neurodegeneration has already become evident in terms of symptoms, neurodegenerative cascades are already initiated and neuro-axonal degeneration cannot be reversed with neuroprotective treatments [1]. Green tea and green tea extracts (GTEs) seem to be a promising and safe alternative for targeted prevention and individualised treatment of neurological diseases. However, data regarding beneficial effects and possible underlying mechanism, specifically in clinical trials, are rare and rather controversial or non-conclusive.
The pathogenesis of these diseases includes a number of different processes such as oxidative stress, inflammation, or simply neuronal dysfunction due to neuronal degeneration.
Neuronal tissue is more prone to oxidative damage than other tissues because of (1) its high content of unsaturated fatty acids that are sensitive targets for free radical attack leading to peroxidation, (2) the brain’s high oxygen consumption (20% of total) despite its relatively small (2% of human body) weight, (3) its low antioxidant defence mechanisms, (4) higher iron levels in certain brain regions, and (5) high ascorbate level [2]. Although ascorbate serves an important role as scavenger of free radicals in the human body [3], it can generate free radicals in the presence of Fe3+ and Cu2+[4].
Tea is the most widely consumed beverage after water. Green tea preparation precludes the oxidation of leaf polyphenols which are thought to contribute to the health-promoting effects. Tea polyphenols, known as catechins, usually account for 30% to 42% of the dry weight of the solids in brewed green tea. The four major catechins (flavan-3-ols) are (−)–epigallocatechin-3-gallate (EGCG), (−)-epigallocatechin (EGC), (−)-epicatechin-3-gallate (ECG), and (−)epicatechin (EC). EGCG represents the most abundant one of tea catechins (50% to 80% of total catechins) [5-7]. These four catechins act as potent antioxidants via direct scavenging of reactive oxygen and nitrogen species (ROS and RNS), induction of defence enzymes and binding and chelating of divalent metals, such as copper and iron [8].
This review provides information for both basic scientists and clinician scientists in order to encourage translational approaches that address the issues discussed. Undertaking efforts to facilitate translational research with respect to predictive, preventive and personalised medicine in neurodegenerative diseases was recommended recently [1]. The review should meet both: (1) outlining the existing evidence from preclinical studies (cell and tissue cultures and animal models) and clinical trials regarding preventive and therapeutic effects of EGCG in neurodegenerative diseases (Table 1, Figure 1) and (2) considering antioxidative vs. pro-oxidative properties of EGCG important for dosage recommendations. The most important neurodegenerative disorders will be covered.
Table 1.
Effects of epigallocatechin-3-gallate
| Neurological disease | Preclinical studies | Epidemiological and clinical studies |
|---|---|---|
| Multiple sclerosis |
Inflammation, proliferation and TNF-α secretion of T cells ↓ in EAE mice [9] |
Effects on T2 lesions in brain MRI in RRMS? (NCT00525668) |
| Combination with glatiramer acetate: cell death ↓ and neuronal outgrowth ↑ in primary neurons, disease severity ↓ in EAE mice [10] |
Effects on brain atrophy in progressive forms? (NCT00799890) |
|
| Th1 and Th17 ↓, Treg ↑ in EAE mice [11] |
Metabolic effects in MS patients? (NCT01417312) |
|
| Alzheimer’s disease |
Aβ-induced death of hippocampal cells ↓ [12] |
Effects on the course of AD in 50 early stage patients? (NCT00951834) |
| α-secretase activity in Alzheimer transgenic mice ↑ [13] | ||
| Recovery of Aβ-induced memory dysfunction in mice [14] | ||
| Protection of microglia cells from Aβ-induced ↑ iNOS expression and NO production [15] | ||
| Direct conversion of fibrillar species into benign protein aggregates [16] | ||
| Parkinson’s disease |
Lipopolysaccharide-induced microglial activation ↓ [17] |
Inverse relation of tea drinking and PD in Chinese [18], Western Washington [19], Finish [20], and Israelian [21] cohorts |
| Protection of PC12 cells against 6-hydroxydopamine-induced apoptosis [22] | ||
| Attenuation of MPP+-induced ROS production in PC12 cells [23] |
Safe and efficient in de novo PD patients? (NCT00461942) |
|
| Protection against striatal dopamine depletion and neuronal loss in MPTP mice [24] | ||
| Loss of dopaminergic neurons ↓, nNOS expression ↓ in MPTP mice [25] | ||
| No behavioural improvements in 6-hydroxydopamine-lesioned rats [26] | ||
| Attenuation of increased iNOS expression in MPTP mice [27] | ||
| Inhibition of levodopa methylation by catechol-O-methyltransferase [28] | ||
| Huntington’s disease |
Inhibition of huntingtin protein aggregation in yeast and fly models of HD [29] |
Efficient (changes in cognitive decline) and tolerable (1,200 mg/day for 12 months) in HD patients? (NCT01357681) |
| Memory impairment ↓ in 3-NP-treated rats, glutathione level of neuronal cells ↑ [30] | ||
| Duchenne muscular dystrophy |
Elongator digitorum longus muscle necrosis ↓ in mdx mice [31] |
Safe and tolerable in DMD boys? (NCT01183767) |
| Hind limb muscle necrosis ↓, muscle force and fatigue resistance ↑ in mdx mice [32] |
|
|
| Creatine kinase and oxidative stress ↓, improved histology, utrophin ↑ in mdx mice [33] | ||
| Glutathione synthesis ↑ in muscle cells cultured from mdx mice [34] | ||
| Muscle aerobic metabolism ↑ in combination with endurance training in mdx mice [35] | ||
| Muscle pathology in regenerating fibres ↓ in mdx mice [36] | ||
| Amyotrophic lateral sclerosis |
Delayed symptom onset, prolonged life span, attenuated death signals in ALS mice [37] |
|
| Protection of motor neurons, microglial activation ↓ in ALS mice [38] | ||
| Protection of motor neurons against THA-induced toxicity in rat spinal cord explants [39] | ||
| Cerebral ischaemia | Ischaemia-reperfusion brain injury↓ in gerbils [40-42], rats [43-51] and mice [52,53] | ≥3 Cups of tea/day decrease the risk of stroke [54] |
↓ decreased, ↑ increased.
Figure 1.
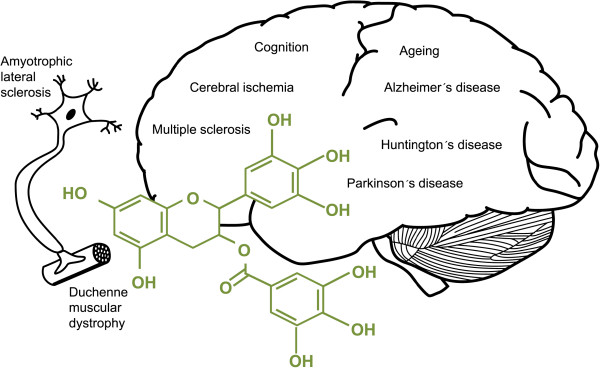
Neurological conditions positively affected by epigallocatechin-3-gallate.
EGCG: bioavailability and possible therapeutic targets
There is only limited data on the bioavailability of EGCG in the brain, an important prerequisite for its neuroprotective effects. A single, very high oral EGCG dose (500 mg/kg body weight) to rats yielded EGCG concentrations of 12.3 nmol/mL in plasma and 0.5 nmol/g in brain (measured by CL-HPLC) [55]. A single administration of 3H] EGCG into the stomach of mice leads to significant amounts of radioactivity in the brain - thought to consist of EGCG itself and its metabolites in free and protein-bound forms. A second administration 6 h later increased brain’s radioactivity above the level measured after the first dose. This finding suggests that EGCG obviously accumulates in the brain when given repeatedly [56]. However, after drinking 250 ml green tea by six human subjects, HPLC-MS analysis revealed that flavan-3-ol methyl-glucuronide and sulfate metabolites appeared in the bloodstream but did not pass through the blood-cerebrospinal fluid barrier [57]. The discrepancy between animal and human data might originate from (1) the very high dose of EGCG applied in the animal model versus the single, rather small dose contained in one cup of tea, (2) the time point (2 h after drinking) chosen for the lumbar puncture in the clinical study, and (3) an apparently more rapid catechin metabolism in humans. Catechins and epicatechin pass the blood brain barrier as shown by in vivo microdialysis of rat hippocampus following intravenous application of these basic monomer units of the flavanols [58].
Once in the brain, EGCG can influence numerous processes. Possible underlying mechanisms derived from in vitro and in vivo studies have been reviewed by Mandel et al. [59]. Briefly, EGCG acts as a powerful hydrogen-donating radical scavenger of ROS and RNS and chelates divalent transition metal ions (Cu2+, Zn2+ and Fe2+), thereby preventing the Fe2+-induced formation of free radicals in vitro. Among 12 polyphenolic compounds, EGCG most potently inhibited Fe2+-mediated DNA damage and iron ascorbate-promoted lipid peroxidation of brain mitochondrial membranes. In vivo, EGCG increased expression and activity of antioxidant enzymes, such as glutathione peroxidase, glutathione reductase, superoxide dismutase (SOD) and catalase but inhibits pro-oxidative ones, such as monoamine oxidase (MAO)-B and nitric oxide synthase (NOS) [59].
ROS and RNS are well recognised for being both deleterious and beneficial. Overproduction of ROS results in oxidative stress that damages membrane lipids, proteins and DNA. In contrast, beneficial effects occurring at low/moderate concentrations of ROS/RNS involve anti-infective processes, various cellular signalling pathways and induction of a mitogenic response [60]. ROS are by-products of cellular, mainly mitochondrial, oxidative metabolism. They are formed by enzymes that reduce oxygen, such as MAO, xanthine oxidase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and NOS. The ‘primary’ oxygen-derived ROS is the superoxide anion radical (O2·−) capable of further reactions with other molecules to generate ‘secondary’ ROS, such as hydrogen peroxide (H2O2), hydroxyl radical, and singlet oxygen [61,62] or RNS by reacting with nitric oxide (NO·) to peroxynitrite anion (ONOO−). Within the brain, NO is produced by cell type-specific NO synxthases, i.e. neuronal (nNOS), endothelial (eNOS) and inducible (iNOS) - the latter being specific for glial cells. NO is essential for controlling cerebral blood flow and neurotransmission and is involved in synaptic plasticity, modulation of neuroendocrine functions, memory formation and behavioural activity. However, NO does not only exert protective effects in the central nervous system (CNS) but also mediates tissue damage [63] because of its reaction with O2·−-yielding OONO−[64], an ion that can nitrosylate tyrosine or cysteine residues in proteins [62]. The hydroxyl and trihydroxyl (gallate) groups of EGCG appear important for scavenging physiologically relevant ROS/RNS (Figure 2) [65].
Figure 2.
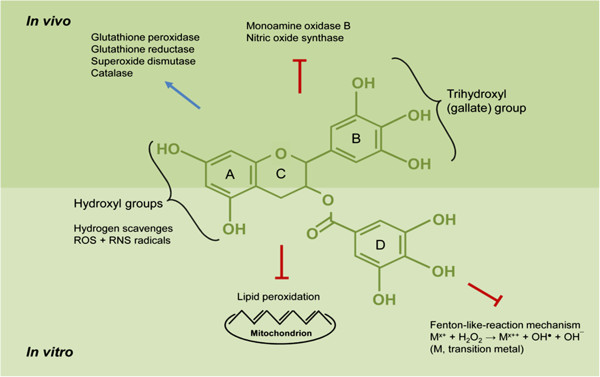
Proposed neuroprotective mechanisms of epigallocatechin-3-gallate.
Ferric ion (Fe2+) is an essential cofactor for many proteins involved in neuronal function. However, increasing evidence suggests that Fe2+ accumulation in the brain is pathologically relevant in CNS disorders. During ageing, total Fe2+ concentration increases in some brain regions that are involved in the pathogenesis of degenerative diseases, such as Alzheimer’s, Parkinson’s and Huntington’s disease. This Fe2+ accumulation obviously fosters the production of ROS [66], namely the highly reactive OH·, which attacks a large number of functional groups of the biomolecules in neurons. By chelating redox-active transition metal ions, the gallate groups of EGCG are thought to inhibit the Fenton-like-reaction mechanism [62]. Thus, the formation of highly reactive hydroxyl radicals (OH·) is inhibited. Consequently, polyunsaturated fatty acids in, for example, mitochondrial membranes are protected from lipid peroxidation (Figure 2).
To maintain normal cell physiology, enzymatic antioxidant mechanisms counteract ROS formation. Key players within these mechanisms are SOD, catalase and glutathione peroxidase, as well as enzymes involved in the recycling of oxidised glutathione, such as glutathione reductase and glucose-6-phosphate dehydrogenase which is a key enzyme within the pentose phosphate pathway for generating NADPH [67]. In aged rat and adult mouse brain, EGCG augmented the activities of these antioxidative enzymes (Figure 2) [24,68]. EGCG also inhibited the activity of pro-oxidative enzymes such as MAO-B and NOS. MAO–B catalyses the oxidative deamination of many amine neurotransmitters in the brain thereby producing H2O2. Lin et al. first reported that EGCG supplementation decreases brain MAO-B activity in adult rats [69]. Furthermore, EGCG inhibited iNOS in lipopolysaccharide-treated microglia cells [17] and in a mouse model of Parkinson’s disease [27] and suppressed nNOS in the striatum of mice [25].
There are two major subgroups of glial cells in the brain: the macroglia (astrocytes, oligodendrocytes and ependymal cells) and the microglia which are resident macrophages of the brain [70]. Chronic neurodegenerative disorders are characterised by activation of microglia in the affected neural pathways. Activated microglia is a source of oxidants, prostanoids and inflammatory cytokines that promote neuronal damage which in turn sustains microglial activation [71]. Thus, agents that down-regulate microglial activation could have favourable effects on the course of diseases accompanied by neurodegeneration and inflammation. There is evidence that EGCG counteracts microglial activation in Parkinson’s [17,72,73] and Alzheimer’s diseases [13] and amyotrophic lateral sclerosis [38].
Therapeutic efficacy of EGCG in neurological disorders and ageing
Multiple sclerosis
An important mechanism that underlies the major health benefits of consuming EGCG, such as its anticancer and anti-inflammatory properties, is its suppressive effect on the growth of different cell types. T cells are highly active cells and an effective T cell-mediated immune response depends on rapid T cell expansion. Physiological levels (0.5 to 10 μM) of EGCG inhibited the proliferation of primary T cells from mice. This effect was mediated by inhibition of cell cycle progression and cell division [74]. Thus, EGCG may exert an effect on autoimmune and inflammatory diseases that involve excessive T cell activation, such as multiple sclerosis (MS). MS is an autoimmune inflammatory disorder of the CNS in which focal lymphocytic infiltration not only leads to demyelination but also axonal and neuronal damage in both the grey and the white matter [75,76], resulting in accrual of neurological disability during the course of the disease and measurable atrophy of the brain, the spinal cord and the retina [77-87]. Women who are more frequently affected than men mainly experience disease onset during their childbearing age [88,89]. The cause of the disease is unknown; however, both genetic susceptibility and environmental factors, such as vitamin D deficiency, ultraviolet B radiation exposure, smoking and Epstein-Barr virus infection, have been shown to play a role in disease aetiopathogenesis [90-103]. Neurological disability can occur in any functional system of the CNS and manifests with impaired vision, muscle weakness, ataxia, spasticity, gait abnormalities, sensory disturbances and bowel and bladder dysfunction. A substantial number of patients also suffer from fatigue, cognitive impairment and sleep disorders, all of which may have a considerable negative impact on the quality of life and vocational status [104-108]. There is no cure for MS; injectable disease-modifying drugs such as beta-interferons and glatiramer acetate have a moderate impact on relapse rate and measures of disease activity on brain magnetic resonance imaging (MRI) with a favourable safety profile [109,110]. Newer drugs such as natalizumab and fingolimod are more effective, however, at a price of more severe side effects such as progressive multifocal leukoencephalopathy, macular oedema [111-116] or cardiac side effects [117-119]. Also mitoxantrone, an immunosuppressive compound given as escalation therapy in relapsing-remitting or progressive MS, exhibits potentially life-threatening side effects such as leukaemia or cardiotoxicity that limit its application in MS patients [120-125]. Moreover, none of the aforementioned drugs has proven neuroprotective properties. In an animal model of human MS with experimental autoimmune encephalomyelitis (EAE) [126-129], oral application of EGCG prevented and reversed the disability and inhibited myelin-specific inflammatory responses. This effective protection against relapsing CNS autoimmune disease resulted in a favourable long-term clinical outcome. Furthermore, EGCG reduced axonal damage and neuronal cell death by directly targeting ROS formation. Decreased proliferation of human CD4+ T cells incubated with EGCG could be linked to its interference with cell cycle, NF-κB activation and protein degradation pathway (proteasome) [9]. Combined application of EGCG and glatiramer acetate [130-133] synergistically reduced cell death and promoted axonal outgrowth of primary neurons. These effects could be translated into the EAE model in which diminished clinical disease severity was associated with reduced CNS inflammation. Since no unexpected adverse events occurred when the two substances were applied together [10], their use seems promising in clinical trials with MS patients. These findings were recently confirmed and expanded by showing that EGCG dose-dependently ameliorated clinical symptoms and delayed disease onset in mice with EAE, which was well associated with reduced inflammatory infiltration and demyelination in the CNS. The mechanism underlying these effects was altered regulation of CD4+ T cell subsets (Th1/Th17 downregulation, Treg up-regulation) [11].
Meanwhile, clinical trials investigate the effects of EGCG on (1) the number of T2 lesions in brain MRI in patients with relapsing-remitting MS (ClinicalTrials.gov identifier: NCT00525668) and (2) brain atrophy in progressive forms of MS (ClinicalTrials.gov identifier: NCT00799890).
Muscle weakness and excessive muscle fatigue [134-136], commonly experienced by MS patients, lead to a reduced physical activity level in the course of the disease. Sufficient energy production and/or supply are mandatory for efficient muscle performance. Owing to the fact that EGCG increases postprandial lipid oxidation in overweight/obese male volunteers [137] and the importance of lipid oxidation to fuel muscle’s energy metabolism, metabolic effects of EGCG in MS patients are currently being investigated (ClinicalTrials.gov identifier: NCT01417312).
Neuromyelitis optica
For a long time, neuromyelitis optica (NMO, Devic’s syndrome) has been considered as a rare variant of MS. However, with the detection of a highly specific serum biomarker, an autoantibody targeting the most abundant astrocytic water channel aquaporin-4 (AQP4), in patients with NMO but not MS or other inflammatory or neurological diseases, it has become clear that NMO is a disease entity distinct from MS despite a considerable overlap in clinical presentation and paraclinical findings [138-148]. Although commercial availability of tests for AQP4 antibodies has facilitated differential diagnosis, a considerable number of NMO patients are still misdiagnosed with MS despite emerging evidence for differences in cerebrospinal fluid, MRI and OCT findings [149-156]. A timely and correct diagnosis, however, is of utmost relevance, as some of the classic immunomodulatory drugs given in MS such as beta-interferons, natalizumab and presumably also fingolimod, are ineffective or even harmful in NMO [157-165]. NMO is usually treated with azathioprine, rituximab, mycophenolate mofetil or mitoxantrone [166-171]. Very recently, the role of T cells in the autoimmune disease process in NMO has come into focus of research [172-174]. In addition, neutrophils are also apparently a major player in the pathogenesis of tissue damage [175,176]. Thus, from a pathophysiological standpoint, EGCG with its inhibitory effect on proliferation of T cells and neutrophils [177,178] could theoretically serve as therapeutic agent in NMO, preferably in combination with established immunosuppressive drugs. However, clinical trials are lacking.
Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disease characterised by progressive impairment of cognitive function and loss of memory in association with widespread neuronal death [179]. Neuronal loss in AD is accompanied by the deposition of amyloid beta protein (Aβ) in senile plaques. ROS-induced lipid peroxidation has been suggested to play an important role in Aβ-mediated neurotoxicity. In cultured hippocampal neuronal cells, EGCG prevented Aβ-induced cell death and lipid peroxidation through its antioxidant property. The fundamental process of Aβ peptide formation is the proteolysis of amyloid precursor protein (APP). APP proteolytic products arise from the coordinated action of α-, β-, and γ-secretases. In the amyloidogenic pathway, Aβ peptides are produced by the initial action of α-secretase. Conversely, the non-amyloidogenic pathway involves events indicative of α-secretase activity. Because intracellular APP is limited, these pathways are believed to compete for their substrate. In cell culture, EGCG promotes the generation of the non-amyloidogenic soluble form of APP via a protein kinase C-dependent activation of α-secretase [12,179,180] and in Alzheimer transgenic mice; it reduced cerebral amyloidosis through promotion of α–secretase activity [13]. Furthermore, it was found that EGCG markedly reduced secreted Aβ levels in the conditioned medium of Chinese hamster ovarian cells, overexpressing the ‘Swedish’ mutated APP (CHO/ΔNL) [181] and in primary neuronal cells derived from transgenic mice bearing the APP Swedish mutation [13]. Consequently, compounds that alter proteolytic cleavage of APP (by enhancing α-, or inhibiting β- and γ-secretase activities) thereby reducing Aβ peptides are interesting therapeutic options for AD treatment [182]. EGCG administration via drinking water recovered Aβ-induced memory dysfunction in mice. The underlying Aβ reduction was facilitated through increased α- and decreased β- and γ–secretase activities (via ERK/NF-κB pathway inhibition) respectively [14].
Oxidative stress has been suggested to play a causative role in the pathogenesis and progression of AD [183]. Aβ accelerates neurodegeneration by activating microglia, the resident macrophages in the CNS, which in turn exert a cytotoxic effect on neurons by releasing ROS and RNS. EGCG protected BV2 microglia cells from Aβ-induced cytotoxicity and apoptotic cell death by suppressing iNOS expression and subsequent NO production, inhibiting intracellular peroxynitrite accumulation and augmenting intracellular glutathione levels [15].
However, EGCG does not only influence the amyloid formation process via modulation of cellular signal transduction pathways and ROS production but also directly converts fibrillar species into benign protein aggregates [16].
To date, these experimental data have not been confirmed in patients with AD. However, investigations of drug and non-drug interventions are needed. They should be specifically designed to meet the needs of people in the early stages of dementia [1]. An ongoing clinical trial is investigating if EGCG positively affects the course of AD as assessed by Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) in early state AD patients co-medicated with acetylcholine esterase inhibitors (ClinicalTrials.gov identifier: NCT00951834).
Parkinson’s disease
Parkinson’s disease (PD) is an age-related disorder characterised by progressive degeneration of dopaminergic neurons in the substantia nigra leading to resting tremor, rigidity, bradykinesia or slowness, gait disturbance and postural instability [184].
Microglial activation is believed to play a pivotal role in the selective neuronal injury associated with PD [72,73,185-188]. In an in vitro study using dopaminergic mesencephalic cells from rat brain, EGCG potently inhibited NO production and tumour necrosis factor-α (TNF-α) release from lipopolysaccharide-activated microglia. Since activated microglia is the cardinal donor of free radicals and inflammatory factors in the brain, this in vitro inhibition could be a reasonable explanation for EGCG-mediated neuroprotection in vivo[17].
Pheochromocytoma (PC12) cells from rat adrenal medulla, characterised by catecholamine synthesis, metabolism and transport [189], are used as a cellular model of PD. A GTE [190] or EGCG (at an extreme high concentration of 200 μM) [22] exerted protective effects against 6-hydroxydopamine (OHDA)-induced PC12 cell apoptosis and death. A recent study also used PC12 cells to investigate the effects of EGCG on 1-methyl-4-phenyl-pyridine (MPP+)-induced cell injury. The authors found that EGCG improved cell viability and attenuated MPP+-induced intracellular ROS formation via the SIRT1/PGC-1α signalling pathway [23].
Neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) produces an irreversible and severe Parkinsonian syndrome that replicates almost all features of PD [191]. Therefore, MPTP is used in animal models to specifically induce dopaminergic neurodegeneration via oxidative stress. In 2001, it was first reported that GTE and EGCG prevented MPTP-induced loss of striatal tyrosine hydroxylase (TH) positive neurons, increased dopamine (DA) turnover and TH activity in mice. As a result of decreased oxidative stress, increased activities of antioxidative enzymes (SOD, catalase) were attenuated by EGCG. In mouse brain homogenates, the pro-oxidative activity of MAO-B was inhibited [24]. NO is thought to play an important role in mediating MPTP-induced oxidative stress. Therefore, another study investigated whether NOS underlies the protection of tea and EGCG against MPTP-induced dopaminergic neurodegeneration in mice. The authors attributed the prevention of dopaminergic neuron loss, DA and DA metabolite depletion and the preservation of striatal TH activity to the inhibition of nNOS expression [25]. Contrary to the MPTP mouse model of PD, in which 2 mg/kg EGCG afforded neuroprotection [24], oral pretreatment with either 1 or 2 mg/kg EGCG did not attenuate dopaminergic neuron loss in the 6-OHDA rat model of PD. In addition, there were no consistent improvements of behavioural measures due to EGCG treatment. This lack of neuroprotection was assumed to be due to different dose and/or length of EGCG administration in the studies. Furthermore, the mechanisms by which MPTP and 6-OHDA exert their neurotoxic effects are different. Unlike MPTP, 6-OHDA did not require glial cells or the dopamine transporter for its activation but spontaneously auto-oxidised to produce damaging ROS [26]. Apart from amounts used and duration of administration, these conflicting results could be due to the different species used. The pharmacokinetic parameters of EGCG in the mouse are more similar to humans in terms of EGCG plasma availability and biotransformation than those of the rat [192], suggesting that findings from mice are more relevant for human studies. Also, the poor bioavailability of oral EGCG in rats [193] could be a reason why similar doses led to different results in the animal models of PD. Higher but non-toxic doses (10 and 50 mg/kg) of EGCG reduced neuronal death and iNOS expression in MPTP model mice giving further evidence for its neuroprotection via NO reduction [27]. Another proposed mechanism, by which green tea polyphenols could protect dopaminergic neurons against MPTP-induced injury, is by their inhibitory effect on the DA-transporter which blocks the uptake of MPTPs active metabolite (MPP+) [194].
Levodopa, a precursor in the biosynthesis of dopamine, is considered to be the most effective drug to alleviate motor symptoms in PD patients. Computational molecular modelling showed that EGCG is a potent, non-competitive inhibitor but a poor substrate of catechol-O-methyltransferase (COMT) [195]. In vitro, EGCG inhibited human liver COMT and, therefore, the metabolic conversion of levodopa to 3-O-methyldopa. Rats receiving oral EGCG displayed lower 3-O-methyldopa level in plasma and striatum [28].
Owing to the fact that PD is one of the most common neurological diseases, there are a number of epidemiological studies on the influence of environmental factors, such as tea drinking. In 1998, a case-control study with Chinese PD patients and controls showed that regular tea drinking protects against PD [18]. This was confirmed in PD cases and controls from Washington State in whom reduced risk for PD was related with tea consumption (two cups/day) [19]. A large prospective study in the Finish population, which usually drinks little tea, showed a reduced risk of incident PD in subjects who habitually drank three or more cups of tea per day [20]. However, no distinction between green and black tea was made in the questionnaire making it difficult to associate this finding with EGCG. Of note, Western populations are more likely to consume black tea which is fermented and therefore contains less catechins than green tea. The potential relevance was investigated in the Singapore Chinese Health Study. In this prospective cohort study, black tea drinking was inversely associated with PD risk while green tea drinking was unrelated [196]. A retrospective study associated drinking of more than three cups of tea per day with a delayed onset of motor symptoms in Israeli PD patients [21]. In this study, no distinction was made between black and green tea.
Whether these encouraging results from cellular and animal models apply to humans has not been sufficiently investigated in clinical trials yet. However, the application of early interventions at premotor phases of PD is desirable for patients and health care providers. Authors of a Chinese study sought to determine whether green tea polyphenols are effective and safe in the treatment of de novo PD patients (who took no antiparkinsonism drugs). Outcome measures were progression of motor dysfunction and cognition, mood and quality of daily life (ClinicalTrials.gov identifier: NCT00461942).
Huntington’s disease
Huntington’s disease (HD) is a dominantly inherited neurodegenerative disorder that is caused by an unstable expansion of a CAG repeat within the coding region of the gene that encodes for the protein huntingtin (htt). The mutation results in an elongated stretch of glutamine near the NH2 terminus of the protein. HD symptoms mainly occur as adult-onset HD between age 35 and 50 and comprise personality changes, generalised motor dysfunctions, cognitive decline and neuroendocrine disturbances [197,198].
Growing evidence suggests that the misfolding and aggregation of htt is central to HD pathogenesis. The screening of a library of natural compounds identified EGCG and related polyphenols as potent inhibitors of mutant htt exon 1 protein aggregation in vitro. Additionally, EGCG modulated misfolding as well as the assembly of oligomers in cell-free assays and reduced both toxicity and aggregate formation in yeast and fly models of HD [29].
3-Nitropropionic acid (3-NP), a fungal-derived neurotoxin that irreversibly inhibits succinate dehydrogenase, is a well-known model to study the pathogenesis of HD. Memory disturbances in rats caused by 3-NP treatment were attenuated by chronic EGCG treatment. Striatal, cortical and hippocampal glutathione levels of the rats were significantly reduced by 3-NP treatment that was reversed by chronic EGCG treatment. These effects were attributed to NOS inhibition as evidenced by the application of NO modulators (l-arginine and l-NG-nitroarginine methyl ester) [30].
A multicenter trial evaluating the efficiency and tolerability of EGCG in HD patients is currently ongoing. Primary outcome measures are changes of cognitive functions (as measured by UHDRS-Cognition composite score of Stroop test, verbal fluency and Symbol Digit Modalities Test) after 12 months maximal daily intake of 1,200 mg EGCG compared to baseline intake (ClinicalTrials.gov identifier: NCT01357681).
Duchenne muscular dystrophy
Another disease, in which the generation of ROS seems to play an important pathogenic role, is Duchenne muscular dystrophy (DMD) [199-201]. DMD is a severe X-linked congenital disorder characterised by progressive muscle wasting caused by the absence of the structural protein dystrophin. The pathogenesis of DMD is frequently studied in the dystrophic mdx mouse model. Dietary GTE supplementation preferentially protected the elongator digitorum longus muscle rather than the soleus muscle of mdx mice from necrosis [31]. Histological examination of leg muscles and functional recordings of the triceps surae muscle contraction showed that GTE and EGCG protected the hindlimb muscle of dystrophic mice from massive necrosis and greatly improved muscle force and resistance to fatigue [32]. Subcutaneous EGCG injections into the backs of mdx mice starting immediately after birth (1) reduced the activity of serum creatine kinase to almost normal levels, (2) decreased oxidative stress indicated by the number of lipofuscine granules in muscle fibres, (3) improved the histology of both the fast-contracting diaphragm muscle and the slow-contracting soleus, (4) increased the mean time for the maximum tetanic force to fall by a half (T1/2max) in mdx soleus muscles, and (5) increased the amount of utrophin (partially compensating for the lack of dystrophin) formed in mdx diaphragm muscle [33]. A study on cultured muscle cells from mdx dystrophic mice showed that green tea polyphenols and EGCG offer both immediate antioxidant effects and long-term prevention by stimulating glutathione synthesis and protect against H2O2 toxicity [34]. The above mentioned findings were recently extended to study different administration routes and dosages of EGCG to find the most effective for limiting the onset of dystrophic lesions in both the same strain of mdx mice and assessment methods. Oral administration of 180 mg/kg EGCG daily in the diet for 5 weeks most effectively reduced muscular dystrophy [202].
Endurance training (i.e. voluntary wheel running) and 0.5% GTE in the diet synergistically improved skeletal and cardiac muscle aerobic metabolism and serum antioxidant capacity and decreased lipid peroxidation and cardiac hypertrophy in young mdx mice [35]. Prenatal and early dietary intervention with GTE decreased dystrophic muscle pathology potentially by suppressing NF-κB activity in regenerating fibres of mdx mice [36].
In light of this considerable body of evidence, clinical studies in DMD patients should be performed. One registered multicenter, prospective, double blind, placebo-controlled randomised pilot study is investigating safety and tolerance of EGCG in boys with muscular dystrophy of the Duchenne type (ClinicalTrials.gov identifier: NCT01183767).
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder in which degeneration of upper and lower motor neurons leads to progressive muscle weakness and atrophy [203,204]. In recent years, non-motor involvement has been increasingly reported in ALS [205-212]; a clinical, pathological and genetic continuum with frontotemporal dementia has been acknowledged [213,214]. Moreover, it became evident that inflammatory mechanisms, including microglial activation, contribute not only to neuronal cell death but may also promote neuronal survival [210,215-218]. Drugs targeting inflammatory pathways showed beneficial effects on survival in transgenic ALS mouse models [219]. The first in vivo study on EGCG’s effects on ALS was performed on ALS model (SOD1-G93A) mice. Oral EGCG treatment (>2.9 μg/g body weight) significantly delayed symptom onset, prolonged life span, preserved survival signals and attenuated death signals [37]. The disease onset delaying and survival prolonging effects of oral EGCG treatment in ALS mice were confirmed by another study. EGCG protected the motor neurons and reduced microglia activation and associated iNOS expression, such as NF-κB and caspase-3 [38]. Another widely used model to study ALS employs threohydroxyaspartate (THA), an inhibitor of glutamate transport, to induce motor neuron death in cultured rat spinal cord explants. In this model, EGCG protected motor neurons against THA-induced toxicity, which was accompanied by regulation of glutamate levels in the synaptic cleft, and decreased tissue levels of lipid peroxides [39].
Cerebral ischaemia
Stroke is the second most common cause of death and major cause of disability worldwide [220]. Temporary or permanent reduction of blood flow deprives the brain of oxygen and glucose which causes structural damage during ischaemia and reperfusion [43,221]. Neuronal cell death is delayed for several days after ischaemic insult and is restricted to sensitive areas of the brain, such as the hippocampus and the striatum [52]. Possible neuroprotective effects of EGCG on cerebral ischaemia have been studied extensively in animal models. Administration of GTE/EGCG in ischaemia-reperfusion brain injury decreased the extent of neuronal injury in experimental models of stroke in gerbils [40-42], rats [43-51] and mice [52,53].
Proposed protective actions of GTE/EGCG are (1) increase of nNOS and eNOS but decrease of iNOS expression [43,47,49], (2) inhibition of matrix metalloproteinase-9 activity (which decreases degradation of matrix components of basement membranes thereby maintaining cerebral vasculature integrity) [52,53], (3) amelioration of synaptic transmission [51], and (4) stimulation of 67LR, increased generation of ROS via NADPH oxidase and subsequent activation of PKCε [221]. One of the above mentioned animal studies observed a >50% increase of intracerebral haemorrhages after EGCG treatment. Consequently, EGCG might not be an appropriate intervention for the acute treatment of ischaemic stroke [46].
A meta-analysis revealed that, regardless of their country of origin (China, Japan, Finland, The Netherlands, Australia and USA), individuals consuming ≥3 cups of tea per day had a 21% lower risk of stroke than those consuming <1 cup per day [54]. To the best of our knowledge, there are no clinical studies addressing effects of EGCG in human stroke.
Cognitive behaviour
Some evidence from animal and human studies indicates a possible contribution of EGCG in the prevention of cognitive decline. Increased release of glutamate from CNS neurons is considered to be an indicator of maintenance of cognitive functions, such as learning and memory [222,223]. On isolated nerve terminals prepared from rat cerebral cortex, EGCG potently facilitated evoked glutamate release by enhancing Ca2+ entry through voltage-dependent Ca2+ channels, rather than any upstream effect on nerve terminal excitability. This release was linked to the activation of protein kinase C (PKC) and cytoskeleton disassembly [224].
Long-term (26 weeks) administration of green tea catechins to rats improved their performance in radial maze tasks and hippocampal levels of lipid peroxides. Consequently, green tea catechins may be involved in protecting against neuronal degenerative stress and in the accumulation of lipid peroxides and ROS [225]. EGCG treatment of rats before and after traumatic brain injury eliminated and/or absorbed free radicals induced by brain injury. As a result, neuronal cell degeneration and apoptotic cell death around the damaged area were inhibited, and brain dysfunction was improved [226]. Fatigue is an overwhelming sense of tiredness or lack of energy, affecting both mental and physical domains. In a rat model of load-induced chronic fatigue syndrome, EGCG restored all behavioural and biochemical alterations produced by chronic fatigue [227].
There are, albeit limited, data on the effects of EGCG on human cognition. A cross-sectional analysis of a self-administered questionnaire showed that higher green tea consumption is associated with less prevalent cognitive impairment in elderly Japanese subjects [228]. In a double-blind, placebo-controlled, balanced crossover study, the effects of oral doses of EGCG on cerebral blood flow in the frontal cortex during tasks that activate this brain region and cognitive performance were investigated. A single dose of 135 mg EGCG reduced cerebral blood flow during task performance. However, this was not associated with any significant modulation of either cognitive performance or mood [229].
Ageing
Ageing is the progressive accumulation of changes over time that is associated with, or responsible for, increasing susceptibility to disease and death [230]. Oxidative stress has been associated with both the ageing process and the development of age-dependent tissue degenerative pathologies.
The nematode Caenorhabditis elegans is a tiny roundworm of 1 to 2 mm in length that colonises various microbe-rich habitats, in particular, decaying plant matter. Due to its rapid lifecycle, it is one of the major model organisms in ageing, genetic, molecular and other biological observations [231-234]. EGCG attenuated age-related pharyngeal contraction decline, moderately alleviated Aβ-induced behavioural pathologies and sustained an increased chemotactic behaviour. However, there were no significant increases in mean or maximal life span due to EGCG feeding [235]. This was confirmed by another study that found no significant longevity-extending effects of EGCG in C. elegans under normal culture conditions but with extension of life span by EGCG under heat and oxidative stress [236]. In another investigation on wild-type N2 and transgenic strains of C. elegans, EGCG administration increased the mean lifespan, inhibited heat shock protein expression and decreased intracellular H2O2 levels [237].
A systematic study on the effects of EGCG on ageing and ageing promoting factors, such as ROS accumulation, mitochondrial integrity and antioxidative enzyme activity in human fibroblasts, directly linked mitochondrial integrity to the efficiency of the antioxidant defence system in the ageing process [238]. In mice in which ageing had been induced by d–galactose, EGCG improved learning and memory functions, increased SOD and glutathione peroxidase activities and decreased the hippocampal malondialdehyde content and neuronal apoptosis [239].
Several neuronal systems, such as dopaminergic, cholinergic and serotoninergic ones, undergo alterations during ageing. Rats supplemented with EGCG had increased cortical neurotransmitter levels (dopamine, acetylcholine and serotonin) and acetylcholine esterase activity and performed better in radial maze experiments when compared to aged-matched controls [240].
Dose regimen: antioxidative versus pro-oxidative
As green tea/EGCG are becoming more and more popular due to their proposed health benefits, persons with different pathological conditions might consume commercially available supplements that contain catechin concentrations exceeding those of tea preparations by far. Owing to their reducing ability, antioxidant compounds can activate transition metal ions (e.g. Fe3+ to Fe2+ or Cu2+ to Cu+), making them behave as pro-oxidants. It has been demonstrated that four common dietary antioxidants (cysteine > ascorbate > EGCG > glutathione), in the presence of copper (cupric sulfate and cupric gluconate) and physiologically relevant levels of H2O2, can also act as pro-oxidants by producing hydroxyl radicals [61]. Yoshioka et al. reported that tea catechins advanced DNA damage and lipid peroxidation in the presence of Cu2+. Tea catechins formed a complex with Cu2+ resulting in the formation of hydroxyl radical that, in the case of EGCG, was scavenged by the additional gallate group [241]. Interestingly, a mixture of the four tea catechins (Polyphenon 100) and individual tea catechins differently modulated human cytochrome P450 1A expression. The underlying mechanisms of increased effectiveness of green tea and a catechin mixture compared to single catechins were not elucidated in this study [242].
Under in vitro conditions, catechins exerted antioxidant and antiapoptotic properties at low concentrations (1 to 50 μM), whereas at higher concentrations (100 to 500 μM) the reverse was observed [243]. It was demonstrated that GTE and EGCG inhibit cell growth and induce cell death at concentrations of 10 to 20 μM. In murine macrophage and human leukemic cell lines, EGCG increased H2O2-induced oxidative stress and DNA damage. The oxidant activity of EGCG exceeded that of H2O2. Therefore, excessive EGCG concentrations could induce toxic levels of ROS in vivo[244]. EGCG dose-dependently inhibited the growth of H1299 cells in culture and in xenograft tumours and induced ROS formation both in vitro and in vivo[245].
In murine and human plasma, EGCG concentrations up to 4 μM were reported after single doses [246-248]. However, repeated doses might lead to higher concentrations with possible deleterious effects. In healthy adult mice fed with diets containing none, moderate (0.15% and 0.3%) or high (1%) w/w EGCG for 6 weeks, inflammatory responses were studied. While no influence of moderate EGCG levels was observed, the high dose significantly elevated several pro-inflammatory markers. This was accompanied by significant weight loss without visible toxicity as assessed by the histological examination of several key organs [249].
For several reasons, extrapolation of these animal data to the human situation is difficult, if not impossible. It has to be ensured that patients treated with EGCG benefit from its antioxidative effects and, at the same time, are not harmed by pro-oxidative effects. Factors that could lead to conflicting results between in vitro and in vivo studies are varying degrees of responsiveness to EGCG in patients (responder vs. non-responder), poor bioavailability (cave:blood sample collection) that is influenced by autoxidation (air contact) or metal ions in food and water (calcium, magnesium and iron), extensive biotransformation in the liver and variations in serum albumin levels. To optimise EGCG bioavailability, capsules should be stored cool and dry, taken at least 30 min before breakfast/dinner with soft water and possibly combined with ascorbic acid, sucrose or fish oil [250]. To avoid potential side effects, clinical trials should include close attention to patients, control of liver enzymes and regular determination of outcome measures.
Conclusions
EGCG is thought to interfere with several pathways in numerous neurological functions in health and disease. Despite a considerable body of evidence from cell and animal models, there is a lack of epidemiological and clinical studies on potential health benefits of EGCG in patients with the neurological conditions discussed. Within the clinical studies, the number of subjects is mostly rather small and patients are not well phenotyped or standardised regarding disease severity and duration. Green tea preparations and GTEs are also not standardised. Therefore, optimum dose of EGCG for preventing or treating a disease is still a matter of debate. Before EGCG can be recommended as a targeted prevention and individualised treatment to patients, the plasma and brain bioavailability, dose-response effects, safety, tolerability, efficacy and possible interactions with other drugs have to be studied in more detail and in a disease-specific manner. For a number of clinical effects the underlying mechanisms are still unclear. Because EGCG is acting on so many different routes (neuro-endocrine, metabolic, defence and others), studies with an integrated approach are strongly needed. This will be, however, a cost-intensive and time consuming endeavour.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AM, MB, and FP drafted the manuscript. SM, ML, UR, and EW completed and critically revised the manuscript. All authors read and approved the manuscript.
Contributor Information
Anja Mähler, Email: anja.maehler@charite.de.
Silvia Mandel, Email: mandel@tx.technion.ac.il.
Mario Lorenz, Email: mario.lorenz@charite.de.
Urs Ruegg, Email: Urs.Ruegg@unige.ch.
Erich E Wanker, Email: ewanker@mdc-berlin.de.
Michael Boschmann, Email: michael.boschmann@charite.de.
Friedemann Paul, Email: friedemann.paul@charite.de.
Acknowledgement
This work was supported by the German Research Council (DFG Exc 257 to FP) and other grants.
References
- Golubnitschaja O, Costigliola V. EPMA. General report & recommendations in predictive, preventive and personalised medicine 2012: white paper of the European Association for Predictive, Preventive and Personalised Medicine. EPMA J. 2012;3:14. doi: 10.1186/1878-5085-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose RC, Bode AM. Biology of free radical scavengers: an evaluation of ascorbate. FASEB J. 1993;7:1135–1142. [PubMed] [Google Scholar]
- Letelier ME, Sanchez-Jofre S, Peredo-Silva L, Cortes-Troncoso J, Aracena-Parks P. Mechanisms underlying iron and copper ions toxicity in biological systems: pro-oxidant activity and protein-binding effects. Chem Biol Interact. 2010;188:220–227. doi: 10.1016/j.cbi.2010.06.013. [DOI] [PubMed] [Google Scholar]
- Graham HN. Green tea composition, consumption, and polyphenol chemistry. Prev Med. 1992;21:334–350. doi: 10.1016/0091-7435(92)90041-f. [DOI] [PubMed] [Google Scholar]
- Sang S, Lambert JD, Ho CT, Yang CS. The chemistry and biotransformation of tea constituents. Pharmacol Res. 2011;64:87–99. doi: 10.1016/j.phrs.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Balentine DA, Wiseman SA, Bouwens LC. The chemistry of tea flavonoids. Crit Rev Food Sci Nutr. 1997;37:693–704. doi: 10.1080/10408399709527797. [DOI] [PubMed] [Google Scholar]
- Mandel SA, Youdim MB. In the rush for green gold: can green tea delay age-progressive brain neurodegeneration? Recent Pat CNS Drug Discov. 2012;7:205–217. doi: 10.2174/157488912803252005. [DOI] [PubMed] [Google Scholar]
- Aktas O, Prozorovski T, Smorodchenko A, Savaskan NE, Lauster R, Kloetzel PM, Infante-Duarte C, Brocke S, Zipp F. Green tea epigallocatechin-3-gallate mediates T cellular NF-kappa B inhibition and exerts neuroprotection in autoimmune encephalomyelitis. J Immunol. 2004;173:5794–5800. doi: 10.4049/jimmunol.173.9.5794. [DOI] [PubMed] [Google Scholar]
- Herges K, Millward JM, Hentschel N, Infante-Duarte C, Aktas O, Zipp F. Neuroprotective effect of combination therapy of glatiramer acetate and epigallocatechin-3-gallate in neuroinflammation. PLoS One. 2011;6:e25456. doi: 10.1371/journal.pone.0025456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ren Z, Xu Y, Xiao S, Meydani SN, Wu D. Epigallocatechin-3-gallate ameliorates experimental autoimmune encephalomyelitis by altering balance among CD4+ T-cell subsets. Am J Pathol. 2012;180:221–234. doi: 10.1016/j.ajpath.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YT, Jung CH, Lee SR, Bae JH, Baek WK, Suh MH. The green tea polyphenol (−)-epigallocatechin gallate attenuates beta-amyloid-induced neurotoxicity in cultured hippocampal neurons. Life Sci. 2001;70:603–614. doi: 10.1016/s0024-3205(01)01438-2. [DOI] [PubMed] [Google Scholar]
- Rezai-Zadeh K, Shytle D, Sun N, Mori T, Hou H, Jeanniton D, Ehrhart J, Townsend K, Zeng J, Morgan D, Hardy J, Town T, Tan J. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci. 2005;25:8807–8814. doi: 10.1523/JNEUROSCI.1521-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Lee YK, Ban JO, Ha TY, Yun YP, Han SB, Oh KW, Hong JT. Green tea (−)-epigallocatechin-3-gallate inhibits beta-amyloid-induced cognitive dysfunction through modification of secretase activity via inhibition of ERK and NF-kappaB pathways in mice. J Nutr. 2009;139:1987–1993. doi: 10.3945/jn.109.109785. [DOI] [PubMed] [Google Scholar]
- Kim CY, Lee C, Park GH, Jang JH. Neuroprotective effect of epigallocatechin-3-gallate against beta-amyloid-induced oxidative and nitrosative cell death via augmentation of antioxidant defense capacity. Arch Pharm Res. 2009;32:869–881. doi: 10.1007/s12272-009-1609-z. [DOI] [PubMed] [Google Scholar]
- Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci U S A. 2010;107:7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Huang YG, Fang D, Le WD. (−)-Epigallocatechin gallate inhibits lipopolysaccharide-induced microglial activation and protects against inflammation-mediated dopaminergic neuronal injury. J Neurosci Res. 2004;78:723–731. doi: 10.1002/jnr.20315. [DOI] [PubMed] [Google Scholar]
- Chan DK, Woo J, Ho SC, Pang CP, Law LK, Ng PW, Hung WT, Kwok T, Hui E, Orr K, Leung MF, Kay R. Genetic and environmental risk factors for Parkinson's disease in a Chinese population. J Neurol Neurosurg Psychiatry. 1998;65:781–784. doi: 10.1136/jnnp.65.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkoway H, Powers K, Smith-Weller T, Franklin GM, Longstreth WT Jr, Swanson PD. Parkinson's disease risks associated with cigarette smoking, alcohol consumption, and caffeine intake. Am J Epidemiol. 2002;155:732–738. doi: 10.1093/aje/155.8.732. [DOI] [PubMed] [Google Scholar]
- Hu G, Bidel S, Jousilahti P, Antikainen R, Tuomilehto J. Coffee and tea consumption and the risk of Parkinson's disease. Mov Disord. 2007;22:2242–2248. doi: 10.1002/mds.21706. [DOI] [PubMed] [Google Scholar]
- Kandinov B, Giladi N, Korczyn AD. Smoking and tea consumption delay onset of Parkinson's disease. Parkinsonism Relat Disord. 2009;15:41–46. doi: 10.1016/j.parkreldis.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Nie G, Cao Y, Zhao B. Protective effects of green tea polyphenols and their major component, (−)-epigallocatechin-3-gallate (EGCG), on 6-hydroxydopamine-induced apoptosis in PC12 cells. Redox Rep. 2002;7:171–177. doi: 10.1179/135100002125000424. [DOI] [PubMed] [Google Scholar]
- Ye Q, Ye L, Xu X, Huang B, Zhang X, Zhu Y, Chen X. Epigallocatechin-3-gallate suppresses 1-methyl-4-phenyl-pyridine-induced oxidative stress in PC12 cells via the SIRT1/PGC-1alpha signaling pathway. BMC Complement Altern Med. 2012;12:82. doi: 10.1186/1472-6882-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Weinreb O, Maor G, Youdim MB, Mandel S. Green tea polyphenol (−)-epigallocatechin-3-gallate prevents N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration. J Neurochem. 2001;78:1073–1082. doi: 10.1046/j.1471-4159.2001.00490.x. [DOI] [PubMed] [Google Scholar]
- Choi JY, Park CS, Kim DJ, Cho MH, Jin BK, Pie JE, Chung WG. Prevention of nitric oxide-mediated 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease in mice by tea phenolic epigallocatechin 3-gallate. Neurotoxicology. 2002;23:367–374. doi: 10.1016/s0161-813x(02)00079-7. [DOI] [PubMed] [Google Scholar]
- Leaver KR, Allbutt HN, Creber NJ, Kassiou M, Henderson JM. Oral pre-treatment with epigallocatechin gallate in 6-OHDA lesioned rats produces subtle symptomatic relief but not neuroprotection. Brain Res Bull. 2009;80:397–402. doi: 10.1016/j.brainresbull.2009.08.013. [DOI] [PubMed] [Google Scholar]
- Kim JS, Kim JM, JJ O, Jeon BS. Inhibition of inducible nitric oxide synthase expression and cell death by (−)-epigallocatechin-3-gallate, a green tea catechin, in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. J Clin Neurosci. 2010;17:1165–1168. doi: 10.1016/j.jocn.2010.01.042. [DOI] [PubMed] [Google Scholar]
- Kang KS, Wen Y, Yamabe N, Fukui M, Bishop SC, Zhu BT. Dual beneficial effects of (−)-epigallocatechin-3-gallate on levodopa methylation and hippocampal neurodegeneration: in vitro and in vivo studies. PLoS One. 2010;5:e11951. doi: 10.1371/journal.pone.0011951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnhoefer DE, Duennwald M, Markovic P, Wacker JL, Engemann S, Roark M, Legleiter J, Marsh JL, Thompson LM, Lindquist S, Muchowski PJ, Wanker EE. Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington's disease models. Hum Mol Genet. 2006;15:2743–2751. doi: 10.1093/hmg/ddl210. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kumar A. Effect of lycopene and epigallocatechin-3-gallate against 3-nitropropionic acid induced cognitive dysfunction and glutathione depletion in rat: a novel nitric oxide mechanism. Food Chem Toxicol. 2009;47:2522–2530. doi: 10.1016/j.fct.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Buetler TM, Renard M, Offord EA, Schneider H, Ruegg UT. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am J Clin Nutr. 2002;75:749–753. doi: 10.1093/ajcn/75.4.749. [DOI] [PubMed] [Google Scholar]
- Dorchies OM, Wagner S, Vuadens O, Waldhauser K, Buetler TM, Kucera P, Ruegg UT. Green tea extract and its major polyphenol (−)-epigallocatechin gallate improve muscle function in a mouse model for Duchenne muscular dystrophy. Am J Physiol Cell Physiol. 2006;290:C616–C625. doi: 10.1152/ajpcell.00425.2005. [DOI] [PubMed] [Google Scholar]
- Nakae Y, Hirasaka K, Goto J, Nikawa T, Shono M, Yoshida M, Stoward PJ. Subcutaneous injection, from birth, of epigallocatechin-3-gallate, a component of green tea, limits the onset of muscular dystrophy in mdx mice: a quantitative histological, immunohistochemical and electrophysiological study. Histochem Cell Biol. 2008;129:489–501. doi: 10.1007/s00418-008-0390-2. [DOI] [PubMed] [Google Scholar]
- Dorchies OM, Wagner S, Buetler TM, Ruegg UT. Protection of dystrophic muscle cells with polyphenols from green tea correlates with improved glutathione balance and increased expression of 67LR, a receptor for (−)-epigallocatechin gallate. Biofactors. 2009;35:279–294. doi: 10.1002/biof.34. [DOI] [PubMed] [Google Scholar]
- Call JA, Voelker KA, Wolff AV, McMillan RP, Evans NP, Hulver MW, Talmadge RJ, Grange RW. Endurance capacity in maturing mdx mice is markedly enhanced by combined voluntary wheel running and green tea extract. J Appl Physiol. 2008;105:923–932. doi: 10.1152/japplphysiol.00028.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans NP, Call JA, Bassaganya-Riera J, Robertson JL, Grange RW. Green tea extract decreases muscle pathology and NF-kappaB immunostaining in regenerating muscle fibers of mdx mice. Clin Nutr. 2010;29:391–398. doi: 10.1016/j.clnu.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SH, Lee SM, Kim HY, Lee KY, Lee YJ, Kim HT, Kim MH, Hwang MS, Song C, Yang KW, Lee KW, Kim SH, Kim OH. The effect of epigallocatechin gallate on suppressing disease progression of ALS model mice. Neurosci Lett. 2006;395:103–107. doi: 10.1016/j.neulet.2005.10.056. [DOI] [PubMed] [Google Scholar]
- Xu Z, Chen S, Li X, Luo G, Li L, Le W. Neuroprotective effects of (−)-epigallocatechin-3-gallate in a transgenic mouse model of amyotrophic lateral sclerosis. Neurochem Res. 2006;31:1263–1269. doi: 10.1007/s11064-006-9166-z. [DOI] [PubMed] [Google Scholar]
- Yu J, Jia Y, Guo Y, Chang G, Duan W, Sun M, Li B, Li C. Epigallocatechin-3-gallate protects motor neurons and regulates glutamate level. FEBS Lett. 2010;584:2921–2925. doi: 10.1016/j.febslet.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Lee H, Bae JH, Lee SR. Protective effect of green tea polyphenol EGCG against neuronal damage and brain edema after unilateral cerebral ischemia in gerbils. J Neurosci Res. 2004;77:892–900. doi: 10.1002/jnr.20193. [DOI] [PubMed] [Google Scholar]
- Lee S, Suh S, Kim S. Protective effects of the green tea polyphenol (−)-epigallocatechin gallate against hippocampal neuronal damage after transient global ischemia in gerbils. Neurosci Lett. 2000;287:191–194. doi: 10.1016/s0304-3940(00)01159-9. [DOI] [PubMed] [Google Scholar]
- Lee SY, Kim CY, Lee JJ, Jung JG, Lee SR. Effects of delayed administration of (−)-epigallocatechin gallate, a green tea polyphenol on the changes in polyamine levels and neuronal damage after transient forebrain ischemia in gerbils. Brain Res Bull. 2003;61:399–406. doi: 10.1016/s0361-9230(03)00139-4. [DOI] [PubMed] [Google Scholar]
- Wu KJ, Hsieh MT, Wu CR, Wood WG, Chen YF. Green tea extract ameliorates learning and memory deficits in ischemic rats via its active component polyphenol epigallocatechin-3-gallate by modulation of oxidative stress and neuroinflammation. Evid Based Complement Alternat Med. 2012;201(2):163106. doi: 10.1155/2012/163106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YB, Kim YI, Lee KS, Kim BS, Kim DJ. Protective effect of epigallocatechin gallate on brain damage after transient middle cerebral artery occlusion in rats. Brain Res. 2004;1019:47–54. doi: 10.1016/j.brainres.2004.05.079. [DOI] [PubMed] [Google Scholar]
- Nagai K, Jiang MH, Hada J, Nagata T, Yajima Y, Yamamoto S, Nishizaki T. (−)-Epigallocatechin gallate protects against NO stress-induced neuronal damage after ischemia by acting as an anti-oxidant. Brain Res. 2002;956:319–322. doi: 10.1016/s0006-8993(02)03564-3. [DOI] [PubMed] [Google Scholar]
- Rahman RM, Nair SM, Helps SC, Shaw OM, Sims NR, Rosengren RJ, Appleton I, Appleton I. (−)-Epigallocatechin gallate as an intervention for the acute treatment of cerebral ischemia. Neurosci Lett. 2005;382:227–230. doi: 10.1016/j.neulet.2005.03.018. [DOI] [PubMed] [Google Scholar]
- Sutherland BA, Shaw OM, Clarkson AN, Jackson DN, Sammut IA, Appleton I. Neuroprotective effects of (−)-epigallocatechin gallate following hypoxia-ischemia-induced brain damage: novel mechanisms of action. FASEB J. 2005;19:258–260. doi: 10.1096/fj.04-2806fje. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Tabuchi M, Ikeda M, Umegaki K, Tomita T. Protective effects of green tea catechins on cerebral ischemic damage. Med Sci Monit. 2004;10:BR166–BR174. [PubMed] [Google Scholar]
- Wei IH, Wu YC, Wen CY, Shieh JY. Green tea polyphenol (−)-epigallocatechin gallate attenuates the neuronal NADPH-d/nNOS expression in the nodose ganglion of acute hypoxic rats. Brain Res. 2004;999:73–80. doi: 10.1016/j.brainres.2003.11.056. [DOI] [PubMed] [Google Scholar]
- Hong JT, Ryu SR, Kim HJ, Lee JK, Lee SH, Kim DB, Yun YP, Ryu JH, Lee BM, Kim PY. Neuroprotective effect of green tea extract in experimental ischemia-reperfusion brain injury. Brain Res Bull. 2000;53:743–749. doi: 10.1016/s0361-9230(00)00348-8. [DOI] [PubMed] [Google Scholar]
- Ding J, Fu G, Zhao Y, Cheng Z, Chen Y, Zhao B, He W, Guo LJ. EGCG ameliorates the suppression of long-term potentiation induced by ischemia at the Schaffer collateral-CA1 synapse in the rat. Cell Mol Neurobiol. 2012;32:267–277. doi: 10.1007/s10571-011-9758-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Jang YH, Kim JM, Lee H, Park WK, Lim MB, Chu YK, Lo EH, Lee SR. Green tea polyphenol (−)-epigallocatechin gallate reduces neuronal cell damage and up-regulation of MMP-9 activity in hippocampal CA1 and CA2 areas following transient global cerebral ischemia. J Neurosci Res. 2009;87:567–575. doi: 10.1002/jnr.21847. [DOI] [PubMed] [Google Scholar]
- Park JW, Hong JS, Lee KS, Kim HY, Lee JJ, Lee SR. Green tea polyphenol (−)-epigallocatechin gallate reduces matrix metalloproteinase-9 activity following transient focal cerebral ischemia. J Nutr Biochem. 2010;21:1038–1044. doi: 10.1016/j.jnutbio.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Arab L, Liu W, Elashoff D. Green and black tea consumption and risk of stroke: a meta-analysis. Stroke. 2009;40:1786–1792. doi: 10.1161/STROKEAHA.108.538470. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Miyazawa T. Absorption and distribution of tea catechin, (−)-epigallocatechin-3-gallate, in the rat. J Nutr Sci Vitaminol (Tokyo) 1997;43:679–684. doi: 10.3177/jnsv.43.679. [DOI] [PubMed] [Google Scholar]
- Suganuma M, Okabe S, Oniyama M, Tada Y, Ito H, Fujiki H. Wide distribution of [3H](−)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis. 1998;19:1771–1776. doi: 10.1093/carcin/19.10.1771. [DOI] [PubMed] [Google Scholar]
- Zini A, Del RD, Stewart AJ, Mandrioli J, Merelli E, Sola P, Nichelli P, Serafini M, Brighenti F, Edwards CA, Crozier A. Do flavan-3-ols from green tea reach the human brain? Nutr Neurosci. 2006;9:57–61. doi: 10.1080/10284150600637739. [DOI] [PubMed] [Google Scholar]
- Wu L, Zhang QL, Zhang XY, Lv C, Li J, Yuan Y, Yin FX. Pharmacokinetics and blood–brain barrier penetration of (+)-catechin and (−)-epicatechin in rats by microdialysis sampling coupled to high-performance liquid chromatography with chemiluminescence detection. J Agric Food Chem. 2012;60:9377–9383. doi: 10.1021/jf301787f. [DOI] [PubMed] [Google Scholar]
- Mandel SA, Amit T, Weinreb O, Youdim MB. Understanding the broad-spectrum neuroprotective action profile of green tea polyphenols in aging and neurodegenerative diseases. J Alzheimers Dis. 2011;25:187–208. doi: 10.3233/JAD-2011-101803. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Yin JJ, Fu PP, Lutterodt H, Zhou YT, Antholine WE, Wamer W. Dual role of selected antioxidants found in dietary supplements: crossover between anti- and pro-oxidant activities in the presence of copper. J Agric Food Chem. 2012;60:2554–2561. doi: 10.1021/jf204724w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomova K, Vondrakova D, Lawson M, Valko M. Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem. 2010;345:91–104. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- Frei B, Higdon JV. Antioxidant activity of tea polyphenols in vivo: evidence from animal studies. J Nutr. 2003;133:3275S–3284S. doi: 10.1093/jn/133.10.3275S. [DOI] [PubMed] [Google Scholar]
- Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- Ho YS, Magnenat JL, Gargano M, Cao J. The nature of antioxidant defense mechanisms: a lesson from transgenic studies. Environ Health Perspect. 1998;106(Suppl 5):1219–1228. doi: 10.1289/ehp.98106s51219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srividhya R, Jyothilakshmi V, Arulmathi K, Senthilkumaran V, Kalaiselvi P. Attenuation of senescence-induced oxidative exacerbations in aged rat brain by (−)-epigallocatechin-3-gallate. Int J Dev Neurosci. 2008;26:217–223. doi: 10.1016/j.ijdevneu.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Lin SM, Wang SW, Ho SC, Tang YL. Protective effect of green tea (−)-epigallocatechin-3-gallate against the monoamine oxidase B enzyme activity increase in adult rat brains. Nutrition. 2010;26:1195–1200. doi: 10.1016/j.nut.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Benveniste EN. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med (Berl) 1997;75:165–173. doi: 10.1007/s001090050101. [DOI] [PubMed] [Google Scholar]
- McCarty MF. Down-regulation of microglial activation may represent a practical strategy for combating neurodegenerative disorders. Med Hypotheses. 2006;67:251–269. doi: 10.1016/j.mehy.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Hunot S, Boissiere F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, Hirsch EC. Nitric oxide synthase and neuronal vulnerability in Parkinson's disease. Neuroscience. 1996;72:355–363. doi: 10.1016/0306-4522(95)00578-1. [DOI] [PubMed] [Google Scholar]
- Le W, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson's disease. J Neurosci. 2001;21:8447–8455. doi: 10.1523/JNEUROSCI.21-21-08447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Guo Z, Ren Z, Guo W, Meydani SN. Green tea EGCG suppresses T cell proliferation through impairment of IL-2/IL-2 receptor signaling. Free Radic Biol Med. 2009;47:636–643. doi: 10.1016/j.freeradbiomed.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Sinnecker T, Mittelstaedt P, Dorr J, Pfueller CF, Harms L, Niendorf T, Paul F, Wuerfel J. Multiple sclerosis lesions and irreversible brain tissue damage: a comparative ultrahigh-field strength magnetic resonance imaging study. Arch Neurol. 2012;69:739–745. doi: 10.1001/archneurol.2011.2450. [DOI] [PubMed] [Google Scholar]
- Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M, Schmidbauer M, Parisi JE, Lassmann H. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–2712. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- Bock M, Brandt AU, Dorr J, Kraft H, Weinges-Evers N, Gaede G, Pfueller CF, Herges K, Radbruch H, Ohlraun S, Bellmann-Strobl J, Kuchenbecker J, Zipp F, Paul F. Patterns of retinal nerve fiber layer loss in multiple sclerosis patients with or without optic neuritis and glaucoma patients. Clin Neurol Neurosurg. 2010;112:647–652. doi: 10.1016/j.clineuro.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Bock M, Brandt AU, Dorr J, Pfueller CF, Ohlraun S, Zipp F, Paul F. Time domain and spectral domain optical coherence tomography in multiple sclerosis: a comparative cross-sectional study. Mult Scler. 2010;16:893–896. doi: 10.1177/1352458510365156. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Dorr J, Wernecke KD, Bock M, Gaede G, Wuerfel JT, Pfueller CF, Bellmann-Strobl J, Freing A, Brandt AU, Friedemann P. Association of retinal and macular damage with brain atrophy in multiple sclerosis. PLoS One. 2011;6:e18132. doi: 10.1371/journal.pone.0018132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Trapp BD. Pathogenesis of axonal and neuronal damage in multiple sclerosis. Neurology. 2007;68:S22–S31. doi: 10.1212/01.wnl.0000275229.13012.32. [DOI] [PubMed] [Google Scholar]
- Gordon-Lipkin E, Chodkowski B, Reich DS, Smith SA, Pulicken M, Balcer LJ, Frohman EM, Cutter G, Calabresi PA. Retinal nerve fiber layer is associated with brain atrophy in multiple sclerosis. Neurology. 2007;69:1603–1609. doi: 10.1212/01.wnl.0000295995.46586.ae. [DOI] [PubMed] [Google Scholar]
- Oberwahrenbrock T, Schippling S, Ringelstein M, Kaufhold F, Zimmermann H, Keser N, Young KL, Harmel J, Hartung H-P, Martin R, Paul F, Aktas O, Brandt AU. Retinal damage in multiple sclerosis disease subtypes measured by high-resolution optical coherence tomography. Mult Scler Int. 2012;201(2):530305. doi: 10.1155/2012/530305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfueller CF, Brandt AU, Schubert F, Bock M, Walaszek B, Waiczies H, Young KL, Harmel J, Hartung HP, Martin R, Paul F, Aktas O, Brandt AU. Metabolic changes in the visual cortex are linked to retinal nerve fiber layer thinning in multiple sclerosis. PLoS One. 2011;6:e18019. doi: 10.1371/journal.pone.0018019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J, Paul F, Aktas O, Muller-Wielsch K, Dorr J, Dorr S, Bharathi BS, Glumm R, Schmitz C, Steinbusch H, Raine CS, Tsokos M, Nitsch R, Zipp F. Lower motor neuron loss in multiple sclerosis and experimental autoimmune encephalomyelitis. Ann Neurol. 2009;66:310–322. doi: 10.1002/ana.21719. [DOI] [PubMed] [Google Scholar]
- Brandt AU, Oberwahrenbrock T, Ringelstein M, Young KL, Tiede M, Hartung HP, Martin R, Aktas O, Paul F, Schippling S. Primary retinal pathology in multiple sclerosis as detected by optical coherence tomography. Brain. 2011;134:e193. doi: 10.1093/brain/awr095. [DOI] [PubMed] [Google Scholar]
- Zimmermann H, Freing A, Kaufhold F, Gaede G, Bohn E, Bock M, Oberwahrenbrock T, Young KL, Dörr J, Wuerfel JT, Schippling S, Paul F, Brandt AU. Optic neuritis interferes with optical coherence tomography and magnetic resonance imaging correlations. Mult Scler. 2012. [DOI] [PubMed]
- Borisow N, Doring A, Pfueller CF, Paul F, Dorr J, Hellwig K. Expert recommendations to personalization of medical approaches in treatment of multiple sclerosis: an overview of family planning and pregnancy. EPMA J. 2012;3:9. doi: 10.1186/1878-5085-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel AE, Jarvis L, McLaughlin R, Fries A, Ebers GC, Ramagopalan SV. The epidemiology of multiple sclerosis in Scotland: inferences from hospital admissions. PLoS One. 2011;6:e14606. doi: 10.1371/journal.pone.0014606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons RD, Tribe KL, McDonald EA. Living with multiple sclerosis: longitudinal changes in employment and the importance of symptom management. J Neurol. 2010;257:926–936. doi: 10.1007/s00415-009-5441-7. [DOI] [PubMed] [Google Scholar]
- Sundstrom P, Nystrom L. Smoking worsens the prognosis in multiple sclerosis. Mult Scler. 2008;14:1031–1035. doi: 10.1177/1352458508093615. [DOI] [PubMed] [Google Scholar]
- Hawkes CH. Smoking is a risk factor for multiple sclerosis: a metanalysis. Mult Scler. 2007;13:610–615. doi: 10.1177/1352458506073501. [DOI] [PubMed] [Google Scholar]
- Doring A, Paul F, Dorr J. Vitamin D and multiple sclerosis: the role for risk of disease and treatment. Nervenarzt. 2013;84(2):173–189. doi: 10.1007/s00115-012-3645-z. [DOI] [PubMed] [Google Scholar]
- D'hooghe MB, Haentjens P, Nagels G, Garmyn M, De KJ. Sunlight exposure and sun sensitivity associated with disability progression in multiple sclerosis. Mult Scler. 2012;18:451–459. doi: 10.1177/1352458511423778. [DOI] [PubMed] [Google Scholar]
- Munger KL, Ascherio A. Prevention and treatment of MS: studying the effects of vitamin D. Mult Scler. 2011;17:1405–1411. doi: 10.1177/1352458511425366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel AE, Ramagopalan SV. Vitamin D and multiple sclerosis: an interaction between genes and environment. Mult Scler. 2012;18:2–4. doi: 10.1177/1352458511418353. [DOI] [PubMed] [Google Scholar]
- Goodin DS. The causal cascade to multiple sclerosis: a model for MS pathogenesis. PLoS One. 2009;4:e4565. doi: 10.1371/journal.pone.0004565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel AE, Williamson AJ, Disanto G, Dobson R, Giovannoni G, Ramagopalan SV. Smoking and multiple sclerosis: an updated meta-analysis. PLoS One. 2011;6:e16149. doi: 10.1371/journal.pone.0016149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel AE, Williamson AJ, Disanto G, Handunnetthi L, Giovannoni G, Ramagopalan SV. An updated meta-analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolders J, Thewissen M, Peelen E, Menheere P, Tervaert JW, Damoiseaux J, Hupperts R. Vitamin D status is positively correlated with regulatory T cell function in patients with multiple sclerosis. PLoS One. 2009;4:e6635. doi: 10.1371/journal.pone.0006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas RM, Ponsonby AL, Dear K, Valery PC, Pender MP, Taylor BV, Kilpatrick TJ, Dwyer T, Coulthard A, Chapman C, van der Mei I, Williams D, McMichael AJ. Sun exposure and vitamin D are independent risk factors for CNS demyelination. Neurology. 2011;76:540–548. doi: 10.1212/WNL.0b013e31820af93d. [DOI] [PubMed] [Google Scholar]
- Otto C, Oltmann A, Stein A, Frenzel K, Schroeter J, Habbel P, Gärtner B, Hofmann J, Ruprecht K. Intrathecal EBV antibodies are part of the polyspecific immune response in multiple sclerosis. Neurology. 2011;76:1316–1321. doi: 10.1212/WNL.0b013e318215286d. [DOI] [PubMed] [Google Scholar]
- De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, Raychaudhuri S, Tran D, Aubin C, Briskin R, Romano S, International MS Genetics C, Baranzini SE, McCauley JL, Pericak-Vance MA, Haines JL, Gibson RA, Naeglin Y, Uitdehaag B, Matthews PM, Kappos L, Polman C, McArdle WL, Strachan DP, Evans D, Cross AH. et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke C, Pech LM, Sommer C, Schlichting J, Stricker S, Endres M, Ostendorf F, Ploner CJ, Brandt AU, Paul F. Dynamics of saccade parameters in multiple sclerosis patients with fatigue. J Neurol. 2012;259:2656–2663. doi: 10.1007/s00415-012-6565-8. [DOI] [PubMed] [Google Scholar]
- Veauthier C, Paul F. Fatigue in multiple sclerosis: which patient should be referred to a sleep specialist? Mult Scler. 2012;18:248–249. doi: 10.1177/1352458511411229. [DOI] [PubMed] [Google Scholar]
- Veauthier C, Radbruch H, Gaede G, Pfueller CF, Dorr J, Bellmann-Strobl J, Wernecke KD, Zipp F, Paul F, Sieb JP. Fatigue in multiple sclerosis is closely related to sleep disorders: a polysomnographic cross-sectional study. Mult Scler. 2011;17:613–622. doi: 10.1177/1352458510393772. [DOI] [PubMed] [Google Scholar]
- Urbanek C, Weinges-Evers N, Bellmann-Strobl J, Bock M, Dorr J, Hahn E, Neuhaus AH, Opgen-Rhein C, Ta TM, Herges K, Pfueller CF, Radbruch H, Wernecke KD, Ohlraun S, Zipp F, Dettling M, Paul F. Attention Network Test reveals alerting network dysfunction in multiple sclerosis. Mult Scler. 2010;16:93–99. doi: 10.1177/1352458509350308. [DOI] [PubMed] [Google Scholar]
- Weinges-Evers N, Brandt AU, Bock M, Pfueller CF, Dorr J, Bellmann-Strobl J, Scherer P, Urbanek C, Boers C, Ohlraun S, Zipp F, Paul F. Correlation of self-assessed fatigue and alertness in multiple sclerosis. Mult Scler. 2010;16:1134–1140. doi: 10.1177/1352458510374202. [DOI] [PubMed] [Google Scholar]
- Bates D. Treatment effects of immunomodulatory therapies at different stages of multiple sclerosis in short-term trials. Neurology. 2011;76:S14–S25. doi: 10.1212/WNL.0b013e3182050388. [DOI] [PubMed] [Google Scholar]
- Wiendl H, Toyka KV, Rieckmann P, Gold R, Hartung HP, Hohlfeld R. Basic and escalating immunomodulatory treatments in multiple sclerosis: current therapeutic recommendations. J Neurol. 2008;255:1449–1463. doi: 10.1007/s00415-008-0061-1. [DOI] [PubMed] [Google Scholar]
- Jain N, Bhatti MT. Fingolimod-associated macular edema: incidence, detection, and management. Neurology. 2012;78:672–680. doi: 10.1212/WNL.0b013e318248deea. [DOI] [PubMed] [Google Scholar]
- Turaka K, Bryan JS. Does fingolimod in multiple sclerosis patients cause macular edema? J Neurol. 2012;259:386–388. doi: 10.1007/s00415-011-6367-4. [DOI] [PubMed] [Google Scholar]
- Sorensen PS, Bertolotto A, Edan G, Giovannoni G, Gold R, Havrdova E, Kappos L, Kieseier BC, Montalban X, Olsson T. Risk stratification for progressive multifocal leukoencephalopathy in patients treated with natalizumab. Mult Scler. 2012;18:143. doi: 10.1177/1352458511435105. [DOI] [PubMed] [Google Scholar]
- Castillo-Trivino T, Mowry EM, Gajofatto A, Chabas D, Crabtree-Hartman E, Cree BA, Goodin DS, Green AJ, Okuda DT, Pelletier D, Zamvil SS, Vittinghoff E, Waubant E. Switching multiple sclerosis patients with breakthrough disease to second-line therapy. PLoS One. 2011;6:e16664. doi: 10.1371/journal.pone.0016664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwig K, Gold R. Progressive multifocal leukoencephalopathy and natalizumab. J Neurol. 2011;258:1920–1928. doi: 10.1007/s00415-011-6116-8. [DOI] [PubMed] [Google Scholar]
- Dinkin M, Paul F. Higher macular volume in patients with MS receiving fingolimod: positive outcome or side effect. Neurology. 2012;80:128–129. doi: 10.1212/WNL.0b013e31827ccf4a. [DOI] [PubMed] [Google Scholar]
- Faber H, Fischer HJ, Weber F. Prolonged and symptomatic bradycardia following a single dose of fingolimod. Mult Scler. 2012;19:126–128. doi: 10.1177/1352458512447596. [DOI] [PubMed] [Google Scholar]
- Lindsey J, Haden-Pinneri K, Memon N, Buja L. Sudden unexpected death on fingolimod. Mult Scler. 2012;18:1507–1508. doi: 10.1177/1352458512438456. [DOI] [PubMed] [Google Scholar]
- Espinosa PS, Berger JR. Delayed fingolimod-associated asystole. Mult Scler. 2011;17:1387–1389. doi: 10.1177/1352458511410344. [DOI] [PubMed] [Google Scholar]
- Le PE, Leray E, Edan G. Long-term safety profile of mitoxantrone in a French cohort of 802 multiple sclerosis patients: a 5-year prospective study. Mult Scler. 2011;17:867–875. doi: 10.1177/1352458511398371. [DOI] [PubMed] [Google Scholar]
- Goffette S, Van PV, Vanoverschelde JL, Morandini E, Sindic CJ. Severe delayed heart failure in three multiple sclerosis patients previously treated with mitoxantrone. J Neurol. 2005;252:1217–1222. doi: 10.1007/s00415-005-0839-3. [DOI] [PubMed] [Google Scholar]
- Chanvillard C, Millward JM, Lozano M, Hamann I, Paul F, Zipp F, Dörr J, Infante-Duarte C. Mitoxantrone induces natural killer cell maturation in patients with secondary progressive multiple sclerosis. PLoS One. 2012;7:e39625. doi: 10.1371/journal.pone.0039625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroet A, Hemmelmann C, Starck M, Zettl U, Dorr J, Friedemann P, Flachenecker P, Fleischer V, Zipp F, Nückel H, Kieseier BC, Ziegler A, Gold R, Chan A. Incidence of therapy-related acute leukaemia in mitoxantrone-treated multiple sclerosis patients in Germany. Ther Adv Neurol Disord. 2012;5:75–79. doi: 10.1177/1756285611433318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörr J, Bitsch A, Schmailzl KJ, Chan A, von Ahsen N, Hummel M, Varon R, Lill CM, Vogel HP, Zipp F, Paul F. Severe cardiac failure in a patient with multiple sclerosis following low-dose mitoxantrone treatment. Neurology. 2009;73:991–993. doi: 10.1212/WNL.0b013e3181b878f6. [DOI] [PubMed] [Google Scholar]
- Paul F, Dorr J, Wurfel J, Vogel HP, Zipp F. Early mitoxantrone-induced cardiotoxicity in secondary progressive multiple sclerosis. J Neurol Neurosurg Psychiatry. 2007;78:198–200. doi: 10.1136/jnnp.2006.091033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- Mix E, Meyer-Rienecker H, Zettl UK. Animal models of multiple sclerosis for the development and validation of novel therapies - potential and limitations. J Neurol. 2008;255(Suppl 6):7–14. doi: 10.1007/s00415-008-6003-0. [DOI] [PubMed] [Google Scholar]
- Vesterinen HM, Sena ES, Ffrench-Constant C, Williams A, Chandran S, Macleod MR. Improving the translational hit of experimental treatments in multiple sclerosis. Mult Scler. 2010;16:1044–1055. doi: 10.1177/1352458510379612. [DOI] [PubMed] [Google Scholar]
- Baker D, Gerritsen W, Rundle J, Amor S. Critical appraisal of animal models of multiple sclerosis. Mult Scler. 2011;17:647–657. doi: 10.1177/1352458511398885. [DOI] [PubMed] [Google Scholar]
- Racke MK, Lovett-Racke AE, Karandikar NJ. The mechanism of action of glatiramer acetate treatment in multiple sclerosis. Neurology. 2010;74(Suppl 1):S25–S30. doi: 10.1212/WNL.0b013e3181c97e39. [DOI] [PubMed] [Google Scholar]
- Stuve O, Kieseier BC, Hemmer B, Hartung HP, Awad A, Frohman EM, Greenberg BM, Racke MK, Zamvil SS, Phillips JT, Gold R, Chan A, Zettl U, Milo R, Marder E, Khan O, Eagar TN. Translational research in neurology and neuroscience 2010: multiple sclerosis. Arch Neurol. 2010;67:1307–1315. doi: 10.1001/archneurol.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comi G, Martinelli V, Rodegher M, Moiola L, Leocani L, Bajenaru O, Carra A, Elovaara I, Fazekas F, Hartung HP, Hillert J, King J, Komoly S, Lubetzki C, Montalban X, Myhr KM, Preziosa P, Ravnborg M, Rieckmann P, Rocca MA, Wynn D, Young C, Filippi M. Effects of early treatment with glatiramer acetate in patients with clinically isolated syndrome. Mult Scler. 2012. [DOI] [PubMed]
- Giovannoni G, Southam E, Waubant E. Systematic review of disease-modifying therapies to assess unmet needs in multiple sclerosis: tolerability and adherence. Mult Scler. 2012;18:932–946. doi: 10.1177/1352458511433302. [DOI] [PubMed] [Google Scholar]
- Trojan DA, Arnold D, Collet JP, Shapiro S, Bar-Or A, Robinson A, Le Cruguel JP, Ducruet T, Narayanan S, Arcelin K, Wong AN, Tartaglia MC, Lapierre Y, Caramanos Z, Da Costa D. Fatigue in multiple sclerosis: association with disease-related, behavioural and psychosocial factors. Mult Scler. 2007;13:985–995. doi: 10.1177/1352458507077175. [DOI] [PubMed] [Google Scholar]
- Doring A, Pfueller CF, Paul F, Dorr J. Exercise in multiple sclerosis – an integral component of disease management. EPMA J. 2011;3:2. doi: 10.1007/s13167-011-0136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen AK, Jakobsen J, Petersen T, Andersen H. Fatigued patients with multiple sclerosis have impaired central muscle activation. Mult Scler. 2009;15:818–827. doi: 10.1177/1352458509105383. [DOI] [PubMed] [Google Scholar]
- Thielecke F, Rahn G, Bohnke J, Adams F, Birkenfeld AL, Jordan J, Boschmann M. Epigallocatechin-3-gallate and postprandial fat oxidation in overweight/obese male volunteers: a pilot study. Eur J Clin Nutr. 2010;64:704–713. doi: 10.1038/ejcn.2010.47. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Nakatsuji Y. Where do AQP4 antibodies fit in the pathogenesis of NMO? Mult Scler Int. 2012;201(2):862169. doi: 10.1155/2012/862169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarius S, Franciotta D, Paul F, Bergamaschi R, Rommer PS, Ruprecht K, Ringelstein M, Aktas O, Kristoferitsch W, Wildemann B. Testing for antibodies to human aquaporin-4 by ELISA: sensitivity, specificity, and direct comparison with immunohistochemistry. J Neurol Sci. 2012;320:32–37. doi: 10.1016/j.jns.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Mader S, Lutterotti A, Di PF, Kuenz B, Schanda K, Aboul-Enein F, Khalil M, Storch MK, Jarius S, Kristoferitsch W, Berger T, Reindl M. Patterns of antibody binding to aquaporin-4 isoforms in neuromyelitis optica. PLoS One. 2010;5:e10455. doi: 10.1371/journal.pone.0010455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratelade J, Bennett JL, Verkman AS. Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One. 2011;6:e27412. doi: 10.1371/journal.pone.0027412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granieri L, Marnetto F, Valentino P, Frau J, Patanella AK, Nytrova P, Sola P, Capobianco M, Jarius S, Bertolotto A. Evaluation of a multiparametric immunofluorescence assay for standardization of neuromyelitis optica serology. PLoS One. 2012;7:e38896. doi: 10.1371/journal.pone.0038896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, Kleiter I, Kleinschnitz C, Berthele A, Brettschneider J, Hellwig K, Hemmer B, Linker RA, Lauda F, Mayer CA, Tumani H, Melms A, Trebst C, Stangel M, Marziniak M, Hoffmann F, Schippling S, Faiss JH, Neuhaus O, Ettrich B, Zentner C, Guthke K, Hofstadt-van Oy U, Reuss R, Pellkofer H, Ziemann U, Kern P. et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14. doi: 10.1186/1742-2094-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarius S, Franciotta D, Paul F, Ruprecht K, Bergamaschi R, Rommer PS, Reuss R, Probst C, Kristoferitsch W, Wandinger KP, Wildemann B. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: frequency, origin, and diagnostic relevance. J Neuroinflammation. 2010;7:52. doi: 10.1186/1742-2094-7-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, Vincent A, Wildemann B. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. 2008;4:202–214. doi: 10.1038/ncpneuro0764. [DOI] [PubMed] [Google Scholar]
- Paul F, Jarius S, Aktas O, Bluthner M, Bauer O, Appelhans H, Franciotta D, Bergamaschi R, Littleton E, Palace J, Seelig HP, Hohlfeld R, Vincent A, Zipp F. Antibody to aquaporin 4 in the diagnosis of neuromyelitis optica. PLoS Med. 2007;4:e133. doi: 10.1371/journal.pmed.0040133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–477. doi: 10.1084/jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- Storoni M, Petzold A, Plant GT. The use of serum glial fibrillary acidic protein measurements in the diagnosis of neuromyelitis optica spectrum optic neuritis. PLoS One. 2011;6:e23489. doi: 10.1371/journal.pone.0023489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noval S, Contreras I, Munoz S, Oreja-Guevara C, Manzano B, Rebolleda G. Optical coherence tomography in multiple sclerosis and neuromyelitis optica: an update. Mult Scler Int. 2011;201(1):472790. doi: 10.1155/2011/472790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfueller CF, Paul F. Imaging the visual pathway in neuromyelitis optica. Mult Scler Int. 2011;201(1):869814. doi: 10.1155/2011/869814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnecker T, Dorr J, Pfueller CF, Harms L, Ruprecht K, Jarius S, Brück W, Niendorf T, Wuerfel J, Paul F. Distinct lesion morphology at 7-T MRI differentiates neuromyelitis optica from multiple sclerosis. Neurology. 2012;79:708–714. doi: 10.1212/WNL.0b013e3182648bc8. [DOI] [PubMed] [Google Scholar]
- Calabrese M, Oh MS, Favaretto A, Rinaldi F, Poretto V, Alessio S, Lee BC, Yu KH, Ma HI, Perini P, Gallo P. No MRI evidence of cortical lesions in neuromyelitis optica. Neurology. 2012;79:1671–1676. doi: 10.1212/WNL.0b013e31826e9a96. [DOI] [PubMed] [Google Scholar]
- Lu Z, Zhang B, Qiu W, Kang Z, Shen L, Long Y, Huang J, Hu X. Comparative brain stem lesions on MRI of acute disseminated encephalomyelitis, neuromyelitis optica, and multiple sclerosis. PLoS One. 2011;6:e22766. doi: 10.1371/journal.pone.0022766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Gomez JA, Kister I. Conventional brain MRI in neuromyelitis optica. Eur J Neurol. 2012;19:812–819. doi: 10.1111/j.1468-1331.2011.03565.x. [DOI] [PubMed] [Google Scholar]
- Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R, Rommer P, Kleiter I, Stich O, Reuss R, Rauer S, Zettl UK, Wandinger KP, Melms A, Aktas O, Kristoferitsch W, Wildemann B. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci. 2011;306:82–90. doi: 10.1016/j.jns.2011.03.038. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW, Buckland ME, Parratt JD, Pollard JD. Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult Scler. 2012;18:108–112. doi: 10.1177/1352458511421185. [DOI] [PubMed] [Google Scholar]
- Shimizu Y, Yokoyama K, Misu T, Takahashi T, Fujihara K, Kikuchi S, Itoyama Y, Iwata M. Development of extensive brain lesions following interferon beta therapy in relapsing neuromyelitis optica and longitudinally extensive myelitis. J Neurol. 2008;255:305–307. doi: 10.1007/s00415-007-0730-5. [DOI] [PubMed] [Google Scholar]
- Shimizu J, Hatanaka Y, Hasegawa M, Iwata A, Sugimoto I, Date H, Goto J, Shimizu T, Takatsu M, Sakurai Y, Nakase H, Uesaka Y, Hashida H, Hashimoto K, Komiya T, Tsuji S. IFNbeta-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology. 2010;75:1423–1427. doi: 10.1212/WNL.0b013e3181f8832e. [DOI] [PubMed] [Google Scholar]
- Papeix C, Vidal JS, De SJ, Pierrot-Deseilligny C, Tourbah A, Stankoff B, Lebrun C, Moreau T, Vermersch P, Fontaine B, Lyon-Caen O, Gout O. Immunosuppressive therapy is more effective than interferon in neuromyelitis optica. Mult Scler. 2007;13:256–259. doi: 10.1177/1352458506070732. [DOI] [PubMed] [Google Scholar]
- Palace J, Leite MI, Nairne A, Vincent A. Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol. 2010;67:1016–1017. doi: 10.1001/archneurol.2010.188. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler. 2012;18:1480–1483. doi: 10.1177/1352458512439439. [DOI] [PubMed] [Google Scholar]
- Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult Scler. 2012;18:113–115. doi: 10.1177/1352458511431973. [DOI] [PubMed] [Google Scholar]
- Jacob A, Hutchinson M, Elsone L, Kelly S, Ali R, Saukans I, Tubridy N, Boggild M. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012;79:1065–1066. doi: 10.1212/WNL.0b013e31826845fe. [DOI] [PubMed] [Google Scholar]
- Kleiter I, Hellwig K, Berthele A, Kumpfel T, Linker RA, Hartung HP, Paul F, Aktas O. Neuromyelitis Optica Study Group. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. 2012;69:239–245. doi: 10.1001/archneurol.2011.216. [DOI] [PubMed] [Google Scholar]
- Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J, Thapa P, McKeon A. Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology. 2011;77:659–666. doi: 10.1212/WNL.0b013e31822a2780. [DOI] [PubMed] [Google Scholar]
- Jacob A, Weinshenker BG, Violich I, McLinskey N, Krupp L, Fox RJ, Wingerchuk DM, Boggild M, Constantinescu CS, Miller A, De Angelis T, Matiello M, Cree BA. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol. 2008;65:1443–1448. doi: 10.1001/archneur.65.11.noc80069. [DOI] [PubMed] [Google Scholar]
- Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, Carter J, Keegan BM, Kantarci OH, Pittock SJ. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol. 2009;66:1128–1133. doi: 10.1001/archneurol.2009.175. [DOI] [PubMed] [Google Scholar]
- Kim SH, Kim W, Park MS, Sohn EH, Li XF, Kim HJ. Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol. 2011;68:473–479. doi: 10.1001/archneurol.2010.322. [DOI] [PubMed] [Google Scholar]
- Greenberg BM, Graves D, Remington G, Hardeman P, Mann M, Karandikar N, Stuve O, Monson N, Frohman E. Rituximab dosing and monitoring strategies in neuromyelitis optica patients: creating strategies for therapeutic success. Mult Scler. 2012;18:1022–1026. doi: 10.1177/1352458511432896. [DOI] [PubMed] [Google Scholar]
- Bichuetti DB, Lobato de Oliveira EM, Oliveira DM, Amorin de Souza N, Gabbai AA. Neuromyelitis optica treatment: analysis of 36 patients. Arch Neurol. 2010;67:1131–1136. doi: 10.1001/archneurol.2010.203. [DOI] [PubMed] [Google Scholar]
- Kalluri SR, Rothhammer V, Staszewski O, Srivastava R, Petermann F, Prinz M, Hemmer B, Korn T. Functional characterization of aquaporin-4 specific T cells: towards a model for neuromyelitis optica. PLoS One. 2011;6:e16083. doi: 10.1371/journal.pone.0016083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PA, Khodadoust M, Prodhomme T, Spencer C, Patarroyo JC, Varrin-Doyer M, Ho JD, Stroud RM, Zamvil SS. Immunodominant T cell determinants of aquaporin-4, the autoantigen associated with neuromyelitis optica. PLoS One. 2010;5:e15050. doi: 10.1371/journal.pone.0015050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, Cree BA, Zamvil SS. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol. 2012;72:53–64. doi: 10.1002/ana.23651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S, Waters P, MacDonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol. 2012;71:323–333. doi: 10.1002/ana.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herges K, de Jong BA, Kolkowitz I, Dunn C, Mandelbaum G, Ko RM, Maini A, Han MH, Killestein J, Polman C, Goodyear AL, Dunn J, Steinman L, Axtell RC. Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult Scler. 2012;18:398–408. doi: 10.1177/1352458512440060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abboud PA, Hake PW, Burroughs TJ, Odoms K, O'Connor M, Mangeshkar P, Wong HR, Zingarelli B. Therapeutic effect of epigallocatechin-3-gallate in a mouse model of colitis. Eur J Pharmacol. 2008;579:411–417. doi: 10.1016/j.ejphar.2007.10.053. [DOI] [PubMed] [Google Scholar]
- Aneja R, Hake PW, Burroughs TJ, Denenberg AG, Wong HR, Zingarelli B. Epigallocatechin, a green tea polyphenol, attenuates myocardial ischemia reperfusion injury in rats. Mol Med. 2004;10:55–62. doi: 10.2119/2004-00032.aneja. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton NM, Shin R, Tajinda K, Heusner CL, Kogan JH, Miyake S, Chen Q, Tamura K, Matsumoto M. Adult neurogenesis transiently generates oxidative stress. PLoS One. 2012;7:e35264. doi: 10.1371/journal.pone.0035264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Amit T, Mandel S, Youdim MB. Neuroprotection and neurorescue against Abeta toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (−)-epigallocatechin-3-gallate. FASEB J. 2003;17:952–954. doi: 10.1096/fj.02-0881fje. [DOI] [PubMed] [Google Scholar]
- Reznichenko L, Amit T, Zheng H, Avramovich-Tirosh Y, Youdim MB, Weinreb O, Mandel S. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (−)-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer's disease. J Neurochem. 2006;97:527–536. doi: 10.1111/j.1471-4159.2006.03770.x. [DOI] [PubMed] [Google Scholar]
- Citron M. Beta-secretase inhibition for the treatment of Alzheimer's disease - promise and challenge. Trends Pharmacol Sci. 2004;25:92–97. doi: 10.1016/j.tips.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Behl C. Alzheimer's disease and oxidative stress: implications for novel therapeutic approaches. Prog Neurobiol. 1999;57:301–323. doi: 10.1016/s0301-0082(98)00055-0. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- Rojanathammanee L, Murphy EJ, Combs CK. Expression of mutant alpha-synuclein modulates microglial phenotype in vitro. J Neuroinflammation. 2011;8:44. doi: 10.1186/1742-2094-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Guajardo V, Febbraro F, Kirik D, Romero-Ramos M. Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson's disease. PLoS One. 2010;5:e8784. doi: 10.1371/journal.pone.0008784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Fernandez-Montesinos R, Caro M, Lachaud CC, Waudby CA, Delgado M, Dobson CM, Pozo D. Glial innate immunity generated by non-aggregated alpha-synuclein in mouse: differences between wild-type and Parkinson's disease-linked mutants. PLoS One. 2010;5:e13481. doi: 10.1371/journal.pone.0013481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabon MM, Jernberg JN, Morganti J, Contreras J, Hudson CE, Klein RL, Bickford PC. A spirulina-enhanced diet provides neuroprotection in an alpha-synuclein model of Parkinson's disease. PLoS One. 2012;7:e45256. doi: 10.1371/journal.pone.0045256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer TJ, Atchison WD. Transmitter, ion channel and receptor properties of pheochromocytoma (PC12) cells: a model for neurotoxicological studies. Neurotoxicology. 1991;12:473–492. [PubMed] [Google Scholar]
- Levites Y, Youdim MB, Maor G, Mandel S. Attenuation of 6-hydroxydopamine (6-OHDA)-induced nuclear factor-kappaB (NF-kappaB) activation and cell death by tea extracts in neuronal cultures. Biochem Pharmacol. 2002;63:21–29. doi: 10.1016/s0006-2952(01)00813-9. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Vila M. The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model: a tool to explore the pathogenesis of Parkinson's disease. Ann N Y Acad Sci. 2003;991:189–198. [PubMed] [Google Scholar]
- Lambert JD, Lee MJ, Lu H, Meng X, Hong JJ, Seril DN, Sturgill MG, Yang CS. Epigallocatechin-3-gallate is absorbed but extensively glucuronidated following oral administration to mice. J Nutr. 2003;133:4172–4177. doi: 10.1093/jn/133.12.4172. [DOI] [PubMed] [Google Scholar]
- Kim S, Lee MJ, Hong J, Li C, Smith TJ, Yang GY. Plasma and tissue levels of tea catechins in rats and mice during chronic consumption of green tea polyphenols. Nutr Cancer. 2000;37:41–48. doi: 10.1207/S15327914NC3701_5. [DOI] [PubMed] [Google Scholar]
- Pan T, Fei J, Zhou X, Jankovic J, Le W. Effects of green tea polyphenols on dopamine uptake and on MPP+-induced dopamine neuron injury. Life Sci. 2003;72:1073–1083. doi: 10.1016/s0024-3205(02)02347-0. [DOI] [PubMed] [Google Scholar]
- Zhu BT, Shim JY, Nagai M, Bai HW. Molecular modelling study of the mechanism of high-potency inhibition of human catechol-O-methyltransferase by (−)-epigallocatechin-3-O-gallate. Xenobiotica. 2008;38:130–146. doi: 10.1080/00498250701744641. [DOI] [PubMed] [Google Scholar]
- Tan LC, Koh WP, Yuan JM, Wang R, Au WL, Tan JH, Tan EK, Yu MC. Differential effects of black versus green tea on risk of Parkinson's disease in the Singapore Chinese Health Study. Am J Epidemiol. 2008;167:553–560. doi: 10.1093/aje/kwm338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
- Saleh N, Moutereau S, Durr A, Krystkowiak P, Azulay JP, Tranchant C, Broussolle E, Morin F, Bachoud-Lévi AC, Maison P. Neuroendocrine disturbances in Huntington's disease. PLoS One. 2009;4:e4962. doi: 10.1371/journal.pone.0004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzone E, Vasco G, Sormani MP, Torrente Y, Berardinelli A, Messina S, D'Amico A, Doglio L, Politano L, Cavallaro F, Frosini S, Bello L, Bonfiglio S, Zucchini E, De Sanctis R, Scutifero M, Bianco F, Rossi F, Motta MC, Sacco A, Donati MA, Mongini T, Pini A, Battini R, Pegoraro E, Pane M, Gasperini S, Previtali S, Napolitano S, Martinelli D, Bruno C. et al. Functional changes in Duchenne muscular dystrophy: a 12-month longitudinal cohort study. Neurology. 2011;77:250–256. doi: 10.1212/WNL.0b013e318225ab2e. [DOI] [PubMed] [Google Scholar]
- Weber MA, Nagel AM, Jurkat-Rott K, Lehmann-Horn F. Sodium (23Na) MRI detects elevated muscular sodium concentration in Duchenne muscular dystrophy. Neurology. 2011;77:2017–2024. doi: 10.1212/WNL.0b013e31823b9c78. [DOI] [PubMed] [Google Scholar]
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- Nakae Y, Dorchies OM, Stoward PJ, Zimmermann BF, Ritter C, Ruegg UT. Quantitative evaluation of the beneficial effects in the mdx mouse of epigallocatechin gallate, an antioxidant polyphenol from green tea. Histochem Cell Biol. 2012;137:811–827. doi: 10.1007/s00418-012-0926-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Ludolph AC, Brettschneider J, Weishaupt JH. Amyotrophic lateral sclerosis. Curr Opin Neurol. 2012;25:530–535. doi: 10.1097/WCO.0b013e328356d328. [DOI] [PubMed] [Google Scholar]
- Quinn C, Elman L, McCluskey L, Hoskins K, Karam C, Woo JH, Poptani H, Wang S, Chawla S, Kasner SE, Grossman M. Frontal lobe abnormalities on MRS correlate with poor letter fluency in ALS. Neurology. 2012;79:583–588. doi: 10.1212/WNL.0b013e3182635720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaphorst J, Beeldman E, Schmand B, Berkhout J, Linssen WH, van den Berg LH, Pijnenburg YA, Grupstra HF, Weikamp JG, Schelhaas HJ, Papma JM, van Swieten JC, de Visser M, de Haan RJ. The ALS-FTD-Q: a new screening tool for behavioral disturbances in ALS. Neurology. 2012;79:1377–1383. doi: 10.1212/WNL.0b013e31826c1aa1. [DOI] [PubMed] [Google Scholar]
- Ellis CM, Suckling J, Amaro E Jr, Bullmore ET, Simmons A, Williams SC, Leigh PN. Volumetric analysis reveals corticospinal tract degeneration and extramotor involvement in ALS. Neurology. 2001;57:1571–1578. doi: 10.1212/wnl.57.9.1571. [DOI] [PubMed] [Google Scholar]
- Kwan JY, Jeong SY, Van GP, Deng HX, Quezado MM, Danielian LE, Butman JA, Chen L, Bayat E, Russell J, Siddique T, Duyn JH, Rouault TA, Floeter MK. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 Tesla MRI and pathology. PLoS One. 2012;7:e35241. doi: 10.1371/journal.pone.0035241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis J, Katona I, Muller-Newen G, Sommer C, Necula G, Hendrich C, Ludolph AC, Sperfeld AD. Small-fiber neuropathy in patients with ALS. Neurology. 2011;76:2024–2029. doi: 10.1212/WNL.0b013e31821e553a. [DOI] [PubMed] [Google Scholar]
- Lloyd CM, Richardson MP, Brooks DJ, Al-Chalabi A, Leigh PN. Extramotor involvement in ALS: PET studies with the GABA(A) ligand [(11)C]flumazenil. Brain. 2000;123(Pt 11):2289–2296. doi: 10.1093/brain/123.11.2289. [DOI] [PubMed] [Google Scholar]
- Agosta F, Valsasina P, Riva N, Copetti M, Messina MJ, Prelle A, Comi G, Filippi M. The cortical signature of amyotrophic lateral sclerosis. PLoS One. 2012;7:e42816. doi: 10.1371/journal.pone.0042816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillo P, Mioshi E, Burrell JR, Kiernan MC, Hodges JR, Hornberger M. Grey and white matter changes across the amyotrophic lateral sclerosis-frontotemporal dementia continuum. PLoS One. 2012;7:e43993. doi: 10.1371/journal.pone.0043993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, Grinberg Y, Isaia G, Calvo A, Gentile S, Bruni AC, St George-Hyslop PH, Scarpini E, Gallone S, Pinessi L. TODEM Study Group. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79:1556–1562. doi: 10.1212/WNL.0b013e31826e25df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SH, Rowland LP. Amyotrophic lateral sclerosis, frontotemporal lobar dementia, and p62: a functional convergence? Neurology. 2012;79:1526–1527. doi: 10.1212/WNL.0b013e31826e26ec. [DOI] [PubMed] [Google Scholar]
- Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. doi: 10.1212/wnl.57.7.1282. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Engelhardt JI, Siklos L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- Ince PG, Evans J, Knopp M, Forster G, Hamdalla HH, Wharton SB, Shaw PJ. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. 2003;60:1252–1258. doi: 10.1212/01.wnl.0000058901.75728.4e. [DOI] [PubMed] [Google Scholar]
- Brettschneider J, Toledo JB, Van Deerlin VM, Elman L, McCluskey L, Lee VM, Trojanowski JQ. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One. 2012;7:e39216. doi: 10.1371/journal.pone.0039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman DB, Frank K, Dykes-Hoberg M, Teismann P, Almer G, Przedborski S, Rothstein JD. Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2002;52:771–778. doi: 10.1002/ana.10374. [DOI] [PubMed] [Google Scholar]
- Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371:1612–1623. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- Gundimeda U, McNeill TH, Elhiani AA, Schiffman JE, Hinton DR, Gopalakrishna R. Green tea polyphenols precondition against cell death induced by oxygen-glucose deprivation via stimulation of laminin receptor, generation of reactive oxygen species, and activation of protein kinase C{epsilon} J Biol Chem. 2012;287:34694–34708. doi: 10.1074/jbc.M112.356899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewski JT, Danysz W. Modulation of glutamate receptors: molecular mechanisms and functional implications. Annu Rev Pharmacol Toxicol. 1989;29:441–474. doi: 10.1146/annurev.pa.29.040189.002301. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Chou CW, Huang WJ, Tien LT, Wang SJ. (−)-Epigallocatechin gallate, the most active polyphenolic catechin in green tea, presynaptically facilitates Ca2+-dependent glutamate release via activation of protein kinase C in rat cerebral cortex. Synapse. 2007;61:889–902. doi: 10.1002/syn.20444. [DOI] [PubMed] [Google Scholar]
- Haque AM, Hashimoto M, Katakura M, Tanabe Y, Hara Y, Shido O. Long-term administration of green tea catechins improves spatial cognition learning ability in rats. J Nutr. 2006;136:1043–1047. doi: 10.1093/jn/136.4.1043. [DOI] [PubMed] [Google Scholar]
- Itoh T, Imano M, Nishida S, Tsubaki M, Hashimoto S, Ito A, Satou T. (−)-Epigallocatechin-3-gallate protects against neuronal cell death and improves cerebral function after traumatic brain injury in rats. Neuromolecular Med. 2011;13:300–309. doi: 10.1007/s12017-011-8162-x. [DOI] [PubMed] [Google Scholar]
- Sachdeva AK, Kuhad A, Chopra K. Epigallocatechin gallate ameliorates behavioral and biochemical deficits in rat model of load-induced chronic fatigue syndrome. Brain Res Bull. 2011;86:165–172. doi: 10.1016/j.brainresbull.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Kuriyama S, Hozawa A, Ohmori K, Shimazu T, Matsui T, Ebihara S, Awata S, Nagatomi R, Arai H, Tsuji I. Green tea consumption and cognitive function: a cross-sectional study from the Tsurugaya Project 1. Am J Clin Nutr. 2006;83:355–361. doi: 10.1093/ajcn/83.2.355. [DOI] [PubMed] [Google Scholar]
- Wightman EL, Haskell CF, Forster JS, Veasey RC, Kennedy DO. Epigallocatechin gallate, cerebral blood flow parameters, cognitive performance and mood in healthy humans: a double-blind, placebo-controlled, crossover investigation. Hum Psychopharmacol. 2012;27:177–186. doi: 10.1002/hup.1263. [DOI] [PubMed] [Google Scholar]
- Harman D. The aging process. Proc Natl Acad Sci U S A. 1981;78:7124–7128. doi: 10.1073/pnas.78.11.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix MA, Braendle C. The natural history of Caenorhabditis elegans. Curr Biol. 2010;20:R965–R969. doi: 10.1016/j.cub.2010.09.050. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Cao P, Smith MA, Kramp K, Huang Y, Hisamoto N, Matsumoto K, Hatzoglou M, Jin H, Feng Z. Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. elegans. PLoS One. 2011;6:e22354. doi: 10.1371/journal.pone.0022354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran R, Castelblanco L, Tang G, Shapiro I, Goncharov A, Jin Y. Motor neuron synapse and axon defects in a C. elegans alpha-tubulin mutant. PLoS One. 2010;5:e9655. doi: 10.1371/journal.pone.0009655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisine C, Varma H, Walker N, Bates EA, Stockwell BR, Hart AC. Identification of potential therapeutic drugs for Huntington's disease using Caenorhabditis elegans. PLoS One. 2007;2:e504. doi: 10.1371/journal.pone.0000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MK, Evans JL, Luo Y. Beneficial effects of natural antioxidants EGCG and alpha-lipoic acid on life span and age-dependent behavioral declines in Caenorhabditis elegans. Pharmacol Biochem Behav. 2006;85:620–628. doi: 10.1016/j.pbb.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Zhang L, Jie G, Zhang J, Zhao B. Significant longevity-extending effects of EGCG on Caenorhabditis elegans under stress. Free Radic Biol Med. 2009;46:414–421. doi: 10.1016/j.freeradbiomed.2008.10.041. [DOI] [PubMed] [Google Scholar]
- Abbas S, Wink M. Epigallocatechin gallate from green tea (Camellia sinensis) increases lifespan and stress resistance in Caenorhabditis elegans. Planta Med. 2009;75:216–221. doi: 10.1055/s-0028-1088378. [DOI] [PubMed] [Google Scholar]
- Meng Q, Velalar CN, Ruan R. Effects of epigallocatechin-3-gallate on mitochondrial integrity and antioxidative enzyme activity in the aging process of human fibroblast. Free Radic Biol Med. 2008;44:1032–1041. doi: 10.1016/j.freeradbiomed.2007.11.023. [DOI] [PubMed] [Google Scholar]
- He M, Zhao L, Wei MJ, Yao WF, Zhao HS, Chen FJ. Neuroprotective effects of (−)-epigallocatechin-3-gallate on aging mice induced by d-galactose. Biol Pharm Bull. 2009;32:55–60. doi: 10.1248/bpb.32.55. [DOI] [PubMed] [Google Scholar]
- Srividhya R, Gayathri R, Kalaiselvi P. Impact of epigallo catechin-3-gallate on acetylcholine-acetylcholine esterase cycle in aged rat brain. Neurochem Int. 2012;60:517–522. doi: 10.1016/j.neuint.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, Senba Y, Saito K, Kimura T, Hayakawa F. Spin-trapping study on the hydroxyl radical formed from a tea catechin-Cu(II) system. Biosci Biotechnol Biochem. 2001;65:1697–1706. doi: 10.1271/bbb.65.1697. [DOI] [PubMed] [Google Scholar]
- Williams SN, Pickwell GV, Quattrochi LC. A combination of tea (Camellia senensis) catechins is required for optimal inhibition of induced CYP1A expression by green tea extract. J Agric Food Chem. 2003;51:6627–6634. doi: 10.1021/jf030181z. [DOI] [PubMed] [Google Scholar]
- Sutherland BA, Rahman RM, Appleton I. Mechanisms of action of green tea catechins, with a focus on ischemia-induced neurodegeneration. J Nutr Biochem. 2006;17:291–306. doi: 10.1016/j.jnutbio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Elbling L, Weiss RM, Teufelhofer O, Uhl M, Knasmueller S, Schulte-Hermann R, Berger W, Micksche M. Green tea extract and (−)-epigallocatechin-3-gallate, the major tea catechin, exert oxidant but lack antioxidant activities. FASEB J. 2005;19:807–809. doi: 10.1096/fj.04-2915fje. [DOI] [PubMed] [Google Scholar]
- Li GX, Chen YK, Hou Z, Xiao H, Jin H, Lu G, Lee MJ, Liu B, Guan F, Yang Z, Yu A, Yang CS. Pro-oxidative activities and dose–response relationship of (−)-epigallocatechin-3-gallate in the inhibition of lung cancer cell growth: a comparative study in vivo and in vitro. Carcinogenesis. 2010;31:902–910. doi: 10.1093/carcin/bgq039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JD, Lee MJ, Diamond L, Ju J, Hong J, Bose M, Newmark HL, Yang CS. Dose-dependent levels of epigallocatechin-3-gallate in human colon cancer cells and mouse plasma and tissues. Drug Metab Dispos. 2006;34:8–11. doi: 10.1124/dmd.104.003434. [DOI] [PubMed] [Google Scholar]
- Ullmann U, Haller J, Decourt JP, Girault N, Girault J, Richard-Caudron AS, Pineau B, Weber P. A single ascending dose study of epigallocatechin gallate in healthy volunteers. J Int Med Res. 2003;31:88–101. doi: 10.1177/147323000303100205. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Okuda S, Miyazawa T. Dose-dependent incorporation of tea catechins, (−)-epigallocatechin-3-gallate and (−)-epigallocatechin, into human plasma. Biosci Biotechnol Biochem. 1997;61:1981–1985. doi: 10.1271/bbb.61.1981. [DOI] [PubMed] [Google Scholar]
- Pae M, Ren Z, Meydani M, Shang F, Smith D, Meydani SN, Wu D. Dietary supplementation with high dose of epigallocatechin-3-gallate promotes inflammatory response in mice. J Nutr Biochem. 2012;23:526–531. doi: 10.1016/j.jnutbio.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Mereles D, Hunstein W. Epigallocatechin-3-gallate (EGCG) for clinical trials: more pitfalls than promises? Int J Mol Sci. 2011;12:5592–5603. doi: 10.3390/ijms12095592. [DOI] [PMC free article] [PubMed] [Google Scholar]