

Abstract
Using model (R)-2-acetyl-2-phenyl acetate esters of (S)- or (R)-α-substituted-p-hydroxybutyrophenones (S,R)-12a and (R,R)-12b, we have shown that a highly efficient photo-Favorskii rearrangement proceeds through a series of intermediates to form racemic rearrangement products. The stereogenic methine on the photoproduct, rac-2-(p-hydroxyphenyl)propanoic acid (rac-9), is formed by closure of a phenoxy-allyloxy intermediate 17 collapsing to a cyclopropanone, the “Favorskii” intermediate 18. These results quantify the intermediacy of a racemized triplet biradical 316 on the major rearrangement pathway elusively to the intermediate 18. Thus, intersystem crossing from the triplet biradical surface to the ground state generates a planar zwitterion prior to formation of a Favorskii cyclopropanone that retains no memory of its stereochemical origin. These results parallel the mechanism of Dewar and Bordwell for the ground state formation of cyclopropanone 3 that proceed through an oxyallyl zwitterionic intermediate. The results are not consistent with the stereospecific SN2 ground state Favorskii mechanism observed by Stork, House, and Bernetti. Interconversion of the diastereomeric starting esters of (S,R)-12a and (R,R)-12b during photolysis did not occur thus ruling out leaving group return prior to rearrangement.
Keywords: Photochemistry, Stereochemistry, Photo-Favorskii rearrangement, triplet biradical, p-Hydroxyphenacyl, Zimmerman paradigm
Introduction
The p-hydroxyphenacyl (pHP) chromophore has been established as a clean, efficient photoremovable protecting group (PPG) for nucleofuges such as phosphates, sulfonates, carboxylates, and certain thiols and phenols.,1,2 The chromophore imparts good hydrophilicity, excellent biological compatibility, and relatively easy installation of the protected moiety. In aqueous media, the photorelease is accompanied by rearrangement of the chromophore, originally described as a photo-Favorskii rearrangement by Reese and Anderson,3 that results in a dramatic hypsochromic shift in its absorption spectrum. The release of the leaving groups occurs rapidly (kr = 108–109 s−1) in quantitative chemical yield and the quantum yields approach 1.0. Together, these properties have encouraged us and others to investigate further applications2 including the new chemical transformations such as ring contraction reactions.4 In turn, there has been a renewed interest in the mechanism of the photo-Favorskii rearrangement, itself.1,5,6
For example, controversy surrounds the extent to which two transient intermediates, the triplet biradical 32 and the spirodienone 3, play a role in the photo-Favorskii rearrangement. The chronological sequence which we embraced for the photo-Favorskii rearrangement (Scheme 1) includes 32 and 5, two detectable transients using time-resolved (TR) transient absorption spectroscopy.5b The earliest transient (λmax 445 and 455 nm) has been assigned the structure of the triplet allyloxy-phenoxy biradical (32), which is formed from 31. It decays with a rate constant of kbirad = 1.5 × 109 s−1 to form cyclopropanone 3, the “Favorskii” intermediate,3 which has eluded detection thus far. Nevertheless, 3 serves as the necessary bridge between the structure of the pHP chromophore in 1 and its photoproducts, p-hydroxyphenylacetic acid (4) and p-hydroxybenzyl alcohol (6). Phenylacetic acid 4 is formed from hydrolysis of 3 whereas decarbonylation forms 5 (a p-methylenequinone) that hydrolyzes to p-hydroxybenzyl alcohol 6.5b, 7 However, evidence for intermediate 2 and its subsequent conversion to 3 is incomplete and circumstantial, leading others to propose alternative mechanisms that either bypassed the biradical 32 intermediate 5c,6a–g and, in at least one report,6h completely disregarded the Favorskii cyclopropanone 3, altogether.
Scheme 1. General Mechanism for the photo-Favorskii Rearrangement.

The stereochemical integrity of the migrating methylene group should serve as a quantitative measure by the degree of racemization resulting from formation of a racemic intermediate such as biradical 2. Indeed, stereochemistry has often been employed in mechanistic investigations for both ground state and photochemical rearrangements as documented, for example, in the earlier work of Zimmerman’s studies of the Type A photorearrangements of dienones.8 Rrelevant examples are studies of the stereochemistry of the ground state Favorskii rearrangement extensively examined by House,9 Sorenson,10 Bernetti,11 and by Bordwell.12 The seminal studies by Loftfield13 on the 1,2-carbon migration later challenged by Dewar’s theoretical treatment14 now serve as the two limiting mechanisms for the base-promoted Favorskii rearrangement. Loftfield’s one-step concerted SN2 process contrasts with Dewar‘s two-step preformation of an oxyallyl zwitterion intermediate prior to cyclization by disrotatory closure15 to generate the Favorskii cyclopropanone intermediate. While both mechanisms involve the intermediacy of a cyclopropanone, the stereochemical consequences of a migrating stereogenic carbon are clearly distinguishable and both limiting mechanisms have experimental support.8–11,16,17,18
With this abundance of precedent of the limiting ground state mechanisms, we were persuaded to pursue the stereochemistry of the photochemically driven Favorskii rearrangement. Our model employs a p-hydroxybutyrophenone equipped with a chiral leaving group thus providing us with a pair of diastereomeric esters to test the stereochemical changes at the migrating carbon.
Results and Discussion
A convenient framework for testing the two limiting Favorskii rearrangement pathways is depicted in Scheme 2 using p-hydroxybutyrophenone 7. The stereochemical integrity of the migrating α-carbon (marked as *) was monitored by HPLC (chiral stationary phase) and by 1H NMR.
Scheme 2.

Loftfield and Dewar Limiting Mechanisms as Applied to Photolysis of pHP Homologues.
Extension of the pHP aliphatic chain to p-hydroxybutyrophenone raises the possibility of photochemical competition by a Norrish Type II fragmentation instead of the photo-Favorskii rearrangement.19 However, when the normally efficient Type II reaction was pitted against the photo-Favorskii rearrangement with a series of o-methyl pHP analogs, the Type II reaction could be completely quenched by photolysis in solutions containing >15% H2O which gave exclusively photo-Favorskii products. Solvents with less than 10% H2O yielded only Type II products.20
Synthesis and assignment of stereochemistry
Enantiomerically pure (R)-2-acetoxyphenylacetic acid ((R)-11) was reacted with α-bromo-p-hydroxybutyrophenone (rac-10) through an SN2 displacement of bromide to provide the pair of diastereomeric esters 12a,b as a 1:1 mixture (Scheme 3).21,22
Scheme 3.

Synthethesis of Diastereomeric Isomers 12a and 12b.
Choice of the leaving group 11 served the dual purpose of creating a pair of separable diastereomeric esters as well as a direct, internal diagnostic for assigning the configuration of the butyrophenone stereogenic center. The assignment of the absolute configuration of each diastereomer23 was based on the shielding effects of the (R)-11 aryl ring on identifiable elements on the butyrophenone fragment. Using chemical shift correlations analogous to those described by Trost for an O-methylmandelate model24 (Figure 1), the configurations of (S)-1-(4-hydroxyphenyl)-1-oxobutan-2-yl (R)-2-acetoxy-2-phenylacetate for 12a and (R)-1-(4-hydroxyphenyl)-1-oxobutan-2-yl (R)-2-acetoxy-2-phenylacetate for 12b were assigned.
Figure 1.

Fragments of 1H NMR chart of a diastereomeric mixture 12a and 12b. The intervening carboxyl groups on structures A and B are omitted in order to emphasize the phenyl-aryl and phenyl-ethyl shielding interactions for the most stable rotamers of (S,R)-12a and (R,R)-12b, the major contributors to the shielding effects for the chemical shift differences. The more shielded component (highlighted in blue) of each diastereomer is shown to be eclipsed by the phenyl ring of 11 (shown in red).
Photochemistry
The 1:1 diastereomer mixture of 12a,b (30 mmol in 0.6 ml of 4:1 CD3CN:D2O) was irradiated in an NMR tube for 49 min. to nearly complete conversion using a Rayonet reactor equipped with two 15 W 300-nm lamps. Two major products were detected by 1H NMR analysis of the crude reaction mixture; the chiral leaving group (R)-11 (100%) and the rearranged 2-(4-hydroxyphenyl)butanoic acid25 (9, 82%). Two minor products, 4-(1-hydroxypropyl)phenol (13, 7%)26 and 2-hydroxy-1-(4-hydroxyphenyl)butan-1-one (14, 3%) were also detected and identified (Fig. 2 and Figure 7, Experimental Section). Hydroxy acid 14 is apparently formed by photohydrolysis whereas the benzyl alcohol 13, a ubiquitous minor product in our studies, is formed from the photo-Favorskii cyclopropanone 18. Decarbonylation of 18 gave 4-propoylidenecyclohexa-2,5-dien-1-one (19), an analog of p-methylenequinone (5, Scheme 2), which hydrolyzes to 13. No evidence for competing Type II photoreaction of the butyrophenone was observed as anticipated when H2O solvents are employed.19.20
Figure 2.

1H NMR spectra during photolysis of 12a. The arrows point to the stereogenic methine protons for the two minor (13 and 14) and the major (9) products. See Figure 6 (Experimental section) for the assignments.
Figure 7.

(A) Reaction mixture A obtained from photolysis of 12a stopped at ~30% conversion (See Figures 2 and 6); (B) Mixture A spiked with an authentic sample of 13; (C) Mixture A spiked with an authentic sample of 14
The stereochemical consequences of the rearrangement were assessed by irradiation of the individual diastereomers. Photolysis of (S,R)-12a produced racemic α-arylbutanoic acid (rac-9: See Supporting Information). Complete racemization was confirmed for the reaction when diastereomeric (R,R)-12b also gave only racemic products (Scheme 5).
Scheme 5.

Photochemical Integrity of the Butyrophenone Stereogenic Center of (S,R)-12a and (R,R)-12b.
In a control reaction monitored by 1H NMR, 12a was not converted to its diastereomer 12b during photolysis ruling out a recombination reaction which would generate the diastereomeric mixture of the esters (Fig. 3), thus assuring the integrity of starting esters (S,R)-12a and (R,R)-12b throughout the photolysis and demonstrating that rac-9 was not the result of an a priori equilibration of the stereogenic center (Scheme 5). Quantum yields are also consistent with irreversible release of the acid (Φdis = 0.54 (± 0.05) for 12a and b) and are among the most efficient pHP arboxylates.
Figure 3.

Portions of 1H NMR spectra of 12b at t = 14 min (top) at 40% conversion at t = 0 min for 12b, and at t = 0 min. (lower) for 12a. The broadening and added complexity of the 1H NMR signals at t = 14 min surrounding the terminal CH3 group at 0.8 ppm arise from the contributions from the terminal CH3 groups of products 9, 13, and 14. (See Experimental for further details.)
A remaining fundamental questions are the nature of the ring closure to cyclopropanone 3 which is not defined in Scheme 1 and the origin of the hydroxyketone 14 which does not appear in most treatments of the photo-Favorskii reaction mechanism. Here we draw upon the Zimmerman paradigm developed in his early work on the photorearrangement of 2,5-cyclohexadien-1-one to bicycle[3.1.0]hexenone. He suggested that photochemical reactions might be viewed as four common steps: (1) excitation, (2) adiabatic bond alteration and (3) demotion or relaxation to the ground state energy surface where (4) further bond alteration is proceeding along a classical ground state surface to the final photoproducts.8,27 Steps 2 and 4 are classical electron pushing approaches toward defining a mechanism. We have applied the approach to the photorearrangement of 12a,b and to our recent study on photo-Favorskii ring contraction reaction of cyclic analog 20 shown together in Scheme 6.
Scheme 6. Contrasting and Comparing the Cyclic (20) and Acyclic (12a) p-Hydroxybutyrophenone Pathways.

Formation of cyclopropanone 18 (or 24) is a prohibitively high energy process on the triplet excited state surface5b and is spin forbidden; therefore, it must occur from a relaxed singlet oxyallyl biradical (open shell) or closed shell counterpart, the ground state Dewar zwitterion (Scheme 2). In ground state Favorskii rearrangements where the Dewar mechanism obtains, the reaction leads to complete loss of the stereochemical integrity of the α-carbon. For our photo-Favorskii study, the α-carbon is completely racemized at the final productstage, e.g. rac9, thus quantifying the extent of involvement of the oxyallyl biradical 16 intermediate. In our previous study, the transient nature of the oxyallyl biradical limited our assessment of its relative significance as an intermediate in the formation of the phenylacetic acid.5b
The two minor products from the photo-Favorskii rearrangement, hydroxyphenol 13 and hydroxyketone 14 provide additional insight. The hydroxyphenol 13, always seen as a minor product, is formed by decarbonylation of the cyclopropanone 18 followed by hydrolysis4,5b,6 yielding racemic p-hydroxyphenyl propanol ((±)-13; Scheme 6). We previously denoted this product as a signature of the intermediacy of the Favorskii cyclopropanone 18.5,6
The origin of the hydroxyketone 14 is less apparent and less frequently encountered. Corri and Wan5c first reported this minor photohydrolysis product which we have subsequently confirmed.4,5a,b,d Most recent, cycloalkanone 20 (Scheme 6), which may be viewed as having the terminal carbon of the butyl group on butyrophenone bonded directly to the aryl ring, yields a hydroxyketone, e.g. 23, as the major photoproduct (57% vs. 3% for 14) upon photolysis.4 In solvent containing 18O-labeled water, the hydroxyl group was contained the 18O-label. The mechanism that incorporates the previous findings with this stereochemical study suggests that the hydrolysis reaction also results from divergence of the closure pathway for the zwitterion 21 toward reaction with the solvent. The very minor proportion of the bifurcation from Favorskii toward hydrolysis this reaction is the lack of steric or strain interfering with ring closure for 17 vis-a-vis 22. The previous study demonstrated that ring strain was a major factor in the partitioning of the two pathways: smaller cycloalkanone rings favored formation of the hydroxyketone while larger rings and acyclic ketones gave predominantly the rearranged acids.4 The low yield of 14 from 12 accords well with the increase in ring strain of 24. The results further suggest that the butyryl group does not cause sufficient steric congestion to divert zwitterion 17 away from the Favorskii pathway. It is noteworthy that the quantum efficiencies are not diminished, supporting the conclusion that the changes in product ratios are independent of the photorelease of the leaving group, but are subject to the partitioning of a common intermediates, e.g. the zwitterions 17 and 22. Similar results are found for the ground state, ZnCl2-catalyzed Favorskii rearrangements of the corresponding bromides where the formation of the same two products are formed by competition between solvolysis and rearrangement.4,28 These results support a mechanism where the rearrangements emanate from a ground state zwitterion 17, i.e., step 4 in the Zimmerman paradigm.8,27
Conclusions
The most attractive sequence that accomplishes both the rearrangement and complete equilibration of the stereogenic center to form rac-9 begins with the irreversible heterolysis of the leaving group from the chromophore from the chiral triplet, 312a or 312b, forming an achiral, and planar triplet biradical 316. All subsequent products must be racemic. Subsequent relaxation of the triplet biradical 316 to its open shell singlet 116 then to the closed shell Dewar intermediate 1729 followed by ring closure of the oxyallyl-phenoxy intermediate provides a route to the racemic products (Scheme 6). Furthermore, the triplet biradical 32 observed in our earlier TR laser flash absorption studies5b accounts for the complete racemization.
The results do not accord with a stereospecific, SN2-like pathways analogous to those observed for several ground state Favorskii rearrangements, e.g., the work of Stork,16a House,9 Bernetti,11 and Sorenson.10 Instead, the complete racemization of pHP analogs involves an oxyallyl-phenoxy intermediate paralleling the ground state mechanism of Dewar14 and documented by Bordwell.12,17 Alternative routes, e.g., equilibration of the starting esters or a concerted formation of p-hydroxybenzyl acylium intermediates,6h do not occur.
Experimental
General Information
NMR spectra were recorded on a 400 MHz instrument, equipped with a quadruple-band gradient probe (H/C/P/F QNP) or a 500 MHz with a dual carbon/proton cryoprobe (CPDUL). 13C NMR spectra were recorded with broad-band decoupling. Chiral HPLC was performed using Chiralcel OD-H column (250 × 4.6 mm, 10% IPA in hexanes) using a UV-VIS detector tuned to 254 nm. Photolysis studies were carried out in a rotating merry-go-round apparatus fitted into a Rayonet reactor equipped with two 15 W 300-nm lamps.
Anhydrous acetonitrile was obtained by passing degassed HPLC-grade consecutively through two columns filled with activated alumina. Anhydrous DMF was obtained by distillation of ACS-grade DMF over calcium hydride under a nitrogen atmosphere. α-Bromo-p-hydroxybutyrophenone 10,30 4-(1-hydroxypropyl) phenol (13)26, and racemic 2-(4-hydroxyphenyl)butanoic acid (9)25 have been described previously. (R)-(−)-O-Acetylmandelic acid ((R)-11) is commercially available.
Synthesis of α-bromo-p-hydroxybutyrophenone (10)

A 50 mL RBF was charged with 1.00 g (6.09 mmol) of 1-(4-hydroxyphenyl)butan-1-one, 2.99 g of CuBr2 (13.4 mmol) and 30 mL of 1:1 EtOAc/CHCl3. The heterogeneous solution was brought to reflux and, with vigorous stirring, preceded for 12 h. After this time, the solution was cooled to room temperature, and supervening filtration through a plug of silica afforded, after inspissation, 1.50 g of a white, crystalline material that, through 1H NMR analysis (CDCl3) adduced 2-bromo-1-(4-hydroxyphenyl)butan-1-one (9) as the primary product with trace amounts of the starting material and the dibrominated analog. Isolation of 10 was not essential for the next reaction step; however, could be achieved by flash chromatography with EtOAc/Hex (1:2) as the eluent.
Synthesis of 1-(4-hydroxyphenyl)-1-oxobutan-2-yl (R)-2-acetoxy-2-phenylacetate (12a,b)
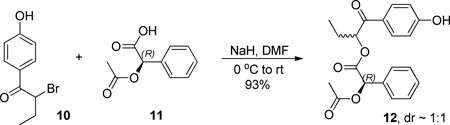
A flame-dried 50 ml RBF was charged with 1.00 g (5.15 mmol) of (R)-2-acetoxy-2-phenylacetic acid 11 and 20 ml of dry DMF. The corresponding solution was cooled to −10 °C with an ice/saturated, aqueous NaCl bath, when, while vigorously stirring, 206 mg of NaH (60% w/w in mineral oil, 5.15 mmol) was added. The suspension brought to room temperature with further stirring for 30 minutes before 1.04 g (4.29 mmol) of 9 (2-bromo-1-(4-hydroxyphenyl)butan-1-one, racemic) was added. The reaction proceeded overnight, after which the solution was diluted with EtOAc and washed profusely with H2O. Flash column chromatography (1:1 EtOAc/Hex) afforded 1.71 g of 12a ((R)-1-(4-hydroxyphenyl)-1-oxobutan-2-yl (R)-2-acetoxy-2-phenylacetate (R)-pHP (R)-APAc)) and 12b ((S)-1-(4-hydroxyphenyl)-1-oxobutan-2-yl (R)-2-acetoxy-2-phenylacetate (S)-pHP (R)-APAc)) as a ~1:1 mixture, in 93% yield, as white crystalline solid.
Resolution of the diastereomeric mixture was accomplished as follows: a 1:1 mixture of diastereomers 12a,b was dissolved in minimum amount of hot ether and dichloromethane (~3:1), and refluxed until all solid was dissolved. Cyclohexane was added gradually, until crystals began to form. The mixture was then cooled in a refrigerator overnight. Crystals were collected by vacuum filtration to give (R,R)-12b with 90% dr. Mother liquor was concentrated to give (S,R)-12a with 85% dr. Additional recrystallization of the diastereomers from ethyl acetate provided (R,R)-12b and (S,R)-12a with >98% dr as determined by 1H NMR analysis. The absolute configurations of the resolved esters were established by 1H NMR using chemical shift correlations analogous to the Trost’s model developed for O-methylmandelates (Figure).24 The extended Newman projections A and B with omitted intervening ester linkage represent the most stable conformations, which contribute the most to the observed chemical shifts of diastereomers (S,R)-12a and (R,R)-12b. The substituents shown in blue, eclipsing the mandelate phenyl rings (shown in red) are expected to be shifted upfield as a result of anisotropic shielding interaction. Accordingly, the absolute configuration (S,R) was assigned to diastereomer 12a based on the observed upfield shift of a triplet component of the A3X2 system corresponding to the ethyl group (0.75 ppm). Analogously, shielding of both doublet components (6.79 and 7.78 ppm) of the AA’XX’ system corresponding to the para-substituted aromatic ring allowed for assigning the (R,R)-configuration to diastereomer 12b (Figure).
(S,R)-12a: m.p. 154–155 °C. 1H NMR (500.13 MHz, CD3CN) δ 7.94-7.76 (m, 2H), 7.56 (dd, J = 6.6 Hz, 2.8 Hz, 2H), 7.49-7.43 (m, 3H), 6.90 (d, J = 8.8 Hz, 2H), 6.02 (s, 1H), 5.76 (dd, J = 8.7 Hz, 3.9 Hz, 1H), 2.15 (s, 3H), 1.93-1.83 (m, 1H), 1.86-1.71 (m, 1H), 0.84 (t, J = 7.4 Hz, 3H); 13C NMR (125.76 MHz, CD3CN) δ 195.1, 171.4, 169.8, 163.3, 135.4, 132.3 (2C), 130.7, 130.1 (2C), 129.2 (2C), 127.9, 116.8 (2C), 78.6, 75.7, 26.1, 21.2, 10.3; FT-IR (KBr, cm−1): 3202, 2980, 1757, 1742, 1726, 1686, 1659, 1603, 1570, 1518, 1458, 1441, 1373, 1267, 1227, 1215, 1173, 1097, 1055, 1041, 933, 901, 856, 843, 750, 698, 685, 623, 513; HRMS (TOF ESI) calculated for M+Li = C20H20O6Li: 363.1420; Found: 363.1423.
(R,R)-12b: m.p. 166–167 °C. 1H NMR (500.13 MHz, CD3CN) δ 7.76 (d, J = 8.8 Hz, 2H), 7.79 (br. s., 1H), 7.49 (dd, J = 6.5 Hz, 3.0 Hz, 2H), 7.45-7.40 (m, 3H), 6.82 (d, J = 8.8 Hz, 2H), 6.01 (s, 1H), 5.80 (dd, J = 8.0 Hz, 4.3 Hz, 1H), 2.11 (s, 3H), 1.98-1.88 (m, 1H), 1.82-1.72 (m, 1H), 0.95 (t, J = 7.4 Hz, 3H); 13C NMR (125.76 MHz, CD3CN) δ 194.7, 171.2, 169.4, 162.9, 134.7, 131.9 (2C), 130.3, 129.7 (2C), 129.0 (2C), 127.6, 116.4 (2C), 78.3, 75.2, 25.7, 20.9, 9.9; FT-IR (KBr, cm−1): 3364, 2984, 1747, 1730, 1688, 1599, 1585, 1512, 1441, 1375, 1350, 1281, 1265, 1231, 1213, 1173, 1097, 1057, 935, 901, 856, 843, 750, 696, 685, 669, 660, 615, 532, 513, 401. HRMS (TOF ESI) calculated for M+H = C20H21O6: 357.1338; Found: 357.1339.
Synthesis of 2-hydroxy-1-(4-hydroxyphenyl)butan-1-one (14)

2-Hydroxy-1-(4-hydroxyphenyl)butan-1-one was obtained from bromide 10 following the previously reported procedure.6d Sodium formate (3.82 g, 112.2 mmol) was dissolved in ethanol (150 mL) and bromide 10 (2.124 g, 8.74 mmol) was added. The resulting mixture was stirred at reflux (78 °C) for 1 day, after which time the mixture was filtered hot and concentrated in vacuo. The crude residue was crystallized from ethyl acetate:hexane to give 2-Hydroxy-1-(4-hydroxyphenyl)butan-1-one (14) (0.757 g, 4.2 mmol, 48%) as white crystalline solid. m.p. 123–124 °C. 1H NMR (500.13 MHz, CD3CN) d 7.89 (d, J = 8.5 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 4.99 (br. s., 1H), 3.69 (d, J = 6.3 Hz, 1H), 2.34 (br. s., 1H), 1.87 (dtd, J = 14.4 Hz, 7.3 Hz, 3.9 Hz, 1H), 1.55 (dq, J = 14.3 Hz, 7.2 Hz, 1H), 0.91 (t, J = 7.4 Hz, 3H); 13C NMR (125.76 MHz, CD3CN) d 201.4, 163.0, 132.2 (2C), 127.3, 116.4 (2C), 74.4, 29.8, 9.4; FT-IR (KBr, cm−1): 3223, 3072, 3026, 2968, 2935, 2912, 2876, 1663, 1603, 1591, 1516, 1445, 1439, 1406, 1313, 1279, 1256, 1238, 1167, 1128, 1113, 1092, 1059, 970, 960, 874, 852, 841, 829, 810, 771, 735, 712, 677, 613, 509, 494; HRMS (TOF ESI) calculated for M+ = C10H12O3: 180.0786; Found: 180.0786.
General procedure for photolysis
Ester 12a (11 mg, 0.03 mmol) was placed in an NMR tube and dissolved in a 4:1 mixture of CD3CN:D2O. The sample was irradiated in a Rayonet reactor equipped with two 15 W RPR3000 lamps for 50 min. The reaction course was monitored by 1H NMR (Figure). The NMR yield of the released (R)-2-acetoxy-2-phenylacetic acid (11) and the rearranged product 9 was found to be 100% and 82%, respectively. Upon completion of the reaction, the crude mixture was concentrated and the main products were isolated by acid-base extraction. The products identity was confirmed by comparison of their 1H and 13C NMR spectra with those of the authentic samples. The identities of the minor side products were established by spiking the reaction mixture with authentic samples of the corresponding compounds (Figure 6).
Figure 6.

Photolysis of 12a at complete conversion to products. See Discussion and Figure 2 for more detail.
Quantum yield measurements
Actinometry: Light output (in mEinsteins/min) was measured by the potassium ferrioxalate method.31 The solutions were prepared and irradiated as described above in the General Procedure. The 1H NMR spectra of the sample were measured at 10 min intervals for up to 40 min. Proton signal of the remaining starting material (singlet at 6.0 ppm) was integrated against signals of DMF as the internal standard. The results are given in the Results and Discussion section.
Supplementary Material
Figure 4.

Chemical shift correlation model for assigning the absolute configurations of (S,R)-pHP (R)-APAc) 12a, b.
Figure 5.

Fragments of 1H NMR charts used for assigning of the absolute configuration of 12a,b: (A) a 63:37 mixture of 12a and 12b. (B) pure 12a. (C) pure 12b.
Scheme 4.

Photochemistry of (S)-1-(4-hydroxyphenyl)-1-oxobutan-2-yl (R)-2-acetoxy-2-phenylacetate ((S,R)-12a).
Acknowledgements
Support for this work was provided by NIH grant R01 GM72910 (RSG).
Footnotes
Supporting Information Available: 1H, 13C NMR, absorption and HRMS spectra are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a. Givens RS, Rubina M, Wirz J. Photochem. Photobiol. Sci. 2012;11:472. doi: 10.1039/c2pp05399c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b. Klán P, Šolomek T, Bochet C, Blanc A, Givens RS, Rubina M, Popik V, Kostikov A, Wirz J. Chem. Rev. 2012;12 doi: 10.1021/cr300177k. 0000 (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]; Klan P, Wirz J. c. Photochemistry of organic compounds: From concepts to practice. 1st ed. Chichester: John Wiley & Sons Ltd.; 2009. [Google Scholar]; Givens RS, Goeldner M, editors. d. Dynamic Studies in Biology: Phototriggers, Photoswitches, and Caged Biomolecules. J. Wiley, and Sons, Inc.; 2005. [Google Scholar]; d. Pelliccioli AP, Wirz J. Photochem. Photobiol. Sci. 2002;1:441. doi: 10.1039/b200777k. [DOI] [PubMed] [Google Scholar]; e. Givens RS, Weber JFW, Jung AH, Park C-H. New photoprotecting groups: desyl and p-hydroxyphenacyl phosphate and carboxylate esters, Methods Enzymol. 1998;Vol. 291:1–29. doi: 10.1016/s0076-6879(98)91004-7. (Caged Compounds) [DOI] [PubMed] [Google Scholar]
- 2.a. C. J. Pickens CJ, Gee KR. Tetrahedron Lett. 2011;52:4989. [Google Scholar]; b. Kötting C, Güldenhaupt J, Gerwert K. Chem.Phys. 2012;396:72. [Google Scholar]; c. Sot B, Kötting C, Deaconescu D, Suveyzdis Y, Gerwert K, Wittinghofer A. EMBO J. 2010;29:1205. doi: 10.1038/emboj.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]; d. Brucker C, Gerwert K, Kötting C. J. Mol.Biol. 2010;401:1–6. doi: 10.1016/j.jmb.2010.05.068. [DOI] [PubMed] [Google Scholar]; e. Kötting C, Gerwert K. Chem. Phys. 2004;307:227. [Google Scholar]; f. Stensrud K, Noh J, Kandler K, Wirz J, Heger D, Givens RS. J. Org. Chem. 2009;74:5219. doi: 10.1021/jo900139h. [DOI] [PMC free article] [PubMed] [Google Scholar]; g. Kötting C, Kallenbach A, Suveyzdis Y, Wittinghofer A, Gerwert K. Proc. Nat. Acad. Sci. 2008;105:6260. doi: 10.1073/pnas.0712095105. [DOI] [PMC free article] [PubMed] [Google Scholar]; h. Warscheid B, Brucker S, Kallenbach A, Meyer HE, Gerwert K, Kötting C. Vibrational Spect. 2008;48:28. [Google Scholar]; i. Kötting C, Kallenbach A, Suveyzdis Y, Eichholz E, Gerwert K. ChemBioChem. 2007;8:781. doi: 10.1002/cbic.200600552. [DOI] [PubMed] [Google Scholar]; j. Kötting C, Gerwert K. Chem. Phys. 2004;307:227. [Google Scholar]; k. Sul J-Y, Orosz G, Givens RS, Haydon PG. Neuron Glia Biology. 2004;1:3. doi: 10.1017/s1740925x04000031. [DOI] [PMC free article] [PubMed] [Google Scholar]; l. Specht A, Loudwig S, Peng L, Goeldner M. Tetrahedron Lett. 2002;43:8947–8950. [Google Scholar]; m. Du X, Frei H, Kim S-H. J. Biol. Chem. 2000;275:8492. doi: 10.1074/jbc.275.12.8492. [DOI] [PubMed] [Google Scholar]
- 3.Anderson JC, Reese CB. Tetrahedron Lett. 1962;1:1. [Google Scholar]
- 4.Kammatah VB, Solomec T, Ngoy BP, Heger D, Klan P, Rubina M, Givens RS. J. Org Chem. 2012;77 doi: 10.1021/jo300850a. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a. Conrad PG, II, Givens RS, Hellrung B, Rajesh CS, Ramseier M, Wirz J. J. Am. Chem. Soc. 2000;122:9346. [Google Scholar]; b. Givens RS, Heger D, Hellrung B, Kamdzhilov Y, Mac M, Conrad PG, Cope E, Lee JI, Mata-Segreda JF, Schowen RL, Wirz J. J. Am. Chem. Soc. 2008;130:3307. doi: 10.1021/ja7109579. [DOI] [PMC free article] [PubMed] [Google Scholar]; c. Zhang K, Corrie JET, Munasinghe VRN, Wan P. J. Am. Chem. Soc. 1999;121:5625. [Google Scholar]; d. Stensrud KF, Heger D, Sebej P, Wirz J, Givens RS. Photochem. Photobiol. Sci. 2008;7:614. doi: 10.1039/b719367j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a. Ma C, Zuo P, Kwok WM, Chan WS, Kan JTZ, Toy PH, Phillips DL. J. Org. Chem. 2004;69:664. doi: 10.1021/jo049331a. [DOI] [PubMed] [Google Scholar]; b. Ma C, Chan WS, Kwok WM, Zuo P, Phillips DL. J. Phys. Chem. B. 2004;108:9264. [Google Scholar]; c. Ma C, Kwok WM, Chan WS, Zuo P, Kan JTW, Toy PH, Phillips DL. J. Am. Chem. Soc. 2005;127:1463. doi: 10.1021/ja0458524. [DOI] [PubMed] [Google Scholar]; d. Ma C, Kwok WM, Chan WS, Du Y, Kan JTW, Phillips DL. J. Am. Chem. Soc. 2006;128:2558. doi: 10.1021/ja0532032. [DOI] [PubMed] [Google Scholar]; e. Chan WS, Kwok WM, Phillips DL. J. Phys. Chem. A. 2005;109:3454–3469. doi: 10.1021/jp044546+. [DOI] [PubMed] [Google Scholar]; f. Zuo P, Ma C, Kwok WM, Chan W, Phillips DL. J. Org. Chem. 2005;70:8661. doi: 10.1021/jo050761q. [DOI] [PubMed] [Google Scholar]; g. Chen XB, Ma C, Kwok WM, Guan X, Du Y, Phillips DL. J. Phys Chem. A. 2006;110:12406. doi: 10.1021/jp064490e. [DOI] [PubMed] [Google Scholar]; h. Cao Q, Guan XG, George MW, Phillips DL, Ma CS, Kwok WM, Li MD, Du Y, Sun XZ, Xue JD. Faraday Discussions. 2010;145 171, and 255 discussion of the paper. [Google Scholar]
- 7.Chiang Y, Kresge AJ, Zhu Y. J. Am. Chem. Soc. 2002;124:6349. doi: 10.1021/ja020020w. [DOI] [PubMed] [Google Scholar]
- 8.Zimmerman HE. Tetrahedron. 1974;30:1617. [Google Scholar]
- 9.House HO, Richey FA., Jr J. Org. Chem. 1967;32:2151. [Google Scholar]
- 10.Sorenson TS, Sun F. Canad. J. Chem. 1997;75:1030. [Google Scholar]
- 11.Ambrosini M, Baricordi N, Benetti S, De Risi C, Pollini GP, Zanirato V. Tetrahedron Asym. 2009;20:2145. [Google Scholar]
- 12.Bordwell FG, Strong JG. J. Org. Chem. 1973;38:579. [Google Scholar]
- 13.a. Loftfield RB. J. Am. Chem. Soc. 1950;72:632. [Google Scholar]; b. Loftfield RB. J. Am. Chem. Soc. 1951;73:4707. [Google Scholar]
- 14.a. Burr JG, Dewar MJS. J. Chem. Soc. 1954:1201. [Google Scholar]; b. Aston JG, Newkirk JD. J. Am. Chem. Soc. 1951;73:3900. [Google Scholar]
- 15.This was later defined in R. B. Woodward and R. Hoffman The Conservation of Orbital Symmetry 1970 Verlag Chemie GmbH, Weinheim.
- 16.a. Stork G, Borowitz IJ. J. Am. Chem. Soc. 1960;82:4307. [Google Scholar]; Kende AS. b. The Favorski rearrangement of haloketones. In: Cope AC, editor. Org. Reactions. Vol. 11. 1960. p. 261. [Google Scholar]
- 17.a. Bordwell FG, Almy J. J. Org. Chem. 1973;38:571. [Google Scholar]; b. Bordwell FG, Scamehorn RG, Springer WR. J. Am. Chem. Soc. 1969;91:2087. [Google Scholar]; c. Bordwell FG, Scamehorn RG. J. Am. Chem. Soc. 1968;90:6751. [Google Scholar]; d. Bordwell FG, Frame RR, Scamehorn RG, Strong JG, Meyerson S. J. Am. Chem. Soc. 1967;89:6704. [Google Scholar]
- 18.Theoretical treatments of the stereochemistry of Favorskii stereochemistry are found in: a. Hamblin GD, Jimenez RP, Sorensen TS. J. Org. Chem. 2007;72:8033–8045. doi: 10.1021/jo701351p. b. Tsuchida N, Yamazaki S, Yamabe S. Org. Biomol. Chem. 2008;6:3109. doi: 10.1039/b806577b.
- 19.a. Wagner PJ. Acc. Chem. Res. 1971;4:168. [Google Scholar]; b. Wagner PJ, Kemppain.Ae, Kochevar IE. J. Am. Chem. Soc. 1972;94:7489. [Google Scholar]
- 20.Šebej P, Lim BH, Park BS, Givens RS, Klán P. Org. Lett. 11:644. doi: 10.1021/ol102887f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King LC, Ostrum GK. J. Org. Chem. 1964;29:3459. [Google Scholar]
- 22.See the Experimental section for more detail on the synthesis and stereochemical assignments and Supporting Information for spectra and details on the photo racemization results.
- 23.Separation of diastereomers was accomplished by fractional crystallization of 12a,b to provide (R,R)-12b and (S,R)-12a each with >98% dr; see Supporting Information for details.
- 24.Trost BM, Belletire JL, Godleski S, McDougal PG, Balkovec JM, Baldwin JJ, Christy ME, Ponticello GS, Varga SL, Springer JP. J. Org. Chem. 1986;51:2370. [Google Scholar]
- 25.Skaddan MB, Katzenellenbogen JA. Bioconjugate Chem. 1999;10:119. doi: 10.1021/bc980094q. [DOI] [PubMed] [Google Scholar]
- 26.Angle SR, Arnaiz DO. J. Org. Chem. 1992;57:5937. [Google Scholar]
- 27.Zimmerman HE. a. In: Adv. Photochem. Noyes WAJ, Hammond GS, Pitts JNJ, editors. Vol. 1. New York: Interscience; 1963. p. 183. [Google Scholar]; b. Zimmerman HE, Schuster DI. J. Am. Chem. Soc. 1962;84:4527. [Google Scholar]
- 28.a. Mandal AN, Bhattacharya S, Raychaudhuri SR, Chatterjee A. J. Chem. Res. 1988;366 [Google Scholar]; b. Maiti SB, Chaudhuri SRR, Chatterjee A. Synthesis. 1987:806. [Google Scholar]
- 29.Two recent studies on the generation of triplet oxyallyl diradicals have established that the ground state is the singlet state. For parent oxyallyl, the ground state singlet is 55 meV more stable than its excited triplet. (Ichino, T.; Villano, S. M.; Gianola, A. J.; Goebbert, D. J.; Verarde, L.; Sanov, A.; Blanksby, S. J.; Zhou, X.; Hrovat, D. A.; Borden, W. T.; Lineberger, W. C., J. Phys. Chem. 2011, 115, 1634). The generation of the biradical by photodecarbonylation of a substituted I,3-cyclobutanedione generated a long-lived singlet ground state oxyallyl intermediate (τ1/2 = 42 min) in the crystalline solid state at 298 K. In solution, the singlet oxyallyl’s barrier to cyclopropanone formation was 2.6 kcal/mol with a lifetime of a few picoseconds. (Kuzmanich, G.; Spanig, F.; Tsai, C-K.; Um, J. M.; Hoekstra, R. M.; Houk, K. N.; Guldi, D. M.; Garcia-Garibay, M. A., J. Am. Chem. Soc. 2011, 133, 2342.)
- 30.Ling C, Zheng Z, Jiang XC, Zhong W, Li S. Bioorg. Med. Chem. Lett. 2010;20:5123. doi: 10.1016/j.bmcl.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 31.Hatchard CG, Parker CA. Proc. R. Soc. London A. 1965;A220:518. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.