

Abstract
The stereochemical course of the amidopalladation of alkenes has important implications for the development of enantioselective Pd-catalyzed “Wacker-type” oxidative amination of alkenes. We have recently shown that the addition of base (Na2CO3) can alter the stereochemical course of amidopalladation in the (IMes)Pd(TFA)2(H2O)-catalyzed aerobic oxidative amidation of alkene. In this study, the mechanism of (IMes)Pd(TFA)2(H2O)-catalyzed oxidative heterocyclization of (Z)-4-hexenyltosylamide was investigated in the presence and absence of exogenous base Na2CO3. The results reveal two parallel pathways in the absence of base: a cis-amidopalladation pathway with turnover-limiting deprotonation of the sulfonamide nucleophile, and a trans-amidopalladation pathway with turnover-limiting nucleophilic attack of sulfonamide on the coordinated alkene. The addition of base (Na2CO3) lowers the energy barrier associated with the proton transfer, leading to an overall faster turnover rate and exclusive cis-amidopalladation of alkene.
Introduction
Palladium-catalyzed “Wacker-type” oxidative heterocyclizations provide efficient access to various classes of nitrogen-containing heterocycles.2 In recent years, we3 and others4 have developed a number of aerobic oxidative cyclization methods with amide-type nucleophiles, including sulfonamides, sulfinamides, and carbamates, for syntheses of pyrrolidines and related heterocycles. In order to facilitate the development of enantioselective applications of these reactions, considerable effort has been directed toward understanding and controlling the stereochemical course of the amidopalladation step.2i Both cis-5 and trans-amidopalladation6 steps (Scheme 1) have been established in catalytic reactions, and various experimental approaches have been developed to probe these two pathways.7–9 In a previous study, we employed the deuterium-labeled substrate probe 1 to analyze the stereochemical course of alkene amidopalladation with a number of different catalyst systems (Scheme 2A).7a Nearly all of the catalyst systems evaluated in this study showed exclusive preference for a cis-amidopalladation pathway. The only exception was (IMes)Pd(TFA)2(OH2). When this catalyst was employed in the absence of a basic additive, the reaction exhibited a mixture of products arising from both cis- and trans-amidopalladation pathways (cis/trans ~ 2:1). Very recently, we used another deuterium-labeled substrate probe 2 to examine the amidopalladation pathway with a Pd(TFA)2 catalyst bearing a chiral pyridine-oxazoline ligand 3 (Scheme 2B).7d In this case, trans-amidopalladation is highly favored (cis/trans ~ 1:9).
Scheme 1.

Cis- vs. Trans-Amidopalladation of Alkenes.
Scheme 2.

Stereochemical Probe Experiments for the Pd-Catalyzed Oxidative Amidocyclizations.7a,d
The (IMes)Pd(TFA)2(H2O)-catalyzed oxidative heterocyclization reaction provides a unique opportunity to investigate the factors that influence the stereochemical course of the amidopalladation reaction. The balance between cis- and trans-amidopalladation observed in the absence of additives contrasts exclusive cis-amidopalladation when the reaction is carried out in the presence of Na2CO3 (Scheme 2A, entries 5 and 6). In order to understand the basis for this switch in selectivity, we have carried out systematic mechanistic investigations of (IMes)Pd(TFA)2(H2O)-catalyzed aerobic oxidative intramolecular amidation of (Z)-4-hexenyltosylamide under both conditions. Kinetic and isotope-labeling studies of these reactions reveal a number of key differences between the two reaction conditions, and the results highlight the kinetic influence of proton-transfer from the nitrogen nucleophile on the catalytic rate and the stereochemical course of alkene amidopalladation. Comparison between these results and those obtained with a Pd(OAc)2/pyridine catalyst system10 are also presented below.
Results
Kinetic Studies
The intramolecular aerobic oxidative amidation of (Z)-4-hexenyltosylamide 4 catalyzed by (IMes)Pd(TFA)2(H2O) (5 mol%) proceeds to complete conversion in the presence of 2 equiv. Na2CO3 within 12 h at 80 °C (eq 1). A computer-interfaced gas-uptake apparatus was utilized to acquire the kinetics of the catalytic reaction by monitoring the change in oxygen pressure within a sealed, temperature-controlled reaction vessel. The reaction time-course reveals a monotonic decrease in pressure (Figure 1), thereby permitting the kinetic data to be analyzed by initial-rates methods.
Figure 1.

A representative kinetic time course for (IMes)Pd(TFA)2(H2O)-catalyzed intramolecular oxidative amidation of (Z)-4-hexenyltosylamide 4 obtained by gas-uptake methods. Data sampling occurred at a rate of 1 s−1 (not all data are shown). Conditions: [(IMes)Pd(TFA)2(H2O)] = 2.0 mM, [4] = 0.10 M, 0.8 mmol Na2CO3, 4.0 ml of toluene, initial pO2 = 700 torr, 4.0 ml of toluene, 80 °C.
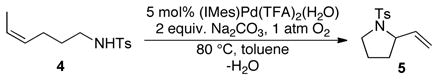 |
(1) |
The initial kinetic study focused on the contribution of the primary reaction components (O2, tosylamide, and catalyst) to the initial turnover rate. Data were acquired for reactions carried out in the presence and absence of exogenous base Na2CO3 (2 equiv). A zero-order rate dependence on the initial O2 pressure was obtained, both in the presence and absence of added base, but the initial turnover rate of the oxidation reaction exhibits a nearly three-fold enhancement with added base (Figure 2). A saturation-like dependence on [4] was observed under both sets of conditions, and the initial oxidation rate plateaus at a lower [4] in the absence of exogenous base (Figure 3). Finally, a first-order dependence on [(IMes)Pd(TFA)2(H2O)] is evident under conditions with and without added base (Figure 4).
Figure 2.

Dependences of the initial rate on the initial oxygen pressure in the presence and absence of 2 equiv. of exogenous base Na2CO3. Conditions: [(IMes)Pd(TFA)2(H2O)] = 2.0 mM, [4] = 0.1 M, 4.0 ml of toluene, initial pO2 = 400 – 900 torr, 80 °C. In the measurement of pO2 dependence in the presence of base, 0.8 mmol Na2CO3 was used.
Figure 3.

Dependences of the initial rate on amide concentration in the presence and absence of 2 equiv. of exogenous base Na2CO3. The curve fit results from a nonlinear least-squares fit to a hyperbolic function of [4]. Conditions: [(IMes)Pd(TFA)2(H2O)] = 2 mM, [4] = 0 – 0.24 M, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C. In the experiments with added base, 0.2 – 1.6 mmol Na2CO3 was used.
Figure 4.

Dependences of the initial rate on catalyst concentration in the presence and absence of 2 equiv. of exogenous base Na2CO3. The curve fit results from a fit to a linear function of [IMesPd(TFA)2(H2O)]. Conditions: [(IMes)Pd(TFA)2(H2O)] = 0 – 16 mM, [4] = 0.1 M, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C. In the experiments with added base, 0.8 mmol Na2CO3 was used.
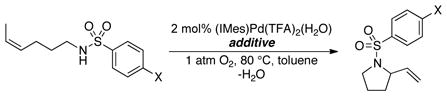
The Hammett plot obtained with a series of para-substituted benzenesulfonamides (eq 2) reveals that there is negligible electronic effect on the catalytic turnover rate in the absence of exogenous base (Figure 5A). In contrast, the non-linear Hammett plot obtained in the presence of 2 equiv of exogenous base (Na2CO3) shows that benzenesulfonamides bearing electron-withdrawing substituents react faster than the ones bearing electron-donating substituents (Figure 5B).
Figure 5.
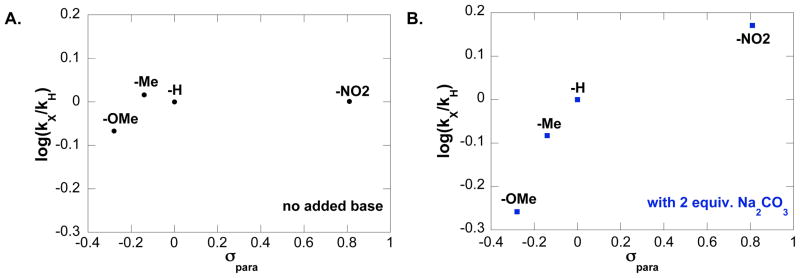
Hammett plots obtained from the relative initial rates of catalytic oxidative amidation conducted with a series of para-substituted benzenesulfonamides. (A) Hammett plot in the absence of exogenous base. Conditions: [(IMes)Pd(TFA)2(H2O)] = 2 mM, [benzenesulfonamide] = 0.1 M, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C. (B) Hammett plot in the presence of 2 equiv. exogenous base Na2CO3. Conditions: [(IMes)Pd(TFA)2(H2O)] = 2 mM, [benzenesulfonamide] = 0.1 M, 4.0 ml of toluene, initial pO2 = 700 torr, 0.8 mmol Na2CO3, 80 °C.
 |
(2) |
The dependence of initial turnover rates on the concentration of added trifluoroacetic acid was examined in the absence of exogenous base. A sharp inhibitory effect of [CF3COOH] on the initial turnover rate was observed (Figure 6).
Figure 6.

Dependence of the initial rate on added CF3COOH concentration in the absence of added base. Conditions: [(IMes)Pd(TFA)2(H2O)] = 2 mM, [4] = 0.1 M, [CF3COOH] = 0 – 1 M, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C.
Kinetic Isotope Effects and Isotopic-labeling Studies
The dependence of the initial rate on [amide], measured with the CD3-labeled tosylamide 4-d3, reveals a slight saturation dependence similar to that observed with the parent tosylamide 4 (eq 3, Figure 7). By comparing the initial rates from independent oxidative amidation reactions of tosylamides 4 and 4-d3 (Figure 7), a negligible kinetic isotope effect (kCH3/kCD3 = 0.95) is evident.
Figure 7.

Dependences of the initial O2 uptake rate on [amide] (amide = 4 or 4-d3). Conditions: [(IMes)Pd(TFA)2(H2O)] = 2 mM, [amide] = 0 – 200 mM, 2 equiv. Na2CO3 relative to amide, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C.
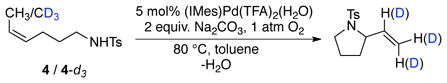 |
(3) |
The mono-deuterium-labeled substrate 4-d1 was utilized to probe the intramolecular selectivity between β-hydride and β-deuteride elimination from the palladium-alkyl intermediate 6 (Scheme 3). Analysis of final products by 1H NMR spectroscopy, however, revealed that deuterium is incorporated into the internal vinyl position, both in the presence and in the absence of exogenous base. These observations suggest that the β-hydride elimination step is reversible (see further discussion below). Experiments carried out with a 1:1 mixture of 4 and the CD3-labeled tosylamide 4-d3 showed the formation of crossover products under both reaction conditions, arising from deuterium incorporation into the product originating from unlabeled 4 (Scheme 4). These observations show that deuterium exchange can take place in an intermolecular, as well as intramolecular, process. These deuterium exchange processes prevent determination of the intrinsic isotope effect for the β-hydride elimination step.
Scheme 3.

Isotopic-Labeling Studies with Substrate Probe 4-d1 and Evidence for Deuterium Exchange.
Scheme 4.
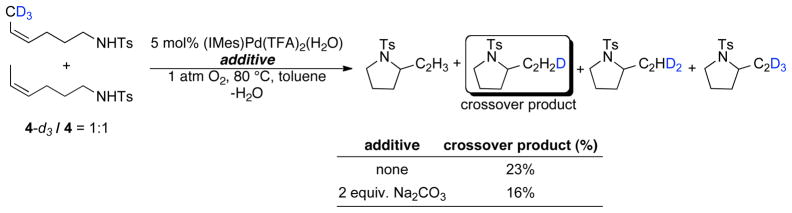
Crossover Experiments with 1:1 Mixture of Tosylamides 4-d3 and 4, Providing Evidence for Intermolecular H/D Exchange.
1H NMR Spectroscopic Studies
1H NMR spectroscopic studies were conducted to gain insights into the palladium speciation under catalytic conditions. Due to the lack of efficient liquid/solid mixing in the NMR tubes, only the homogeneous reaction conditions (without base) were investigated. Elevated pressures of oxygen gas (2–3 atm) were employed to ensure that adequate dissolved O2 was present during the catalytic reaction. The 1H NMR spectroscopic data suggested that (IMes)Pd(TFA)2(L) (L = H2O or tosylamide 4) is the Pd resting state during catalytic turnover (conditions: 17 mol% (IMes)Pd(TFA)2(H2O), 0.0108 M tosylamide 4, 80 °C, toluene-d8). Evidence for coordination of tosylamide 4 to the catalyst (IMes)Pd(TFA)2(H2O) was obtained by mixing the two components at room temperature in toluene-d8. A plot of the 1H NMR chemical shift of the NHC proton as a function of [4] is shown in Figure 8 (solid line).11 To assess whether substrate coordination to PdII takes place via the alkene or the sulfonamide nitrogen lone pair, the same titration experiments were carried out with N-ethyltosylamide and cyclohexene under identical conditions (Figure 8, blue and green dotted lines, respectively). N-Ethyltosylamide induces a shift of the 1H NMR resonances of the NHC ligand similar to that observed with tosylamide 4, whereas no significant change in the NHC ligand resonances was observed upon addition of cyclohexene. These observations suggest that the initial interaction between 4 and the PdII center of the catalyst involves coordination of the sulfonamide nitrogen lone pair rather than the alkene. The addition of heterogeneous base Na2CO3 resulted in no observable change of either the 1H or 19F NMR spectra of (IMes)Pd(TFA)2(H2O) at temperatures ranging from −37 – 40 °C.
Figure 8.

Dependence of 1H NMR chemical shift of the backbone protons of the IMes ligand in (IMes)Pd(TFA)2(L) (L= H2O, 4, N-ethyltosylamide, or cyclohexene) on ligand concentration in the absence of added base Na2CO3. The curve fit results from a nonlinear least-squares fit to a hyperbolic function of [ligand]. Conditions: [(IMes)Pd(TFA)2(H2O)] = 5 mM, [Ligand] = 0 – 0.08 M, in toluene-d8, 24 °C.
Discussion
Proposed Catalytic Mechanism
A general framework for the catalytic mechanism of (IMes)Pd(TFA)2(H2O)-catalyzed oxidative cyclization of 4 is depicted in Scheme 5, which includes both cis- and trans-amidopalladation pathways (cis-AP: 7 → 8 → 9; trans-AP: 10 → 11 → 9). The two amidopalladation pathways converge in the formation of PdII-alkyl intermediate 9. Subsequent β-hydride elimination from 9 gives rise to the vinylpyrrolidine-coordinated PdII-hydride intermediate 12. Product dissociation from the PdII center in 12 generates PdII-hydride intermediate 13, which can undergo HX-reductive elimination. Aerobic oxidation of (IMes)Pd0 regenerates the PdII catalyst.12 The reversibility of the β-hydride elimination (9 → 12) and the vinylpyrrolidine dissociation (12 → 13) were demonstrated in the deuterium labeling study with substrate 4-d1 (Scheme 3) and the crossover experiment with tosylamides 4 and 4-d3 (Scheme 4), respectively.
Scheme 5.

Proposed General Catalytic Mechanism for the (IMes)Pd(TFA)2(H2O)-Catalyzed Oxidative Amidocyclization of 4.
Influence of Na2CO3 on the Catalytic Mechanism
In the presence of Na2CO3 as an added base, the cycle in Scheme 5 can be simplified by omitting the trans-amidopalladation pathway, which does not occur under these conditions.7a In this case, the oxidative amidation reaction is initiated by equilibrium formation of the PdII–amide adduct 7 (K1cis), followed by deprotonation of the sulfonamide nucleophile (k2cis). Insertion of the alkene into the palladium-amidate bond of 8 generates palladium-alkyl intermediate 9.
The presence of heterogeneous Na2CO3 could promote this pathway and enhance the overall catalytic rate by facilitating deprotonation of the coordinated amide in 7. The insolubilty of Na2CO3 in toluene complicates direct investigation of this step, but two possible roles of the carbonate base seem plausible. Preequilibrium proton transfer could take place within the coordination sphere of PdII, from the coordinated sulfonamide to the trifluoroacetate ligand, followed by dissociation of TFAH and irreversible deprotonation by the insoluble carbonate base. Alternatively, small amounts of CO32− could exchange with TFA at the PdII center, and the more basic carbonate anion could facilitate deprotonation of the coordinated sulfonamide. These alternatives cannot be distinguished on the basis of the available data, but both processes could account for the enhanced catalytic rate and corresponding exclusive cis-amidopalladation of the alkene.
A rate law derived for this proposed mechanism (eq 4) rationalizes the saturation dependence on [4] and the first-order dependence on [(IMes)Pd(TFA)2(H2O)] (cf. Figures 3 and 4).13 That the rate exhibits only a slight saturation dependence on [4] suggests that sulfonamide coordination to PdII is not strongly favored, a conclusion consistent with the independent binding studies in Figure 8. Hammett data for the reaction with added Na2CO3 (cf. Figure 5B) indicate that sulfonamides with electron-withdrawing groups react more rapidly. This trend is expected if substrate deprotonation, 7 → 8, is at least partially turnover limiting. The non-linearity of the Hammett plot may reflect a change in mechanism, but we speculate that these data arise from two (or more) steps contributing to the turnover rate (e.g., amide coordination and N–H deprotonation), each of which exhibits a different electronic dependence. Finally, the proposed mechanism accounts for the negligible kinetic isotope effect obtained from the reactions of tosylamides 4 and 4-d3 (cf. Figure 7), because β-hydride elimination step occurs after the turnover-limiting step.
| (4) |
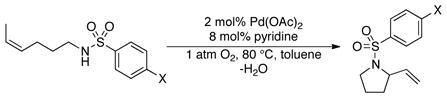
We recently reported a mechanistic study of the oxidative heterocylization of tosylamide 4 with Pd(OAc)2/pyridine as the catalyst (eq 5).10 Both Pd(OAc)2/pyridine and (IMes)Pd(TFA)2(H2O)/Na2CO3 promote exclusive cis-amidopalladation of alkene; however, the reactions exhibit significantly different substrate electronic effects. Hammett analysis of Pd(OAc)2/pyridine-catalyzed oxidative cyclization of 4 revealed a negative slope (ρ = −0.22) (Figure 9). This observation, together with additional kinetic and mechanistic data, are consistent with alkene insertion into a Pd–N bond as the turnover-limiting step in the Pd(OAc)2/pyridine-catalyzed reaction. Briefly summarized, electron-withdrawing substituents enhance the acidity and increase the rate of deprotonation of the sulfonamide ligand with the (IMes)Pd(TFA)2(H2O)/Na2CO3 catalyst, whereas they decrease the nucleophilicity of the sulfonamidate and lower the rate of the nucleophilic attack on the alkene with the Pd(OAc)2/pyridine catalyst. The different turnover-limiting steps with these two catalyst systems can be rationalized by the different relative basicities of the anionic ligands, trifluoroacetate vs. acetate. Specifically, the more basic acetate anion in the Pd(OAc)2/pyridine catalyst results in sufficiently facile (and/or favorable) deprotonation of the amide that alkene insertion into the Pd–N bond becomes the turnover-limiting step.
Figure 9.

Hammett correlation of the Pd(OAc)2/pyridine-catalyzed oxidative heterocyclization (data replotted from ref. 1014). Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 8.0 mM, [amide] = 100 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C.
 |
(5) |
In the absence of base, the (IMes)Pd(TFA)2(H2O)-catalyzed intramolecular oxidative amidation of alkenes proceeds via both cis- and trans-amidopalladation pathways. Kinetic studies suggest the reaction exhibits a similar overall rate law under these conditions (cf. Figures 2–4), but it has a three-fold slower rate. Another noteworthy feature is the lack of a sulfonamide electronic effect on the rate (see Hammett plot in Figure 5B), which differs from the trends observed under the other two reaction conditions (cf. Figures 5A and 9). The result can be rationalized if the cis- and trans-amidopalladation pathways have offsetting electronic effects. Specifically, it is reasonable to expect that the trans-amidopalladation pathway has a negative Hammett slope associated with nucleophilic attack of a neutral sulfonamide on the coordinated alkene (i.e., more electron-rich nucleophiles react more rapidly; Scheme 5, 10 → 11), while the cis-amidopalladation pathway exhibits a positive Hammett trend (Figure 5A). Since the rates of the cis- and trans-amidopalladation pathways are finely balanced, variation of the para-substituent of benzenesulfonamide could simply shift the relative preference of one pathway over the other without significantly altering the overall rate. In support of this hypothesis, (IMes)Pd(TFA)2(H2O)-catalyzed oxidative amidation of p-nitrobenzenesulfonyl-substituted amide yields only the cis-amidopalladation product even in the absence of exogenous base.5a
Conclusion
This mechanistic study of (IMes)Pd(TFA)2(H2O)-catalyzed aerobic oxidative amidation of alkenes highlights the critical influence of “proton management” on the rate and stereochemical course of the catalytic reaction. Addition of an exogenous base (Na2CO3) enhances the turnover rate of the catalytic oxidative amidation reaction and shifts the mechanism to favor exclusively cis-amidopalladation of the alkene. These observations complement recent results obtained in a study of (pyrox)PdX2-catalyzed enantioselective oxidative amidation of alkenes (cf. Scheme 2B).7d Use of the Pd(OAc)2 catalyst (X = OAc) mediates cis-amidopalladation of the alkene, while the Pd catalyst with the less-basic trifluoroactate ligand (X = TFA) mediates trans-amidopalladation of the alkene. These results match those expected from the present study, which shows that basic conditions favor cis-amidopalladation. Collectively, these studies are beginning to provide the basis for rational development of PdII catalysts for oxidative heterofunctionalization of alkenes.
Experimental Section
General considerations
All commercially available compounds were purchased and used as received. Catalyst (IMes)Pd(TFA)2(H2O) was prepared as described previously. 15 (Z)-4-hexenyltosylamide 4, para-substituted benzenesulfonamides, deuterium-labeled tosylamides 4-d1 and 4-d3 were all synthesized according to literature procedure.10 1H NMR spectra were recorded on 300 and 500 MHz spectrometers. 1H NMR chemical shifts (δ) are given in part per million relative to internal TMS (0.00 ppm). Ethylene glycol was used for temperature calibration of the NMR spectrometer for variable temperature measurements.
Representative procedure for gas-uptake kinetics
A typical reaction was conducted as follows: a 25 mL round-bottom flask with a stirbar was attached to an apparatus with a calibrated volume and a pressure transducer designed to measure the gas pressure within the sealed reaction vessel. The apparatus was evacuated to 10 torr and filled with oxygen to 800 torr and this cycle was repeated 10 times. The final pressure was established at 675 torr. After the pressure stablilzed within the apparatus, a stock solution of (IMes)Pd(TFA)2(H2O) (2.5 mM, in 3.6 mL toluene) was added via syringe through a septum. The flask was heated to 80 °C. After the temperature stabilized, a stock solution of tosylamide substrate (1.0 M, in 0.4 mL toluene) was added via syringe through a septum. Data was acquired using custom software written within LabVIEW. Correlations between oxygen uptake and conversions were made by 1H NMR analysis with 1,3,5-trimethoxybenzene as an internal standard.
The procedure for the (IMes)Pd(TFA)2(H2O)-catalyzed oxidative amidation of 4 in the presence of Na2CO3 is similar to the above procedure, except that the 25 mL round-bottom flask was pre-charged with anhydrous Na2CO3 (e.g. 0.8 mmol).
Representative procedure for (IMes)Pd(TFA)2(H2O)-catalyzed aerobic oxidative cyclization of deuterium-labeled tosylamide 4-d1
(IMes)Pd(TFA)2(H2O) (3 mg, 5 μmol) was placed in 13×100 mm disposable culture tubes. The reaction tubes were placed into a custom 48-well parallel reactor mounted on a large capacity mixer and the headspace was purged with molecular oxygen for ca. 15 min. A solution of substrate probe 4-d1 (0.1 mmol) in toluene (1 mL) was added to tubes. The reactions were carried out for 24 h under an oxygen atmosphere (1 atm) at 80 °C. Following removal of the solvent under vacuum, the crude oxidative amination product was purified via column chromatography with hexanes/ethyl acetate and analyzed by 1H NMR spectroscopy.
The procedure for the (IMes)Pd(TFA)2(H2O)-catalyzed oxidative amidation of 4-d1 in the presence of Na2CO3 is similar to the above procedure, except that the culture tube was pre-charged with both anhydrous Na2CO3 (0.2 mmol) and the catalyst.
Representative procedure for crossover experiments
(IMes)Pd(TFA)2(H2O) (3 mg, 5 μmol) was placed in 13×100 mm disposable culture tubes. The reaction tubes were placed into a custom 48-well parallel reactor mounted on a large capacity mixer and the headspace was purged with molecular oxygen for ca. 15 min. Solutions of a 1:1 mixture of tosylamides 4 and 4-d3 (0.1 mmol total) in toluene (1 mL) were added to tubes. The reactions were carried out for 24 h under an oxygen atmosphere (1 atm) at 80 °C. Following removal of the solvent under vacuum, the crude oxidative amination product was purified via column chromatography with hexanes/ethyl acetate. The purified product was dissolved in CH2Cl2 and submitted for mass spectroscopy measurement (ESI). The MS detector was set to avoid saturation in order to get accurate relative peak intensities in these experiments.
The procedure for the crossover experiment performed in the presence of Na2CO3 is similar to the above procedure, except that the culture tube was pre-charged with both anhydrous Na2CO3 (0.2 mmol) and the catalyst.
Catalytic reaction monitored by 1H NMR spectroscopy
A freshly prepared solution of (IMes)Pd(TFA)2(H2O) and 4 (1.8 mM palladium, 10.8 mM 4, 6.0 mM 1,3,5-trimethoxybenzene, in toluene-d8, 0.7 mL) was added to a medium-wall NMR tube attached to a sealed 14/20 ground glass joint. The solution was frozen in liquid nitrogen. The NMR tube was connected to a gas manifold attached to a mercury monometer, both of which were calibrated for volume. The solution was degassed three times and then oxygen (0.263 mmol) was condensed in the tube to achieve a final pressure of 2.6 atm in the headspace above the solution. The solution was kept cold in a bath of dry ice/acetone until it was inserted into the spectrometer probe, preheated to 80 °C.
Supplementary Material
Acknowledgments
We are grateful to the NIH for financial support of this work (R01 GM67163).
Footnotes
Dedicated to the memory of Prof. Howard E. Zimmerman (July 5, 1926 – February 12, 2012), a revered colleague and pioneer in the field of physical organic chemistry, including organic photochemistry, applications of molecular orbital theory to organic chemical reactivity, and stereochemical implications of kinetically-controlled protonation steps.1
Supporting Information Available: Experimental details, supplemental rate dependences and 1H NMR spectra, and mathematical derivation of the rate law. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Schuster DI. Angew Chem Int Ed. 2012;51:5286–5288. doi: 10.1002/anie.201202970. [DOI] [PubMed] [Google Scholar]; (b) Hixson SS, Mariano PS, Zimmerman HE. Chem Rev. 1973;73:531–551. [Google Scholar]; (c) Zimmerman HE. Acc Chem Res. 1987;20:263–268. [Google Scholar]; (d) Zimmerman HE, Nesterov EE. Acc Chem Res. 2002;35:77–85. doi: 10.1021/ar000210g. [DOI] [PubMed] [Google Scholar]
- 2.For reviews, see: Hegedus LS. In: Comprehensive Organic Synthesis. Semmelhack MF, editor. Vol. 4. Pergamon Press, Inc; Elmsford: 1991. pp. 551–569.Hosokawa T, Murahashi S-I. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, de Meijere A, editors. Vol. 2. John Wiley and Sons, Inc; New York: 2002. pp. 2169–2192.Hosokawa T. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E, de Meijere A, editors. Vol. 2. John Wiley and Sons, Inc; New York: 2002. pp. 2211–2225.Zeni G, Larock RC. Chem Rev. 2004;104:2285–2309. doi: 10.1021/cr020085h.Stahl SS. Angew Chem, Int Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630.Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem Rev. 2007;107:5318–5365. doi: 10.1021/cr068006f.Minatti A, Muñiz K. Chem Soc Rev. 2007;36:1142–1152. doi: 10.1039/b607474j.Kotov V, Scarborough CC, Stahl SS. Inorg Chem. 2007;46:1910–1923. doi: 10.1021/ic061997v.McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y.
- 3.(a) Fix SR, Brice JL, Stahl SS. Angew Chem, Int Ed. 2002;41:164–166. doi: 10.1002/1521-3773(20020104)41:1<164::aid-anie164>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]; (b) Rogers MM, Wendlandt JE, Guzei IA, Stahl SS. Org Lett. 2006;8:2257–2260. doi: 10.1021/ol060327q. [DOI] [PubMed] [Google Scholar]; (c) Scarborough CC, Bergant A, Sazama GT, Guzei IA, Spencer LC, Stahl SS. Tetrahedron. 2009;65:5084–5092. doi: 10.1016/j.tet.2009.04.072. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org Lett. 2011;13:2830–2833. doi: 10.1021/ol200784y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lu Z, Stahl SS. Org Lett. 2012;14:1234–1237. doi: 10.1021/ol300030w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Redford JE, McDonald RI, Rigsby ML, Wiensch JD, Stahl SS. Org Lett. 2012;14:1242–1245. doi: 10.1021/ol3000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Larock RC, Hightower TR, Hasvold LA, Peterson KP. J Org Chem. 1996;61:3584–3584. doi: 10.1021/jo952088i. [DOI] [PubMed] [Google Scholar]; (b) Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew Chem, Int Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196. [DOI] [PubMed] [Google Scholar]; (c) Yip KT, Yang M, Law KL, Zhu NY, Yang D. J Am Chem Soc. 2006;128:3130–3131. doi: 10.1021/ja060291x. [DOI] [PubMed] [Google Scholar]; (d) He W, Yip KT, Zhu NY, Yang D. Org Lett. 2009;11:5626–5628. doi: 10.1021/ol902348t. [DOI] [PubMed] [Google Scholar]; (e) Jiang F, Wu Z, Zhang W. Tetrahedron Lett. 2010;51:5124–5126. [Google Scholar]; (f) Jana R, Pathak TP, Jensen KH, Sigman MS. Org Lett. 2012;14:4074–4077. doi: 10.1021/ol3016989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Representative examples of cis-amidopalladation: Brice JL, Harang JE, Timokhin VI, Anastasi NR, Stahl SS. J Am Chem Soc. 2005;127:2868–2869. doi: 10.1021/ja0433020.Liu G, Stahl SS. J Am Chem Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h.Isomura K, Okada N, Saruwatari M, Yamasaki H, Taniguchi H. Chem Lett. 1985:385–388.Ney JE, Wolfe JP. Angew Chem, Int Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060.Ney JE, Wolfe JP. J Am Chem Soc. 2005;127:8644–8651. doi: 10.1021/ja0430346.Nakhla JS, Kampf JW, Wolfe JP. J Am Chem Soc. 2006;128:2893–2901. doi: 10.1021/ja057489m.Muñiz K, Hovelmann CH, Streuff J. J Am Chem Soc. 2007;130:763–773. doi: 10.1021/ja075041a.
- 6.Representative examples of trans-amidopalladation: Åkermark B, Bäckvall JE, Siirala-Hansén K, Sjöberg K, Zetterberg K. Tetrahedron Lett. 1974;15:1363–1366.Åkermark B, Bäckvall JE, Hegedus LS, Zetterberg K, Siirala-Hansén K, Sjöberg K. J Organomet Chem. 1974;72:127–138.Hegedus LS, Allen GF, Waterman EL. J Am Chem Soc. 1976;98:2674–2676.Bäckvall JE. Acc Chem Res. 1983;16:335–342.Bäckvall JE, Björkman EE. Acta Chem Scand B. 1984;38:91–93.Watson MP, Overman LE, Bergman RG. J Am Chem Soc. 2007;129:5031. doi: 10.1021/ja0676962.Cochran BM, Michael FE. J Am Chem Soc. 2008;130:2786–2792. doi: 10.1021/ja0734997.Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w.
- 7.For studies employing stereospecifically deuterated substrates, see: Liu GS, Stahl SS. J Am Chem Soc. 2007;129:6328–6335. doi: 10.1021/ja070424u.Bertrand MB, Neukom JD, Wolfe JP. J Org Chem. 2008;73:8851–8860. doi: 10.1021/jo801631v.Mai DN, Wolfe JP. J Am Chem Soc. 2010;132:12157–12159. doi: 10.1021/ja106989h.Weinstein AB, Stahl SS. Angew Chem, Int Ed. 2012;51:11505–11509. doi: 10.1002/anie.201206702.
- 8.For studies of stoichiometric alkene insertion into Pd-N bonds of well-defined Pd-anilide or -amidate complexes, see: Hanley PS, Markovic D, Hartwig JF. J Am Chem Soc. 2010;132:6302–6303. doi: 10.1021/ja102172m.Neukom JD, Perch NS, Wolfe JP. J Am Chem Soc. 2011;132:6276–6277. doi: 10.1021/ja9102259.Hanley PS, Hartwig JF. J Am Chem Soc. 2011;133:15661–15673. doi: 10.1021/ja205722f.Neukom JD, Perch NS, Wolfe JP. Organometallics. 2011;30:1269–1277.White PB, Stahl SS. J Am Chem Soc. 2011;133:18594–18597. doi: 10.1021/ja208560h.
- 9.For trapping of the amidopalladation intermediate with an exogenous ligand, see: Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w.
- 10.Ye X, Liu G, Popp BV, Stahl SS. J Org Chem. 2011;76:1031–1044. doi: 10.1021/jo102338a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.See Supporting Information for the corresponding NMR spectra.
- 12.For mechanistic studies of the reaction of O2 with IMes-ligated PdII-hydride complexes, see Konnick MM, Guzei IA, Stahl SS. J Am Chem Soc. 2004;126:10212–10213. doi: 10.1021/ja046884u.Konnick MM, Gandhi BA, Guzei IA, Stahl SS. Angew Chem, Int Ed. 2006;45:2904–2907. doi: 10.1002/anie.200600532.Popp BV, Stahl SS. J Am Chem Soc. 2007;129:4410–4422. doi: 10.1021/ja069037v.Konnick MM, Stahl SS. J Am Chem Soc. 2008;130:5753–5762. doi: 10.1021/ja7112504.Konnick MM, Decharin N, Popp BV, Stahl SS. Chem Sci. 2011;2:326–330.
- 13.See Supporting Information for the mathematical derivation of rate laws.
- 14.Note: The original Hammett plot reported in ref. 10 featured incorrect y-axis labels; however, the reported ρ value (−0.22) is correct. The corrected axes are included in Figure 9.
- 15.Sigman and coworkers reported the use of (IMes)Pd(TFA)2(H2O) and related complexes as catalysts for aerobic alcohol oxidation. See: Jensen DR, Schultz MJ, Mueller JA, Sigman MS. Angew Chem, Int Ed. 2003;42:3810–3813. doi: 10.1002/anie.200351997.Mueller JA, Goller CP, Sigman MS. J Am Chem Soc. 2004;126:9724–9734. doi: 10.1021/ja047794s.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.