

Abstract
Chagas disease, caused by Trypanosoma cruzi, is an important cause of morbidity and mortality primarily resulting from cardiac dysfunction, although T. cruzi infection results in inflammation and cell destruction in many organs. We found that T. cruzi (Brazil strain) infection of mice results in pancreatic inflammation and parasitism within pancreatic β-cells with apparent sparing of α cells and leads to the disruption of pancreatic islet architecture, β-cell dysfunction, and surprisingly, hypoglycemia. Blood glucose and insulin levels were reduced in infected mice during acute infection and insulin levels remained low into the chronic phase. In response to the hypoglycemia, glucagon levels 30 days postinfection were elevated, indicating normal α-cell function. Administration of L-arginine and a β-adrenergic receptor agonist (CL316, 243, respectively) resulted in a diminished insulin response during the acute and chronic phases. Insulin granules were docked, but the lack of insulin secretion suggested an inability of granules to fuse at the plasma membrane of pancreatic β-cells. In the liver, there was a concomitant reduced expression of glucose-6-phosphatase mRNA and glucose production from pyruvate (pyruvate tolerance test), demonstrating defective hepatic gluconeogenesis as a cause for the T. cruzi-induced hypoglycemia, despite reduced insulin, but elevated glucagon levels. The data establishes a complex, multi-tissue relationship between T. cruzi infection, Chagas disease, and host glucose homeostasis.
Chagas disease or American trypanosomiasis, a neglected tropical disease, is the result of a persistent infection with Trypanosoma cruzi.1,2 Not only does it remain an important cause of morbidity and mortality in Latin America, but it is now also a global disease due to immigration to non-endemic areas.3 Furthermore, T. cruzi causes opportunistic infection in the setting of immunosuppression (eg, HIV/AIDS).2 Infection of humans and experimental animals results in an intense inflammatory reaction accompanied by an upregulation of inflammatory mediators.4,5 In the heart, this results in acute myocarditis and some patients eventually develop a chronic dilated cardiomyopathy accompanied by arrhythmias, congestive heart failure, and stroke.2 T. cruzi infection also results in mega syndromes predominantly observed in the gastrointestinal tract.2
In the 1980s, we became interested in the interface of host glucose homeostasis and parasitic diseases and observed that T. cruzi infection of mice pre-treated with streptozotocin, which destroys the insulin producing pancreatic β-cells, displayed increased parasitemia.6 We also demonstrated that obese diabetic mice, null for the leptin receptor (db/db)7 infected with T. cruzi displayed a high parasitemia and increased mortality. It has been reported that acute T. cruzi infection of mice resulted in hypoglycemia, which in some cases was predictive of increased mortality,8,9 however, the etiology of T.cruzi-associated hypoglycemia remains unclear.
There are also reports suggesting that diabetes may be more prevalent in individuals with Chagas disease, but these are based on small clinical case series10–14 and often there are other confounding factors such as obesity, hyperlipidemia, and poverty.15–17 The possibility that T. cruzi infection could contribute to the diabetic state is not surprising because pathological examination of the pancreas obtained from infected mice and humans revealed morphological and physiological alterations.18–21 In addition, because of the interrelationship between the adipocyte and glucose metabolism, we examined the contributions of adipose tissue and the adipocyte in the pathogenesis of T. cruzi infection.8,22,23 In this regard, we established that adipose tissue and adipocytes are important early targets, as well as a reservoir site for the parasite.8,23,24 Infection of mice with T. cruzi resulted in inflammation of adipose tissue and an upregulation of inflammatory mediators,8,22,23 suggesting that the adipocyte-pancreatic axis (ie, the adipo-insular axis) could also be impaired by T. cruzi infection.
The role of the pancreas in T. cruzi infection has received limited attention in Chagas disease pathogenesis.18,19,21 Herein, we report that T. cruzi-infection induces inflammation and parasitism of the pancreas, including the pancreatic β-cell. Furthermore, physiological studies strongly suggest that there is an impairment of both pancreatic function and hepatic gluconeogenesis. Alterations in glucose homeostasis in the setting of T. cruzi infection appear to be multifactorial and complex.
Materials and Methods
Parasitology and Pathology
Six- to eight-week-old male CD-1 mice (Charles River, Wilmington, MA) were infected with 5 × 104 trypomastigotes of the Brazil strain of T. cruzi. The parasitemia was assessed by counting trypomastigotes in tail blood using a hemocytometer. Pancreata and white adipose tissue obtained from the epididymal fat pad were removed and processed for either routine H&E staining, transmission electron microscopy or immunohistochemical analysis or immunofluorescence. In addition, the liver was removed for quantitative real-time PCR (RT-qPCR) studies.
Immunohistochemical Analysis of Adipose Tissue
Adipose tissue was fixed in 10% neutral buffered formalin and embedded in paraffin wax. Sections (5 μm thick) were deparaffinized and boiled at 95°C for 20 minutes in sodium citrate solution (DAKO, Carpentaria, CA) for antigen retrieval. Macrophage activity was assessed using a rabbit antibody to ionized calcium-binding adaptor molecule-1 (Iba-1) (Wako Chemicals, Richmond, VA). This polypeptide is selectively expressed in cells of monocytic origin. Sections were incubated overnight at 4°C with Iba-1 at a dilution of 1:300. A standard avidin-biotin complex method (Vector Laboratories, Burlingame, CA) was applied for the secondary antibody (anti-rabbit) by using a 1:200 dilution and a 1-hour incubation. Slides were developed using a peroxidase detection kit (Vector Laboratories) and counterstained with H&E (Sigma-Aldrich, St. Louis, MO) after immunolabeling.
Transmission Electron Microscopy
Pancreata were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, postfixed with 1% osmium tetroxide, followed by 1% uranyl acetate, dehydrated through a graded series of ethanol and embedded in LX112 resin (Ladd Research Industries, Burlington VT). Ultrathin sections were cut on a Reichert Ultracut E, ultramicrotome (Reichert-Jung, Vienna, Austria) stained with uranyl acetate, followed by lead citrate, and viewed on a JEOL 1200EX transmission electron microscope at 80 kv (Tokyo, Japan). Images were obtained by transmission electron microscopy and analyzed using ImageJ version 1.46 (NIH, Bethesda, MD). Secretory granule density per unit area and relative distribution within cells was assessed in infected and uninfected pancreatic β-cells. For granule distribution measurements, the cortical region was defined by a distance of 1 granule diameter, or approximately 350 nmol/L from the plasma membrane.
Immunofluorescence of Pancreatic Tissue
Pancreata were dissected and fixed in 10% neutral buffered formalin overnight, then processed by paraffin embedding and sectioning (5 μm). For immunofluorescence, sections were incubated with primary antibodies for 24 hours and subsequently labeled with fluorescenated secondary antibodies for 1 hour at room temperature. A guinea pig anti-swine insulin antibody (1:500; DAKO) was used for insulin staining and a rabbit anti-human glucagon antibody (1:250; Zymed, Grand Island, NY) was used for glucagon staining. Images were then taken on a Zeiss Axio Observer Wide-field Epi-fluorescence microscope (Carl Zeiss, Jena, Germany) at ×20 magnification. Specimens were obtained from uninfected control mice and from mice 15, 30, and 100 days postinfection (dpi).
Physiological Studies
L-arginine, the β-adrenergic receptor agonist, CL316, 243, and sodium pyruvate were all obtained from Sigma-Aldrich and were dissolved in PBS, pH 7.2. In these experiments, infected and uninfected age-matched controls were used. In all of the experiments, mice were fasted for 7 hours before beginning the experimental protocols. In the control group, mice were injected with PBS (pH 7.2). Insulin levels were determined by enzyme-linked immunosorbent assay according to the manufacturer’s protocol (ALPCO Diagnostics, Salem, NH). Glucagon was determined by radioimmunoassay according to the manufacturer’s protocol (Millipore, St. Charles, MO). Blood glucose was measured with a One Touch Ultra Glucometer (Life Scan Inc., Burnaby, BC, Canada).
L-Arginine Experiments
Injection of L-arginine stimulates the secretion of insulin by pancreatic β-cells.25 Mice at 15, 30, and 100 dpi were administered L-arginine by the i.p. route at a dose of 1 g/kg of body weight and insulin levels in the peripheral blood were determined 3 minutes after injection using the multiplex system. For controls, we used age- and sex-matched mice.
Adipocyte β-3-Adrenergic Receptor Agonist Experiments
The β-3-adrenergic receptor is found on the surface of adipocytes and it is believed that activation of this receptor results in the activation of the adipo-insular axis, thereby resulting in increased secretion of insulin by the pancreas mediated, perhaps, by free fatty acids.26 We injected 1 mg/kg of body weight of the β-adrenergic receptor agonist (CL316, 243 i.p.) into the infected and uninfected age-matched mice and insulin levels were determined 10 minutes later.
Gluconeogenesis
Infected and uninfected mice were injected with sodium pyruvate (2 g/kg i.p.). Plasma glucose was determined at 0, 5, 15, 30, 60, 90, and 120 minutes after injection. We performed real-time PCR for expression of glucose-6-phosphatase. The primers used were: forward,5′-GGCGCAGCAGGTGTATACTA-3′, reverse, 5′-ATGCCTGACAAGACTCCAGC-3′; 18S forward: 5′-AGGGTTCGATTCCCGGAGAGG-3′, reverse, 5′-CAACTTTAATATACGCTATTGG-3′. RT-qPCR was performed as previously described by our laboratory.8
Statistical Analysis
Graphics and statistical analyses were performed using GraphPad Prism version 5 (GraphPad Software, San Diego, CA). Statistical analyses were performed on paired data and two-tailed P values were obtained. The data are presented as the means ± SEM for all experiments. Significance was at P < 0.05.
Results
Parasitology
Infected CD-1 mice had an overall mortality of 60% by 40 dpi. The parasitemia peaked at 5 × 105 trypomastigotes/mL 30 to 35 dpi. Thereafter, the parasitemia waned and by 60 dpi there were no detectable parasites in the blood. Previously, we demonstrated that the surviving mice displayed a dilated cardiomyopathy as determined by pathology and cardiac imaging by 90 to 100 days postinfection.27–29
Histopathology
The histopathology of the pancreas during the course of infection was determined. At 15 dpi, there was intense inflammation surrounding the blood vessels and ducts and destruction of the acinar cells of the pancreas (Figure 1A–C). There were scattered amastigotes in acinar cells and marked inflammation and parasitism of the peri-pancreatic fat (Figure 1D). At 30 and 100 dpi, there was persistent inflammation (data not shown). The examination of the adipose tissue was consistent with our previous observations in that as early as 15 dpi onward there was inflammation and a marked infiltration of macrophages, which was determined by Iba-1 staining (Figure 2).
Figure 1.

Representative H&E stained sections of pancreata obtained from control and T. cruzi-infected mice. The degree of inflammation and parasitism of the pancreas was examined. A: Normal pancreas (20×). B: Pancreas obtained from an infected mouse 15 days postinfection; note the inflammation within the islet area (arrows) (20×). C: Infected pancreas; note the inflammation (black arrows) and rare intracellular amastigotes in an acinar cell (white arrow) (40×). D: Inflammation in the peripancreatic fat (arrows).
Figure 2.
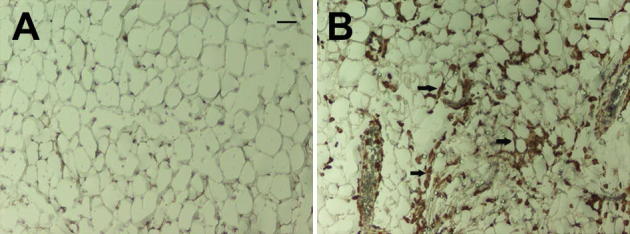
Representative white adipose tissue obtained from infected and control mice. The alterations in adipose tissue at the early stage of infection. A and B: Control mice (A) and T. cruzi-infected mice (B), 15 days postinfection. There is infiltration Iba-1 positive cells (arrows) (macrophages) into the adipose tissue obtained from infected mice.
Transmission Electron Microscopy of the Pancreas
Next we correlated the histopathological studies of the pancreas with ultrastructural studies. During acute infection, amastigotes were observed within the pancreatic β-cells (Figure 3I). However, the α-cells were not parasitized and the glucagon granules appeared to be intact (Figure 3I). The integrity of insulin granules appeared morphologically normal (Figure 3, A–D, and I). These observations were based on the examination of at least 50 fields under low power (5,000×). The total number of insulin granules was reduced in pancreata obtained from mice 30 dpi compared with tissues obtained from uninfected mice (Figure 3G). In the infected pancreas, there was a significant increase in the number of insulin granules localized within 1 granule diameter from the plasma membrane compared with the uninfected pancreas (Figure 3, E, F, and H). However, granule distributions 2 diameters away from the plasma membrane did not reach statistically significant difference between the control and infected mice (Figure 3H). These observations suggest a defect late in the secretory pathway and the L-arginine studies (see as follows) support an impaired release of granules involved in the first phase of insulin secretion.
Figure 3.

Electron micrographs of control and infected islets 30 days postinfection. Correlation of histopathological studies of the pancreas with ultrastructural studies. Electron micrographs (EM) of T. cruzi-infected or uninfected islets were analyzed for changes in granule number or distribution. Representative images and the quantitative evaluations using ImageJ version 1.46 are shown. Granule numbers (A–D): The unique morphology of insulin granules, electron-dense core surrounded by a halo and membrane, remained unchanged in both normal (A and C) and infected mice (B and D). C and D are magnified from the insets (A and B, respectively). Secretory granule density per unit area was used as a measure of total insulin content and appeared minimally reduced with T. cruzi infection (EM, A and B; quantitation, G). Controls 3.98 ± 0.32 (SEM) (n = 8); infected 2.56 ± 0.45 (SEM) (n = 14). (P < 0.05). Granule distribution (E and F): Arrowheads pinpoint the plasma membrane. The cortical region was defined as a distance from the plasma membrane corresponding to 1 granule diameter (1 D), or ∼350 nmol/L (EM, E and F; quantitation, H). One granule diameter, 0.80 ± 0.16 (SEM), n = 14; infected 2.00 ± 0.18 (SEM), (n = 20); P < 0.05. Two granule diameters, control 1.46 ± 0.20 (SEM), n = 14; infected = 1.15 ± 0.15 (SEM) n = 20. Increased secretory granule association with the cortical region (ie, docked granule pool) is observed in infected β cells (EM, E and F; quantitation, H). Nests of parasites were observed in β-cells (I, white arrows, T. cruzi). Morphological appearance of insulin β-granules (I, black arrows) and glucagon containing α-granules (I, arrowheads) seemed normal.
Immunofluorescence of the Pancreas
Figure 4 illustrates representative immunofluorescence studies of the islets in the pancreas that were stained for insulin and glucagon. β-Cells can be recognized by the green insulin staining, and the red glucagon staining identifies the α-cells. Figure 4A shows the typical morphology of normal pancreatic islets. There was a dramatic disruption of islet integrity at 15 dpi (Figure 4B). The disruption of islet integrity continued at 30 dpi. The glucagon-secreting α-cells were disorganized (Figure 4C). These cells are usually located in the periphery of mouse islets. There appeared to be partial recovery of islet integrity at 100 dpi (Figure 4D).
Figure 4.
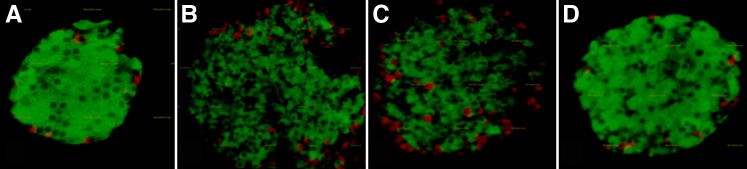
Pancreatic islet morphology at different stages of infection. Immunohistochemical analysis of pancreatic islets. Insulin-stained β-cells are green, and glucagon-stained α-cells are red. A: Classical morphology at baseline. B: There was a dramatic disruption of islet integrity at day 15 postinfection. C: The disruption of islet integrity continued at day 30 postinfection. Note the disorganized structure of the α-cells on day 30, which are usually located in the periphery of mouse islets. D: There appears to be partial recovery of islet integrity at day 100 postinfection.
Physiological Studies
Throughout the course of infection, basal serum insulin levels were reduced as compared to uninfected control mice (Figure 5A). As noted, there is a β-3-adrenergic receptor population on the surface of the adipocyte that, when stimulated, releases mediators resulting in the secretion of insulin from the pancreatic β-cell. During both acute and chronic infection, after the administration of the β-3-adrenergic receptor agonist, the insulin response was significantly reduced in infected mice at all time points tested compared with uninfected controls (Figure 5B). The administration of L-arginine results in the release of insulin from the pancreas. In the current experiment, 3 minutes after L-arginine administration, uninfected control mice displayed the expected increase in insulin levels. However, the insulin response to L-arginine was significantly reduced at all time points in the infected mice (Figure 5C). Based on the data, it is strongly suggested that there is a defect in the secretion of insulin in infected mice.
Figure 5.

Serum insulin levels in infected and control mice. A: Insulin levels in infected and control mice during the course of infection. B: Insulin levels after injection of beta-adrenergic agonist (Ar). Note the reduced insulin response after stimulation of the adipocyte β-adrenergic receptor in infected mice. C: Insulin levels after injection of L-arginine (LR). Note the reduced insulin response after LR stimulation in infected mice. In all experiments: n = 4; P < 0.05. All determinations were made on mice that were fasted for at least 7 hours.
Insulin, Glucose, and Glucagon Levels
At 30 dpi, glucose and insulin levels were significantly reduced in infected mice compared with uninfected mice (Figures 5A and 6A). Furthermore, the glucagon levels were elevated proportional to the reduction in glucose (Figure 6B) and the insulin levels remained significantly reduced into the chronic phase (Figure 5A). Thus, hypoglycemia appears not to be the result of a defect in glucagon synthesis and secretion.
Figure 6.

Glucose, glucagon, hepatic gluconeogenesis in control and infected mice 30 days postinfection. A: Infected mice had an average blood sugar of 70 mg/dL compared with 150 mg/dL for control (P < 0.05) (n = 5 each group). B: In response to the hypoglycemia and hypoinsulinemia, there were increased glucagon levels in the sera of infected mice (P < 0.05) (n = 3 each group). C: Pyruvate tolerance test: infected and uninfected mice 30 days postinfection were injected i.p. with 2 g/kg sodium pyruvate. Blood glucose was determined at 0, 5, 15, 30, 60, 90, and 120 minutes after injection. There was a significant reduction in glucose at every time point (P < 0.05) (n = 5 each group). D: Pyruvate tolerance test: reduction of mRNA expression of glucose-6-phosphatase in livers of infected mice compared with livers obtained from control mice. (P < 0.05) (n = 3 each group). All determinations were made on mice that were fasted for at least 7 hours.
Gluconeogenesis
Glucagon stimulates the synthesis of glucose via hepatic gluconeogenesis. To examine hepatic gluconeogenesis in the setting of T. cruzi infection, we used the pyruvate tolerance test. This test was performed at 30 and 70 dpi. The results of this test are shown in Figure 6C. When compared to the expected pattern in the uninfected control mice, there was a significant impairment of gluconeogenesis, as demonstrated by the level of glucose production in the infected mice. Similar results were observed 70 dpi. RT-qPCR analyses for the expression of glucose-6-phosphatase, a critical enzyme in the gluconeogenesis pathway, demonstrated a significant reduction in mRNA expression of gluocose-6-posphatase in the infected mice compared with uninfected controls (Figure 6D), thus validating the pyruvate kinase tolerance tests results, which indicated the impairment of hepatic gluconeogenesis. All determinations were made on mice fasted for at least 7 hours.
Discussion
T. cruzi infects many organs and thus it is not surprising that alterations in glucose homeostasis are multifactorial. Acute infection is accompanied by high parasitemia and intense inflammation in many organs, including the pancreas and adipose tissue. During this period, there is both hypoglycemia and hypoinsulinemia, and hypoglycemia has been predictive of increased mortality in some studies.8,9 The mechanism of the hypoglycemia has been unclear and may be due to a combination of a cytokine storm, resulting in reduced food intake7 and/or enhanced glucose uptake by the parasite. During chronic infection, the reduction in insulin persists, as does infiltration of macrophages into adipose tissue.8 Herein, we observed inflammation and parasitism of the pancreas as early as 15 dpi, a time characterized by the absence of peripheral parasitemia. The infected mice were clinically well and the inflammation persisted into the chronic phase. Ultrastructural studies revealed amastigotes within pancreatic β-cells, whereas the glucagon producing α-cells appeared normal. These observations are consistent with the expected increase in glucagon in response to the hypoglycemia. Because the glucagon response was intact, we examined effects of infection on hepatic gluconeogenesis. The results of the pyruvate tolerance test strongly suggested an impairment in that pathway, which was confirmed by an analysis of glucose-6-phosphatase, an enzyme critical for that pathway. By RT-qPCR, we could demonstrate a reduction in message levels for this enzyme. Although a reduction in hepatic mRNA levels for glucose-6-phospatase is not conclusive proof of reduced gluconeogenesis, the combined results from the pyruvate tolerance tests and the mRNA expression profile provide support for an impaired hepatic glucose output. The immunofluorescence studies using antibodies to insulin and glucagon clearly indicated that infection caused a disruption in the architecture of the islets.
The relationship of the adipocyte and the pancreas (the adipo-insular axis) is only partially understood. We demonstrated that in the adipose tissue in T. cruzi-infected mice, there was a marked increase in the inflammatory response including an increase in macrophages, as shown by Iba-1 staining (Figure 2) and an upregulation of cytokines and chemokines.8,22,30
Next we examined the adipo-insular axis and T. cruzi infection. The β-3-adrenergic receptor is found on the surface of the adipocyte,26 and when activated, it ultimately stimulates insulin secretion.26 The highly selective receptor agonist (CL 316, 243) increases plasma insulin levels by 10- to 100-fold within 15 minutes of administration. Mice lacking this receptor fail to display β-3 agonist-stimulated insulin secretions indicating that the receptor agonist-mediated insulin release requires the expression of β-3 adrenergic receptors on the surface of white adipocytes. The underlying pathways leading to increased release of insulin on stimulation of the β-3 adrenergic receptors in adipose tissue remain unclear. A prominent feature of this receptor activation is stimulation of lipolysis resulting in a release of free fatty acids from adipocytes.26 In this regard, we recently demonstrated upregulation in lipolytic enzymes in the adipose tissue of T. cruzi infected mice.22 Because free fatty acids can stimulate insulin release from β-cells both in vivo and in vitro, it has been assumed but not proven that the β-3 adrenergic agonist-induced effect on insulin release is caused by a transient elevation of free fatty acids, thus linking the adipocyte to the control of insulin secretion from the β-cells. This effect remains a point of controversy in that it has also been shown to be independent of the free fatty acid release (P.E. Scherer, unpublished data). The administration of the β-3 adrenergic agonist, CL316, 243, into mice during acute and chronic infection resulted in impaired insulin secretion. The administration of L-arginine resulted in an increase in insulin secretion by the normal pancreas.25 However, injection of CL316, 243 into acutely and chronically infected mice caused a significant reduction in insulin response. There was no significant difference in total pancreatic insulin at 30 dpi between infected and control mice (data not shown). Collectively, these data indicate a defect in insulin secretion in T. cruzi infected mice.
To understand the mechanistic basis of the reduced insulin secretion, we examined whether infection resulted in changes in insulin granule number and/or distribution. The stages of secretory granule (SG) exocytosis are docking, priming, and fusion at the plasma membrane. Docking describes the tethering of SGs with the plasma membrane in close proximity of the exocytic site. Priming is an ATP-dependent step that is not sufficient to achieve exocytosis. After priming, the only requirement for SG fusion with the plasma membrane is Ca2+,31–35 and Ca2+ sensors regulate SG release by preventing SG fusion until the arrival of a Ca2+ signal. Thus, the immediate and first phase of insulin secretion entails the fusion of primed β-cell SG pre-docked in the cortical region of the plasma membrane (the ready releasable pool).31–35 We observed that SGs were docked in the cortical region of the infected but not uninfected β-cells. Insulin secretion in response to L-arginine stimulation, however, expected to release the ready releasable pool, was attenuated in T. cruzi infected β-cells. There was also a small a decrease of SG granules in the infected pancreata. To our knowledge, these are the first observations suggesting a late event in granule/plasma membrane fusion as a mechanistic basis for reduced insulin secretion in T. cruzi infection.
Obesity, diabetes, and the metabolic syndrome are becoming increasingly prevalent in the tropical world and there are reports that they are being observed in individuals with Chagas disease.36–38 For example, Saldanha et al21 demonstrated alterations in insulin and glucose responses after oral glucose tolerance tests in patients with Chagas disease. Those patients with mega syndromes had increased size of pancreatic islets and there was inflammation in the pancreatic ganglia. Vieira and Hadler39 reported that patients with chronic Chagas disease had severe fibrosis, which they termed pancreatic cirrhosis. In Brazil, which ranks very high (ie, fifth) for diabetes prevalence, recent calculations indicate there could be as many as 300,000 people with both Chagas disease and diabetes. In the United States of America and Europe, there has been an increase in the recognition of Chagas disease among Latin American immigrants and many of these individuals suffer from obesity and diabetes.16 The consequences of the interaction of Chagas disease, obesity, and the diabetic state are poorly understood. These clinical relationships need to be explored and our pre-clinical studies may provide a guide.
dos Santos et al19 conducted studies of pancreatic function in T. cruzi-infected hamsters and similar to our observations, found lower than normal insulin levels in infected hamsters. The authors concluded that the reduction in insulin levels was likely to be multifactorial. It should be noted that due to variations in parasite strains and animal models used, a direct comparison of these experimental approaches of T. cruzi infection is difficult.
A variety of pathogens have been associated with alterations in host glucose homeostasis and specifically in the etiology of diabetes,40–58 and T. cruzi infection is not unique in causing alterations in glucose homeostasis. During bacterial sepsis, for example, hyperglycemia is well-documented. Many bacterial infections result in an increased demand for insulin and, paradoxically, increased insulin release may be accompanied by an increase in glucagon secretion. These observations suggest that a robust pancreatic output of both hormones in the early stages of certain infections is possible.
As we have clearly demonstrated in the current studies, there are several possible mechanisms to explain the interrelationship between T. cruzi infection and glucose metabolism. T. cruzi invades the brain, affecting the central nervous system endocrine pathways,59–61 which may possibly perturb insulin secretion. Some or all of these factors may result in T. cruzi-infection-related diabetes. It is important to conduct human observational studies in Chagasic populations regarding the prevalence of diabetes, but our experimental approaches provide an opportunity to examine the role of T. cruzi infection in the absence of other confounding factors that may be found in human clinical studies.
Acknowledgments
We thank Steven Connell (Touchstone Diabetes Center) and the University of Texas Southwestern Medical Center, (Dallas, TX) and Robin Sgueglia (Einstein Diabetes Center Core Facility, Bronx, NY) for insulin measurements and Geoff Perumal (Analytical Imaging Facility, Albert Einstein College of Medicine, Bronx, NY) for assistance with electron microscopy.
Footnotes
Supported by NIH grant HL-112099 (F.N.), Einstein Nathan Shock Center’s Cellular and Tissue Aging Core (P30AG038072 to R.K.), R01-DK55758 and P01-DK088761 (P.E.S.), AI-76248 (H.B.T), FAPEMIG and CNPq (F.S.M.), DK-033823 (J.E.P.), DK087776 (R.S.), NS069577 (M.S.D.), CA-129003 (C.A.), and the Einstein Diabetes Research CenterDK020541 (J.E.P and G.S.), a fellowship from the Juvenile Diabetes Association (JDRF-3-2008-130 to C.M.K.); and a grant from the National Cancer Institute (P30CA013330 to C.A.) partially supports all work conducted through shared facilities.
F.N, R.K., P.E.S., and H.B.T. contributed equally to this work.
A guest editor acted as editor-in-chief for this manuscript. No person at Thomas Jefferson University or Albert Einstein College of Medicine was involved in the peer review process or final disposition for this article.
References
- 1.Machado F.S., Jelicks L.A., Kirchhoff L.V., Shirani J., Nagajyothi F., Mukherjee S., Nelson R., Coyle C.M., Spray D.C., de Carvalho A.C., Guan F., Prado C.M., Lisanti M.P., Weiss L.M., Montgomery S.P., Tanowitz H.B. Chagas heart disease: report on recent developments. Cardiol Rev. 2012;20:53–65. doi: 10.1097/CRD.0b013e31823efde2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagajyothi F., Machado F.S., Burleigh B.A., Jelicks L.A., Scherer P.E., Mukherjee S., Lisanti M.P., Weiss L.M., Garg N.J., Tanowitz H.B. Mechanisms of Trypanosoma cruzi persistence in Chagas disease. Cell Microbiol. 2012;14:634–643. doi: 10.1111/j.1462-5822.2012.01764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanowitz H.B., Weiss L.M., Montgomery S.P. Chagas disease has now gone global. PLoS Negl Trop Dis. 2011;5:e1136. doi: 10.1371/journal.pntd.0001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang H., Chan J., Wittner M., Jelicks L.A., Morris S.A., Factor S.M., Weiss L.M., Braunstein V.L., Bacchi C.J., Yarlett N., Chandra M., Shirani J., Tanowitz H.B. Expression of cardiac cytokines and inducible form of nitric oxide synthase (NOS2) in Trypanosoma cruzi-infected mice. J Mol Cell Cardiol. 1999;31:75–88. doi: 10.1006/jmcc.1998.0848. [DOI] [PubMed] [Google Scholar]
- 5.Machado F.S., Souto J.T., Rossi M.A., Esper L., Tanowitz H.B., Aliberti J., Silva J.S. Nitric oxide synthase-2 modulates chemokine production by Trypanosoma cruzi-infected cardiac myocytes. Microbes Infect. 2008;10:1558–1566. doi: 10.1016/j.micinf.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanowitz H.B., Amole B., Hewlett D., Wittner M. Trypanosoma cruzi infection in diabetic mice. Trans R Soc Trop Med Hyg. 1988;82:90–93. [PubMed] [Google Scholar]
- 7.Nagajyothi F., Zhao D., Machado F.S., Weiss L.M., Schwartz G.J., Desruisseaux M.S., Zhao Y., Factor S.M., Huang H., Albanese C., Teixeira M.M., Scherer P.E., Chua S.C., Jr., Tanowitz H.B. Crucial role of the central leptin receptor in murine Trypanosoma cruzi (Brazil strain) infection. J Infect Dis. 2010;202:1104–1113. doi: 10.1086/656189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Combs T.P., Nagajyothi, Mukherjee S., de Almeida C.J., Jelicks L.A., Schubert W., Lin Y., Jayabalan D.S., Zhao D., Braunstein V.L., Landskroner-Eiger S., Cordero A., Factor S.M., Weiss L.M., Lisanti M.P., Tanowitz H.B., Scherer P.E. The adipocyte as an important target cell for Trypanosoma cruzi infection. J Biol Chem. 2005;280:24085–24094. doi: 10.1074/jbc.M412802200. [DOI] [PubMed] [Google Scholar]
- 9.Holscher C., Mohrs M., Dai W.J., Kohler G., Ryffel B., Schaub G.A., Mossmann H., Brombacher F. Tumor necrosis factor alpha-mediated toxic shock in Trypanosoma cruzi-infected interleukin 10-deficient mice. Infect Immun. 2000;68:4075–4083. doi: 10.1128/iai.68.7.4075-4083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.dos Santos V.M., da Cunha S.F., Teixeira Vde P., Monteiro J.P., dos Santos J.A., dos Santos T.A., dos Santos L.A., da Cunha D.F. [Frequency of diabetes mellitus and hyperglycemia in chagasic and non-chagasic women] Rev Soc Bras Med Trop. 1999;32:489–496. doi: 10.1590/s0037-86821999000500004. [DOI] [PubMed] [Google Scholar]
- 11.dos Santos V.M., Teixeira Vde P., da Cunha D.F., da Cunha S.F., Monteiro J.P., dos Santos J.A., dos Santos T.A., dos Santos L.A. [Pancreatic anatomopathologic changes in chronic chagasic women. Preliminary data] Arq Gastroenterol. 1999;36:127–132. [PubMed] [Google Scholar]
- 12.Guariento M.E., Saad M.J., Muscelli E.O., Gontijo J.A. Heterogenous insulin response to an oral glucose load by patients with the indeterminate clinical form of Chagas’ disease. Braz J Med Biol Res. 1993;26:491–495. [PubMed] [Google Scholar]
- 13.Silva C.C., Santos C.A., Mostarda C., Krieger E.M., Lopes H.F. Blood pressure, metabolic and autonomic responses to insulin and intralipid(R) infusion in chagasic patients. Arq Bras Cardiol. 2012;98:225–233. doi: 10.1590/s0066-782x2012005000018. [DOI] [PubMed] [Google Scholar]
- 14.Oliveira L.C., Juliano Y., Novo N.F., Neves M.M. Blood glucose and insulin response to intravenous glucose by patients with chronic Chagas’ disease and alcoholism. Braz J Med Biol Res. 1993;26:1187–1190. [PubMed] [Google Scholar]
- 15.Hotez P.J., Gurwith M. Europe’s neglected infections of poverty. Int J Infect Dis. 2011;15:e611–e619. doi: 10.1016/j.ijid.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Jackson Y., Castillo S., Hammond P., Besson M., Brawand-Bron A., Urzola D., Gaspoz J.M., Chappuis F. Metabolic, mental health, behavioural and socioeconomic characteristics of migrants with Chagas disease in a non-endemic country. Trop Med Int Health. 2012;17:595–603. doi: 10.1111/j.1365-3156.2012.02965.x. [DOI] [PubMed] [Google Scholar]
- 17.Yacoub S., Kotit S., Yacoub M.H. Disease appearance and evolution against a background of climate change and reduced resources. Philos Transact A Math Phys Eng Sci. 2011;369:1719–1729. doi: 10.1098/rsta.2011.0013. [DOI] [PubMed] [Google Scholar]
- 18.Corbett C.E., Scremin L.H., Lombardi R.A., Gama-Rodrigues J.J., Okumura M. Pancreatic lesions in acute experimental Chagas’ disease. Rev Hosp Clin Fac Med Sao Paulo. 2002;57:63–66. doi: 10.1590/s0041-87812002000200003. [DOI] [PubMed] [Google Scholar]
- 19.dos Santos V.M., de Lima M.A., Cabrine-Santos M., de Stefani Marquez D., de Araujo Pereira G., Lages-Silva E., Ramirez L.E. Functional and histopathological study of the pancreas in hamsters (Mesocricetus auratus) infected and reinfected with Trypanosoma cruzi. Parasitol Res. 2004;94:125–133. doi: 10.1007/s00436-004-1183-8. [DOI] [PubMed] [Google Scholar]
- 20.Novaes R.D., Goncalves R.V., Penitente A.R., Talvani A., Neves C.A., Natali A.J., Maldonado I.R. Trypanosoma cruzi infection alters glucose metabolism at rest and during exercise without modifying the morphology of pancreatic islets in rats. Pathol Res Pract. 2012;208:480–488. doi: 10.1016/j.prp.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 21.Saldanha J.C., dos Santos V.M., dos Reis M.A., da Cunha D.F., Antunes Teixeira V.P. Morphologic and morphometric evaluation of pancreatic islets in chronic Chagas’ disease. Rev Hosp Clin Fac Med Sao Paulo. 2001;56:131–138. doi: 10.1590/s0041-87812001000500001. [DOI] [PubMed] [Google Scholar]
- 22.Nagajyothi F., Desruisseaux M.S., Machado F.S., Upadhya R., Zhao D., Schwartz G.J., Teixeira M.M., Albanese C., Lisanti M.P., Chua S.C., Jr., Weiss L.M., Scherer P.E., Tanowitz H.B. Response of adipose tissue to early infection with Trypanosoma cruzi (Brazil strain) J Infect Dis. 2012;205:830–840. doi: 10.1093/infdis/jir840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagajyothi F., Desruisseaux M.S., Thiruvur N., Weiss L.M., Braunstein V.L., Albanese C., Teixeira M.M., de Almeida C.J., Lisanti M.P., Scherer P.E., Tanowitz H.B. Trypanosoma cruzi infection of cultured adipocytes results in an inflammatory phenotype. Obesity (Silver Spring) 2008;16:1992–1997. doi: 10.1038/oby.2008.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreira A.V., Segatto M., Menezes Z., Macedo A.M., Gelape C., de Oliveira Andrade L., Nagajyothi F., Scherer P.E., Teixeira M.M., Tanowitz H.B. Evidence for Trypanosoma cruzi in adipose tissue in human chronic Chagas disease. Microbes Infect. 2011;13:1002–1005. doi: 10.1016/j.micinf.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holland W.L., Miller R.A., Wang Z.V., Sun K., Barth B.M., Bui H.H., Davis K.E., Bikman B.T., Halberg N., Rutkowski J.M., Wade M.R., Tenorio V.M., Kuo M.S., Brozinick J.T., Zhang B.B., Birnbaum M.J., Summers S.A., Scherer P.E. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55–63. doi: 10.1038/nm.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Susulic V.S., Frederich R.C., Lawitts J., Tozzo E., Kahn B.B., Harper M.E., Himms-Hagen J., Flier J.S., Lowell B.B. Targeted disruption of the beta 3-adrenergic receptor gene. J Biol Chem. 1995;270:29483–29492. doi: 10.1074/jbc.270.49.29483. [DOI] [PubMed] [Google Scholar]
- 27.Chandra M., Shirani J., Shtutin V., Weiss L.M., Factor S.M., Petkova S.B., Rojkind M., Dominguez-Rosales J.A., Jelicks L.A., Morris S.A., Wittner M., Tanowitz H.B. Cardioprotective effects of verapamil on myocardial structure and function in a murine model of chronic Trypanosoma cruzi infection (Brazil Strain): an echocardiographic study. Int J Parasitol. 2002;32:207–215. doi: 10.1016/s0020-7519(01)00320-4. [DOI] [PubMed] [Google Scholar]
- 28.Jelicks L.A., Shirani J., Wittner M., Chandra M., Weiss L.M., Factor S.M., Bekirov I., Braunstein V.L., Chan J., Huang H., Tanowitz H.B. Application of cardiac gated magnetic resonance imaging in murine Chagas’ disease. Am J Trop Med Hyg. 1999;61:207–214. doi: 10.4269/ajtmh.1999.61.207. [DOI] [PubMed] [Google Scholar]
- 29.Prado C.M., Fine E.J., Koba W., Zhao D., Rossi M.A., Tanowitz H.B., Jelicks L.A. Micro-positron emission tomography in the evaluation of Trypanosoma cruzi-induced heart disease: comparison with other modalities. Am J Trop Med Hyg. 2009;81:900–905. doi: 10.4269/ajtmh.2009.09-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagajyothi F., Weiss L.M., Silver D.L., Desruisseaux M.S., Scherer P.E., Herz J., Tanowitz H.B. Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl Trop Dis. 2011;5:e953. doi: 10.1371/journal.pntd.0000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henquin J.C. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia. 2009;52:739–751. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- 32.Henquin J.C., Ravier M.A., Nenquin M., Jonas J.C., Gilon P. Hierarchy of the beta-cell signals controlling insulin secretion. Eur J Clin Invest. 2003;33:742–750. doi: 10.1046/j.1365-2362.2003.01207.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z., Thurmond D.C. Mechanisms of biphasic insulin-granule exocytosis — roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122:893–903. doi: 10.1242/jcs.034355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rorsman P., Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- 35.Juhl K., Hutton J. Stimulus-secretion coupling in the pancreatic beta-cell. Adv Exp Med Biol. 2004;552:66–90. [PubMed] [Google Scholar]
- 36.Geraix J., Ardisson L.P., Marcondes-Machado J., Pereira P.C. Clinical and nutritional profile of individuals with Chagas disease. Braz J Infect Dis. 2007;11:411–414. doi: 10.1590/s1413-86702007000400008. [DOI] [PubMed] [Google Scholar]
- 37.Tanowitz H.B., Jelicks L.A., Machado F.S., Esper L., Qi X., Desruisseaux M.S., Chua S.C., Scherer P.E., Nagajyothi F. Adipose tissue, diabetes and Chagas disease. Adv Parasitol. 2011;76:235–250. doi: 10.1016/B978-0-12-385895-5.00010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yudkin J.S. Inflammation, obesity, and the metabolic syndrome. Horm Metab Res. 2007;39:707–709. doi: 10.1055/s-2007-985898. [DOI] [PubMed] [Google Scholar]
- 39.Vieira C.B., Hadler W.A. [Histological study of the parotid gland and of the pancreas in megaesophagus]. Portuguese. Rev Assoc Med Bras. 1961;7:89–96. [PubMed] [Google Scholar]
- 40.Elased K., Playfair J.H. Hypoglycemia and hyperinsulinemia in rodent models of severe malaria infection. Infect Immun. 1994;62:5157–5160. doi: 10.1128/iai.62.11.5157-5160.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elased K.M., Playfair J.H. Reversal of hypoglycaemia in murine malaria by drugs that inhibit insulin secretion. Parasitology. 1996;112(Pt 6):515–521. doi: 10.1017/s0031182000066087. [DOI] [PubMed] [Google Scholar]
- 42.Goldberg E., Krause I. Infection and type 1 diabetes mellitus — a two edged sword? Autoimmun Rev. 2009;8:682–686. doi: 10.1016/j.autrev.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 43.Graham K.L., Sanders N., Tan Y., Allison J., Kay T.W., Coulson B.S. Rotavirus infection accelerates type 1 diabetes in mice with established insulitis. J Virol. 2008;82:6139–6149. doi: 10.1128/JVI.00597-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jun H.S., Yoon J.W. A new look at viruses in type 1 diabetes. Diabetes Metab Res Rev. 2003;19:8–31. doi: 10.1002/dmrr.337. [DOI] [PubMed] [Google Scholar]
- 45.Pino S.C., Kruger A.J., Bortell R. The role of innate immune pathways in type 1 diabetes pathogenesis. Curr Opin Endocrinol Diabetes Obes. 2010;17:126–130. doi: 10.1097/MED.0b013e3283372819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richer M.J., Horwitz M.S. Viral infections in the pathogenesis of autoimmune diseases: focus on type 1 diabetes. Front Biosci. 2008;13:4241–4257. doi: 10.2741/3002. [DOI] [PubMed] [Google Scholar]
- 47.Richer M.J., Horwitz M.S. Coxsackievirus infection as an environmental factor in the etiology of type 1 diabetes. Autoimmun Rev. 2009;8:611–615. doi: 10.1016/j.autrev.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Sano H., Terasaki J., Tsutsumi C., Imagawa A., Hanafusa T. A case of fulminant type 1 diabetes mellitus after influenza B infection. Diabetes Res Clin Pract. 2008;79:e8–e9. doi: 10.1016/j.diabres.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 49.von Herrath M.G., Holz A., Homann D., Oldstone M.B. Role of viruses in type I diabetes. Semin Immunol. 1998;10:87–100. doi: 10.1006/smim.1997.0108. [DOI] [PubMed] [Google Scholar]
- 50.Curnow R.T., Rayfield E.J., George D.T., Zenser T.V., DeRubertis F.R. Altered hepatic glycogen metabolism and glucoregulatory hormones during sepsis. Am J Physiol. 1976;230:1296–1301. doi: 10.1152/ajplegacy.1976.230.5.1296. [DOI] [PubMed] [Google Scholar]
- 51.George D.T., Rayfield E.J., Wannemacher R.W., Jr. Altered glucoregulatory hormones during acute pneumococcal sepsis in the rhesus monkey. Diabetes. 1974;23:544–549. doi: 10.2337/diab.23.6.544. [DOI] [PubMed] [Google Scholar]
- 52.Meszaros K., Lang C.H., Bagby G.J., Spitzer J.J. Contribution of different organs to increased glucose consumption after endotoxin administration. J Biol Chem. 1987;262:10965–10970. [PubMed] [Google Scholar]
- 53.Mizock B.A. Alterations in carbohydrate metabolism during stress: a review of the literature. Am J Med. 1995;98:75–84. doi: 10.1016/S0002-9343(99)80083-7. [DOI] [PubMed] [Google Scholar]
- 54.Preissig C.M., Rigby M.R. Pediatric critical illness hyperglycemia: risk factors associated with development and severity of hyperglycemia in critically ill children. J Pediatr. 2009;155:734–739. doi: 10.1016/j.jpeds.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 55.Rattanataweeboon P., Vilaichone W., Vannasaeng S. Stress hyperglycemia in patients with sepsis. J Med Assoc Thai. 2009;92(Suppl 2):S88–S94. [PubMed] [Google Scholar]
- 56.Rocha D.M., Santeusanio F., Faloona G.R., Unger R.H. Abnormal pancreatic alpha-cell function in bacterial infections. N Engl J Med. 1973;288:700–703b. doi: 10.1056/NEJM197304052881402. [DOI] [PubMed] [Google Scholar]
- 57.Zenser T.V., DeRubertis F.R., George D.T., Rayfield E.J. Infection-induced hyperglucagonemia and altered hepatic response to glucagon in the rat. Am J Physiol. 1974;227:1299–1305. doi: 10.1152/ajplegacy.1974.227.6.1299. [DOI] [PubMed] [Google Scholar]
- 58.Capua I., Mercalli A., Pizzuto M.S., Romero-Tejeda A., Kasloff S., De Battisti C., Bonfante F., Patrono L.V., Vicenzi E., Zappulli V., Lampasona V., Stefani A., Doglioni C., Terregino C., Cattoli G., Piemonti L. Influenza A viruses grow in human pancreatic cells and cause pancreatitis and diabetes in an animal model. J Virol. 2013;87:597–610. doi: 10.1128/JVI.00714-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Correa-de-Santana E., Paez-Pereda M., Theodoropoulou M., Kenji Nihei O., Gruebler Y., Bozza M., Arzt E., Villa-Verde D.M., Renner U., Stalla J., Stalla G.K., Savino W. Hypothalamus-pituitary-adrenal axis during Trypanosoma cruzi acute infection in mice. J Neuroimmunol. 2006;173:12–22. doi: 10.1016/j.jneuroim.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 60.da Silva A., Pereira G., de Souza A., Silva R., Rocha M., Lannes-Vieira J. Trypanosoma cruzi-induced central nervous system alterations: from the entry of inflammatory cells to potential cognitive and psychiatric abnormalities. J Neuroparasitology. 2010 [Google Scholar]
- 61.Roggero E., Perez A.R., Bottasso O.A., Besedovsky H.O., Del Rey A. Neuroendocrine-immunology of experimental Chagas’ disease. Ann NY Acad Sci. 2009;1153:264–271. doi: 10.1111/j.1749-6632.2008.03982.x. [DOI] [PubMed] [Google Scholar]