

Abstract
One of the most rapidly evolving genes in humans, PRDM9, is a key determinant of the distribution of meiotic recombination events. Mutations in this meiotic-specific gene have previously been associated with male infertility in humans and recent studies suggest that PRDM9 may be involved in pathological genomic rearrangements. In studying genomes from families with children affected by B-cell precursor acute lymphoblastic leukemia (B-ALL), we characterized meiotic recombination patterns within a family with two siblings having hyperdiploid childhood B-ALL and observed unusual localization of maternal recombination events. The mother of the family carries a rare PRDM9 allele, potentially explaining the unusual patterns found. From exomes sequenced in 44 additional parents of children affected with B-ALL, we discovered a substantial and significant excess of rare allelic forms of PRDM9. The rare PRDM9 alleles are transmitted to the affected children in half the cases; nonetheless there remains a significant excess of rare alleles among patients relative to controls. We successfully replicated this latter observation in an independent cohort of 50 children with B-ALL, where we found an excess of rare PRDM9 alleles in aneuploid and infant B-ALL patients. PRDM9 variability in humans is thought to influence genomic instability, and these data support a potential role for PRDM9 variation in risk of acquiring aneuploidies or genomic rearrangements associated with childhood leukemogenesis.
Most of the effort in cancer genomics has focused on capturing somatic mutations from the screening of tumor and normal somatic tissue genomes, to identify factors mutated somatically during tumor progression. Genetic approaches aim to find genomic regions predisposing individuals to cancer, to capture inherited predisposing mutations segregating in the population by using genetic linkage or association studies. For late-onset cancers, such as breast and colorectal cancers (Turnbull et al. 2010; Peters et al. 2012), many predisposing allelic variants have been described, supporting a polygenic model of susceptibility (Easton and Eeles 2008), but only a few genetic risk factors for pediatric cancer have been established (Healy et al. 2007; Sherborne et al. 2010). Dominant mutations causing cancer early in life are likely to be rapidly eliminated from the population, and as a result, it is unlikely that affected children will share inherited mutations. Parental germline events may play a role in pediatric cancer development with early evidence for epigenetically marking of imprinted genes during meiosis (Joyce and Schofield 1998), which may be involved directly in tumorigenesis for cancers of embryonal origin, such as Wilms' tumors, rhabdomyosarcoma, adrenocortical carcinoma, and hepatoblastoma. Besides this, little is known about the contribution of meiotic events to the genetic instability driving the early onset of childhood cancer. In particular, novel genomic changes that occur during meiosis will not be detectable using standard genetic mapping approaches. However, interrogating normal and tumor genomes from families of patients provides an ideal framework to study de novo genomic events potentially linked to childhood malignancies.
Recent genomic studies using family data have shown that many early onset diseases arise from defects caused by de novo genetic aberrations, be they point mutations (Awadalla et al. 2010), copy number variants (Greenway et al. 2009), structural rearrangements (Kloosterman et al. 2011), or aneuploidies (Hassold et al. 2007). Recombination rates in children correlate negatively with maternal age at birth (Hussin et al. 2011), which may have implications for understanding aneuploid conceptions. Intriguingly, children born with constitutional aneuploidies and rearrangements are at an increased risk for various malignancies (Ganmore et al. 2009). For example, children with Down syndrome have nearly a 20-fold increased risk for acute leukemia (Ross et al. 2005), suggesting that carcinogenesis and congenital anomalies may have a common basis for some pediatric cancers (Bjorge et al. 2008). Known recombination associated factors, such as DNA repair and histone modifications, are associated with genomic instabilities and cancers (Fernandez-Capetillo et al. 2004; Helleday 2010), and congenital genomic rearrangements and aneuploidies have been associated with errors in meiotic recombination (Hassold and Hunt 2001; Sasaki et al. 2010). Such gross genomic events are frequent in pediatric cancers.
Cancer is the leading cause of death by disease among children in western countries, and the overall incidence rate continues to rise steadily. The most common pediatric cancer, acute lymphoblastic leukemia (ALL), is a hematological malignancy resulting from chromosomal alterations and mutations affecting molecular pathways that disrupt lymphoid progenitor cell differentiation (Greaves 1999). Childhood ALL is likely explained by a combination of genetic predisposition and environmental exposure during early development, in fetal life and in infancy. However, genetic association studies for childhood ALL have been hampered by insufficient sample sizes. Furthermore, ALL is a heterogenous disease presenting many molecular subtypes, with different populations having different incidence rates, such that the power of stratified analyses will be limited due to a small number of cases in each subgroup. Finally, there is well-established evidence for prenatal initiation of the leukemogenesis process in children (Wiemels et al. 1999; Greaves 2006), and focusing exclusively on child genetic material in ALL association studies may be insufficient for understanding disease etiology.
To characterize the importance of parental germline events in susceptibility to childhood ALL, we first set out to determine whether meiotic recombination patterns can lead to factors associated with the development of childhood ALL. From exome sequencing and genotyping data, we characterized meiotic recombination patterns in a unique family (referred herein as the ALL quartet) with two siblings having hyperdiploid B-cell precursor ALL (B-ALL). We observed unusual localization of maternal meiotic recombination events, with a small number of crossovers taking place in previously well-characterized population recombination hotspots. Such hotspots are short segments (1–2 kb) identified to be highly recombinogenic in the human genome (Myers et al. 2005). The mother of the family carries a rare PRDM9 allele, potentially explaining the unusual placement of recombination events observed (Berg et al. 2011). PRDM9 is a meiosis-specific histone H3 methyltransferase that controls the activation of recombination hotspots via its zinc finger (ZnF) DNA binding domain recognizing a short sequence motif, which then triggers hotspot activity through modification of the chromatin state (Grey et al. 2011). Analyses of next-generation sequencing and Sanger re-sequencing read data from a cohort of parents with B-ALL affected children of French-Canadian descent revealed a substantial excess of rare allelic forms of PRDM9. This association was successfully replicated in an independent cohort of children with B-ALL diagnosed in Tennessee, USA, where the effect was found particularly in aneuploid and infant B-ALL patients. Not only has PRDM9 variability been associated with hotspot activation, but in humans it has been suggested to influence genomic instability (Berg et al. 2010; McVean and Myers 2010). The results presented here point to rare PRDM9 allelic forms of involvement in the development of preleukemic clones in B-ALL patients and we propose that PRDM9 histone H3K4 methyltransferase activity in the parental germline could lead to the genomic instability associated with childhood ALL, a plausible mechanism consistent with the current understanding of molecular pathways of leukemogenesis and the disease.
Results
The ALL family quartet
To study germline processes such as recombination, data from families with at least two siblings are required. Within the Quebec Childhood ALL cohort, we identified a family with two siblings having hyperdiploid B-ALL diagnosed at Sainte-Justine University Hospital. Families with two cases of childhood ALL in a sibship are rare and it is not clear whether siblings of children with ALL have an increased risk of developing ALL themselves (Draper et al. 1996; Winther et al. 2001). From studies published between 1951 and 2009 and registry-based childhood ALL data, an international collaboration only identified three sibships that were concordant for hyperdiploid ALL (Schmiegelow et al. 2012). However, the high concordance rate in ALL subtype within sibships is somewhat incompatible with a scenario where all cases in sibships occur randomly through independent events.
Sampling from the family included six biological samples from four family members: the mother and father, sampled once, and their two sons, patients 383 and 610, sampled at diagnosis and in remission (Fig. 1). The brothers were both diagnosed with B-ALL with FAB-L1 morphology, at the age of 2 for patient 383 and 3 yr later, at 14 yr of age, for patient 610. At diagnosis, both siblings showed hyperdiploid leukemia clones (>50 chromosomes, Supplemental Results), a childhood B-ALL subtype that is very likely to be prenatally initiated (Gruhn et al. 2008). However, chromosomal instabilities found in preleukemic clones are generated prenatally in the normal population at ∼100 times the rate of overt ALL (Mori et al. 2002), thus a second hit is required to trigger ALL during childhood and results from other genetic and/or environmental factors. Because the patients were both diagnosed within a 3-yr time period, it is likely that the second hit is due to environmental exposure (Greaves 2006).
Figure 1.

The ALL quartet family pedigree. The ALL quartet is composed of the two parents and two brothers (patients 383 and 610) affected by hyperdiploid B-cell precursor childhood ALL, sampled prior to and after chemotherapy treatment. The brothers were diagnosed within a 3-yr time period. The parents report Moroccan origins. Both maternal and paternal grandfathers are deceased from cancer. One of the father's sisters had children with poly-malformation syndromes, likely due to the high degree of consanguinity reported. Age at death is shown for deceased individuals.
De novo mutation and recombination events
Entire exomes were sequenced at high coverage using the SOLiD platform (Supplemental Table S1A) to allow the full interrogation of the mutational and recombinational landscape occurring in coding regions (Methods). Among variable positions discovered in the patients' exome, we looked for de novo mutations (Supplemental Methods). Given the human mutation rate, no more than one de novo point mutation is expected in a normal exome (Conrad et al. 2011). We only identified a single coding de novo point mutation in one of the patients (patient 383, Supplemental Fig. S1), which suggests that the parental germline mutation rate in this family is not higher than expected. Nevertheless, the putative de novo mutation identified is predicted to affect the structure and function of SMAD6. SMAD6 functions as an inhibitor of TGF-beta family signaling and was found to be a ligand-specific inhibitor of growth arrest and apoptosis in mouse B-cells (Ishisaki et al. 1999). Furthermore, SMAD6 is required for HL-60 myeloid leukemia cell line differentiation (Glesne and Huberman 2006) and is a key determinant of hematopoietic stem cell development (Pimanda et al. 2007).
Combining the exome sequencing with genotyping data obtained from Illumina Omni2.5 arrays, we identified over 816,000 high-confidence variable genomic positions within the ALL quartet exomes that showed no aberration in allele inheritance (Supplemental Table S1B). We performed fine-scale dissection of meiotic recombination events on autosomes and located a total of 102 and 47 crossovers in maternal and paternal meioses, respectively (Fig. 2A; Supplemental Methods). We also identified nine short tracts flanked by crossover events, possibly indicating gene conversion events (Supplemental Results; Supplemental Table S2). The maternal and paternal mean numbers of recombination per meiosis for the ALL quartet were compared with the distribution of maternal and paternal means in a control cohort of French-Canadian families (FC family cohort, Methods). The mother of the ALL quartet exhibits a high recombination rate with respect to the FC family cohort, with her mean number of crossovers per gamete found to be at the end of the spectrum (Supplemental Fig. S2). She carries two copies of the haplotype at the RNF212 locus associated with high recombination rates in females (haplotype [T, C] at SNP rs3796619 and rs1670533) (Kong et al. 2008). The father carries one copy of the rs3796619 T allele, which was estimated to decrease male genome-wide recombination rate by 2.62% per copy. This is consistent with the lower recombination rate seen in the father of the ALL quartet (Supplemental Fig. S2).
Figure 2.

Map of recombination events and hotspot usage in the ALL quartet. (A) Single and double crossovers in the two meioses that give rise to the patients, determined from analyses of SNPs from exome sequencing and genotyping data. Analyses were performed using pre- and post-treatment samples and only kept crossovers inferred in both. Using two somatic tissues allowed us to remove genotyping errors and double recombination events resulting from errors. All crossovers displayed are supported by at least three informative markers and high-resolution events are localized between informative markers <30 kb apart. (B) Fraction of high-resolution crossover intervals overlapping population hotspots in the FC family cohort and in the ALL quartet. Mothers (triangles) and fathers (circles) are ordered according to their proportion of overlap. We estimate that 11.78% (10.56–13.24 CI 95%) of these crossover intervals are expected to overlap population hotpots by chance.
We next evaluated hotspot usage by computing the proportion of high-resolution recombination events (localized between informative markers <30 kb apart) overlapping known population hotspots inferred from HapMap2 data (Myers et al. 2005). This measure does not directly evaluate hotspot usage, since a fraction of crossovers is expected to map in population hotspots by chance alone, but it is a good proxy to compare relative hotspot usage. Among recombination events identified in the ALL quartet, 85 maternal events and 34 paternal events were localized between informative markers <30 kb apart. Fifty-four percent (46/85) of maternal events and 79% (27/34) of paternal events overlapped with HapMap2 recombination hotspots. These proportions are significantly different between the parents (P = 0.0124, Fisher's exact test). Since it was reported that, on average, 70% of events are expected to overlap population hotspots (Coop et al. 2008; Hussin et al. 2011), this result suggests that the mother has a lack of recombination in population hotspots. To validate this result, we further derived a null distribution of the proportion of recombination events expected to overlap with HapMap2 recombination hotspots in the FC family cohort (Methods). The paternal crossovers show the expected enrichment in population hotspots inferred from HapMap2 data, while the maternal recombination landscape is unusual with a particularly low proportion of meiotic recombination events occurring in HapMap2 hotspots (Fig. 2B).
Characterizing PRDM9 in the ALL family quartet
Variability in recombination hotspot usage correlates with variation in PRDM9, a gene identified as a major hotspot determinant in mammals (Baudat et al. 2010; Berg et al. 2010; Myers et al. 2010; Parvanov et al. 2010) and the only locus known to be involved in hotspot specification in humans (Kong et al. 2010; Hinch et al. 2011). Allelic variation at the PRDM9 locus consists of variable repeating units, encoded by a minisatellite formed by tandem-repeat C2H2 zinc finger (ZnF), and has a strong effect on recombination hotspots positioning and activity (Berg et al. 2010, 2011). The reduced proportion of recombination events overlapping population hotspots in the mother of the ALL quartet led us to investigate genetic variation at the PRDM9 gene. The sequencing data revealed that the father of the two affected brothers is homozygote for the most common 13-repeat allele (allele A) whereas the mother carries an allele A and a 14-repeat allele C, inherited by one of the two brothers (Supplemental Results). We further validated the presence of the C allele, which encodes major changes in the PRDM9 ZnF array (Baudat et al. 2010), by Sanger re-sequencing (Methods).
Although this allele is rare in populations of European ancestry (∼1%), it is more frequent in individuals of African descent (∼13%) (Berg et al. 2010). Because the parents have Moroccan Arab ancestry (Supplemental Results; Supplemental Fig. S3), we studied PRDM9 diversity in 27 Moroccan individuals to establish whether the presence of the C allele in the mother reflects a different distribution of Moroccan PRDM9 alleles relative to populations of European descent. Among 54 Moroccan PRDM9 alleles sequenced, no C allele was found (Supplemental Fig. S4), suggesting that the frequency of the C allele in the Moroccan population is similar to the observed frequency in populations of European descent (Supplemental Results).
Motifs overrepresented in recombination hotspots in European and African-American individuals have been inferred from population studies (Myers et al. 2008; Hinch et al. 2011): A 13-mer motif is enriched in linkage disequilibrium-based hotspots inferred from HapMap2 data, whereas a 17-mer motif is overrepresented in African-enriched hotpots. The A allele has been shown to bind to the 13-mer CCNCCNTNNCCNC motif, whereas the C allele has demonstrated inability to activate recombination hotspots presenting this motif (Berg et al. 2010). The C allele is predicted to specifically bind to a 17-mer motif CCNCNNTNNNCNTNNCC (Berg et al. 2011), very close to the motif found to occur at an increased frequency in African-enriched hotspots (Hinch et al. 2011). Compared with the distribution of these motifs seen at recombination events in the FC family cohort, we observed that the 17-mer motif is highly represented in the ALL mother's recombination events whereas the 13-mer motif is underrepresented (Supplemental Fig. S5; Methods). These observations confirm that the presence of the C allele in the mother is likely to have caused the genome-wide shift from HapMap2 hotspots observed in the maternal recombination landscape (Berg et al. 2011; Hinch et al. 2011).
Association between PRDM9 and ALL in parents
Recent studies suggest that PRDM9 is implicated in genomic rearrangements leading to congenital diseases (Myers et al. 2008; Berg et al. 2010; Borel et al. 2012). Because large-scale genomic rearrangements are common events in childhood leukemia, we sought to type PRDM9 alleles in the parents of the B-ALL cohort. We assayed PRDM9 ZnF alleles in a panel of 44 additional parents from 22 French-Canadian families with children affected by B-ALL (FCALL cohort) by analyzing reads aligning to the PRDM9 ZnF array, in the parental trios where exome sequencing was previously performed (Methods). The read data allow for the detection of all zinc finger repeats present in the common allele A, along with two rarer repeats, k and l, present in the C allele (Supplemental Fig. S6). Around one fourth of the parents (12/46) (Supplemental Tables S3, S4) have alleles with k finger repeats (k-finger alleles), usually rare in populations of European descent (Berg et al. 2010), suggesting that k-finger alleles are in excess in the FCALL cohort (P = 0.0181) (Supplemental Results). To validate this result, Sanger re-sequencing of the PRDM9 ZnF alleles was further performed in 13 pairs of parents from the FCALL cohort and in 76 parents from the ethnically matched FC family cohort (Supplemental Fig. S7; Supplemental Results). Among the 26 B-ALL parents re-sequenced, evidence for the presence of rare alleles in the read data had been found in nine parents from eight families, and all of them were confirmed. Through re-sequencing, we discovered rare alleles in two additional families, which were not originally detected in the exome sequencing reads. In contrast, only five parents from the FC family cohort carried k-finger alleles (Supplemental Table S5). This confirms the excess of rare alleles in the FCALL cohort with respect to controls (P = 3.22 × 10−3) (Fig. 3), with 76.9% of B-ALL families with at least one parent carrying a rare PRDM9 allele. The rare alleles were preferentially carried by the mother (P = 0.0235, Fisher's exact test), although this maternal effect is not observed for k-finger alleles alone. Furthermore, the observation is not restricted to families with children having hyperdiploid B-ALL, since alternative PRDM9 ZnF alleles are also found in parents of children presenting translocations and as yet uncharacterized genetic defects (Supplemental Table S6). Finally, we found no significant evidence for transmission distortion, as k-finger parental alleles were transmitted to the affected child in six out of 11 cases with carrier parents (Supplemental Tables S3, S4), resulting in 25% of the children of the FCALL cohort (6/24) carrying a k-finger allele.
Figure 3.

Excess of rare PRDM9 alleles in parents from the FCALL cohort. (A) Pie charts showing frequencies of PRDM9 zinc-finger (ZnF) alleles obtained through Sanger sequencing of 26 parents of patients with B-ALL and 76 parents from the FC family cohort (controls). Alleles labeled as “Others” are population-specific alleles. Individuals' alleles are detailed in Supplemental Tables S3, S5. (B) Differences in allele frequencies between parents of patients and controls. The P-value in parentheses was calculated by including alleles inferred from exome sequencing reads for the 20 ALL parents for which PRDM9 ZnF arrays were not re-sequenced by Sanger (Supplemental Table S4). Applying a Bonferroni correction for performing the same test in two subsets of alleles (non-A and k-fingers alleles) would yield α = 0.025, although this correction is particularly conservative since subsets are correlated. (C) Allelic structure of PRDM9 ZnF array for alleles found in these cohorts (population-specific alleles not shown).
Replication in a B-ALL patient cohort
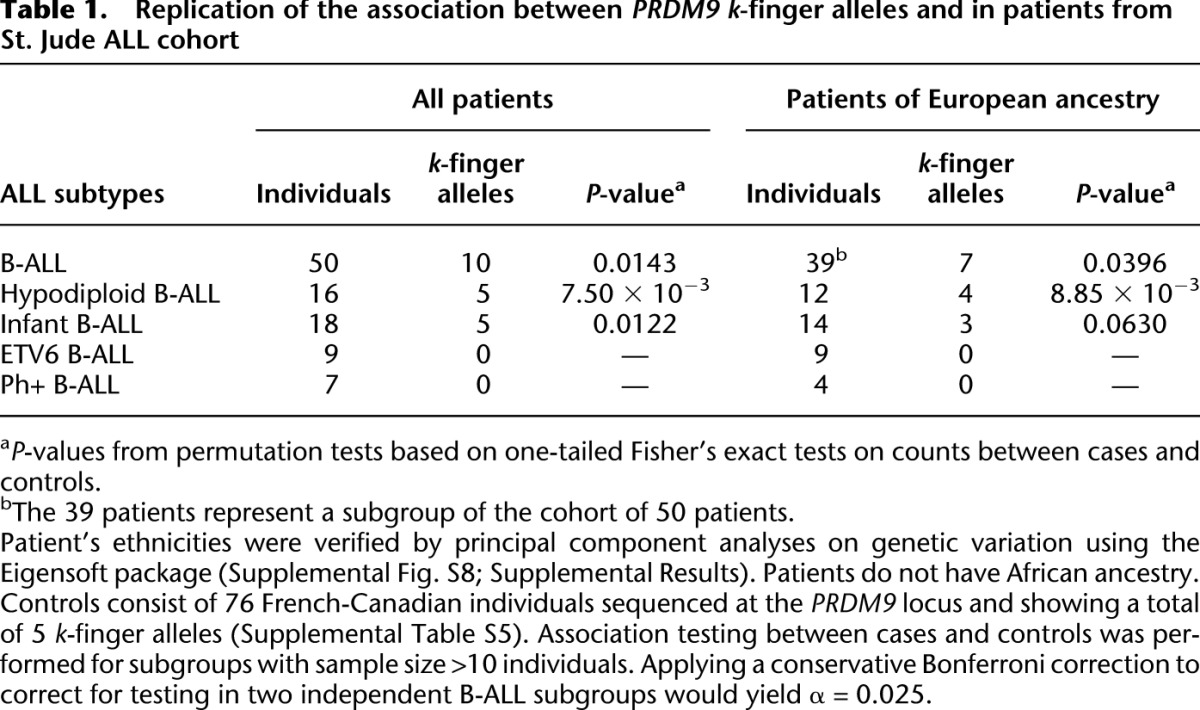
The association was also detectable in the patients themselves (P = 0.0123). We replicated this latter association in an independent cohort of 50 children, sequenced whole genome, from St. Jude Children's Research Hospital. The children were affected by B-ALL from four subtypes: ETV6-rearranged, Phildelphia chromosome-positive, hypodiploid, and infant B-ALL. We observed an excess of B-ALL patients with rare alleles in the St. Jude ALL cohort, with read data showing evidence for the k and/or l fingers in 10 children (Supplemental Table S7). This excess is significant with respect to the French-Canadian controls (P = 0.0143) (Table 1) and to 1000 Genomes Project controls from the CEU population (P = 0.0353) (Supplemental Results). No k-finger alleles were detected in B-ALL patients from Philadelphia chromosome-positive and ETV6-rearranged subtypes. In hypodiploid and infant B-ALL subtypes, the excess of k-finger alleles is significant (Table 1). The children have no African ancestry, and 39 of them ethnically cluster with the controls of European ancestry, whereas the other children have different levels of Hispanic, Asian, and Native American ancestry (Supplemental Results; Supplemental Fig. S8). Although the frequencies of PRDM9 k-finger alleles in Chinese and Mexican individuals are likely similar to the ones observed in Europeans (Parvanov et al. 2010), to be conservative we also tested for the association between k-finger alleles and B-ALL when only non-admixed white patients were included. The association remains significant overall and in the hypodiploid subtype, although the effect becomes marginal in the infant B-ALL subtype (Table 1).
Table 1.
Replication of the association between PRDM9 k-finger alleles and in patients from St. Jude ALL cohort
PRDM9 binding motifs and ALL translocations
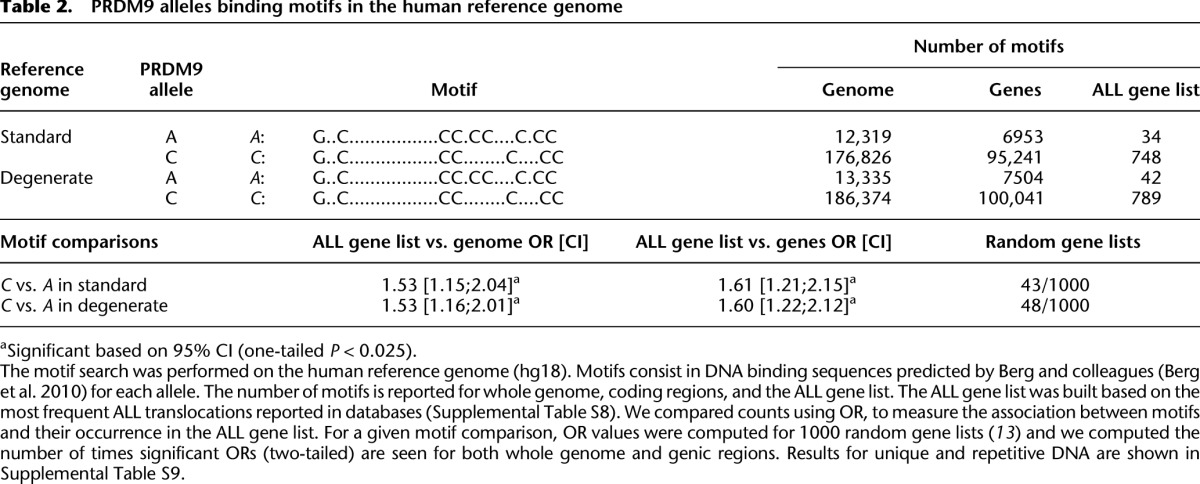
Chromosome-number abnormalities and chromosomal rearrangements have been associated with altered recombination (Hassold and Hunt 2001; Sasaki et al. 2010). Given that PRDM9 may be responsible for causing recombination-associated pathological genomic rearrangements (Myers et al. 2008; Berg et al. 2010; Borel et al. 2012), the high frequency of translocations and aneuploidies in leukemia raises the question of whether PRDM9 is implicated in the generation of preleukemic cells early in development. In particular, the C allele accounts for an important fraction of the rare alleles detected in the ALL cohorts, and its binding motif has been predicted and validated (Baudat et al. 2010; Berg et al. 2011). To assess whether the C PRDM9 allele was more likely than the common A allele to bind to genes known to be involved in ALL translocations (ALL gene list, Supplemental Table S8), we performed a motif search to identify putative A and C binding sequences in the human reference genome. We found an enrichment of sequences potentially recognized by allele C relative to allele A in the ALL gene list in comparison to the rest of the reference genome (OR = 1.53 [1.15;2.04], Table 2) and to other coding regions (OR = 1.61 [1.21;2.15], Table 2). The excess of C binding motif relative to A binding motif within the ALL gene list was found to be significant in unique DNA (OR = 2.39 [1.50–3.81 CI 95%], Supplemental Table S9) but not in repetitive DNA, except for segmental duplications. The C motif is highly overrepresented relative to the A motif in segmental duplications in the ALL gene list compared with segmental duplication in other genes. Indeed, after correcting for the higher proportion of sequences matching the C motif than the A motif in the genome, the C binding motif is more than four times more likely to be found in a segmental duplication within the ALL genes than the A motif (OR = 4.78 [1.83–12.46 CI 95%], Supplemental Table S9).
Table 2.
PRDM9 alleles binding motifs in the human reference genome
Infant B-ALL patients show an excess of C PRDM9 alleles in the St. Jude ALL cohort and harbor leukemic clones with translocations involving the MLL gene on chromosome band 11q23. We therefore scanned the nucleotide sequence of MLL and found a motif matching the C putative binding sequence occurring within the breakpoint cluster region (Fig. 4A) whereas no A binding motif was present in MLL. However, 75%–90% of genomic DNA sequences are packaged into nucleosome particles, blocking the DNA from interacting with DNA binding proteins (Segal et al. 2006). Studies suggest that the sequence itself is highly predictive of nucleosome positioning, we thus used an in silico approach (Xi et al. 2010) to predict nucleosome positioning within the MLL breakpoint cluster region (Methods). The tool predicts a potential starting position of the nucleosome at the end of the motif identified (Fig. 4B), suggesting that the motif might be accessible in a stretch of unwrapped linker DNA. It follows that PRDM9 C allele can potentially bind the MLL breakpoint cluster region, although this needs to be demonstrated in vitro and in vivo. Additionally, we reanalyzed translocation data from sperm cells in men with known PRDM9 alleles (Berg et al. 2010). The t(11;22)(q23;q11) translocations, often resulting in MLL rearrangements, occur at significantly higher frequencies in European males with k-finger alleles compared with those without k-finger alleles (P = 0.0436, Kruskal-Wallis test). However, no significant difference in translocation frequencies was observed between individuals of African descent with and without a k-finger allele (P = 0. 7998, Kruskal-Wallis test).
Figure 4.

PRDM9 C binding motif in the MLL breakpoint cluster region. (A) Logo plot of the C allele binding motif (Baudat et al. 2010), predicted based on the three indicated residues forming the binding unit of the ZnF repeats (positions −1, 3, and 6 of the ZnF alpha helices) and the consensus sequence motif simplified showing the most strongly predicted bases (in lowercase for >80% consensus for a specific base and in uppercase for >95% consensus; Berg et al. 2010). (B) Presence of a motif at chr11:117857908–117857950 (hg18), within the breakpoint cluster region of MLL, matching the predicted PRDM9 C allele binding motif for the seven strongly predicted bases shown in uppercase in the consensus sequence presented in A, and three predicted bases shown in lowercase. (Light blue) Intronic regions. (Black arrows) Positions of all occurrences of sequences matching the motif at uppercase characters. No PRDM9 A allele binding motif was found in MLL. (C) Nucleosome starting positions predicted by NuPoP (Xi et al. 2010). (Red line) Position of the predicted C binding motif.
Discussion
In this study, we examined germline processes in families with children having childhood pre-B acute lymphoblastic leukemia (B-ALL). With exome sequencing and dense genotyping data, we were able to capture parental germline recombination events in a unique family with two affected siblings. We identified PRDM9 as being associated with unusual recombination patterns and discovered a substantial excess of rare allelic forms of PRDM9 in two independent ALL cohorts. In both the initial and replication ALL cohorts, care has been taken in controlling for population structure (Supplemental Results), the cause of many false-positive genetic associations. These data support the hypothesis that PRDM9 rare allelic variants are associated with ALL in children, but represent a relatively small data set and the findings require further support in independent cohorts. The association should also be investigated in other types of childhood leukemias, such as T-lineage ALL and acute myeloid leukemia, as well as in parents of children with constitutional aneuploidies (Ganmore et al. 2009) or in woman experiencing molar pregnancies (Roman et al. 2006). The minisatellite alleles of PRDM9 have to be carefully typed from sequencing read data and rare alleles should be validated through re-sequencing. These alleles are known to cause functional biological variation, as variants in the PRDM9 gene influence recombination locations, although they have little effect on the total genome-wide recombination rate (Kong et al. 2010). If confirmed, this novel association suggests additional biological function for PRDM9 allelic variation that might impact other processes than meiotic recombination.
These findings raise many important questions about both leukemogenesis and PRDM9 function. First of all, PRDM9 activity is likely exclusive to parental germ cells but it remains unclear if it acts in the patient somatic cells. The parents carrying rare PRDM9 alleles only transmit the susceptibility allele to half of their affected children in the FCALL cohort, indicating that these alleles may act during meiosis, giving rise to gametes that predispose the offspring to B-ALL. Furthermore, PRDM9 specific expression and function at early stages of meiosis (Hayashi et al. 2005) support the parental model and point to a germline mechanism of ALL development. However, PRDM9 expression has been observed, albeit at low levels, in several hematopoetic tissues including leukemia cell lines (Johnson et al. 2003). Additional family data will also be informative with respect to the sex specificity of the effect. Indeed, the higher frequency of maternal rather than paternal carriers of rare alleles among parents in the FCALL cohort suggests a strong sex-specific effect, at least for some alleles. Maternal-specific effects on recombination with implications for child health have been demonstrated in humans, such as maternal age effect on recombination (Hussin et al. 2011) and a maternal origin of most division errors leading to trisomies (Hassold and Hunt 2009). The sex-specific effect of PRDM9 alleles on B-ALL risk could result from the differences in recombination patterns along chromosomes and in hotspot usage between male and female mammals (Paigen et al. 2008; Kong et al. 2010) or from sexual dimorphism in the regulation of the meiotic process (Cohen et al. 2006).
The mechanism underlying this novel association is not known, yet previous evidence suggests that PRDM9 may be responsible for chromosomal translocations due to its ability to determine sites of genetic crossing over (Myers et al. 2008; Berg et al. 2010; Borel et al. 2012). Therefore, PRDM9 could be implicated in the generation of some chromosomal rearrangements found in leukemia, a hypothesis supported by analysis of putative binding motifs of PRDM9 alleles occurring in a subset of genes involved in chromosomal rearrangements frequently found in ALL (Supplemental Table S8). In these analyses, we used the in silico predicted binding motifs for the PRDM9 C and A alleles as predictors for the binding activity of PRDM9 ZnF array to DNA sequences. PRDM9 binding properties are still mysterious (Segurel et al. 2011), and PRDM9 in silico–derived binding sites are not necessarily reliable for predicting PRDM9 binding activity. This is because in silico predictions using zinc finger databases and the algorithm developed by Persikov and colleagues (Persikov et al. 2009) give a vast excess of sites compared with those actually bound by PRDM9. Nevertheless, the binding predictions for PRDM9 common allele A led to the discovery of the role of this gene in human recombination (Myers et al. 2010) and the predicted C binding motif matches almost perfectly the DNA motif found in excess in African-enriched hotspots (Hinch et al. 2011). Therefore, it appears that human alleles A and C are able to bind to at least a subset of these genomic motifs. It follows that the significant excess of sequences matching the C motif compared with A motif in the ALL translocated genes, although not a demonstration that C binds these sequences, suggests that the C allele is more likely than the A allele to bind in these fragile regions. In particular, we identified a motif matching the C allele binding motif occurring within the breakpoint cluster region of MLL (Fig. 4), a gene translocated in infant B-ALL patients; however, in vitro and in vivo binding of PRDM9 C allele in this region remains to be demonstrated.
Importantly, translocations in ALL patients generally occur somatically. In humans, perturbation of the H3K4me3 dynamics at early stages of development specifically leads to inappropriate differentiation of haematopoietic progenitor cells (Chi et al. 2010). Furthermore, the H3K4me3 mark, specifically placed by PRDM9 (Hayashi et al. 2005), is a histone methylation event misregulated in many pediatric cancers (Schwartzentruber et al. 2012; Wu et al. 2012; Zhang et al. 2012) and has been linked to leukemia initiation (Chi et al. 2010). In particular, deregulation of factors that mediate H3K4me3 interferes with RAG-mediated V(D)J recombination, crucial for B-cell maturation, and affects hematopoietic cell populations. For example, local accumulation of H3K4me3 has recently been shown to occur within the breakpoint cluster region of BTG1, a driver gene affected by deletions that result from aberrant somatic recombination events in B-ALL (Waanders et al. 2012). Aberrant histone methylation in germline may therefore help establish tumor-initiating cell populations in early leukemogenesis. However, this hypothesis implies that H3K4me3 marks would be passed on to the child and maintained until early development. Transgenerational epigenetic inheritance has been recently reported for the H3K4me3 mark in Caenorhabditis elegans (Greer et al. 2011) and depends on chromatin modifiers but also on the H3K4me3 demethylase RBR-2 acting in germline, suggesting that other contributors, acting in concert with PRDM9, would be required to disrupt normal H3K4me3 patterns.
As these results potentially link allelic variation at PRDM9 with childhood ALL risk, it is reasonable to expect that higher frequencies of alternative alleles in individuals of African descent (Berg et al. 2010; Hinch et al. 2011) would indicate a potentially higher incidence of childhood leukemia in African populations. Incidence of childhood leukemia in sub-saharian Africa is not well documented; however, the incidence rate among admixed African-American children is approximately half the rate in children of European descent (Gurney et al. 1995). Therefore, the potential role of PRDM9 in ALL will likely involve multi-locus interactions arising in specific genomic backgrounds. The significant difference in frequency of t(11;22)(q23;q11) translocations between men with and without PRDM9 k-finger alleles found in Europeans but not in Africans suggests that different alleles, or combinations of alleles in a heterozygote, may not have the same impact on different genetic backgrounds. These differences could arise due to the existence of variation between European and African individuals in factors implicated in regulating H3K4me3 in meiosis after DBS repair. Moreover, since PRDM9 interacts with specific binding motifs to regulate histone methylation and recombination, these loci, if mutated, could modify downstream PRDM9 deleterious functions. Two studies (Jeffreys and Neumann 2002; Myers et al. 2010) have shown that a self-destructive drive due to biased gene conversion disrupts the common-allele binding motifs and, in the African population, the same process appears to be eliminating binding motifs for alleles at higher frequencies (Berg et al. 2011). Furthermore, a recent study argued that genetic ancestry is critical to understand ALL risk and failure to go into remission, an indicator of relapse risk in ALL (Yang et al. 2011). In this context, it makes sense to consider that not only are PRDM9 alleles critical, but also that the ancestral background of the patients is a key factor for the role of PRDM9 in leukemogenesis.
While PRDM9 is known to be involved in sterility in humans (Irie et al. 2009) and mice (Hayashi et al. 2005), this is the first study to specifically implicate PRDM9 in human disease and this novel association will hopefully inspire further investigation. PRDM9 clearly interacts with multiple factors to facilitate proper histone methylation and recruit recombination, but its molecular partners remain largely unidentified and the biological importance of PRDM9 is not fully understood. Finally, if PRDM9 is implicated in a germline mechanism of ALL development, it would mean that risk factors for ALL could be established as early as meiosis in the parental germline. Therefore, the results reported here raise the intriguing possibility that germline events and recombination processes play a role in the susceptibility to pediatric cancer, which have considerable implications for mapping strategies as well as prognosis and treatment of childhood leukemia.
Methods
Data sets
The initial French-Canadian B-cell precursor acute lymphoblastic leukemia cohort (FCALL cohort) includes blood samples from 23 French-Canadian nuclear families, which include 22 parental trios (both parents and an affected child) and one family composed of both parents and two affected brothers, referred herein as the ALL quartet. DNA from each sample was extracted from peripheral blood cells or bone marrow as previously described (Baccichet et al. 1997). Exome sequencing using the Applied Biosystems SOLiD 4.0 System, read mapping, and variant calling were performed as outlined below. Study subjects are from the established Quebec Childhood ALL cohort (Healy et al. 2010) diagnosed in the Hematology-Oncology Unit of Sainte-Justine University Hospital, Montreal, Canada, between October 1985 and November 2006.
The replication cohort (St. Jude ALL cohort) is composed of 50 unrelated children affected with B-ALL treated at St. Jude Children's Research Hospital, Memphis, Tennessee, USA. Whole-genome DNA sequencing was performed using Illumina HiSeq paired-end sequencing at a coverage of 30×, aligned using BWA (0.5.5) aligner to the human NCBI Build 36 reference sequence. The Institutional Review Board of the respective hospitals approved the research protocol and informed consent was obtained from all participants and/or their parents.
Exome sequencing data from two control cohorts were used in this study. The first one is a French-Canadian exome data set (FCEXOME) consisting of 68 French-Canadian controls for whom exome sequencing reads aligning to PRDM9 ZnF array were available. The second exome data control cohort comprises 99 individuals of European descent from the CEU population sequenced in the 1000 Genomes Project (The 1000 Genomes Project Consortium 2010). The read data from these control cohorts are further described in Supplemental Results. PRDM9 re-sequencing data and genotyping data from two additional control cohorts were included: a cohort of families of self-declared French-Canadian origin (FC family cohort) (Hussin et al. 2011) and a cohort of unrelated Moroccan individuals (Moroccan cohort) (Idaghdour et al. 2010). Supplemental Table S10 summarizes cohorts' composition, data generated, and analyses performed on each data set.
We also used SNP data from the publicly available HGDP (Cann et al. 2002) and HapMap3 (The International HapMap Consortium 2005) data sets as controls in some ancestry and association analyses.
Exome sequencing in the FCALL cohort
Exome capture was performed with the SureSelect Target Enrichment System from Agilent Technologies using the protocol optimized for Applied Biosystems' SOLiD sequencing. For each sample of the FCALL, single-end exome sequencing cohort was performed and generated ∼5 Gb of mappable sequence data per sample. Color space reads were mapped to the NCBI Build 36 reference sequence with BioScope v1.2 (Ondov et al. 2008). Base quality recalibration was performed using GATK and BAQ (Li et al. 2009; McKenna et al. 2010). PCR duplicates were removed using Picard implemented in Samtools (Li et al. 2009). In the ALL quartet, SNP calling from exome data was performed as described in Supplemental Methods. De novo mutation discovery was performed using the DND software (Cartwright et al. 2012). In the 22 trios, we called SNPs in parents using Samtools and only SNPs within targeted regions with Phred score above 30 were kept.
Genome-wide SNP arrays
Genotyping data were available for all individuals included in this study except for the 22 trios from the FCALL cohort. The ALL quartet samples were genotyped using the Illumina HumanOmni 2.5-Quad BeadChips and the Affymetrix 6.0 arrays (Supplemental Methods). Normal samples from children in the St. Jude ALL cohort were genotyped on Affymetrix SNP 6.0 (48 individuals) or 250k Nsp and 250k Sty (two individuals). The Moroccan cohort comprises 163 unrelated individuals genotyped on the Illumina's Human 610-Quad SNP Beadchip (Idaghdour et al. 2010). The FC family cohort comprises 69 nuclear families with at least two offsprings, genotyped on the Affymetrix 6.0 array (Hussin et al. 2011).
Recombination analyses
Recombination events were called in two data sets using the algorithm described previously (Hussin et al. 2011) available at www.iro.umontreal.ca/∼hussinju/NucFamTools.html, on two data sets:
(1) The ALL quartet recombination SNP data set, which is obtained by combining the exome and the Illumina SNPs (Supplemental Methods). The recombination events were called separately for the pre-treatment and post-treatment samples. All double recombination events separated by one and two informative markers were ignored. The pre-treatment and post-treatment sets of recombination events were subsequently compared and only events seen in both were kept. All events excluded were double recombinants separated by less than five informative markers. A full description of the recombination landscape in the parents of the ALL quartet is provided in Supplemental Results.
(2) The Affymetrix SNP data set for the 69 families from the FC family cohort and the ALL quartet. These samples were genotyped on the same chip, allowing direct comparison of the ALL quartet recombination landscape with recombination patterns observed in the FC family cohort. The 355,271 SNPs used in Hussin et al. (2011) were selected and 20,118 SNPs were removed because of missing genotypes or Mendelian errors in the ALL quartet data. Recombination events were located using two children in each family: For families with three offspring or more in the FC family cohort, two of the latter were chosen at random to match the ALL quartet family structure. We removed all double recombination events separated by less than five informative SNPs.
The congruence of HapMap2 recombination hotspots (The International HapMap Consortium 2005) was assessed using events localized between informative markers <30 kb apart. We estimated the proportion of recombination events that overlap a HapMap2 hotspot by chance using the approach described in Coop et al. (2008). We also investigated the overlap between the inferred maternal and paternal recombination events and the putative PRDM9 alleles A and C binding motif. Again, we only considered events localized between informative markers <30 kb apart. We also excluded recombination events overlapping a DNA sequence matching both motifs and, for each individual, we computed the proportion of events overlapping sequences matching exclusively A or C predicted binding motifs.
PRDM9 zinc finger typing in short read data
To identify PRDM9 zinc finger (ZnF) repeat types present in an individual, we analyzed sequencing reads that aligned to the PRDM9 ZnF array (exon 11) of the human NCBI Build 36 reference sequence (hg18), corresponding to the region chr5:23562605-23563612. The reads were extracted from BAM files before removing PCR duplicates, i.e., multiple reads starting at the same reference coordinate. This is because applying this filter would cause the removal of reads sampling additional ZnF repeats, absent from the reference genome, that will align to the repeats present in the reference genome. Each read was aligned to the known human PRDM9 ZnF types identified in previous studies (zinc finger repeat types a to t; Supplemental Fig. S6). We computed the proportion of reads that aligned uniquely and without mismatch to each ZnF type. Given the proportion of reads aligning to types b, c, d, and f, included in all PRDM9 ZnF alleles reported so far (Berg et al. 2010), we determine an inclusion criteria of 1% to infer the presence of a ZnF in a sample. Validation experiments by Sanger sequencing allowed us to confirm the accuracy of the 1% empirical criteria, since ZnF types predicted using this approach were present in the re-sequenced PRDM9 alleles.
Sanger sequencing of PRDM9 ZnF alleles
We sequenced the ZnF array of PRDM9 in 26 parents from the ALL cohort (including the ALL quartet parents), 76 parents from the FC family cohort, and 27 unrelated individuals from the Moroccan cohort, resulting in a total of 258 alleles sequenced. PRDM9 ZnF alleles were amplified from 5 to 20 ng of genomic DNA using the primers HsPrdm9-F3 and HsPrdm9-R1 (Baudat et al. 2010), designed to discriminate PRDM9 from his paralogous copy PRDM7. Alleles were sequenced from diploid PCR products with primers 214F, 731F, 1742R, and 1992R (Supplemental Fig. S9). Nonmixed sequence traces matching the A allele of PRDM9, indicating A/A homozygosity, were identified. We subsequently used the web-based tool Mutliple SeqDoc (Crowe 2005) to compare mixed traces with nonmixed A/A traces. This algorithm produces aligned images of a reference and a test chromatogram together with a subtracted trace showing differences between chromatograms. These difference profiles allow rapid visual identification of base substitutions, insertions, and deletions in the test sequence. The differences highlighted by the algorithm are then visually checked and interpreted to avoid potential artifact calls often introduced by automatic base-calling software. This procedure allowed us to determine allele status for all individuals (Supplemental Tables S3, S5; Supplemental Fig. S4). Most of the individuals were homozygotes A/A (64%), and all remaining individuals were heterozygotes. We identified 10 previously characterized alleles (B, C, D, E, L1, L3, L9, L20, L24, L14) (Baudat et al. 2010; Berg et al. 2010) and seven novel alleles found only in this study (L32–38). The novel alleles are described in Supplemental Results.
Association testing and ancestry analyses
Association between PRDM9 alleles and ALL in the FCALL cohort was evaluated using randomization inference based on two-tailed Fisher's exact test with 10,000 permutations. For replication in the SJDALL cohort, one-tailed Fisher-based permutation tests were performed. To test whether rare alleles of PRDM9 were overrepresented in ALL subtypes, we performed a permutation test where we permuted the 50 children from the St. Jude cohort across subtypes 10,000 times and computed how many times patients with k-finger alleles were only seen in hypodiploid and infant B-ALL subtypes. The FC family cohort was used as control cohort for association between PRDM9 ZnF alleles and disease, and HapMap3 CEU individuals were considered as controls for association between SNP rs12153202 and disease (Supplemental Results). Ancestry was studied by Principal Component Analyses of genotyped genetic variation of subjects and controls using the smartpca module from the Eigensoft package (Price et al. 2006). Detailed ancestry analyses can be found in Supplemental Results.
Genomic motif search
A motif search was performed to identify PRDM9 ZnF allele A and C binding sequences in the human genome. The motif search was performed on the human reference genome (hg18) and on a custom-made degenerate reference genome, constructed using biallelic SNPs in dbSNP v134 that were validated (VLD flag, set if the SNP has at least two minor allele counts). At each position where a SNP was reported, the nucleotide in the reference genome was replaced by the IUPAC code corresponding to allele variation. We then scan degenerate or nondegenerate genomes to search for specific degenerate motifs, denoted A and C (Table 2), representing DNA binding motifs predicted by Berg and colleagues (Berg et al. 2010) for PRDM9 ZnF allele A and C, containing only nucleotides predicted with >95% accuracy, based on the algorithm by Persikov and colleagues (Persikov et al. 2009). The sequences found were then annotated based on their starting position using a list of coordinates for regions of interest. We counted the number of nonoverlapping motifs occurring whole-genome, in genes and in segmental duplications within genes. Coordinates of all annotated human genes and segmental duplications were obtained from UCSC tables.
Mapping PRDM9 binding motifs within the ALL gene list
We built an ALL gene list using an unbiased strategy based on Mitelman and dbCRID databases (Kong et al. 2011; Mitelman et al. 2011) as of July 2011. Translocations were selected only if they were reported in more than 10 entries for ALL in these databases (Supplemental Table S8A). We next retrieved fusion genes involved in these translocations from the databases and a final total of 38 genes were kept, following a literature search performed to verify that they have been implicated in ALL in peer-reviewed publications (Supplemental Table S8B). We computed the number of sequences matching the A and C motifs occurring within and outside of selected ALL genes. Chi-square tests are sensitive to sample size and will tend to reject the null when the sample becomes sufficiently large. Because we have huge numbers for the motif counts, we used odds ratios (OR) to compare the frequencies between motifs:
with Pm the ratio between the number of motifs m in the ALL gene list and the total number of motifs m in a particular genomic data set, such as the whole genome, genic regions (repetitive and unique DNA, Supplemental Table S9), and in segmental duplication occurring in genes. This provides a measure of the strength of nonindependence between the motifs and their occurrence in the ALL gene list. Confidence intervals were calculated following the procedure described in Morris and Gardner (1988). We generated 1000 lists of randomly chosen genes, with the inclusion criteria being a gene length of 3 kb or more. The experiments described above were repeated with each random gene list in place of the ALL gene list and we computed the number of time significant OR were seen to obtain a two-tailed P-value. We used the program NuPoP (Xi et al. 2010) to predict nucleosome positioning within the MLL breakpoint cluster region (chr11:117857800-117858100, hg18).
Translocation data
We used t(11;22)(q23;q11) translocation frequencies previously published by Berg and colleagues (Berg et al. 2010), based on de novo detection of translocations in sperm from men with different PRDM9 alleles. We separated African from European men and performed a Kruskal-Wallis test to compare men carrying k-finger alleles and men carrying other alleles in each population. For Africans, the k-finger alleles (C, L4, L6, L14-19) showed no significant influence on translocation frequency (χ2 = 0.0643, P = 0.7998). However, in Europeans, men with k-finger alleles (C, L20) showed significantly increased translocation frequencies (χ2 = 4.0735, P = 0.04356).
Data access
Sequences of the PRDM9 novel alleles have been deposited in GenBank under accession numbers JQ044371–JQ044377. Genomic reads aligning to PRDM9 exon 12 from all individuals in the FCALL and control cohorts are deposited in the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA060797. Whole-exome sequencing reads and genotyping data for the ALL quartet will be made available through the Quebec childhood leukemia web portal (http://childhoodleukemiagenomics.org). Genomic sequence data from the SJALL cohort is available through the European Genome-Phenome Archive (https://www.ebi.ac.uk/ega/) under accession number EGAC00001000044 and can be accessed by application to the Pediatric Cancer Genome Project (PCGP) Data Access Committee. More information can be found at http://explore.pediatriccancergenomeproject.org.
Acknowledgments
We thank all patients and their parents from Sainte-Justine University Hospital and the St. Jude Children's Research Hospital for participating in this study. We acknowledge P. Legendre and G. Bourret from the Genome Quebec Innovation Center sequencing platform; J. Langdon and M. Hurles for providing PRDM9 sequencing primer sequences; B. Ge and T. Pastinen for the genotyping data of the ALL quartet; T. Sontag, S. Leclerc, and D. Arafat for preparing DNA samples; and B. Li and G. Abecasis for the polymutt executable. We thank C. Bherer, M. Capredon, P. Donnelly, E. Kritikou, and J. Quinlan for helpful discussions. This work was supported by MDEIE of Quebec, Canadian Foundation for Innovation, NSERC, The Cole Foundation, Terry Fox Foundation CIHR, St. Jude Children's Research Hospital—Washington University Pediatric Cancer Genome Project, the American Lebanese and Syrian Associated Charities of St. Jude Children's Research Hospital. P.A. holds a Genome-Quebec Award for Population and Medical Genomics and an FRSQ research award.
Author contributions: J. Hussin and P.A. designed the study. D.S., Y.I., J. Healy, G.G., A.A., L.H., G.A., J.R.D., C.G.M., and P.A. contributed reagents, patient materials, and samples. J. Hussin, F.C., J.-F.S., M.L., and S.B. performed experimental work. J. Hussin, V.B., V.S., J.-C.G., T.d.M., and P.A. performed computational data analyses of the Sainte-Justine cohort data. J. Hussin, J.M., L.W., and J.Z. performed computational data analyses of the St. Jude cohort data. J. Hussin and P.A. wrote the paper.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.144188.112.
Freely available online through the Genome Research Open Access option.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR, Cote M, Henrion E, Spiegelman D, Tarabeux J, et al. 2010. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet 87: 316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccichet A, Qualman SK, Sinnett D 1997. Allelic loss in childhood acute lymphoblastic leukemia. Leuk Res 21: 817–823 [DOI] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg IL, Neumann R, Lam KW, Sarbajna S, Odenthal-Hesse L, May CA, Jeffreys AJ 2010. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet 42: 859–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg IL, Neumann R, Sarbajna S, Odenthal-Hesse L, Butler NJ, Jeffreys AJ 2011. Variants of the protein PRDM9 differentially regulate a set of human meiotic recombination hotspots highly active in African populations. Proc Natl Acad Sci 108: 12378–12383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorge T, Cnattingius S, Lie RT, Tretli S, Engeland A 2008. Cancer risk in children with birth defects and in their families: A population based cohort study of 5.2 million children from Norway and Sweden. Cancer Epidemiol Biomarkers Prev 17: 500–506 [DOI] [PubMed] [Google Scholar]
- Borel C, Cheung F, Stewart H, Koolen DA, Phillips C, Thomas NS, Jacobs PA, Eliez S, Sharp AJ 2012. Evaluation of PRDM9 variation as a risk factor for recurrent genomic disorders and chromosomal non-disjunction. Hum Genet 131: 1519–1524 [DOI] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Cambon-Thomsen A, et al. 2002. A human genome diversity cell line panel. Science 296: 261–262 [DOI] [PubMed] [Google Scholar]
- Cartwright RA, Hussin J, Keebler JE, Stone EA, Awadalla P 2012. A family-based probabilistic method for capturing de novo mutations from high-throughput short-read sequencing data. Stat Appl Genet Mol Biol 11 doi: 10.2202/1544-6115.1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG 2010. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10: 457–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PE, Pollack SE, Pollard JW 2006. Genetic analysis of chromosome pairing, recombination, and cell cycle control during first meiotic prophase in mammals. Endocr Rev 27: 398–426 [DOI] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Wen X, Ober C, Pritchard JK, Przeworski M 2008. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science 319: 1395–1398 [DOI] [PubMed] [Google Scholar]
- Crowe ML 2005. SeqDoC: Rapid SNP and mutation detection by direct comparison of DNA sequence chromatograms. BMC Bioinformatics 6: 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper GJ, Sanders BM, Lennox EL, Brownbill PA 1996. Patterns of childhood cancer among siblings. Br J Cancer 74: 152–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DF, Eeles RA 2008. Genome-wide association studies in cancer. Hum Mol Genet 17: R109–R115 [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A 2004. H2AX: The histone guardian of the genome. DNA Repair (Amst) 3: 959–967 [DOI] [PubMed] [Google Scholar]
- Ganmore I, Smooha G, Izraeli S 2009. Constitutional aneuploidy and cancer predisposition. Hum Mol Genet 18: R84–R93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glesne D, Huberman E 2006. Smad6 is a protein kinase X phosphorylation substrate and is required for HL-60 cell differentiation. Oncogene 25: 4086–4098 [DOI] [PubMed] [Google Scholar]
- Greaves M 1999. Molecular genetics, natural history and the demise of childhood leukaemia. Eur J Cancer 35: 1941–1953 [DOI] [PubMed] [Google Scholar]
- Greaves M 2006. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer 6: 193–203 [DOI] [PubMed] [Google Scholar]
- Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, et al. 2009. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet 41: 931–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, Benayoun BA, Shi Y, Brunet A 2011. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479: 365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey C, Barthes P, Chauveau-Le Friec G, Langa F, Baudat F, de Massy B 2011. Mouse PRDM9 DNA-binding specificity determines sites of histone H3 lysine 4 trimethylation for initiation of meiotic recombination. PLoS Biol 9: e1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruhn B, Taub JW, Ge Y, Beck JF, Zell R, Hafer R, Hermann FH, Debatin KM, Steinbach D 2008. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia 22: 1692–1697 [DOI] [PubMed] [Google Scholar]
- Gurney JG, Severson RK, Davis S, Robison LL 1995. Incidence of cancer in children in the United States. Sex-, race-, and 1-year age-specific rates by histologic type. Cancer 75: 2186–2195 [DOI] [PubMed] [Google Scholar]
- Hassold T, Hunt P 2001. To err (meiotically) is human: The genesis of human aneuploidy. Nat Rev Genet 2: 280–291 [DOI] [PubMed] [Google Scholar]
- Hassold T, Hunt P 2009. Maternal age and chromosomally abnormal pregnancies: What we know and what we wish we knew. Curr Opin Pediatr 21: 703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassold T, Hall H, Hunt P 2007. The origin of human aneuploidy: Where we have been, where we are going. Hum Mol Genet 16: R203–R208 [DOI] [PubMed] [Google Scholar]
- Hayashi K, Yoshida K, Matsui Y 2005. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438: 374–378 [DOI] [PubMed] [Google Scholar]
- Healy J, Belanger H, Beaulieu P, Lariviere M, Labuda D, Sinnett D 2007. Promoter SNPs in G1/S checkpoint regulators and their impact on the susceptibility to childhood leukemia. Blood 109: 683–692 [DOI] [PubMed] [Google Scholar]
- Healy J, Richer C, Bourgey M, Kritikou EA, Sinnett D 2010. Replication analysis confirms the association of ARID5B with childhood B-cell acute lymphoblastic leukemia. Haematologica 95: 1608–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T 2010. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 31: 955–960 [DOI] [PubMed] [Google Scholar]
- Hinch AG, Tandon A, Patterson N, Song Y, Rohland N, Palmer CD, Chen GK, Wang K, Buxbaum SG, Akylbekova EL, et al. 2011. The landscape of recombination in African Americans. Nature 476: 170–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussin J, Roy-Gagnon MH, Gendron R, Andelfinger G, Awadalla P 2011. Age-dependent recombination rates in human pedigrees. PLoS Genet 7: e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idaghdour Y, Czika W, Shianna KV, Lee SH, Visscher PM, Martin HC, Miclaus K, Jadallah SJ, Goldstein DB, Wolfinger RD, et al. 2010. Geographical genomics of human leukocyte gene expression variation in southern Morocco. Nat Genet 42: 62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium 2005. A haplotype map of the human genome. Nature 437: 1299–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie S, Tsujimura A, Miyagawa Y, Ueda T, Matsuoka Y, Matsui Y, Okuyama A, Nishimune Y, Tanaka H 2009. Single-nucleotide polymorphisms of the PRDM9 (MEISETZ) gene in patients with nonobstructive azoospermia. J Androl 30: 426–431 [DOI] [PubMed] [Google Scholar]
- Ishisaki A, Yamato K, Hashimoto S, Nakao A, Tamaki K, Nonaka K, ten Dijke P, Sugino H, Nishihara T 1999. Differential inhibition of Smad6 and Smad7 on bone morphogenetic protein- and activin-mediated growth arrest and apoptosis in B cells. J Biol Chem 274: 13637–13642 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Neumann R 2002. Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat Genet 31: 267–271 [DOI] [PubMed] [Google Scholar]
- Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD 2003. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302: 2141–2144 [DOI] [PubMed] [Google Scholar]
- Joyce JA, Schofield PN 1998. Genomic imprinting and cancer. Mol Pathol 51: 185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Guryev V, van Roosmalen M, Duran KJ, de Bruijn E, Bakker SC, Letteboer T, van Nesselrooij B, Hochstenbach R, Poot M, et al. 2011. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum Mol Genet 20: 1916–1924 [DOI] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Stefansson H, Masson G, Helgason A, Gudbjartsson DF, Jonsdottir GM, Gudjonsson SA, Sverrisson S, Thorlacius T, et al. 2008. Sequence variants in the RNF212 gene associate with genome-wide recombination rate. Science 319: 1398–1401 [DOI] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Gylfason A, Kristinsson KT, Gudjonsson SA, et al. 2010. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467: 1099–1103 [DOI] [PubMed] [Google Scholar]
- Kong F, Zhu J, Wu J, Peng J, Wang Y, Wang Q, Fu S, Yuan LL, Li T 2011. dbCRID: A database of chromosomal rearrangements in human diseases. Nucleic Acids Res 39: D895–D900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G, Myers S 2010. PRDM9 marks the spot. Nat Genet 42: 821–822 [DOI] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F 2011. Mitelman database of chromosome aberrations and gene fusions in cancer. http://cgap.nci.nih.gov/Chromosomes/Mitelman
- Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C, Greaves M 2002. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci 99: 8242–8247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JA, Gardner MJ 1988. Calculating confidence intervals for relative risks (odds ratios) and standardised ratios and rates. Br Med J (Clin Res Ed) 296: 1313–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324 [DOI] [PubMed] [Google Scholar]
- Myers S, Freeman C, Auton A, Donnelly P, McVean G 2008. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet 40: 1124–1129 [DOI] [PubMed] [Google Scholar]
- Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, Donnelly P 2010. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327: 876–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondov BD, Varadarajan A, Passalacqua KD, Bergman NH 2008. Efficient mapping of Applied Biosystems SOLiD sequence data to a reference genome for functional genomic applications. Bioinformatics 24: 2776–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paigen K, Szatkiewicz JP, Sawyer K, Leahy N, Parvanov ED, Ng SH, Graber JH, Broman KW, Petkov PM 2008. The recombinational anatomy of a mouse chromosome. PLoS Genet 4: e1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K 2010. Prdm9 controls activation of mammalian recombination hotspots. Science 327: 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persikov AV, Osada R, Singh M 2009. Predicting DNA recognition by Cys2His2 zinc finger proteins. Bioinformatics 25: 22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters U, Hutter CM, Hsu L, Schumacher FR, Conti DV, Carlson CS, Edlund CK, Haile RW, Gallinger S, Zanke BW, et al. 2012. Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum Genet 131: 217–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimanda JE, Donaldson IJ, de Bruijn MF, Kinston S, Knezevic K, Huckle L, Piltz S, Landry JR, Green AR, Tannahill D, et al. 2007. The SCL transcriptional network and BMP signaling pathway interact to regulate RUNX1 activity. Proc Natl Acad Sci 104: 840–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909 [DOI] [PubMed] [Google Scholar]
- Roman E, Doyle P, Lightfoot T, Ansell P, Simpson J, Allan JM, Kinsey S, Eden TO 2006. Molar pregnancy, childhood cancer and genomic imprinting—is there a link? Hum Fertil (Camb) 9: 171–174 [DOI] [PubMed] [Google Scholar]
- Ross JA, Spector LG, Robison LL, Olshan AF 2005. Epidemiology of leukemia in children with Down syndrome. Pediatr Blood Cancer 44: 8–12 [DOI] [PubMed] [Google Scholar]
- Sasaki M, Lange J, Keeney S 2010. Genome destabilization by homologous recombination in the germ line. Nat Rev Mol Cell Biol 11: 182–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiegelow K, Lausten Thomsen U, Baruchel A, Pacheco CE, Pieters R, Pombo-de-Oliveira MS, Andersen EW, Rostgaard K, Hjalgrim H, Pui CH 2012. High concordance of subtypes of childhood acute lymphoblastic leukemia within families: Lessons from sibships with multiple cases of leukemia. Leukemia 26: 675–681 [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482: 226–231 [DOI] [PubMed] [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J 2006. A genomic code for nucleosome positioning. Nature 442: 772–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segurel L, Leffler EM, Przeworski M 2011. The case of the fickle fingers: How the PRDM9 zinc finger protein specifies meiotic recombination hotspots in humans. PLoS Biol 9: e1001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherborne AL, Hosking FJ, Prasad RB, Kumar R, Koehler R, Vijayakrishnan J, Papaemmanuil E, Bartram CR, Stanulla M, Schrappe M, et al. 2010. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet 42: 492–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, Seal S, Ghoussaini M, Hines S, Healey CS, et al. 2010. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet 42: 504–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waanders E, Scheijen B, van der Meer LT, van Reijmersdal SV, van Emst L, Kroeze Y, Sonneveld E, Hoogerbrugge PM, Geurts van Kessel A, van Leeuwen FN, et al. 2012. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet 8: e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, Masera G, Saha V, Biondi A, Greaves MF 1999. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet 354: 1499–1503 [DOI] [PubMed] [Google Scholar]
- Winther JF, Sankila R, Boice JD, Tulinius H, Bautz A, Barlow L, Glattre E, Langmark F, Moller TR, Mulvihill JJ, et al. 2001. Cancer in siblings of children with cancer in the Nordic countries: A population-based cohort study. Lancet 358: 711–717 [DOI] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. 2012. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44: 251–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi L, Fondufe-Mittendorf Y, Xia L, Flatow J, Widom J, Wang JP 2010. Predicting nucleosome positioning using a duration hidden Markov model. BMC Bioinformatics 11: 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JJ, Cheng C, Devidas M, Cao X, Fan Y, Campana D, Yang W, Neale G, Cox NJ, Scheet P, et al. 2011. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet 43: 237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. 2012. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481: 157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]