

Abstract
FSH and IGF-I synergistically stimulate gonadal steroid production; conversely, silencing the FSH or the IGF-I genes leads to infertility and hypogonadism. To determine the molecular link between these hormones, we examined the signaling cross talk downstream of their receptors. In human and rodent granulosa cells (GCs), IGF-I potentiated the stimulatory effects of FSH and cAMP on the expression of steroidogenic genes. In contrast, inhibition of IGF-I receptor (IGF-IR) activity or expression using pharmacological, genetic, or biochemical approaches prevented the FSH- and cAMP-induced expression of steroidogenic genes and estradiol production. In vivo experiments demonstrated that IGF-IR inactivation reduces the stimulation of steroidogenic genes and follicle growth by gonadotropins. FSH or IGF-I alone stimulated protein kinase B (PKB), which is also known as AKT and in combination synergistically increased AKT phosphorylation. Remarkably, blocking IGF-IR expression or activity decreased AKT basal activity and abolished AKT activation by FSH. In GCs lacking IGF-IR activity, FSH stimulation of Cyp19 expression was rescued by overexpression of constitutively active AKT. Our findings demonstrate, for the first time, that in human, mouse, and rat GCs, the well-known stimulatory effect of FSH on Cyp19 and AKT depends on IGF-I and on the expression and activation of the IGF-IR.
Insulin-like growth factor 1 (IGF-I) regulates differentiation and proliferation of normal and neoplastic cells both as an endocrine and auto/paracrine signal (1). In addition, the infertility of male and female IGF-I–knockout mice (2) demonstrates an essential role for IGF-I in reproduction. IGF-I influences fertility at different levels within the hypothalamic–pituitary-gonadal axis (3). In the hypothalamus and pituitary, IGF-I regulates GnRH, LH, and prolactin secretion (4–6). In the gonads, IGF-I and its receptor (IGF-IR) are selectively expressed in granulosa cells (GCs) of healthy and growing ovarian follicles and in testicular Sertoli cells (7–10). Consequently, IGF-I–null mice of both sexes have hypoplastic gonads resulting in anovulation in females and delayed testis development in males (2).
In the ovary, IGF-I stimulates follicular steroidogenesis, either alone or in synergy with gonadotropins. In porcine and human ovarian cells, IGF-I alone increases proliferation and the production of estradiol and progesterone; in addition, it potentiates the effects of FSH on GC differentiation (11). In contrast, in rodents, IGF-I alone does not impact gene expression, but it enhances the stimulatory effects of FSH on progesterone production (12), cytochrome P450 aromatase (Cyp19) activity (13), LH receptor expression (14), and inhibin-α secretion (15). IGF-I–knockout mice have a normal complement of oocytes and primordial follicles; however, follicle development stops at the preantral stage most probably due to a decrease on FSH receptor (FSHR) expression, which preclude the rescue of folliculogenesis by gonadotropin administration (2, 16). This evidence clearly illustrates the essential role of IGF-I in ovarian function; however, the molecular mechanisms mediating these important effects of IGF-I remain largely unknown.
Similarly, FSHR and FSH-deficient females are infertile due to a block in folliculogenesis before antral follicle formation (17, 18). The lack of follicle development in the absence of IGF-I or FSH and their synergistic effects on GC differentiation and steroidogenesis indicate that these hormones interact to maintain ovarian function. This premise is supported by evidence that suggests some overlap of the FSHR and the IGF-IR intracellular signaling pathways. For example, although FSHR signals mainly via the cAMP/protein kinase A pathway (19), it also activates the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) and MAPK/ERK pathways (19, 20), which are the major pathways mediating IGF-I actions (21). Moreover, Gonzalez-Robayna et al (22) showed that FSH and IGF-I synergistically activate AKT. In addition, Zeleznik et al (23) demonstrated that AKT amplifies the effects of FSH on steroid production. These reports suggest that the intracellular pathways activated by the FSHR and the IGF-IR cooperate closely to maintain normal GC function. We postulate that cross talk between IGF-IR and FSHR signaling is crucial for promoting follicle development.
Here, we explore the interactions between the IGF-IR and FSHR signaling pathways in human, mouse, and rat GCs and demonstrate that FSH actions on steroidogenic gene expression depend on IGF-IR activation. Our results suggest that IGF-I signaling is obligatory for FSH stimulation of AKT and subsequent induction of ovarian cell differentiation.
Materials and Methods
Cell isolation and culture
Immature female Sprague Dawley rats were purchased from Charles River Laboratories Inc (Wilmington, Massachusetts) and treated in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals following protocols approved by the University of Illinois at Chicago Animal Care Committee. Rats were treated with estradiol (1.5 mg for each rat) for 3 days followed by euthanasia and dissection of the ovaries. GCs were isolated and cultured as previously described (24, 25). Cells were seeded into extracellular matrix-coated (BD Biosciences, San Jose, California) plates at a density of 2.5 × 105/ml in serum-free and antibiotic-supplemented DMEM/F12 medium for 2 hours before initiation of treatments.
Primary human cumulus GCs were isolated from patients undergoing follicular aspiration at University of Illinois Hospital. Human GCs were seeded into extracellular matrix-coated plates at a density of 2 × 105/ml in serum-free DMEM/F12 medium (Invitrogen, Carlsbad, California). A mouse Sertoli TM4 cell line (ATCC CRL-1715) was seeded at a density of 1 × 105/ml and cultured in DMEM/F12 medium supplemented with 5% horse serum, 2.5% fetal bovine serum, and antibiotics. Cells were serum starved for at least 16 hours before the initiation of treatments.
mRNA quantitation
Total RNA was isolated using Trizol reagent (Invitrogen) following the manufacturer's instructions. Total RNA (1 μg) was reverse transcribed at 42°C using Moloney murine leukemia virus (Epicenter Tech, Madison, Wisconsin). Standard curves ranging from 10 × 102 to 6 × 106 copies/μl were prepared for each gene using purified PCR products. Aliquots of standards or cDNA were combined with PCR buffer containing SYBR Green I (Sigma Chemical Co, St. Louis, Missouri), Taq polymerase (Genscript, Piscataway, NJ), and primers specific for the gene of interest. Only intron-spanning primers were used. DNA amplification and quantification of PCR products was done in an iQcycler (Bio-Rad, Hercules, California). Melting curves were routinely determined to ascertain that only the expected products had been generated. For each gene of interest, the number of mRNA molecules was calculated and expressed as the relative expression to the reference ribosomal L19 mRNA.
Western blot analysis
GCs were harvested in ice-cold RIPA lysis buffer containing protease inhibitor cocktail (Sigma), 1mM NaF, and 1mM Na3VO4. After determination of protein concentration by using a bicinchoninic acid protein assay kit (Thermo Scientific, Rockford, Illinois), denatured extracts were subjected to Western blot analysis following standard procedures with the following conditions: blocking with 5% nonfat dry milk for 2 hours, primary antibodies against Cyp19 (1:500; Serotec, Ltd, Oxford, United Kingdom), FSHR (1:500; Epitomics, Inc, Burlingame, California), Akt (1:1000), phospho-Akt(Ser473) (1:1000), Erk1/2 (1:1000), phospho-Erk1/2(Thr202/Tyr204-Thr185/Tyr187) (1:1000), cAMP response element-binding protein (CREB) (1:500), or phospho-CREB(Ser133) (1:500; Cell Signaling Technology, Inc, Danvers, Massachusetts), and horseradish peroxidase-conjugated secondary antibodies. Detection was done by using Immobilion Western chemiluminescent substrate (Millipore, Billerica, Massachusetts). The band intensities from each blot were quantified using ImageJ software, and the relative expression was plotted. Columns represent mean ± SEM.
Recombinant DNA and cell transfection
GCs were transfected with pGL3 reporter constructs containing the rat Cyp19 proximal promoter by using FuGene 6 (Roche, Indianapolis, Indiana) according to manufacturer's instructions. After 42 hours of transfection, cells were treated with either FSH (50 ng/ml) or vehicle for 6 hours. Cell lysates were prepared using 100 μl of passive lysis buffer (Promega, Madison, Wisconsin). Luciferase activity was determined in 50 μl of lysate/sample using the dual luciferase reporter assay (Promega). The IGF-binding protein 2 (IGFBP2) expression construct or an empty vector (pcDNA) was transfected by using Fugene 6.
Short hairpin RNAs (shRNAs) under the control of the H1 promoter were used to specifically knock down the expression of IGF-I and IGF-IR. The target recognition sequence for rat IGF-I was gca ttt ccc tca atg aaat and for IGF-IR was cca acg gat tga ttc taat. A shRNA against luciferase, gca ctc tga ttg aca aata, was used as control. Oligonucleotides containing inverted repeats connected with the rat mir-25 loop sequence (26) were chemically synthesized (Integrated DNA Technologies, Inc, Coralville, Iowa) and cloned into a lentiviral transfer plasmid to produce shRNA expression vectors (24). Viral stocks were generated in HEK293 cells (Invitrogen) and concentrated by ultracentrifugation and used at a multiplicity of infection of 10.
Lentiviral constructs expressing constitutively activated (CA), dominant-negative (DN), or null AKT were kindly provided by Michael Robinson (Children's Hospital of Philadelphia, Philadelphia, Pennsylvania). All the constructs were subcloned into lentiviral transfer plasmid pTY-CMV. Lentivirus were generated in HEK293 cells and concentrated by ultracentrifugation. Lentiviruses were added directly to the GCs at the time of plating and cells treated 24 hours after infection.
Intraovarian bursa injection
Rat ovaries were exposed through lateral incisions under 30% isoflurane anesthesia. Each ovary received 30 μL of vehicle (3% methylcellulose in saline) or NVP-AEW541 (AEW) (Cayman Chemical, Ann Arbor, Michigan) diluted in the same vehicle. Administration was done into the ovarian bursa through the fat pad as previously described (27).
IGF-I and estradiol determination
IGF-I and estradiol levels were determined by using IGF-I or estradiol immunoassay ELISA kit (R&D Systems, Inc, Minneapolis, Minnesota) following the manufacturer's instructions.
Hematoxylin and eosin stain
Dewaxed and rehydrated sections of paraffin-embedded ovaries were dipped in Harris hematoxylin (Thermo Scientific) followed by rinsing in running water. Slides were next dipped in Define reagent (Fisher, Rockford, Illinois) followed by rinsing. Bluing reagent (Fisher) was used to sharpen the staining of the nucleus. Slides were then placed in 95% ethanol followed by staining in eosin-Y and phloxine (Thermo Scientific) to stain the cytoplasm.
Statistics
One-way ANOVA followed by the Tukey test was used for the statistical analysis of protein expression, relative mRNA expression, and luciferase activity data using Prism software (GraphPad Software, Inc, San Diego, California). Values were considered statistically significant at P < .05.
Results
IGF-I Enhances FSH stimulation of gene expression
To determine the effect of IGF-I on the transcription of steroidogenic genes across species, we cultured GCs from rats, mice, and humans in serum-free medium in the presence of either FSH or IGF-I alone or combined. In rat GCs, FSH stimulated Cyp19, P450scc, and StAR mRNA expression. IGF-I alone had no effect; however, it enhanced the stimulatory effect of FSH on Cyp19 expression in a dose-dependent manner (Figure 1A). IGF-I also potentiated FSH stimulation of Cyp19 promoter activity and protein expression (Figure 1, B and C). Accordingly, estradiol production was significantly higher in GCs treated with FSH plus IGF-I (468.5 ± 73.76 pg/mL) when compared with FSH (234.9 ± 32.09 pg/mL; P < .01). Estradiol levels were not different between control (37.40 ± 7.1 pg/mL) and IGF-I-treated cells (42.76 ± 2.5 pg/mL). IGF-I also potentiated the stimulatory effect of FSH on StAR and P450scc (Figure 1D). FSH and IGF-I combined, but not alone, stimulated 3β-hydroxysteroid dehydrogenase (Figure 1D).
Figure 1.
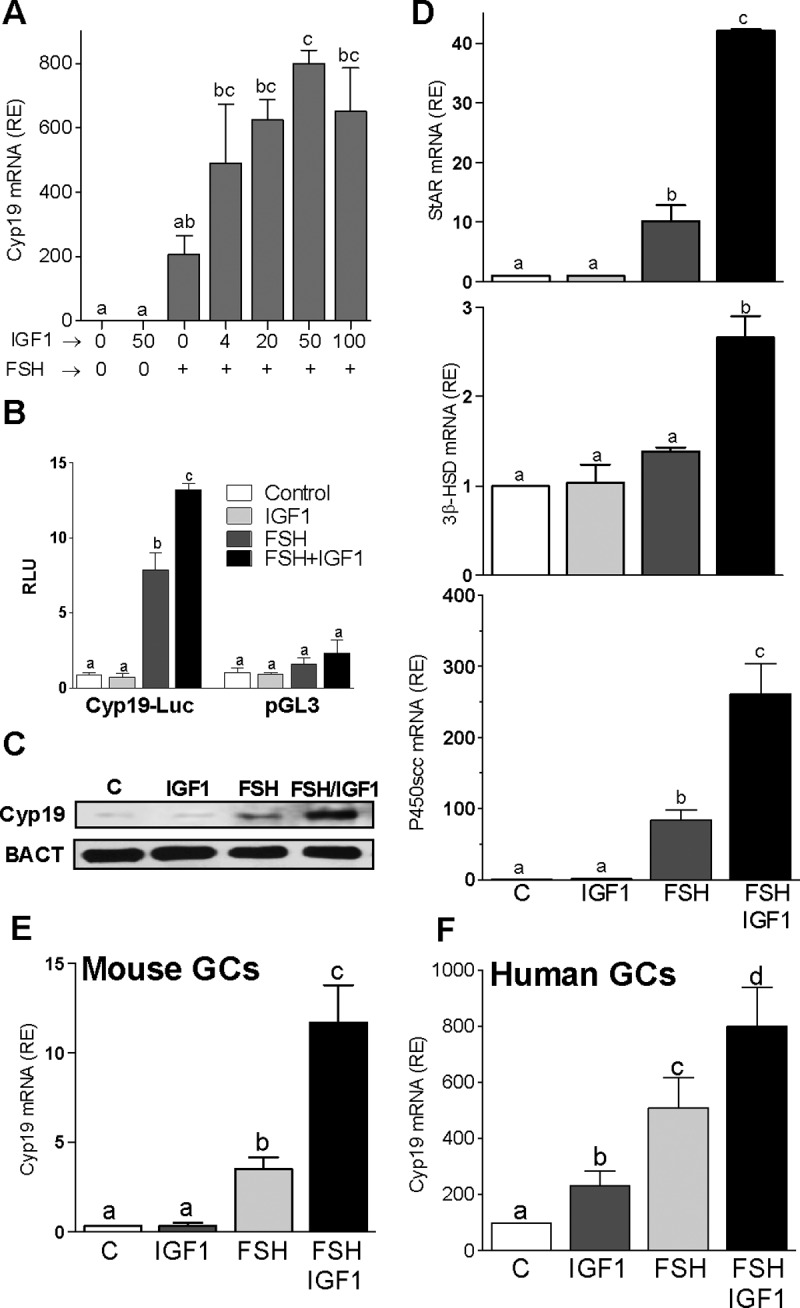
IGF-I synergizes with FSH to stimulate steroidogenic genes. A–F, Rat (A and D), mouse (E), or human (F) primary GCs were cultured in the presence of 50 ng/ml FSH with increasing concentrations (A) or 50 ng/ml (D–F) of IGF-I. Relative mRNA levels for Cyp19 (A, E, and F) and StAR, 3β-HSD, and P450scc (D) expression are shown. B, Rat GCs were transfected with a Cyp19 promoter reporter construct or an empty vector (pGL3) and treated with 50 ng/ml FSH and/or 50 ng/ml IGF-I. C, GCs were treated with 50 ng/ml FSH and/or 50 ng/ml IGF-I for 48 hours, and Cyp19 protein and β-actin (BACT) levels were determined by Western blot. Each experiment was repeated at least three times. Columns represent the mean ± SEM; n = 3. Columns with different letters differ significantly (P < .05). RE, relative expression.
A synergistic effect between FSH and IGF-I on Cyp19 expression was also observed in mouse GCs (Figure 1E). In human GCs, Cyp19 expression was significantly higher in cells treated with FSH plus IGF-I when compared with cells treated with FSH alone (Figure 1F). However, in contrast to rat and mouse GCs, human cells responded to IGF-I with an increase in Cyp19 expression, although the stimulatory effect of IGF-I was significantly lower than that evoked by FSH. These results confirm the strong amplifying effect of IGF-I on FSH-induced gene expression in GCs across species.
Low FSHR expression has been proposed to cause a reduction in follicle growth in IGF-I–knockout mice (28); therefore, we next examined whether IGF-I increases FSHR expression. As shown in Figure 2A, FSHR protein expression remained unchanged after treatment with IGF-I. Moreover, IGF-I did not affect FSHR mRNA levels at any time during treatment (Figure 2B). Similarly, FSH had no effect on IGF-IR (Figure 2C) or IGF-I mRNA levels (Figure 2D). In contrast, FSH decreased the production of IGF-I by GCs (Figure 2E). These findings indicate that the synergistic interaction between IGF-I and FSH cannot be explained by an enhanced expression of their receptors.
Figure 2.
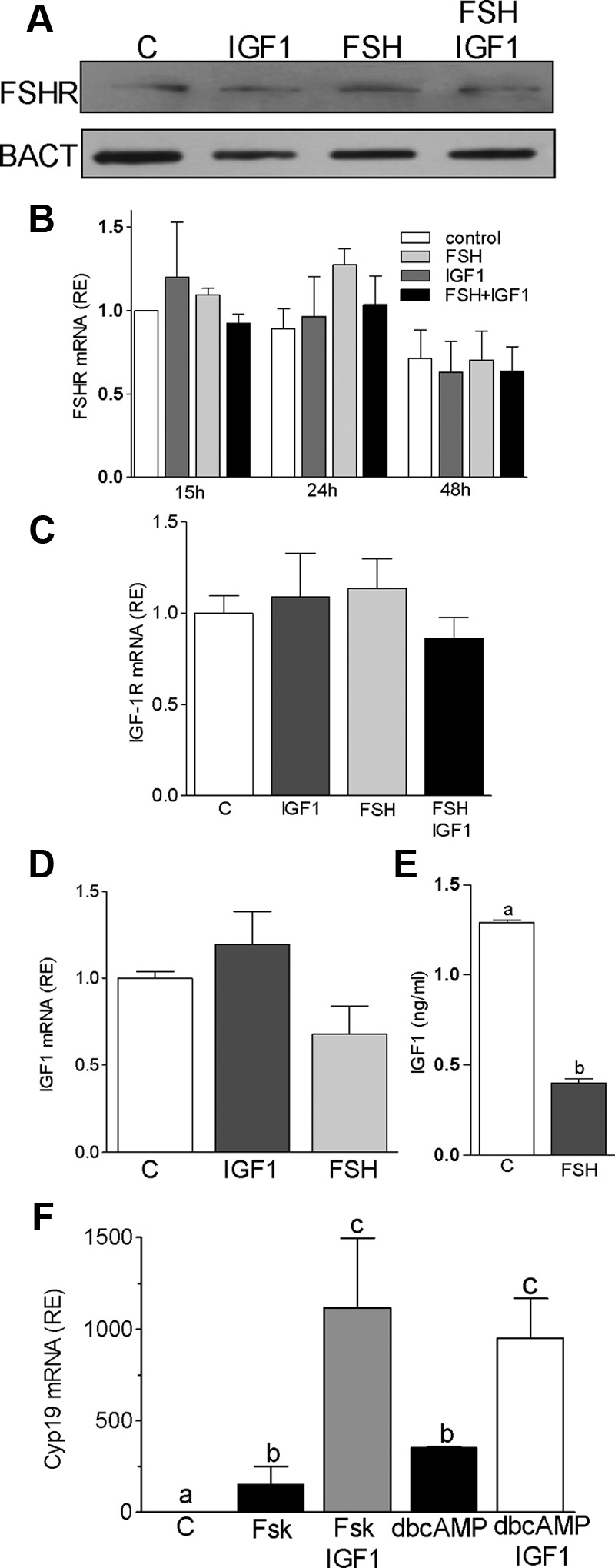
IGF-I does not affect FSHR expression in GCs in culture. Rat GCs were cultured in the presence of FSH (50 ng/ml) and/or IGF-I (50 ng/ml) for 48 hours. Cells were harvested for protein or RNA isolation, and the culture medium was collected for IGF-I determination. A, FSHR and β-actin (BACT) protein levels were quantified by using Western blot. B, Rat GCs were cultured for 15, 24, or 48 hours before FSHR mRNA quantification. C and D, IGF-IR (C) and IGF-I (D) mRNA expression levels after treatment for 48 hours as indicated in the graph. E, IGF-I levels were quantified by ELISA after 48 hours treatment with FSH. F, Rat GCs were cultured in the presence of forskolin (Fsk, 5μM) or dbcAMP (2mM) with or without IGF-I (50ng/ml) for 48 hours before Cyp19 mRNA quantification. Columns with different letters differ significantly (P < .05). Error bars are ±SEM; n = 3.
Additional experiments demonstrated that the stimulation of Cyp19 expression by forskolin, which increases intracellular cAMP via activation of adenylate cyclase, or by dibutyryl cAMP (dbcAMP), a cell-permeable cAMP analog, was potentiated by cotreatment with IGF-I (Figure 2F). These findings demonstrated that IGF-I amplification of FSH actions occurs downstream of cAMP.
IGF-I and IGF-IR are required for FSH actions
The IGF-IR is a transmembrane tyrosine kinase. To explore the involvement of IGF-IR on the amplification of FSH actions, we targeted 1) the kinase activity, 2) the activation by IGF-I, and 3) the expression of IGF-IR.
Pretreatment with AG490, a broad-spectrum tyrosine kinase inhibitor, prevented the FSH-induced expression of Cyp19 (Figure 3A). Next, we used AEW and picropodophyllotoxin (PPP), two selective inhibitors of IGF-IR activity (21, 29). Both inhibitors blocked FSH stimulation of Cyp19 (Figure 3, B and C) and P450scc (Figure 3D and data not shown) in a dose-dependent manner. It is noteworthy that none of the inhibitors affected FSHR expression (Figure 3E and data not shown). These results demonstrate that the actions of FSH in GCs require an active IGF-IR.
Figure 3.

Inhibition of IGF-IR activation blocks FSH-induced differentiation. Rat GCs were cultured with FSH plus AG490 (25μM, panel A) or different concentrations of AEW (panels B, D, and E) or PPP (panel C). mRNA levels for Cyp19 (panels A–C), P450scc (panel D), or FSHR (panel E) were determined after 48 hours of treatment. Panels F and G, GCs transfected with a plasmid expressing IGFBP2 (BP2) or the empty vector (EV) were treated with FSH (50 ng/ml) or vehicle for 48 hours before Cyp19 and P450scc mRNA level quantification. The inset in panel F shows IGFBP2 expression levels in cells transfected with EV or BP2. Panels H and I, GCs were infected with a lentivirus carrying a control shRNA (shLUC or C), anti–IGF-I shRNA (shIGF-I), or anti–IGF-IR shRNA (shIGF-IR). At 24 hours after infection, cells were treated with FSH (50 ng/ml) and/or IGF-I (50 ng/ml), and Cyp19 mRNA levels were quantified 48 hours later. The inset shows IGF-I (panel H) or IGF-IR (panel I) mRNA levels in cells infected with shLUC, shIGF-I, or shIGF-IR. Each experiment was performed three times, and mean and SEM are shown. Columns with different letters differ significantly (a-b and b-c P < .05; a-c P < .01).
The essential role of IGF-IR activation on FSH actions was further confirmed by the overexpression of IGFBP2 (Figure 3F, inset), which specifically binds IGF-I and prevents IGF-IR activation (30). As shown in Figure 3, F and G, FSH stimulation of Cyp19 and P450scc was abolished by IGFBP2. The inhibitory effect of IGFBP2 was reversed by the addition of an excess (50 ng/ml) of IGF-I to the medium (last column in Figure 3, F and G). Similarly, shRNA-driven knockdown of IGF-I (Figure 3H, inset) decreased the induction of Cyp19 (Figure 3H). As expected, the addition of IGF-I rescued FSH stimulation of Cyp19 in the presence of anti–IGF-I shRNA (Figure 3H, last column). These findings show that IGF-IR activation by locally produced IGF-I is needed for the stimulation of Cyp19 and P450scc by FSH.
Finally, the expression of the IGF-IR was silenced using shRNA. In agreement with the results obtained by inhibiting the activity or the activation of IGF-IR, knockdown of the IGF-IR (shIGF-IR, inset in Figure 3I) prevented the stimulation of Cyp19 by FSH (Figure 3I). These results prove that IGF-IR expression is required for the induction of Cyp19 expression by FSH.
In vivo inhibition of IGF-IR activity decreases follicle growth
To determine whether FSH actions in vivo also depend on the simultaneous activation of the IGF-IR, we administered AEW intrabursally (50 μg per ovary) to immature rats. Two hours after AEW administration, animals were treated with equine chorionic gonadotropin (eCG) to stimulate follicular development. Forty-eight hours later, ovaries were processed for histology and gene expression studies. In vivo administration of AEW into the ovarian bursa blunted follicle growth as indicated by a decrease in ovary and follicle size (Figure 4). Pretreatment with AEW also significantly decreased the induction of Cyp19, P450scc, and StAR expression induced by eCG (Figure 4, A–C). Taken together, these in vitro and in vivo studies suggest that FSH-induced differentiation of GCs and follicle growth are significantly attenuated in the absence of IGF-IR signaling.
Figure 4.

In vivo IGF-IR inhibition blocks FSH-induced follicle growth. AEW (50 μg per ovary) or vehicle (control) was injected into the ovarian bursa of immature rats 2 hours before the administration of 10 IU eCG, and 48 hours later, one ovary was processed for hematoxylin and eosin staining and the other was used to isolate total RNA for mRNA quantification. A, Hhistology of a representative ovary (bar, 0.5 mm). B, Mean and SEM of relative mRNA levels. *** P < .01 vs control. Three animals were included in each group.
FSH reliance on IGF-IR activation is species and cell-type independent
Next, we examined whether IGF-I activity is required for the stimulation of steroidogenic genes by FSH in human and mouse GCs. FSH-induced expression of Cyp19, P450scc, StAR, and LHR in human and mouse cells was prevented by the inhibition of IGF-IR activity with AEW (Figure 5). We also examined whether IGF-I is involved in the stimulation of Cyp19 expression in Sertoli cells, which are known to respond to FSH and to produce IGF-I (10, 31). As shown in Figure 5, FSH stimulation of Cyp19 in Sertoli cells was abolished by the inhibition of IGF-IR activity. In addition, IGF-IR activity was also required for the induction of Cyp19 by IGF-I alone in human granulosa cells (relative Cyp19 expression: control = 12.2 ± 2.0a; IGF-I = 24.9 ± 3.2b; AEW = 6.2 ± 0.9c; IGF-I+AEW = 6.1 ± 1.1c; groups with different superscript letters differ significantly, P < .01). These results demonstrate the requirement of IGF-IR activation for FSH actions in ovarian and testicular cells.
Figure 5.

FSH reliance on IGF-IR activation is species and cell-type independent. Human or mouse GCs and Sertoli cells were cultured in presence of FSH (50 ng/ml) with or without AEW (0.5μM) for 48 hours before the quantification of Cyp19, P450scc, StAR, and LHR mRNA levels (n = 4). Controls (C) received vehicle. Columns with different letters differ significantly (P < .05).
FSH and IGF-I signaling cross talk
Next, we examined the effect of either IGF-I or FSH alone or combined on the activation of AKT, ERK1/2, and CREB, all of which are known to mediate FSH effects in the gonads (20, 32). The levels of the phosphorylated form of AKT (S473), ERK1/2 (T202/Y204/T185/Y187), and CREB (S133) were low in untreated GCs but increased after treatment with FSH for 1 hour (Figure 6, A–C). In contrast, although IGF-I stimulated AKT phosphorylation after 1 hour of incubation, it had no effect on ERK1/2 or CREB activation. In cells treated with IGF-I and FSH for 1 hour, the levels of phosphorylated AKT were significantly higher than in those treated with either FSH or IGF-I alone (Figure 6A). No significant enhancement of FSH-induced ERK1/2 (Figure 6B) or CREB (Figure 6C) phosphorylation was observed after cotreatment with IGF-I.
Figure 6.

FSH and IGF-I signaling cross talk. Rat primary GCs were treated with FSH (50 ng/ml) and/or IGF-I (50 ng/ml) for 1 hour. A–C, Phosphorylated and total forms of AKT (panel A), ERK1/2 (panel B), and CREB (panel C) levels were determined by Western blot. The bar graphs under each blot show the mean ± SEM of the ratio of phosphorylated to total protein of three or more experiments. Panels D and E, Rat GCs were pretreated with wortmannin (100nM), an inhibitor of PI3K; MK2206 (1μM), an inhibitor of AKT; or UO126 (5μM), which inhibits ERK1/2, for 1 hour followed by treatment with FSH and/or IGF-I for 48 hours. Cyp19 levels were quantified by real-time PCR. Panel F, GCs were pretreated for 1 hour with wortmannin, MK2206, or UO126 followed by 1 hour treatment with vehicle or FSH and/or IGF-I before protein isolation. Phosphorylated and total AKT protein levels were evaluated by Western blot. Each experiment was repeated 6 times. Columns with different letters differ significantly (a-b and b-c P < .05; a-c P < .01). Abbreviations: C, control; F, FSH; I, IGF-I; p, phospho.
To test whether AKT phosphorylation mediates the synergistic effect of FSH and IGF-I on the expression of steroidogenic genes in GCs, the activation and activity of AKT were targeted by pretreating GCs for 1 hour with wortmannin, a PI3K inhibitor, or MK2206, a highly selective inhibitor of AKT. The stimulation of Cyp19 by FSH or FSH and IGF-I was significantly decreased in cells treated with wortmannin or MK2206 (Figure 6D). However, in the presence of wortmannin or MK2206, Cyp19 expression in FSH/IGF-I-treated cells was significantly higher than in cells treated with FSH alone. Treatment with UO126, an inhibitor of ERK1/2 kinases, prevented FSH and FSH/IGF-I stimulation of Cyp19 (Figure 6E). Treatment with each inhibitor alone did not affect Cyp19 expression when compared with control (Figure 6, D and E).
Next, we examined the mechanisms involved in the synergistic activation of AKT by FSH and IGF-I. For this experiment, cells were treated with specific inhibitors or vehicle (dimethylsulfoxide [DMSO]) for 1 hour and then treated with buffer or FSH and/or IGF-I for another hour. As expected, wortmannin prevented AKT phosphorylation by FSH, whereas MK2206 treatment significantly reduced FSH-induced expression of phospho-AKT (Figure 6F). However, inhibition of ERK1/2 activity did not prevent AKT activation by FSH and had no impact on the synergistic effect of FSH and IGF-I on AKT phosphorylation. In summary, IGF-I and FSH act synergistically on the stimulation of Cyp19 in cells treated with PI3K/AKT inhibitors and FSH potentiates the activation of AKT by IGF-I in cells treated with an ERK1/2 inhibitor, suggesting that these hormones interact to activate multiple signaling pathways.
IGF-IR activity is obligatory for FSH-induced phosphorylation of AKT but not for CREB and ERK1/2 activation
To test the possibility that FSH and IGF-I signaling may converge upstream of AKT and ERK1/2, we investigated whether FSH enhances IGF-IR phosphorylation and whether the inhibition of IGF-IR activity affects the phosphorylation of AKT and ERK1/2 by FSH. Low levels of phospho–IGF-IR were found in vehicle-treated cells and were not affected by FSH treatment (Figure 7A, first row). Supporting previous reports (29, 33), IGF-I increased phospho–IGF-IR, whereas treatment with FSH plus IGF-I did not increase IGF-IR activation above the levels found with IGF-I (not shown). In the presence or absence of FSH, AEW (0.2μM) significantly decreased IGF-IR phosphorylation when compared with vehicle (DMSO)-treated cells but did not affect total IGF-IR expression (Figure 7A, second row from top).
Figure 7.

IGF-IR expression and activity are obligatory for FSH activation of AKT. Panel A, Rat GCs were pretreated with vehicle (DMSO) or with AEW (0.2μM) for 1 hour followed by treatment with FSH (50 ng/ml) or buffer (control [C]) for 1 hour. Protein extracts were subjected to Western blot for phosphorylated (P-) IGF-IR, AKT, ERK1/2, and CREB. Total (T-) IGF-IR, AKT, and ERK were used as a loading control. The graphs show the ratio of phosphorylated to total (P/T) for each band as the mean ± SEM of the densitometry quantification of three separated experiments. Panel B, GCs were pretreated with vehicle (DMSO) or AEW (0.2μM or 0.5μM) for 30 minutes before the addition of IGF-I (20 ng/mL) or insulin (20 ng/mL). Controls (C) were treated with buffer. Total and phospho-AKT were determined 1 hour later. This experiment was performed three times with identical results. Panel C, GCs were infected with a lentivirus carrying a control shRNA (shLUC) or anti–IGF-IR shRNAs (shIGF-IR). At 24 hours after infection, cells were treated with FSH (50 ng/ml) for 1 hour and total IGF-IR and phosphorylated and total AKT levels were measured by Western blot. On the right, the ratio of phosphorylated to total AKT is shown as the mean ± SEM of three different experiments. Columns with different letters differ significantly (ab P < .05; a P < .01).
Remarkably, the IGF-IR inhibitor not only prevented AKT activation by FSH but also reduced AKT phosphorylation to undetectable levels (Figure 7A, third row). In marked contrast, lack of IGF-IR activity had no effects on the phosphorylation of CREB or ERK1/2 by FSH (Figure 7A, fifth and sixth rows, respectively). AEW did not affect the expression of total AKT, ERK1/2, or CREB (Figure 7A, fourth and last rows and data not shown). These findings suggest that basal IGF-IR and AKT activation are probably stimulated by locally produced IGF-I and that FSH stimulation of AKT needs an active IGF-IR.
Because of the strong effect of IGF-IR inhibition on AKT phosphorylation, we tested the specificity of AEW by examining its effect on the activation of AKT by insulin and IGF-I. The insulin receptor and the IGF-IR are both tyrosine kinases that stimulate the pyruvate dehydrogenase lipoamide kinase isozyme 1/PI3K/AKT pathway. As expected and in good agreement with previous reports (29, 33), IGF-I increased the expression of phospho-AKT in cells pretreated with vehicle (DMSO), whereas pretreatment with AEW (0.2μM) decreased basal AKT phosphorylation and completely prevented the stimulatory effect of IGF-I on AKT (Figure 7B, left panel). In marked contrast, a higher concentration of AEW (0.5μM) had no effect on the increase in AKT phosphorylation induced by insulin (Figure 7B, right panel). These results demonstrate that the block on FSH-induced activation of AKT by AEW is not due to nonspecific effects on upstream kinases but due to the specific inhibition of IGF-IR activity.
To further confirm the requirement of IGF-IR on the stimulation of AKT by FSH, we silenced the expression of IGF-IR by using shRNA. As shown in Figure 7C, the expression of IGF-IR was significantly reduced in GCs infected with a lentivirus carrying anti–IGF-IR shRNAs when compared with a control carrying antiluciferase shRNA (shLUC). Confirming the results obtained by the inhibition of IGF-IR activity with AEW, knockdown of the IGF-IR completely abolished the phosphorylation of AKT by FSH (Figure 7C).
IGF-IR inhibition also prevented the stimulation of estradiol production by FSH or FSH plus IGF-I (Figure 8A). Moreover, inhibition of IGF-IR activity blocked the stimulatory effect of forskolin and dbcAMP on Cyp19 expression (Figure 8B). Taken together, these results demonstrate that the synergistic effect between IGF-I and FSH on AKT phosphorylation and gene expression is mediated, at least in part, by signaling components downstream of cAMP and establish that the lack of IGF-IR activation or expression prevents FSH stimulation of not only steroidogenic genes but also AKT phosphorylation.
Figure 8.

CA-AKT overcomes the inhibitory effect of AEW. Panel A, Cells were left untreated or treated with FSH (50 ng/ml) and/or IGF-I (50 ng/ml) for 48 hours. Androstenedione was added to the medium at a final concentration of 50nM for the last 4 hours of culture. The concentration of estradiol in the medium was measured by ELISA. Panel B, Rat GCs were pretreated with AEW (0.5μM) for 1 hour before the addition of forskolin (Fsk, 5μM) or dbcAMP (2mM), and 48 hours later, Cyp19 mRNA levels were determined. *** P < .001 vs control (C) and AEW (n = 4). Panel C, Rat GCs were infected with an empty virus (emp) or with a virus that expresses CA-AKT at an increasing multiplicity of infection, and 24 hours later, vehicle or FSH was added to the medium. Cyp19 expression was quantified 48 hours after the initiation of FSH treatment. ** P < .01 vs empty (n = 3). Panel D, Rat GCs were infected with lentiviruses expressing CA-AKT, DN-AKT, or null-AKT. Vehicle (control [C]), FSH, and/or AEW were added to the medium 24 hours later. Cyp19 expression was quantified 48 hours after the initiation of FSH treatment. Columns represent the mean ± SEM (n = 4). Columns with different letters differ significantly (a-b and b-c, P < .05). RE, relative expression.
Finally, because AKT is downstream of the IGF-IR and considering that AKT activation is required for the differentiation of granulosa cells (23, 34, 35), we hypothesized that high levels of AKT activity would prevent the inhibitory effect of AEW on Cyp19 expression. To test this hypothesis, we used lentiviruses engineered to express CA, DN, or null variants of AKT (36). First, we examined the effect of CA-AKT on the stimulation of Cyp19 expression by FSH. As shown in Figure 8C, CA-AKT synergized with FSH on the stimulation of Cyp19 in a concentration-dependent manner. This finding confirms a previous report demonstrating that CA-AKT amplifies the stimulatory effects of FSH plus testosterone on both Cyp19 expression and estradiol production (23). In addition, these results indicate that testosterone is not needed for this synergism to occur.
We next investigated whether overexpression of CA-AKT prevents AEW inhibition of FSH-induced stimulation of Cyp19 expression. As expected, cells transfected with an empty virus responded to FSH with an increase in Cyp19 expression, which was prevented by the inhibition of the IGF-IR with AEW (Figure 8D). Alone, CA-AKT, DN-AKT, or null-AKT had no effects on Cyp19; in contrast, FSH stimulation of Cyp19 was potentiated by the CA-AKT construct but significantly decreased by the DN-AKT and null-AKT constructs. Notably, the inhibitory effect of AEW was overcome by the CA-AKT but not by the DN-AKT or null-AKT constructs.
Discussion
These findings demonstrate for the first time that FSH actions on GC differentiation depend on the presence of IGF-I and an active IGF-IR. IGF-I is involved in the expression of FSHR expression in GCs (28). Our results indicate that the role of IGF-I in the ovary goes beyond the stimulation of FSHR expression and that IGF-I is required for the stimulation of AKT and gene expression by FSH. It has previously been suggested that the synergism between FSH and IGF-I on AKT activation might be a crucial mechanism to enhance the expression of differentiation genes (22). In contrast, our results demonstrate that instead of a parallel synergistic interaction, FSH and IGF-I act in tandem to induce GC differentiation. This novel mechanism seems to involve FSH amplification of signaling downstream of the IGF-IR that leads to AKT activation. More studies are needed to determine the molecular mechanism of the interaction between FSH/cAMP and IGF-IR.
In granulosa and Sertoli cells, FSH activates several signaling pathways including ERK1/2, cAMP/protein kinase A/CREB, and PI3K/AKT. Particularly, in undifferentiated GCs, FSH-induced activation of AKT is essential for the stimulation of 3β-HSD, α-inhibin, Cyp19, Lhr, cartilage link protein, and hypoxia-inducible factor-1 (23, 35, 37). The pathways downstream of the FSHR leading to the phosphorylation of AKT are not fully understood. The activation of the Rap/Raf cascade by cAMP-regulated guanine nucleotide exchange factors or of PI3K by the β/γ-subunits of the G protein had been proposed to explain the effects of FSH on AKT (23). Our results indicate that the activation of AKT by FSH needs a fully functional IGF-IR.
Although IGF-I stimulates AKT, activation of this enzyme is not enough to stimulate Cyp19 expression (23), and it seems that the simultaneous activation of the FSHR signaling pathways is required. FSH effects are mainly mediated by the activation of CREB (38, 39). On the other hand, several transcription factors, including Forkhead box protein O1 and β-catenin, may be involved in the regulation of Cyp19 expression downstream of AKT (40, 41). The role that each of these factors plays on the regulation of Cyp19 by AKT remains to be determined.
Because GCs produce IGF-I, it is puzzling that AKT is not highly phosphorylated in unstimulated cells. However, we did observe background activation of IGF-IR and AKT in GCs cultured in serum-free media. Inhibition of IGF-IR with AEW decreases the phosphorylation of AKT to almost undetectable levels, suggesting that activation of IGF-IR by endogenous IGF-I is responsible for the basal activation of AKT. Moreover, we found that phosphorylation of AKT by FSH relies on IGF-IR activation. Therefore, it is possible to suggest that FSH amplifies IGF-IR signaling or removes an inhibitory influence present in unstimulated cells. This hypothesis implies that only those follicles exposed to increasing levels of FSH can benefit from locally produced IGF-I, and vice versa, only follicles with high endogenous IGF-I production are able to fully respond to FSH. Thus, the interaction between the endocrine effect of FSH and the autocrine actions of IGF-I might play an essential role in the establishment of follicle dominance.
Two possible mechanisms may explain why FSH renders GCs able to fully respond to IGF-I. One possibility is that FSH inhibits the activity of a tyrosine phosphatase that dephosphorylates IGF-IR. IGF-I binding to extracellular IGF-IR α-subunits leads to activation of the intrinsic tyrosine kinase activity of the intracellular β-subunits. Transphosphorylation between the β-subunits leads to full receptor activity (42, 43). Protein tyrosine phosphatases (PTPs) such as PTP-1B and Src-homology 2 domain-containing phosphatase 2 (SHP-2) have been shown to dephosphorylate IGF-IR (44–46). Thus, in the absence of FSH, PTP-1B or SHP-2 could dephosphorylate IGF-IR, resulting in decreased IGF-I signaling. Indeed, FSH has been shown to down-regulate the expression of PTP-1B in GCs (47), whereas okadaic acid, a phosphatase inhibitor, enhances FSH-induced Cyp19 expression and augments gonadotropin-stimulated steroidogenesis (48). Similarly, SHP-2 is highly expressed in GCs (49) and decreases IGF-IR phosphorylation (50). In addition, Cottom et al (51) showed that FSH stimulates ERK activity in GCs by relieving an inhibition imposed by an unknown phosphotyrosine phosphatase. Another possibility is that FSH induces the association of chaperone proteins with the IGF-IR, facilitating the activation of downstream targets. For instance, heat-shock protein 90 (HSP90), an ATP-dependent chaperone that is associated with the IGF-IR (52), is involved in the potentiation of IGF-I signals by cAMP in thyroid cells (53). In support of this mechanism, our preliminary results suggest that 17-N-allylamino-17-demethoxygeldanamycin (17AAG), an HSP90 inhibitor, prevents Cyp19 and P450scc induction by FSH. Future studies warrant examining the effect of tyrosine phosphatases and HSP90 on the stimulation of AKT by FSH.
In human GCs, FSH-induced differentiation greatly relies on IGF-I, as revealed by the failure of FSH to increase Cyp19, P450scc, StAR, and LHR expression in the presence of an IGF-IR inhibitor. Human dominant follicles contain higher concentrations of IGF-I and estradiol than less developed follicles in the cohort (54). IGF-I stimulates estradiol production, and it acts synergistically with FSH to maintain maximal levels of this steroid (55). We have shown that IGF-I also potentiates the stimulatory effect of FSH on gene expression in human GCs. A difference between rodent and human GCs is the stimulation of Cyp19 expression by IGF-I alone in human cells, although IGF-I does not fully mimic the effect of FSH. The clinical implication of this finding is supported by the use of GH in in vitro fertilization (IVF) protocols, which has been proposed to increase the probability of live birth in women that respond poorly to gonadotropins (56, 57).
The key role that IGF-I plays in the regulation of steroidogenesis and follicle development by FSH opens the possibility of pharmacological interventions to maximize or moderate FSH actions. Supporting this idea, it has been shown that IVF patients with higher IGF-I concentrations in the follicular fluid need fewer ampoules of FSH and require fewer days of FSH administration to achieve adequate ovarian stimulation (58). On the other hand, inhibition of IGF-IR signaling may provide means to prevent ovarian hyperstimulation syndrome (OHSS). OHSS is a serious complication of IVF and in rare cases can lead to life-threatening blood clots and strokes (59). Although OHSS symptoms are manifested only after the administration of human chorionic gonadotropin, it can be anticipated by monitoring circulating estradiol and follicle growth during the administration of FSH. OHSS is preceded by a marked elevation of estradiol levels (60). Our findings demonstrate that FSH stimulation of estradiol production and follicle growth could be controlled by targeting the IGF-IR; therefore, new therapies to prevent OHSS by inhibiting IGF-IR activity in GCs could be investigated.
Sertoli cells, as GCs, respond to FSH with an increase in Cyp19 expression and estradiol production (31, 32). FSH activates AKT in Sertoli cells leading to the stimulation of cell proliferation (61) and Cyp19 expression (32). Sertoli cells also produce IGF-I (62) and express the IGF-IR (63), suggesting that IGF-I may regulate FSH actions in the testis. Our results confirm the stimulatory effect of FSH on Cyp19 in Sertoli cells and demonstrate that the inhibition of the IGF-IR prevents this effect. Taken together, these results suggest a conserved and crucial role for IGF-I in mediating FSH actions in the gonads.
In summary, our findings demonstrate for the first time that in human, mouse, and rat GCs, the well-known stimulatory effect of FSH on Cyp19 and AKT depends on IGF-I and on the activation/expression of IGF-IR.
Acknowledgments
This work was supported by National Institutes of Health Grants R01HD057110 and R21HD066233.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AEW
- NVP-AEW541
- AKT
- protein kinase B
- CA
- constitutively activated
- CREB
- cAMP response element-binding protein
- Cyp19
- cytochrome P450 aromatase
- dbcAMP
- dibutyryl cAMP
- DMSO
- dimethylsulfoxide
- DN
- dominant-negative
- eCG
- equine chorionic gonadotropin
- FSHR
- FSH receptor
- GC
- granulosa cell
- HSP90
- heat-shock protein 90
- IGFBP2
- IGF-binding protein 2
- IGF-IR
- IGF-I receptor
- IVF
- in vitro fertilization
- OHSS
- ovarian hyperstimulation syndrome
- PI3K
- phosphoinositide 3-kinase
- PTP
- protein tyrosine phosphatase
- shLUC
- antiluciferase shRNA
- SHP-2
- Src-homology 2 domain-containing phosphatase 2
- shRNA
- short hairpin RNA.
References
- 1. Ohlsson C, Mohan S, Sjogren K, et al. The role of liver-derived insulin-like growth factor-I. Endocr Rev. 2009;30:494–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baker J, Hardy MP, Zhou J, et al. Effects of an Igf1 gene null mutation on mouse reproduction. Mol Endocrinol. 1996;10:903–918 [DOI] [PubMed] [Google Scholar]
- 3. Daftary SS, Gore AC. IGF-1 in the brain as a regulator of reproductive neuroendocrine function. Exp Biol Med. 2005;230:292–306 [DOI] [PubMed] [Google Scholar]
- 4. Hikake T, Hayashi S, Iguchi T, Sato T. The role of IGF1 on the differentiation of prolactin secreting cells in the mouse anterior pituitary. J Endocrinol. 2009;203:231–240 [DOI] [PubMed] [Google Scholar]
- 5. Srivastava VK, Hiney JK, Dees WL. Hypothalamic actions and interactions of alcohol and IGF-1 on the expression of glial receptor protein tyrosine phosphatase-β during female pubertal development. Alcohol Clin Exp Res. 2011;35:1812–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hara N, Takizawa I, Isahaya E, et al. Insulin-like growth factor-1 is associated with regulation of the luteinizing hormone production in men receiving androgen deprivation therapy with gonadotropin-releasing hormone analogues for localized prostate cancer. Urol Oncol. 2012;30:596–601 [DOI] [PubMed] [Google Scholar]
- 7. Oliver JE, Aitman TJ, Powell JF, Wilson CA, Clayton RN. Insulin-like growth factor I gene expression in the rat ovary is confined to the granulosa cells of developing follicles. Endocrinology. 1989;124:2671–2679 [DOI] [PubMed] [Google Scholar]
- 8. Zhou J, Chin E, Bondy C. Cellular pattern of insulin-like growth factor-I (IGF-I) and IGF-I receptor gene expression in the developing and mature ovarian follicle. Endocrinology. 1991;129:3281–3288 [DOI] [PubMed] [Google Scholar]
- 9. Hernandez ER, Roberts CT, Jr, LeRoith D, Adashi EY. Rat ovarian insulin-like growth factor I (IGF-I) gene expression is granulosa cell-selective: 5′-untranslated mRNA variant representation and hormonal regulation. Endocrinology. 1989;125:572–574 [DOI] [PubMed] [Google Scholar]
- 10. Villalpando I, Lira E, Medina G, Garcia-Garcia E, Echeverria O. Insulin-like growth factor 1 is expressed in mouse developing testis and regulates somatic cell proliferation. Exp Biol Med. 2008;233:419–426 [DOI] [PubMed] [Google Scholar]
- 11. Giudice LC. Insulin-like growth factors and ovarian follicular development. Endocr Rev. 1992;13:641–669 [DOI] [PubMed] [Google Scholar]
- 12. Adashi EY, Resnick CE, Svoboda ME, Van Wyk JJ. Somatomedin-C synergizes with follicle-stimulating hormone in the acquisition of progestin biosynthetic capacity by cultured rat granulosa cells. Endocrinology. 1985;116:2135–2142 [DOI] [PubMed] [Google Scholar]
- 13. Adashi EY, Resnick CE, Brodie AM, Svoboda ME, Van Wyk JJ. Somatomedin-C-mediated potentiation of follicle-stimulating hormone-induced aromatase activity of cultured rat granulosa cells. Endocrinology. 1985;117:2313–2320 [DOI] [PubMed] [Google Scholar]
- 14. Adashi EY, Resnick CE, Svoboda ME, Van Wyk JJ. Somatomedin-C enhances induction of luteinizing hormone receptors by follicle-stimulating hormone in cultured rat granulosa cells. Endocrinology. 1985;116:2369–2375 [DOI] [PubMed] [Google Scholar]
- 15. Li D, Kubo T, Kim H, Shimasaki S, Erickson GF. Endogenous insulin-like growth factor-I is obligatory for stimulation of rat inhibin α-subunit expression by follicle-stimulating hormone. Biol Reprod. 1998;58:219–225 [DOI] [PubMed] [Google Scholar]
- 16. Cong LN, Chen H, Li Y, et al. Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol Endocrinol. 1997;11:1881–1890 [DOI] [PubMed] [Google Scholar]
- 17. Dierich A, Sairam MR, Monaco L, et al. Impairing follicle-stimulating hormone (FSH) signaling in vivo: targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc Natl Acad Sci U S A. 1998;95:13612–13617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201–204 [DOI] [PubMed] [Google Scholar]
- 19. Richards JS, Russell DL, Ochsner S, et al. Novel signaling pathways that control ovarian follicular development, ovulation, and luteinization. Recent Prog Horm Res. 2002;57:195–220 [DOI] [PubMed] [Google Scholar]
- 20. Hunzicker-Dunn M, Maizels ET. FSH signaling pathways in immature granulosa cells that regulate target gene expression: branching out from protein kinase A. Cell Signal. 2006;18:1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riedemann J, Macaulay VM. IGF1R signalling and its inhibition. Endocr Relat Cancer. 2006;13(Suppl 1):S33–S43 [DOI] [PubMed] [Google Scholar]
- 22. Gonzalez-Robayna IJ, Falender AE, Ochsner S, Firestone GL, Richards JS. Follicle-stimulating hormone (FSH) stimulates phosphorylation and activation of protein kinase B (PKB/Akt) and serum and glucocorticoid-induced kinase (Sgk): evidence for a kinase-independent signaling by FSH in granulosa cells. Mol Endocrinol. 2000;14:1283–1300 [DOI] [PubMed] [Google Scholar]
- 23. Zeleznik AJ, Saxena D, Little-Ihrig L. Protein kinase B is obligatory for follicle-stimulating hormone-induced granulosa cell differentiation. Endocrinology. 2003;144:3985–3994 [DOI] [PubMed] [Google Scholar]
- 24. Wu YG, Bennett J, Talla D, Stocco C. Testosterone, not 5α-dihydrotestosterone, stimulates LRH-1 leading to FSH-independent expression of Cyp19 and P450scc in granulosa cells. Mol Endocrinol. 2011;25:656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kwintkiewicz J, Cai Z, Stocco C. Follicle-stimulating hormone-induced activation of Gata4 contributes in the up-regulation of Cyp19 expression in rat granulosa cells. Mol Endocrinol. 2007;21:933–947 [DOI] [PubMed] [Google Scholar]
- 26. Schopman NC, Liu YP, Konstantinova P, ter Brake O, Berkhout B. Optimization of shRNA inhibitors by variation of the terminal loop sequence. Antiviral Res. 2010;86:204–211 [DOI] [PubMed] [Google Scholar]
- 27. Stocco CO, Deis RP. Luteolytic effect of LH: inhibition of 3β-hydroxysteroid dehydrogenase and stimulation of 20α-hydroxysteroid dehydrogenase luteal activities in late pregnant rats. J Endocrinol. 1996;150:423–429 [DOI] [PubMed] [Google Scholar]
- 28. Zhou J, Kumar TR, Matzuk MM, Bondy C. Insulin-like growth factor I regulates gonadotropin responsiveness in the murine ovary. Mol Endocrinol. 1997;11:1924–1933 [DOI] [PubMed] [Google Scholar]
- 29. Garcia-Echeverria C, Pearson MA, Marti A, et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell. 2004;5:231–239 [DOI] [PubMed] [Google Scholar]
- 30. Poretsky L, Cataldo NA, Rosenwaks Z, Giudice LC. The insulin-related ovarian regulatory system in health and disease. Endocr Rev. 1999;20:535–582 [DOI] [PubMed] [Google Scholar]
- 31. Dorrington JH, Armstrong DT. Follicle-stimulating hormone stimulates estradiol-17β synthesis in cultured Sertoli cells. Proc Natl Acad Sci U S A. 1975;72:2677–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McDonald CA, Millena AC, Reddy S, et al. Follicle-stimulating hormone-induced aromatase in immature rat Sertoli cells requires an active phosphatidylinositol 3-kinase pathway and is inhibited via the mitogen-activated protein kinase signaling pathway. Mol Endocrinol. 2006;20:608–618 [DOI] [PubMed] [Google Scholar]
- 33. Wolf S, Lorenz J, Mossner J, Wiedmann M. Treatment of biliary tract cancer with NVP-AEW541: mechanisms of action and resistance. World J Gastroenterol. 2010;16:156–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Richards JS, Sharma SC, Falender AE, Lo YH. Expression of FKHR, FKHRL1, and AFX genes in the rodent ovary: evidence for regulation by IGF-I, estrogen, and the gonadotropins. Mol Endocrinol. 2002;16:580–599 [DOI] [PubMed] [Google Scholar]
- 35. Alam H, Maizels ET, Park Y, et al. Follicle-stimulating hormone activation of hypoxia-inducible factor-1 by the phosphatidylinositol 3-kinase/AKT/Ras homolog enriched in brain (Rheb)/mammalian target of rapamycin (mTOR) pathway is necessary for induction of select protein markers of follicular differentiation. J Biol Chem. 2004;279:19431–19440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li LB, Toan SV, Zelenaia O, et al. Regulation of astrocytic glutamate transporter expression by Akt: evidence for a selective transcriptional effect on the GLT-1/EAAT2 subtype. J Neurochem. 2006;97:759–771 [DOI] [PubMed] [Google Scholar]
- 37. Sun GW, Kobayashi H, Suzuki M, Kanayama N, Terao T. Follicle-stimulating hormone and insulin-like growth factor I synergistically induce up-regulation of cartilage link protein (Crtl1) via activation of phosphatidylinositol-dependent kinase/Akt in rat granulosa cells. Endocrinology. 2003;144:793–801 [DOI] [PubMed] [Google Scholar]
- 38. Stocco C. Aromatase expression in the ovary: hormonal and molecular regulation. Steroids. 2008;73:473–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Somers JP, DeLoia JA, Zeleznik AJ. Adenovirus-directed expression of a nonphosphorylatable mutant of CREB (cAMP response element-binding protein) adversely affects the survival, but not the differentiation, of rat granulosa cells. Mol Endocrinol. 1999;13:1364–1372 [DOI] [PubMed] [Google Scholar]
- 40. Park Y, Maizels ET, Feiger ZJ, et al. Induction of cyclin D2 in rat granulosa cells requires FSH-dependent relief from FOXO1 repression coupled with positive signals from Smad. J Biol Chem. 2005;280:9135–9148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fan HY, O'Connor A, Shitanaka M, Shimada M, Liu Z, Richards JS. β-Catenin (CTNNB1) promotes preovulatory follicular development but represses LH-mediated ovulation and luteinization. Mol Endocrinol. 2010;24:1529–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47 [DOI] [PubMed] [Google Scholar]
- 43. Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci. 2000;57:1050–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kenner KA, Anyanwu E, Olefsky JM, Kusari J. Protein-tyrosine phosphatase 1B is a negative regulator of insulin- and insulin-like growth factor-I-stimulated signaling. J Biol Chem. 1996;271:19810–19816 [DOI] [PubMed] [Google Scholar]
- 45. Maile LA, Clemmons DR. The αVβ3 integrin regulates insulin-like growth factor I (IGF-I) receptor phosphorylation by altering the rate of recruitment of the Src-homology 2-containing phosphotyrosine phosphatase-2 to the activated IGF-I receptor. Endocrinology. 2002;143:4259–4264 [DOI] [PubMed] [Google Scholar]
- 46. Maile LA, Clemmons DR. Regulation of insulin-like growth factor I receptor dephosphorylation by SHPS-1 and the tyrosine phosphatase SHP-2. J Biol Chem. 2002;277:8955–8960 [DOI] [PubMed] [Google Scholar]
- 47. Sasson R, Dantes A, Tajima K, Amsterdam A. Novel genes modulated by FSH in normal and immortalized FSH-responsive cells: new insights into the mechanism of FSH action. FASEB J. 2003;17:1256–1266 [DOI] [PubMed] [Google Scholar]
- 48. Gonzalez Reyes J, Santana P, Gonzalez Robaina I, et al. Effect of the protein phosphatase inhibitor okadaic acid on FSH-induced granulosa cell steroidogenesis. J Endocrinol. 1997;152:131–139 [DOI] [PubMed] [Google Scholar]
- 49. Russell DL, Richards JS. Differentiation-dependent prolactin responsiveness and stat (signal transducers and activators of transcription) signaling in rat ovarian cells. Mol Endocrinol. 1999;13:2049–2064 [DOI] [PubMed] [Google Scholar]
- 50. Carver KC, Piazza TM, Schuler LA. Prolactin enhances insulin-like growth factor I receptor phosphorylation by decreasing its association with the tyrosine phosphatase SHP-2 in MCF-7 breast cancer cells. J Biol Chem. 2010;285:8003–8012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cottom J, Salvador LM, Maizels ET, et al. Follicle-stimulating hormone activates extracellular signal-regulated kinase but not extracellular signal-regulated kinase kinase through a 100-kDa phosphotyrosine phosphatase. J Biol Chem. 2003;278:7167–7179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bagatell R, Beliakoff J, David CL, Marron MT, Whitesell L. Hsp90 inhibitors deplete key anti-apoptotic proteins in pediatric solid tumor cells and demonstrate synergistic anticancer activity with cisplatin. Int J Cancer. 2005;113:179–188 [DOI] [PubMed] [Google Scholar]
- 53. Fukushima T, Okajima H, Yamanaka D, et al. HSP90 interacting with IRS-2 is involved in cAMP-dependent potentiation of IGF-I signals in FRTL-5 cells. Mol Cell Endocrinol. 2011;344:81–89 [DOI] [PubMed] [Google Scholar]
- 54. Eden JA, Jones J, Carter GD, Alaghband-Zadeh J. A comparison of follicular fluid levels of insulin-like growth factor-1 in normal dominant and cohort follicles, polycystic and multicystic ovaries. Clin Endocrinol (Oxf). 1988;29:327–336 [DOI] [PubMed] [Google Scholar]
- 55. Erickson GF, Garzo VG, Magoffin DA. Insulin-like growth factor-I regulates aromatase activity in human granulosa and granulosa luteal cells. J Clin Endocrinol Metab. 1989;69:716–724 [DOI] [PubMed] [Google Scholar]
- 56. Kyrou D, Kolibianakis EM, Venetis CA, Papanikolaou EG, Bontis J, Tarlatzis BC. How to improve the probability of pregnancy in poor responders undergoing in vitro fertilization: a systematic review and meta-analysis. Fertil Steril. 2009;91:749–766 [DOI] [PubMed] [Google Scholar]
- 57. Duffy JM, Ahmad G, Mohiyiddeen L, Nardo LG, Watson A. Growth hormone for in vitro fertilization. Cochrane Database Syst Rev. 2010;1:CD000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oosterhuis GJ, Vermes I, Lambalk CB, Michgelsen HW, Schoemaker J. Insulin-like growth factor (IGF)-I and IGF binding protein-3 concentrations in fluid from human stimulated follicles. Hum Reprod. 1998;13:285–289 [DOI] [PubMed] [Google Scholar]
- 59. Sansone P, Aurilio C, Pace MC, et al. Intensive care treatment of ovarian hyperstimulation syndrome (OHSS). Ann N Y Acad Sci. 2011;1221:109–118 [DOI] [PubMed] [Google Scholar]
- 60. Genazzani AR, Monteleone P, Papini F, Artini PG. Pharmacotherapy of ovarian hyperstimulation syndrome. Expert Opin Pharmacother. 2010;11:2527–2534 [DOI] [PubMed] [Google Scholar]
- 61. Riera MF, Regueira M, Galardo MN, Pellizzari EH, Meroni SB, Cigorraga SB. Signal transduction pathways in FSH regulation of rat Sertoli cell proliferation. Am J Physiol. 2012;302:E914–E923 [DOI] [PubMed] [Google Scholar]
- 62. Tres LL, Smith EP, Van Wyk JJ, Kierszenbaum AL. Immunoreactive sites and accumulation of somatomedin-C in rat Sertoli-spermatogenic cell co-cultures. Exp Cell Res. 1986;162:33–50 [DOI] [PubMed] [Google Scholar]
- 63. Froment P, Vigier M, Negre D, et al. Inactivation of the IGF-I receptor gene in primary Sertoli cells highlights the autocrine effects of IGF-I. J Endocrinol. 2007;194:557–568 [DOI] [PubMed] [Google Scholar]