

Abstract
α-Dioxygenases (α-DOX) oxygenate fatty acids into 2R-hydroperoxides. Despite low sequence identity, α-DOX share common catalytic features with Cyclooxygenases (COX), including the use of a tyrosyl radical during catalysis. We determined the x-ray crystal structure of Arabidopsis thaliana α-DOX to 1.5Å resolution. The α-DOX structure is monomeric, predominantly α-helical and comprised of two domains. The base domain exhibits low structural homology with the membrane-binding domain of COX but lies in a similar position with respect to the catalytic domain. The catalytic domain shows the highest similarity with the COX catalytic domain, where 21 of the 22 α-helical elements are conserved. Helices H2, H6, H8, and H17 form the heme binding cleft and walls of the active site channel. His-318, Thr-323, and Arg-566 are located near the catalytic tyrosine, Tyr-386, at the apex of the channel, where they interact with a chloride ion. Substitutions at these positions, coupled with kinetic analyses confirm previous hypotheses that implicate these residues as being involved in binding and orienting the carboxylate group of the fatty acid for optimal catalysis. Unique to α-DOX is the presence of two extended inserts on the surface of the enzyme that restrict access to the distal face of the heme, providing an explanation for the observed reduced peroxidase activity of the enzyme. The α-DOX structure represents the first member of the α-DOX subfamily to be structurally characterized within the cyclooxygenase-peroxidase family of heme-containing proteins.
Oxylipins are potent lipid mediators generated from the oxygenation of fatty acids. Lipoxygenase (LOX) and Cyclooxygenase (COX) enzymes are predominantly responsible for the production of hydroperoxy intermediates from arachidonic acid that are further converted into eicosanoids in animals (1). Similarly, LOX enzymes generate hydroperoxy fatty acid intermediates from linoleic acid (LA; 18:2 ω-6) and α-linolenic acid (αLA; 18:3 ω-3) that are converted into jasmonates and other hormones in plants (2). Collectively, these products regulate processes involved in the maintenance, development, and response of the organism to biotic and abiotic stresses (3, 4).
While LOX is found in both plant and animal kingdoms, no protein with cyclooxygenase activity has been identified in plants. In 1998, Castresana and colleagues identified a protein in tobacco leaves and Arabidopsis that was up regulated during the host defense response (5). Further characterization revealed that the protein was a member of the α-dioxygenase (α-DOX) family of heme-containing proteins that incorporate oxygen into fatty acids (6). This subfamily is part of the peroxidase-cyclooxygenase superfamily, whose members developed the ability early in the evolutionary process to generate oxidants as a general defense strategy (7). Characterization of members of this superfamily, which include COX, myeloperoxidase, eosinophil peroxidase, lactoperoxidase, thyroid peroxidase, and linoleate diol synthase (LDS), have provided insight into evolutionary relationships and allowed for critical comparisons to be made between proteins in different subfamilies. Based on its ability to oxygenate fatty acids, α-DOX is related functionally to COX and LDS, despite low sequence similarities to these enzymes.
α-DOX converts LA, αLA, and other fatty acids into 2R-hydroperoxides via the stereospecific removal of the pro-R hydrogen from carbon-2 of the substrate utilizing a tyrosyl radical (Supplemental Figure S1) (8). The 2R-hydroperoxides undergo spontaneous decarboxylation to chain-shortened aldehydes, with minor amounts of 2R-hydroxy and Cn-1 fatty acids produced as well (8). In the absence of a crystal structure, COX has served as a structural surrogate for α-DOX (9, 10). Interestingly, the enzyme differs from LOX and COX by catalyzing a reaction in which oxygen addition takes place at the alpha carbon instead of at an allylic or bis-allylic position. α-DOX does not possess a strong peroxidase activity analogous to that of COX (10).
We report here the 1.5Å x-ray structure of A. thaliana α-DOX (Ath α-DOX). The structure represents the first member of the α-DOX family within the peroxidase-cyclooxygenase superfamily to be characterized at the molecular level. While the domain makeup of Ath α-DOX differs from COX, the active site architecture is similar between enzymes. Mutagenesis, coupled with functional analyses, reveals that His-318, Thr-323, and Arg-566 are critical determinants for oxygenase activity. Defining the location of these residues within the active site has allowed us to propose a model for productive substrate binding that provides insight into the ability of Ath α-DOX to oxygenate a variety of fatty acids. Finally, identification of two, large inserts located near the heme explain the reduced peroxidase enzyme observed for the enzyme.
EXPERIMENTAL PROCEDURES
Expression and Purification
A codon-optimized version of the wild-type Ath α-DOX gene (NCBI database accession number NP_186791) was synthesized and subcloned into the pQE30 expression vector (Qiagen, Valencia, CA) utilizing the commercial services provided by GeneScript Co. Q159N, Q159S, Q159V, H318A, H318Q, T323A, T323L, Y386F, R565A, R565K, R565L, R566A, R566K, and R566L mutant constructs were created with the QuikChange Mutagenesis Kit using the codon-optimized wild-type Ath α-DOX gene as a template and the primers listed in Supplemental Table S1.
Wild type and mutant constructs were transformed into E. coli M15 cells. For large-scale expression, a 10L fermentor containing enriched TB media was inoculated with cells at 37°C and grown to an OD600 of 0.6. The cells were induced by the addition of IPTG to a final concentration of 1mM and the temperature was lowered to 23°C. The heme precursor δ-aminolevulinic acid was added to a final concentration of 250μM. Cells were harvested 16 hours post-induction via centrifugation and frozen at −80°C until further use.
Cell pellet corresponding to 2L of cell growth was resuspended in 100mM sodium phosphate, pH 8.0, 600mM NaCl and lysed using a microfluidizer. C10M was added to the cell lysate to a final concentration of 0.7% (w/v) and the protein was solubilized for 1 hour at 4°C followed by centrifugation at 100,000 x g for 50 minutes. The supernatant was loaded onto a TALON IMAC column (5mL bed volume) utilizing a buffer system containing 50mM sodium phosphate, pH 8.0, 300mM NaCl, 0.7% (w/v) C10M and washed with 10 column-volumes (cv) of buffer. The protein was eluted from the column by adding 100mM EDTA to the wash buffer. Elution fractions were pooled and concentrated using an Ultrafree-15 centrifugal filter with a 50kDa cutoff (Millipore, Bedford, MA). For activity assays, the concentrated sample was applied to a HiLoad 16/60 Superdex 200 PG size-exclusion column equilibrated in 20mM TRIS, pH 8.0, 150mM NaCl, and 0.2% (w/v) C10M. The major peak corresponding to Ath α-DOX was pooled and dialyzed in 20mM TRIS, pH 8.0, 150mM NaCl, and 0.2% (w/v) C10M for one week to remove imidazole that presumably binds to the protein during expression and subsequent cell lysis.
For structural studies, the choice of detergent for crystallization was optimized utilizing size-exclusion chromatography (11), which identified βNG as a detergent that resulted in a single, sharp protein peak. The concentrated sample from the IMAC step was applied to a HiLoad 16/60 Superdex 200 PG size-exclusion column equilibrated in 50mM TRIS, pH 8.0, 150mM NaCl, and 0.5% (w/v) βNG. The major peak was pooled and loaded onto a MonoQ HR 5/5 column. The column was washed with 2 cv of 50mM TRIS, pH 8.0, 150mM NaCl, 0.5% (w/v) βNG and the protein was eluted by increasing the salt concentration to 500mM NaCl. The protein was dialyzed in 20mM TRIS, pH 8.0, 150mM NaCl, and 0.5% (w/v) βNG at 4°C prior to setup for crystallization. Oxygenase activity of wild type and mutant constructs was measured as described previously (9).
Crystallography
Initial crystallization screening was carried out using the 1536 microbatch-under-oil tailored membrane protein screen (12) in the High-Throughput Crystallization Laboratory at the Hauptman-Woodward Medical Research Institute (13). Crystals of the imidazole-bound enzyme were grown at 23°C in sitting-drops by combining 1μL of protein solution at a concentration of 12mg/mL with 3μL of 26% (v/v) Polyethylene glycol 400 (PEG 400), 200mM CaCl2, and 100mM TRIS, pH 8.0, and equilibrating over 500μL reservoir solutions containing 26–32% PEG 400, 100–400mM CaCl2, and 100mM TRIS, pH 8.0. Crystals of the wild type enzyme were grown using the same protein concentration and cocktail components as above, except utilizing 3μL protein solution added to 3μL of drop solution with equilibration over a reservoir solution containing 38% PEG 400.
Multiwavelength anomalous diffraction (MAD) data were collected on GM/CA-CAT beamline 23ID-D at the Advanced Photon Source at Argonne National Laboratory. Data sets were measured for Y386F Ath α-DOX at the iron K-edge peak (1.73866 Å), inflection (1.74061 Å), and remote (1.65312 Å) energy wavelengths. A data set was also collected for wild-type Ath α-DOX complexed with imidazole at a wavelength of 1.033Å. An additional wild-type Ath α-DOX dataset that was not complexed with imidazole was collected on beamline A1 at the Cornell High Energy Synchrotron Source. All data sets were integrated and scaled using HKL2000 (14).
The programs SHELXD and SHELXE (15) were used to locate and refine the heavy atom substructure. The phases from SHELXE were transferred to the 1.51Å wild-type Ath α-DOX data set complexed with imidazole and extended to high-resolution using the program DM in the CCP4 suite of programs (16). The output phases were then used as input to ARP/wARP (17), which successfully built over 90% of the residues into the experimental electron density map. Iterative cycles of manual model building and refinement, using COOT (18) and REFMAC5 (19), were then carried out to fit the remaining residues and to add other ligand molecules. All calcium and chloride ions added to the structure were confirmed to have greater than 5s peak heights by calculating an anomalous difference map using the data collected at the Fe peak wavelength and phases from the final refined model. TLS refinement (20) was carried out during the final rounds of refinement. Molecular replacement methods were subsequently used to determine initial phases for wild-type Ath α-DOX using the imidazole-bound structure as the search model. Refinement of this structure was carried out as described above. Final crystallographic statistics are summarized in Table 1. Model validation was carried out using MOLPROBITY (21). Volume calculations were carried out using VOIDOO (22). Figures were created using PYMOL (23). The coordinates and structure factors for wild type and imidazole-bound Ath α-DOX have been deposited in the Protein Data Bank (PDB entry 4HHS and 4HHR).
Table 1.
Crystallographic statistics for wild type and imidazole bound Ath αDOX.
| Crystallographic Parameter | WT | WT + Imidazole | Y386F Peak | Y386F Inflection | Y386F Remote |
|---|---|---|---|---|---|
|
| |||||
| Space group | P4322 | P4322 | P4322 | P4322 | P4322 |
| No. in asymmetric unit | 1 | 1 | 1 | 1 | 1 |
| Unit cell length (Å) | |||||
| a | 102.150 | 101.935 | 101.880 | 101.929 | 101.932 |
| b | 102.150 | 101.935 | 101.880 | 101.929 | 101.932 |
| c | 137.565 | 137.510 | 137.646 | 137.703 | 137.703 |
| α=β=γ (°) | 90 | 90 | 90 | 90 | 90 |
| Wavelength (Å) | 0.976 | 1.033 | 1.73866 | 1.74061 | 1.65312 |
| Resolution (Å) | 30-1.70 | 50.0-1.51 | 50-2.1 | 50-2.1 | 50-2.1 |
| Highest res. shell (Å)a | 1.73-1.70 | 1.54-1.51 | 2.14-2.1 | 2.14-2.1 | 2.14-2.1 |
| Rmergeb | 7.1 (55.8) | 6.7 (52.7) | 10.0 (65.3) | 9.4 (62.0) | 9.0 (56.1) |
| Total observations | 1066737 | 893125 | 1099250 | 544540 | 591135 |
| Total unique | 79943 | 107022 | 42786 | 42856 | 43069 |
| I/σ(I) | 39.4 (5.0) | 28.4 (3.0) | 44.5 (2.3) | 39.4 (2.1) | 44.0 (3.7) |
| Completeness (%) | 100 (100) | 94.2 (93.5) | 99.5 (93.3) | 99.4 (91.0) | 100.0 (99.9) |
| Multiplicity | 13.4 (12.2) | 8.3 (7.9) | 25.7 (9.9) | 12.7 (4.4) | 13.7 (8.6) |
| Wilson B factor (Å2) | 18.8 | 18.5 | 34.5 | 36.1 | 34.4 |
|
| |||||
| No. atoms in refinement | 6094 | 6321 | |||
| Rwork | 0.159 (0.199) | 0.153 (0.221) | |||
| Rfreec | 0.187 (0.231) | 0.182 (0.250) | |||
| Ave. B factor, protein (Å2) | 19.8 | 20.8 | |||
| Ave. B factor, solvent (Å2) | 26.9 | 31.5 | |||
| Mean positional error (Å)d | 0.095 | 0.069 | |||
| RMSD bond length (Å) | 0.010 | 0.013 | |||
| RMSD bond angle (°) | 1.207 | 1.427 | |||
The values in parentheses represent the values in the outermost resolution shell.
RMERGE as defined in (38).
5.0% of the total reflections were used to generate the test set.
Coordinate error as calculated by Luzatti plot.
RESULTS
Crystal Structure of α-DOX
A codon-optimized version of Ath α-DOX was cloned and overproduced in an E. coli expression system. UV-visible spectroscopic measurements of the purified protein revealed an absorbance maximum between 410–413nm, indicating that heme incorporation occurred during expression (9, 10). Ath α-DOX effectively oxygenates LA, αLA, and oleic acid (OA; 18:1 ω-9), with KM values that are similar to those reported previously (8, 9, 24) (Table 2).
Table 2. Oxygenation of fatty acids by Ath α-DOX.
Oxygenase activity was measured using an oxygen electrode with the fatty acids LA, αLA, and OA. Kcat and KM values were derived for each substrate using triplicate measurements.
| Substrate | kcat (s−1) | KM (μM) | kcat / KM |
|---|---|---|---|
|
| |||
| αLA (18:3 ω-3) | 11 ± 1.1 | 36 ± 3.2 | 0.31 |
| LA (18:2 ω-6) | 12 ± 0.4 | 23 ± 2.0 | 0.52 |
| OA (18:1 ω-9) | 11 ± 0.3 | 15 ± 1.9 | 0.73 |
We determined the crystal structure of Ath α-DOX in the presence and absence of imidazole bound at the heme. The imidazole-bound structure was solved utilizing MAD phasing techniques to 1.5Å resolution. Initial data were collected at the Fe-edge. Phases were determined using contributions from the iron atom in the heme and from bound calcium and chloride atoms. The asymmetric unit contains one molecule of Ath α-DOX with imidazole bound to the heme. The Ath α-DOX structure without imidazole bound was solved using molecular replacement methods to 1.7Å resolution, with one molecule of Ath α-DOX in the asymmetric unit. Despite the presence of imidazole, both structures are identical, given the calculated root mean square difference between Cα atoms of 0.065Å.
The overall fold of Ath α-DOX is predominantly helical (47% of the residues), consisting of 30 α-helices and 2 short β-strands (Figure 1). The structure can be divided into two domains. The first domain is comprised of 8 α-helices and consists of N-terminal residues 1–70, along with residues 330–368 and C-terminal residues 624–639. The second domain consists of 22 α-helices and the antiparallel β-sheet. The α-helices of the first domain can be thought of as a base that the second domain lies upon. This base domain is located in similar space as the amphipathic α-helices that comprise the membrane-binding domain (MBD) of COX (Figure 1). While there is no sequence or structural similarity between the α-helices of the base domain and the MBD in COX (Supplemental Figure S2), we postulate that the base domain associates with the lipid membrane in a similar fashion to that of the MBD. Indeed, three of the eight α-helices are amphipathic in nature. The arrangement of α-helices in the second domain of Ath α-DOX is highly similar to the arrangement of the α-helices in the catalytic domain of COX (Figure 1). Of the 22 α-helices in the catalytic domain, 21 are conserved between Ath α-DOX and COX (Supplemental Figure S2). There are three significant structural differences between Ath α-DOX and COX: 1) the lack of an epidermal growth factor-like domain in Ath α-DOX, which is present in COX; 2) the presence of a calcium binding site in Ath α-DOX that is not present in COX; and 3) the presence of two extended inserts on the surface of Ath α-DOX. The implications of the calcium binding site and extended inserts on the catalytic activity of Ath α-DOX is discussed below.
Figure 1. Comparison of the Ath αDOX and Murine COX-2 Crystal Structures.
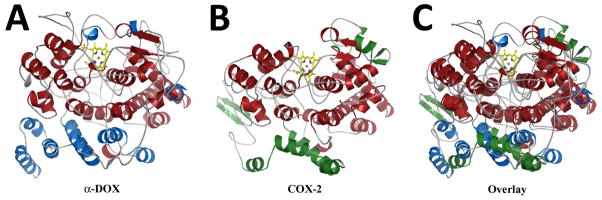
Cartoon depicting the secondary structural elements found in the crystal structures of (A) Ath α-DOX and (B) monomer B of muCOX-2 (PDB ID 3HS5). Secondary structural elements that are unique to Ath α-DOX and muCOX-2 are colored in blue and green, respectively. Secondary structural elements that are common to both structures are colored in red. The Fe3+-protoporphyrin IX moiety in each structure is shown with carbon, nitrogen, oxygen, and iron atoms colored yellow, blue, red, and orange respectively. (C) Superposition of the Ath α-DOX and muCOX-2 crystal structures depicting the similarity of the two structures within the catalytic domains.
The Heme Binding Cleft
The heme of Ath α-DOX is buried within the catalytic domain. Helices H2 and H8 constitute the heme binding cleft and contain the distal and proximal histidine residues, His-163 and His-389, respectively. The heme is liganded to the side chain of His-389, as previously predicted (Supplemental Figure S3) (9, 10, 25). The iron to Nε2 distance is 2.08Å in both the wild type and imidazole-bound Ath α-DOX structures. A water molecule is the sixth ligand to the iron atom, in the wild type structure, while imidazole serves as the sixth ligand to the iron atom, in the imidazole-bound structure (Supplemental Figure S3).
Residues 170–197 and 461–478 loop across the heme binding cleft to restrict access to the distal face of the heme (Figure 2). These two extended inserts, which do not form any secondary structure, are only found within the sequences of members of the α-DOX family and explain why α-DOX lacks significant peroxidase activity (10). Two ionic interactions, between residues Glu-170 and Arg-473 and Gln-174 and Asp-475, tether the extended inserts to each other and to the core of the globular catalytic domain (Figure 2). Ile-463 is the only residue within the inserts to contact the heme, with the cδ1 atom of its side chain participating in two hydrophobic contacts with the protoporphyrin IX moiety. Despite the presence of the two extended inserts, two small channels provide limited access to distal face of the heme (Figure 2). In contrast, the heme in COX is completely accessible to the solvent (26, 27).
Figure 2. The Extended Inserts of Ath α-DOX.
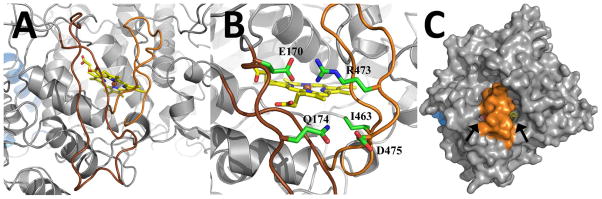
(A) View of the two extended inserts that lie at the surface of Ath α-DOX and cover the heme. The 170–197 loop is colored in brown and the 461–478 loop is colored in orange. (B) View of the ionic interactions between the two inserts that serve to stabilize and rigidify the loops. The side chains of Glu-170, Gln-174, Ile-463, Arg-473, and Asp-475 are labeled accordingly. (C) Surface representation of Ath α-DOX that depicts the location of the two small channels (black arrows) that provide access to the heme. The base and catalytic domain are colored in blue and grey, respectively, while residues 170–197 and 461–478 are colored in orange.
The propionate groups of the heme form six ionic interactions with the side chains of neighboring residues His-168, Arg-486, and Arg-490. In conjunction with the two extended inserts, these interactions act to firmly entrench the heme within the catalytic domain of Ath α-DOX. These observations support biochemical analyses whose results indicate that the heme of Ath α-DOX cannot be easily removed and exchanged with Mn3+-protoporphyrin IX or Co3+-protoporphyrin IX without denaturing the protein (10). This is in stark contrast to COX, whose heme can easily be replaced by protoporphyrin IX groups containing Co3+ and Mn3+ (27, 28).
Calcium Binding Site
The wild type and imidazole-bound Ath α-DOX structures contain eight and five bound calcium ions, respectively. In each structure, one of the calcium ions is bound to the enzyme with pentagonal bipyramidal coordination in a manner similar to that observed in myeloperoxidase and other mammalian peroxidase structures (29). Specifically, residues Thr-216 through Ser-222 form a loop that is responsible for the majority of the residues that participate in the interactions with the calcium ion, while the side chain of Asp-164 is responsible for the other contact. The side chains of Asp-164, Thr-216, Asp-220, and Ser-222 participate in four of the seven interactions, while the carbonyl oxygen atoms of Thr-216, Trp-218, and Asp-164 are responsible for the remaining three interactions. Asp-164, Thr-216, Asp-220, and Ser-222 are conserved within sequences of other α-DOX family members as well as within the sequences of mammalian peroxidase family members (29). COX enzymes do not contain a calcium-binding site, as the residues at the equivalent positions of Asp-164, Thr-216, and Ser-222 are glutamine, glycine, and asparagine, respectively. Interestingly, calcium binding in myeloperoxidase and other mammalian peroxidases plays a structural role, where it properly positions the distal histidine for subsequent catalysis (29). As Ath α-DOX has a significantly reduced peroxidase activity, it is unclear as to what role the binding of calcium has in the α-DOX family.
Active Site Architecture
Four α-helices, H2, H6, H8, and H17 (as defined in (30); Supplemental Figure S2) comprise the walls and ceiling of the fatty acid binding channel within the catalytic domain of Ath α-DOX (Figure 3). The fatty acid binding channel stretches ~20Å from the base domain opening to the catalytic tyrosine, Tyr-386, at the apex, with a calculated volume of 361Å3. The channel is bound by helices H6 and H17 and is predominantly hydrophobic in nature. The side chain of Tyr-386 is stabilized by π-π stacking interactions with the side chains of Phe-382 and Phe-559, located in helices H8 and H17, respectively. The observed interactions between Phe-382 and Tyr-386 in the Ath α-DOX structure are strikingly similar to those seen between Phe-381 and Tyr-385 in COX (26, 31). The phenolic oxygen of Tyr-386 points into the channel and interacts with a chloride ion derived from the crystallization cocktail (Figure 3). The observed structural arrangement of the four helices is highly conserved with the architecture of the heme binding cleft and cyclooxygenase channel of COX (9, 26, 27) (Figure 3).
Figure 3. Active Site Architecture of Ath α-DOX.
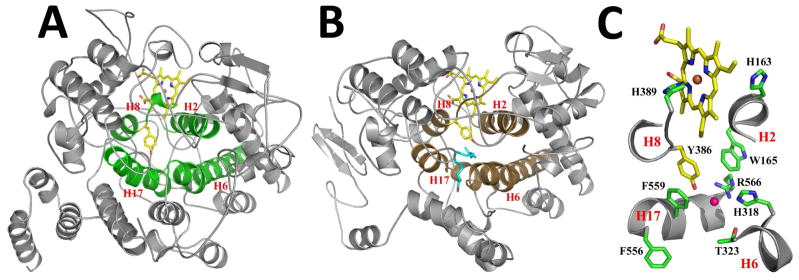
Cartoon representation of the crystal structures of (A) Ath α-DOX and (B) murine COX-2 (PDB id 3HS5), highlighting the four conserved helices (H2, H6, H8, and H17) that make up the fatty acid binding channel (green) and the cyclooxygenase channel (brown) in each structure. The side chain of the catalytic tyrosine and the heme moiety within each active site are depicted in stick form, with carbon, oxygen, nitrogen, and iron atoms colored yellow, red, blue, and orange, respectively. Arachidonic acid is shown in stick form (blue carbon atoms and red oxygen atoms) bound within the cyclooxygenase channel of COX-2 in B. (C) Close up of the active site residues in Ath α-DOX. The view is zoomed and rotated ~90° from the orientation depicted in A. Portions of helix H2, H6, H8, and H17 have been removed for clarity. Residues His-163, Trp-165, His-318, Thr-323, Tyr-386, His-389, Phe-556, Phe-559, and Arg-566 are labeled accordingly. The chloride ion (pink) is shown bound in the channel of Ath α-DOX.
We previously identified highly conserved histidine and arginine residues, predicted to lie near the catalytic tyrosine, as molecular determinants involved in substrate orientation and binding (9). The equivalent residues in Ath α-DOX are His-318 and Arg-566. His-318 and Arg-566, which lie below Tyr-386, are the only two hydrophilic residues in an otherwise hydrophobic channel (Figure 3). A cluster of four tryptophan residues (Trp-157, Trp-165, Trp-219, and Trp-322) encompasses their side chains in order to provide stabilization. Interestingly, Trp-165 and Trp-322 are equivalent to Phe-209 and Tyr-348 in COX, where they stabilize the catalytic tyrosine (26, 31, 32).
Molecular Determinants of the α-Dioxygenase Reaction
Site directed mutagenesis was carried out on Gln-159, His-318, Thr-323, Arg-565, and Arg-566, residues. All mutant constructs were expressed at levels equivalent to wild type enzyme and were judged to be greater than 85% pure based on SDS-PAGE gel analysis (Supplemental Figure S4). UV-visible absorbance spectra of wild type and mutant constructs were indistinguishable from one another indicating that heme was properly incorporated into the mutant constructs. Wild type and mutant constructs were functionally characterized by measuring their oxygenase activity using LA as the substrate. Table 3 summarizes the relative oxygenase activity of the mutant constructs with respect to the wild type enzyme and also lists the oxygenase activities of mutant constructs determined previously for O. sativa α-DOX (Osa α-DOX) (9).
Table 3. Relative activity of wild type and mutant Ath α-DOX constructs.
Oxygenase activity was measured with 10μg of protein construct and 200μM LA as the substrate using an oxygen electrode as described under “Experimental Procedures”. A value of 100% is assigned for the oxygenase activity of wild-type Ath α-DOX, with mutant activity normalized to the wild-type construct. Values represent the average of 3 trials. Values for Osa α-DOX are from (9). The residue numbering equivalences between Ath and Osa α-DOX are as follows: Q159:Q153; H318:H311; T323:T316; Y386:Y379; R565:R558; R566:R559. ND, not determined.
| Construct | Relative Activity (%)
|
|
|---|---|---|
| Ath α-DOX | Osa α-DOX | |
|
| ||
| wild type | 100 | 100 |
| Q159N | 85.5 | ND |
| Q159S | 100 | ND |
| Q159V | 75.4 | ND |
| H318A | 6.9 | 2.6 |
| H318Q | 25.3 | 11.5 |
| T323A | 7.0 | ND |
| T323L | 0.0 | ND |
| Y386F | 0.0 | 0.0 |
| R565A | 76.0 | 46.7 |
| R565K | 32.0 | 18.4 |
| R565L | 55.0 | 74.2 |
| R566A | 0.0 | 0.4 |
| R566K | 2.8 | 0.9 |
| R566L | 0.0 | 0.0 |
Overall, the equivalent substitutions made at His-318, Tyr-386, Arg-565, and Arg-566 resulted in similar reductions in relative oxygenase activity with respect to wild type enzyme when compared to the values obtained for Osa α-DOX. A H318Q construct retained ~25% oxygenase activity compared to wild type enzyme and the KM value calculated for this mutant was 18.2 ± 2.4 μM, which is similar to the wild type enzyme (Table 2). These values are consistent with His-318 not being required for high affinity substrate binding. All substitutions at Arg-566 were detrimental to the oxygenase activity of Ath α-DOX, indicating that this side chain is required for high affinity binding of substrate within the channel. Taken together, the functional data corresponds well with the location of these residues within the fatty acid channel of Ath α-DOX structure.
We had previously suggested that Arg-565 did not orient itself into the fatty acid channel or interact with substrate given that there were only modest reductions in oxygenase activity observed for mutant constructs compared to its neighboring residue, Arg-566. Determination of the structure of Ath α-DOX confirms that Arg-565 does not face the fatty acid channel. Instead, the side chain of Arg-565 points towards the surface of the enzyme where it forms five hydrogen bonds with the main chain carbonyl oxygen atoms of Asn-371, Val-374, and Pro-375, and two water molecules. Mutant constructs of Arg-565 had only modest effects on the oxygenase activity of Ath α-DOX when compared to wild type enzyme. Disruption of the hydrogen-bonding network likely explains the observed reductions in oxygenase activity.
Analysis of the Ath α-DOX structure identified two other residues, Gln-159 and Thr-323, which are located near the heme and within the active site channel, respectively. Along with the distal and proximal histidines, Gln-159 is highly conserved in the α-DOX family. Gln-159 is homologous to Gln-203 in COX, where it has been shown to be important for peroxidase catalysis by serving as a charge-stabilizing residue (33). The side chain of Gln-159 lies below His-163 and within 3.5Å of the distal face of the heme moiety in the Ath α-DOX structure. Although Ath α-DOX does not possess a strong peroxidase activity, substitution of Gln-159 with arginine resulted in a mutant construct that was devoid of oxygenase activity when the products of the reaction were analyzed utilizing tritium-labeled LA as the substrate (25). We determined the oxygenase activity of Q159N, Q159S, and Q159V mutant constructs utilizing LA as the substrate. Interestingly, all three substitutions had little to no effect on oxygenase activity, compared to wild type enzyme (Table 3). Thus, in contrast to previous studies, Gln-159 is not essential for the oxygenation of fatty acids by Ath α-DOX. Thr-323, which is located in helix H6 comprising one of the channel walls, is also highly conserved within the α-DOX family (9) (Figure 3). The side chain of Thr-323 is located ~3.5Å away from the side chain of His-318 and forms a hydrogen bond with a water molecule near the chloride ion bound in the channel. Substitution of Thr-323 with alanine resulted in a mutant construct that retained only 7% activity compared to wild type enzyme, while substitution to leucine completely abolished oxygenase activity (Table 3). These results suggest that the hydroxyl group of the side chain plays a significant role in the oxygenase reaction.
Inhibition by Imidazole
Earlier studies identified imidazole as a weak inhibitor of the oxygenase activity of both Ath α-DOX and Osa α-DOX, with calculated Ki values of 121μM and 46μM, respectively (10, 34). In the imidazole-bound Ath α-DOX structure, one of the ring nitrogen atoms of imidazole ligands to the iron atom on the distal side of the heme, while the other nitrogen atom forms a hydrogen bond with the Nε2 atom of His-163. When, 1mM imidazole was added to the protein solution and incubated for 60 minutes prior to measurement of oxygenase activity, we observed a 50% reduction in activity relative to untreated enzyme, further confirming that imidazole inhibits the activity of Ath α-DOX. The KM value for LA was decreased 1.6-fold to 14.2 ± 1.5 μM when determined in the presence of 1mM imidazole, indicating that imidazole does not significantly influence substrate binding in the channel, but rather affects the specific activity of the enzyme. These results agree with the Ath α-DOX structures presented here, which have virtually identical structural features within the active site channel.
DISCUSSION
The structure of Ath α-DOX presented here provides the first structural snapshot for a member of the α-DOX subfamily within the peroxidase-cyclooxygenase superfamily. The structure provides for a detailed comparison of the active site architectures of Ath α-DOX and COX, coupled with functional characterization of residues important for catalysis. Inspection of the structure in the context of our previously derived homology model of Osa α-DOX bound with LA provides new insight into the mode of substrate binding and explains the observed promiscuous oxygenation of a variety of fatty acids by the enzyme.
There are significant structural similarities between Ath α-DOX and COX, despite the 15% identity observed between their primary sequences. While there is no observed epidermal growth factor-like domain in the Ath α-DOX structure, the domain makeup and spatial layout of the base and catalytic domains are strikingly similar to those observed for the MBD and catalytic domains of COX. The four, amphipathic α-helices of the MBD tether COX to one leaflet of the membrane and serve as the access point for substrate entry into the catalytic domain. Three of the eight α-helices in the base domain of Ath α-DOX are amphipathic in nature. The amphipathic nature of the α-helices, coupled with the spatial location of the base domain compared to the MBD in the structures of COX suggests that this domain is responsible for membrane association. As the catalytic domain of Ath α-DOX is situated on top of the base domain, the arrangement would also allow the base domain to serve as an access point for substrate entry into the active site channel. Deleting the first 74-, 118- or 131-residues at the N-terminus of Ath α-DOX is deleterious to the activity of the enzyme (10), providing further support for this idea.
The catalytic domains of Ath α-DOX and COX show the highest sequence and secondary structure similarities. Twenty-one of the twenty-two α-helices within the catalytic domains of α-DOX and COX-2 are conserved, with a calculated RMSD between α-helices of 1.68Å (207 Cα atoms). As is the case with COX, helices H2 and H8, which house the distal and proximal histidine residues, form the binding cleft for the heme. His-389 is coordinated to the iron atom within the heme, analogous to His-388 in COX. Below the heme binding cleft, Helices H6 and H17 form the walls of the active site channel. The catalytic tyrosine, Tyr-386, also points into the active site channel analogous to its counterpart in COX, Tyr-385.
Abstraction of the 2-pro-R hydrogen from carbon-2 of the fatty acid substrate by Tyr-386 initiates the oxygenase reaction. As such, α-DOX must bind fatty acid with the carboxylate group placed deep within the active site channel near Tyr-386. This is in contrast to COX, which buries the ω-end of the fatty acid deep within the cyclooxygenase channel and places the carboxylate near the opening of the channel (26, 27). Previous studies showed that Ath α-DOX and Osa α-DOX did not oxygenate fatty acids that contained modifications at the carboxylate end (9, 24). Rather than simply being misaligned, the tight volume constraints near His-318, Tyr-386, and Arg-566 within the active site channel, coupled with the increase in size of the modified fatty acids preclude binding of these substrates for oxygenation.
Our previous work identified a histidine and arginine residue, which are highly conserved within the α-DOX family, as determinants involved in the high affinity binding and positioning of substrates for optimal catalysis. In Ath α-DOX, His-318 and Arg-566 are located just below Tyr-386 in the active site channel. Mutation of Arg-566 completely abolishes oxygenase activity, which is consistent with both kinetic analyses and substrate binding assays carried out using Osa α-DOX (9). Substitutions at His-318 also resulted in mutant constructs that had significantly reduced oxygenase activity. However, there was no appreciable decrease in KM, indicating that His-318 is not required for high-affinity binding of substrate. The Nε2 atom of His-318 is 5.7Å away from the phenolic oxygen of Tyr-386 in the Ath α-DOX structure. As such, it is not likely that His-318 influences radical formation as previously hypothesized (35). Instead, we postulate that His-318 is involved in proper alignment of carbon-2 of the fatty acid below Tyr-386 for optimal catalysis, consistent with our earlier studies using Osa α-DOX (9). The current study also shows that substitutions of the side chain of Thr-323, another conserved residue within the α-DOX family, result in a significant loss of oxygenase activity. Given its location within the channel, we speculate that the side chain of Thr-323 also interacts with the carboxylate of the substrate to properly orient carbon-2 for catalysis. The observed interactions of the side chains of His-318 and Arg-566 with the bound chloride ion within the active site channel of the Ath α-DOX crystal structure may mimic the interaction that these residues have with the carboxylate group of the substrate.
OA, LA, and αLA are the predominant substrates utilized by Ath α-DOX. However, α-DOX is quite promiscuous, given their ability to oxygenate fatty acids with carbon lengths ranging from 14–20 carbons and varying degrees of unsaturation (9, 24). In general, fatty acid substrates that do not contain a double bond within the first seven carbon atoms are effectively oxygenated by Ath α-DOX and Osa α-DOX. Moreover, fatty acids are effectively oxygenated when one, two, or three unsaturated bonds are present in the chain beyond carbon seven. Collectively, these results suggest that the carboxylate interaction, along with interactions between the enzyme and substrate within the first seven carbons of the fatty acid chain are most important for binding and catalysis. Analysis of the Ath α-DOX structure provides insight into the conformation of the fatty acid within the active site channel required for productive oxygenation. Using LA as an example, we hypothesize that the carboxylate of LA forms at least one salt bridge with the side chain of Arg-566 and hydrogen bonds with the side chains of His-318 and Thr-323 to anchor and position carbon-2 below Tyr-386 (Supplemental Figure S5). Carbon-3 through carbon-8 would then take on a more linear conformation that allows the chain to pass between helices H6 and H17, near the side chains of Phe-556 and Phe-559, which could form stabilizing hydrophobic contacts. This is a revision to our previous model, which suggested a bent conformation for the substrate that had carbons 1–8 running parallel to helices H6 and H17 (9). The remainder of the fatty acid chain would then pass into the base domain, providing more space to accommodate the two unsaturated double bonds.
Members of the α-DOX family are distinct with respect to COX in that they exhibit a significantly reduced peroxidase activity, which generates a tyrosyl radical on Tyr-385 to initiate the cyclooxygenase reaction and reduce the developing hydroperoxide in COX. As the distal face of the heme in COX is surface accessible, large ligands such as 15-hydroperoxyeicosatetraenoic acid and prostaglandin G2, can serve as peroxidase substrates (36). In α-DOX, the tyrosyl radical on Tyr-386 is produced as a result of oxidizing the heme with H2O2 (10, 35). Liu and colleagues speculated that the reduced peroxidase activity was due to steric hindrance by residues inherent to Ath α-DOX that block access to the distal face of the heme. Inspection of the Ath α-DOX structure confirms this hypothesis. Two extended inserts, consisting of residues 170–197 and 461–478, loop across the heme-binding cleft to restrict access to the distal face of the heme. While there are no covalent linkages between the enzyme and the heme, the ionic interactions between Glu-170 and Arg-473 and Gln-174 and Asp-475 serve to lock the heme within the globular catalytic domain. The structural rigidity of the extended inserts further support earlier studies related to extracting the heme from Ath α-DOX. While the use of butanone under acidic conditions allowed for complete removal of the heme, the resulting protein could not be reconstituted to restore oxygenase activity (10). Despite the significant restrictions imposed by the extended inserts, two small channels exist on the surface of the enzyme that facilitate access of H2O2 to the distal face of the heme. The restricted access may protect the enzyme from overoxidation when high concentrations of H2O2 are present (35).
Previous studies that utilized size-exclusion chromatography to analyze the oligomeric state of various fusion constructs of Ath α-DOX suggested that the protein formed multimeric complexes consisting of 6–10 monomers (24). In our hands, Ath α-DOX formed only monomers in solution. It is not clear why there is such a discrepancy, however, it may be related to the detergent systems utilized during the chromatographic runs. COX behaves as a homodimer in solution (27, 28, 36). Although Ath α-DOX and COX have similar structural features related to catalysis, it is not clear from an evolutionary standpoint why COX forms dimers and Ath α-DOX exists as a monomer. The recent findings that COX exhibits half-of-sites activity suggests that dimer formation may be a mechanism utilized by COX to regulate its activity in an allosteric manner (37).
In summary, the structure of Ath α-DOX presented here represents the first member of the α-DOX family to be structurally characterized at the molecular level. Despite the low sequence identity, residues critical for the oxygenation of fatty acids, including the proximal and distal histidines involved in heme binding, and the catalytic tyrosine, are highly conserved between α-DOX and COX. Thus it is no surprise that the active site architecture of α-DOX most resembles that of COX. The side chains of His-318, Thr-323, and Arg-566 are critical determinants for the oxygenase activity, serving to bind and optimally align the substrate in the active site channel. Defining the location of these residues in the context of the active site channel allow us to propose a model for productive substrate binding within the channel and provides insight into the ability of α-DOX to oxygenate a variety of fatty acids. Moreover, structural characterization of the extended inserts present in the Ath α-DOX crystal structure detail why α-DOX enzymes exhibit a reduced peroxidase activity.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM077176. X-ray diffraction experiments were conducted at the Cornell High Energy Synchrotron Source (CHESS) and the Advanced Photon Source (APS). Use of the APS was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. DE-AC02-06CH11357. GM/CA CAT has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). CHESS is supported by NSF award DMR-0225180, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, supported by NIH award RR-01646.
The abbreviations used are
- LA
linoleic acid
- αLA
α-linolenic acid
- OA
oleic acid
- C10M
decyl maltoside
- IMAC
immobilized metal affinity chromatography
- βNG
β-nonylglucoside
- α-DOX
α-dioxygenase
- MAD
multiwavelength anomalous diffraction
- Ath
Arabidopsis thaliana
- Osa
Oryza sativa
- cv
column-volumes
Footnotes
The coordinates and structure factors for wild type and imidazole-bound Ath α-DOX have been deposited in the Protein Data Bank (PDB entry 4HHS and 4HHR).
Supporting Information Available
Additional figures and tables are provided as supporting information. Supporting figure 1 depicts the cyclooxygenase and α-dioxygenase reaction mechanisms. Supporting figure 2 depicts the sequence and secondary structure alignment of Ath α-DOX with murine COX-2, while supporting figure 3 depicts electron density for the imidazole and heme moiety. Supporting table 1 and figure 4 depicts the primers and utilized to generate the mutant constructs and the purity of wild type and mutant constructs of Ath α-DOX used in this study, respectively. Supporting figure 5 depicts LA modeled into the active site channel of Ath α-DOX. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schneider C, Pratt DA, Porter NA, Brash AR. Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem Biol. 2007;14:473–488. doi: 10.1016/j.chembiol.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lopez MA, Bannenberg G, Castresana C. Controlling hormone signaling is a plant and pathogen challenge for growth and survival. Curr Opin Plant Biol. 2008;11:420–427. doi: 10.1016/j.pbi.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Blee E. Impact of phyto-oxylipins in plant defense. Trends Plant Sci. 2002;7:315–322. doi: 10.1016/s1360-1385(02)02290-2. [DOI] [PubMed] [Google Scholar]
- 4.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 5.Sanz A, Moreno JI, Castresana C. PIOX, a new pathogen-induced oxygenase with homology to animal cyclooxygenase. The Plant Cell. 1998;10:1523–1537. doi: 10.1105/tpc.10.9.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamberg M, Ponce de Leon I, Sanz A, Castresana C. Fatty acid alpha-dioxygenases. Prostaglandins Other Lipid Mediat. 2002;68–69:363–374. doi: 10.1016/s0090-6980(02)00040-0. [DOI] [PubMed] [Google Scholar]
- 7.Zamocky M, Jakopitsch C, Furtmuller PG, Dunand C, Obinger C. The peroxidase-cyclooxygenase superfamily: Reconstructed evolution of critical enzymes of the innate immune system. Proteins. 2008;72:589–605. doi: 10.1002/prot.21950. [DOI] [PubMed] [Google Scholar]
- 8.Hamberg M, Sanz A, Castresana C. alpha-oxidation of fatty acids in higher plants. J Biol Chem. 1999;274:24503–24513. doi: 10.1074/jbc.274.35.24503. [DOI] [PubMed] [Google Scholar]
- 9.Koszelak-Rosenblum M, Krol AC, Simmons DM, Goulah CC, Wroblewski L, Malkowski MG. His-311 and Arg-559 are key residues involved in fatty acid oxygenation in pathogen-inducible oxygenase. J Biol Chem. 2008;283:24962–24971. doi: 10.1074/jbc.M804358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu W, Rogge CE, Bambai B, Palmer G, Tsai AL, Kulmacz RJ. Characterization of the heme environment in Arabidopsis thaliana fatty acid alpha-dioxygenase-1. J Biol Chem. 2004;279:29805–29815. doi: 10.1074/jbc.M401779200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mancia F, Love J. High-throughput expression and purification of membrane proteins. J Struct Biol. 2010;172:85–93. doi: 10.1016/j.jsb.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koszelak-Rosenblum M, Krol A, Mozumdar N, Wunsch K, Ferin A, Cook E, Veatch CK, Nagel R, Luft JR, Detitta GT, Malkowski MG. Determination and application of empirically derived detergent phase boundaries to effectively crystallize membrane proteins. Protein Sci. 2009;18:1828–1839. doi: 10.1002/pro.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luft JR, Snell EH, DeTitta GT. Lessons from high-throughput protein crystallization screening: 10 years of practical experience. Expert Opinion on Drug Discovery. 2011;6:465–480. doi: 10.1517/17460441.2011.566857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter JCW, Sweet RM, editors. Methods in Enzymology. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 15.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 16.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 17.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 20.Winn MC, Isupov MN, Murshudov G. Use of TLS parameters to model anisotrophic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2000;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 21.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D Biol Crystallogr. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 23.Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010. [Google Scholar]
- 24.Liu W, Wang LH, Fabian P, Hayashi Y, McGinley CM, van der Donk WA, Kulmacz RJ. Arabidopsis thaliana fatty acid alpha-dioxygenase-1: evaluation of substrates, inhibitors and amino-terminal function. Plant Physiol Biochem. 2006;44:284–293. doi: 10.1016/j.plaphy.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Hamberg M, Sanz A, Castresana C. Alpha-Dioxygenase, a new enzyme in fatty acid metabolism. International Congress Series. 2002;1233:307–317. [Google Scholar]
- 26.Malkowski MG, Ginell SL, Smith WL, Garavito RM. The productive conformation of arachidonic acid bound to prostaglandin synthase. Science. 2000;289:1933–1937. doi: 10.1126/science.289.5486.1933. [DOI] [PubMed] [Google Scholar]
- 27.Vecchio AJ, Simmons DM, Malkowski MG. Structural basis of fatty acid substrate binding to cyclooxygenase-2. J Biol Chem. 2010;285:22152–22163. doi: 10.1074/jbc.M110.119867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malkowski MG, Theisen MJ, Scharmen A, Garavito RM. The formation of stable fatty acid substrate complexes in prostaglandin H(2) synthase-1. Arch Biochem Biophys. 2000;380:39–45. doi: 10.1006/abbi.2000.1906. [DOI] [PubMed] [Google Scholar]
- 29.Furtmuller PG, Zederbauer M, Jantschko W, Helm J, Bogner M, Jakopitsch C, Obinger C. Active site structure and catalytic mechanisms of human peroxidases. Arch Biochem Biophys. 2006;445:199–213. doi: 10.1016/j.abb.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 30.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 31.Vecchio AJ, Orlando BJ, Nandagiri R, Malkowski MG. Investigating substrate promiscuity in cyclooxygenase-2: the role of Arg-120 and residues lining the hydrophobic groove. J Biol Chem. 2012;287:24619–24630. doi: 10.1074/jbc.M112.372243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thuresson ED, Lakkides KM, Rieke CJ, Sun Y, Wingerd BA, Micielli R, Mulichak AM, Malkowski MG, Garavito RM, Smith WL. Prostaglandin endoperoxide H synthase-1: the functions of cyclooxygenase active site residues in the binding, positioning, and oxygenation of arachidonic acid. J Biol Chem. 2001;276:10347–10357. doi: 10.1074/jbc.M009377200. [DOI] [PubMed] [Google Scholar]
- 33.Landino LM, Crews BC, Gierse JK, Hauser SD, Marnett LJ. Mutational analysis of the role of the distal histidine and glutamine residues of prostaglandin-endoperoxide synthase-2 in peroxidase catalysis, hydroperoxide reduction, and cyclooxygenase activation. J Biol Chem. 1997;272:21565–21574. doi: 10.1074/jbc.272.34.21565. [DOI] [PubMed] [Google Scholar]
- 34.Koeduka T, Matsui K, Akakabe Y, Kajiwara T. Catalytic properties of rice alpha-oxygenase. J Biol Chem. 2002;277:22648–22655. doi: 10.1074/jbc.M110420200. [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee A, Angeles-Boza AM, Huff GS, Roth JP. Catalytic mechanism of a heme and tyrosyl radical-containing fatty acid alpha-(di)oxygenase. J Am Chem Soc. 2011;133:227–238. doi: 10.1021/ja104180v. [DOI] [PubMed] [Google Scholar]
- 36.Garavito RM, Malkowski MG, DeWitt DL. The structures of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 2002;68–69:129–152. doi: 10.1016/s0090-6980(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 37.Dong L, Vecchio AJ, Sharma NP, Jurban BJ, Malkowski MG, Smith WL. Human Cyclooxygenase-2 Is a Sequence Homodimer That Functions as a Conformational Heterodimer. J Biol Chem. 2011;286:19035–19046. doi: 10.1074/jbc.M111.231969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.