

Abstract
Spinal muscular atrophy (SMA) is a leading genetic cause of infant mortality. A neurodegenerative disease, it is caused by loss of SMN1, although low, but essential, levels of SMN protein are produced by the nearly identical gene SMN2. While no effective treatment or therapy currently exists, a new wave of therapeutics has rapidly progressed from cell-based and preclinical animal models to the point where clinical trials have initiated for SMA-specific compounds. There are several reasons why SMA has moved relatively rapidly towards novel therapeutics, including: SMA is monogenic; the molecular understanding of SMN gene regulation has been building for nearly 20 years; and all SMA patients retain one or more copies of SMN2 that produces low levels of full-length, fully functional SMN protein. This review primarily focuses upon the biology behind the disease and examines SMN1- and SMN2-targeted therapeutics.
Spinal muscular atrophy: disease & clinical manifestations
Spinal muscular atrophy (SMA) is an inherited autosomal recessive neurodegenerative disease. It is the leading genetic cause of infantile mortality worldwide with a disease prevalence of approximately 1:6000–1:10,000 and a carrier frequency of approximately 1:35–1:40 [1,2]. SMA prevalence is static throughout all ethnic groups, with few exceptions, although an isolated population from South Dakota, the Hutterites, possesses a dramatically higher carrier frequency approaching 1:8 [3]. SMA is characterized by the degeneration of motor neurons within the anterior horn of the spinal cord leading to skeletal muscle weakness and atrophy [4]. Muscle weakness and atrophy is symmetrical and progressive, often impacting the legs more so than the arms, eventually leading to a decline in intercostal activity. Respiratory failure and complications account for the majority of premature deaths in SMA patients [4].
Clinically, SMA disease severity is broad and for classification purposes, patients are categorized based upon the severity, the age of onset, and achieving (or failing to achieve) physical milestones [5]. Type 0 is extremely severe and initiates during prenatal development and results in death within weeks. Type I, or Werdnig– Hoffman disease, is a severe form characterized by an infantile onset ranging from birth to 6 months and accounts for approximately 50% of all newly diagnosed cases of SMA [6]. The natural history of all forms of SMA has significantly changed due to supportive measures, including respiratory and physical therapy, however, in the absences of these advances, there is a 32% survival probability at 2 years of age and at no time can type I patients sit upright without support [7]. Type II onset occurs between 6–18 months and initiates with proximal limb weakness, with progressive weakness and respiratory complications, joint contractures and scoliosis appearing in childhood [7]. At some point during childhood, type II patients can sit upright without assistance. Approximately 70% of type II patients live to adulthood. Type III SMA presents past 1 year (>1 year for type IIIa; >3 years for IIIb) and individuals can initially stand/walk without assistance and have a normal lifespan, although many become wheelchair-bound during adolescence [4]. Type IV patients develop proximal leg weakness in adulthood and have a normal lifespan [4]. Even within each disease category, further delineation exists and it could be argued that there is a continuous disease severity spectrum, however, in the age of molecular diagnostics and clinical trial enrollment, this system retains a great deal of utility.
Molecular genetics: SMN1 & SMN2
SMN1 is the SMA-determining gene and is located on chromosome 5q13 [8]. While a generalized region of chromosome 5 had been identified several years earlier as the SMA locus, it was not until 1995 that the SMA-determining gene was conclusively identified as SMN1 [9–11]. However, the genetics and the identification were clearly complicated by the presence of a nearly identical gene also located on chromosome 5q. This gene, called SMN2, is positioned within an approximately 500 Kb duplication that lies in a head-to-head orientation [8]. The duplicated region that contains SMN2 is centromeric to SMN1 and additional genes such as p44, SERF and NAIP are also present in each duplicated region [8,12]. Most SMA cases arise from deletions including SMN1 exons 7 and 8, however, larger deletions were also detected that encompassed adjoining genes, such as NAIP [12]. While there was initially some concern that other genes within this region contributed to SMA development, the identification of SMN intragenic mutations including frameshifts and point mutations further confirmed the role of SMN1 [13].
The nucleotide sequences of SMN1 and SMN2 are 99% identical and the amino acid sequences are 100% identical for the overlapping coding elements. There are a handful of sequence variations in the promoter regions as well as small variations within the genes, mostly within intronic elements [8,14,15]. Seemingly paradoxically, mutation or loss of SMN2 has no clinical consequence provided SMN1 is intact, whereas loss of SMN1 results in SMA. The key to understanding this complex genetic question resides in a single non-polymorphic nucleotide difference at the 5′ end of exon 7 (840C>T). SMN1 transcripts are predominately full-length, spanning exons 1–8, whereas the majority of SMN2-derived transcripts are alternatively spliced producing an isoform that lacks the typical final coding exon (exon 7) [16,17]. Exon-skipped products then terminate early in exon 8 after encoding only four amino acids. Insight into the importance of the 840C>T transition was initially identified by ‘hybrid’ alleles in SMA patients [18]. Likely due to the relatively large genomic duplication and inversion of the SMN locus, the SMN genes appear to be prone to recombination and partial recombination events that can produce small arrays of SMN1 or SMN2 genes on a single chromosome or can result in a single ‘hybrid’ SMN gene that is partially derived from SMN1 and SMN2. Through the analysis of hybrid genes as well as synthetic mini-genes, it was determined that the 840C>T dictated the exon 7 alternative splicing event: an SMN1-derived ‘C’ resulted in a hybrid gene that exhibited a SMN1-like expression profile, whereas a SMN2-derived ‘T’ resulted in a hybrid gene that exhibited a SMN2-like expression profile [15,19].
Importantly, approximately 10%, of SMN2-derived transcripts encodes fully functional, full-length SMN protein. It is important to stress that SMA does not arise from the complete absence of SMN, consistent with observations that a SMN null state appears to be lethal in all organisms [20]. Rather, SMA develops from a severe reduction of SMN. The low levels of full-length SMN produced by SMN2 are sufficient to prevent embryonic lethality, but are insufficient to prevent SMA development [21]. Additionally, murine models suggest that the exon 7-skipped protein product, SMNΔ7, retains a small degree of functionality compared to full-length SMN [22]. One or more copies of SMN2 are retained by all SMA patients and, to date, SMN2 is clearly the most important genetic modifier of disease severity [23–26]. Numerous patient studies have been performed, allowing a relatively straightforward conclusion to be drawn: SMN2 dosage inversely correlates with disease severity. In general, type I patients have two copies; type II patients have 2–3 copies; and type III patients have 3–4 copies [27–29]. Additionally, healthy individuals have been identified that are homozygously deleted for SMN1, but retain 5 or more copies of SMN2 [30]. Genetically, these SMA-carriers could be considered as SMA patients as they lack SMN1, however, the high number of SMN2 copies has provided sufficient SMN protein to fully protect from disease development. Clearly, overlap exists for each of these groups and much like the continuous clinical spectrum, SMN2 dosage covers a similarly broad range as well. At the molecular level, the overwhelming majority of patients are screened by genetic means that are designed to assess the presence or absence of SMN1/SMN2 genes [31,32]. These tests, however, are typically based upon the 840C>T difference and do not discriminate between SMN2 alleles that produce high or low levels of SMN or from those that may be completely dysfunctional. Therefore, while gene dosage is an incredibly important component of an individual’s diagnosis, this cannot be the sole determinant and must be merged with the clinical manifestations. Molecular diagnostics, however, are playing an increasingly important role in SMA clinical trials and the stratification of patients with a particular SMN2 genotype will likely become increasingly more important as SMA-specific compounds are examined in clinical trials.
SMN protein function: general activity versus motor neuron activity
SMN is a multifaceted 38 kDa protein that is ubiquitously expressed throughout development [33]. Early studies suggested that SMN performed an essential function for all cells since genetic ablation of the murine Smn resulted in pre-implantation lethality [34]. Insight into SMN function initially came through yeast two-hybrid studies and SMN’s hallmark nuclear staining pattern [35]. Within many cell types, SMN localizes into discrete nuclear foci termed ‘gemini of coiled bodies’ or ‘gems’ [35,36]. While coiled bodies and gems are enriched for factors involved in various aspects of gene expression, including transcription and RNA processing components, these nuclear structures are not active sites of transcription or splicing. However, this localization pattern suggested a role in a variety of RNA-associated activities. Detailed biochemical studies have subsequently revealed that SMN often functions with a cohort of proteins collectively referred to as Gemins [37]. The core complex consists of SMN, Gemins 2–8 and unrip, and can be found in all tissues and cell types. The SMN/Gemin complex is integral to the assembly of small nuclear ribonuclear proteins (snRNPs) within the cytoplasm and their subsequent transport into the nucleus [37]. snRNPs are composed of a single snRNA and a heptameric ring structure composed of Sm proteins. SMN’s role in this activity is without question, both biochemical studies as well as structural data demonstrate the relevance of the SMN/Gemin complex in snRNP assembly. Perhaps one of the most intriguing questions in the SMA field has centered upon how a dysregulation of an essential, general cellular activity, snRNP assembly, could account for the motor neuron-specific vulnerability observed in SMA [38]. While general snRNP assembly defects have been observed in SMA mice, these defects were observed in a pathway referred to as the major spliceosome or at a relatively late stage of disease [39,40]. In contrast, an alternative class of introns comprising <1% of all introns referred to as U12-dependent or minor introns has been the target of recent speculation [41–44]. The connection between SMA and U12-dependent introns stems from the observation that U12 introns are not uniformly distributed throughout the genome [45,46]. Rather, U12 introns are enriched within voltage-gated ion channels and other genes likely to be involved in neuronal function [46]. The discovery of U12 genes that are aberrantly spliced in SMA contexts would provide mechanistic validity to the role of snRNP assembly in SMA development as well as providing novel drug targets beyond SMN. It is likely, however, too simplistic to assume that correcting one or two improperly spliced mRNAs could completely correct the entirety of the SMA phenotype and a more nuanced balance of U12 introns may be difficult to achieve pharmacologically.
An alternative, non-snRNP function has also been proposed that could account for the motor neuron specific loss observed in SMA [47]. SMN largely exists in snRNP-free protein/RNA complexes within axons and it has been hypothesized that SMN serves as an mRNA chaperone for a subset of mRNAs as they are transported distally to the growth cones [48–50]. SMN has been shown to interact and/or co-localize with several factors involved in RNA transport, including hnRNP-R, hnRNP-Q, HuD, COP1 and with actin and candidate plasticity-related gene 15 mRNA [51–55]. In cultured neurons from SMA mice, the SMN-neuronal complexes are disrupted and the cargoes are poorly localized to the developing termini. The current thought is that localized translation of factors, such as β-actin, would be disrupted leading to an alteration in actin dynamics and subsequent cytoskeleton growth and development at the growth cone, however genetic ablation of β-actin does not result in lower motor neuron defects [56]. While the biochemistry behind the SMN-neuronal function is still developing, research in other fronts has supported a role for SMN in some type of cytoskeletal activity. For example, two recent reports have demonstrated that compounds Fasudil and Y-27632 that modulate actin dynamics by inhibition of the RhoA/ROCK pathway can significantly extend survival in an intermediate mouse model of SMA [57,58]. The protective affect was independent of SMN as SMN levels remained stable and low. Plastin 3 has also been postulated to function as a disease modifier as its overexpression was detected in discordant siblings and shown to correlate with decreased disease severity in female siblings [59]. Plastin 3 was also capable of compensating for SMN-deficient phenotypes in zebrafish and cultured neurons [59].
While there are two primary schools of thought regarding the SMN-specific function that leads to motor neuron loss, the reality is that neither has been conclusively proven or disproven and the picture remains complex. SMN interacts with an ever-expanding list of proteins, totaling over 60 to date [60]. While SMN missense mutations typically fail to bind to each of the protein substrates, it is unclear whether these factors contribute to SMA development. The recent analysis of β-actin null mice was hypothesized to result in neuronal defects, however, the β-actin null mice appeared to be surprisingly normal regarding axonal regeneration and neuronal function, although a more detailed analysis in a CNS-restricted ablation revealed abnormalities within the hippocampus and cerebellum [56,61].
Regulation of SMN pre-mRNA splicing: to skip or not to skip
Eukaryotic pre-mRNA splicing is an intricate balance between cis and trans factors that coordinate the identification and proper excision of intronic sequences (Figure 1). SMN exon 7 is 54 nucleotides and includes the stop codon for full-length SMN [8]. Although SMN1 and SMN2 are nearly identical, the pre-mRNA splicing patterns are dramatically different and underscore the importance of cis- and trans-factors that coordinate the complex regulatory environment surrounding SMN exon 7 (Figure 2) [16,17]. In the context of SMN1, exon 7 is constitutively included in the majority of transcripts. While SMN1 exon 7 is flanked by relatively weak splice signals including the poly-pyrimidine tract, and the 3′ and 5′ splice sites, the presence of several exonic splice enhancers within exon 7 overcome these non-consensus regulatory elements [62–64]. The exonic splice enhancers serve as binding substrates for regulatory proteins including the serine/arginine-rich (SR) and SR-like proteins. hTra2-β1, an SR-like family member, directly binds the conserved AG-rich central region of the exon (Figure 2) [65]. The presence of hTra2- β1 may facilitate the indirect recruitment of several other splicing factors including SRp30c, hnRNP-G, RMBY, hnRNP-Q1 and TDP-43 [66–69]. In a variety of experimental contexts including the mini-gene system, transient overexpression studies and cell-free extracts, hTra2-β1 and the associated proteins stimulate SMN exon 7 inclusion [65]. At the 5′ end of SMN1 exon 7, a high-affinity binding site is present that is bound by the SR-protein family member, SF2/ASF [70,71]. Intronic regulatory elements exist as well, including the positively acting TIA1 [72], which binds within intron 7. Interestingly, while most of the putative negatively regulating elements that dominate SMN2 exon 7 expression are present within the SMN1 pre-mRNA, the positively acting splicing factors preferentially exert their influence, resulting in the constitutive inclusion of SMN1 exon 7.
Figure 1. Eukaryotic pre-mRNA splicing factors.

(A) Typical conserved cis and trans (B) splicing signals found at the intron/exon junction. (C) Auxiliary regulatory cis elements, including repressors (red) such as ESS and ISS or positive factors, such as ESE. (D) SR proteins binding to ESEs. (E) Negatively regulating factors, such as hnRNP proteins binding their cognate sites, thereby inhibiting SR protein binding and exon inclusion.
ESE: Exonic splice enhancers; ESS: Exonic splice silencers; ISS: Intronic splice silencers; SR: Ser-/Arg-rich.
Figure 2. Regulation of SMN1 and SMN2.
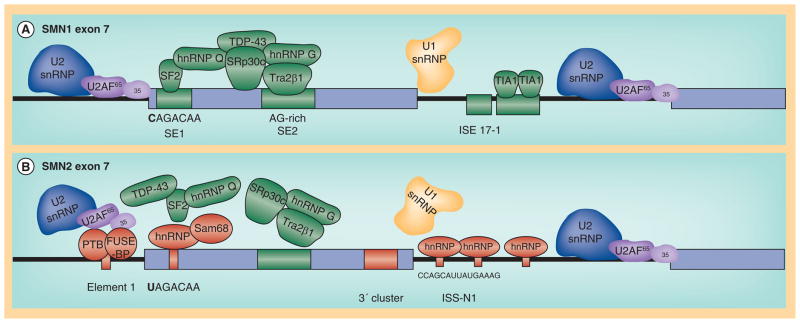
(A) General splicing factors (U2 and U1 snRNP) and auxiallary factors associated with positively acting sequences in and around SMN 1 exon 7. (B) SMN2-associated regulatory factors, including the C/U transition in exon 7, ISS-N1, Element 1 and the 3’ cluster.
While the sequences are nearly identical between SMN1 and SMN2, the SMN2 pre-mRNAs are subjected to a dramatically different set of regulatory constraints [60,64,73]. The presence of the 840C>T nucleotide at the 5′ end of exon 7 confers direct and indirect affects upon the composition of splicing regulatory proteins present at exon 7. The 840C>T transition resides within the high affinity SF2/ASF binding site and based upon SELEX analysis, the ‘U’ greatly reduces this affinity, and, in turn, decreased binding of SF2/ASF correlates with decreased SMN2 exon 7 inclusion [70,71]. Interestingly, not only does the 840C>T reduce the affinity for the positively acting SF2/ASF, but a novel SMN2-specific inhibitory element is created. The C/U transition allows for the formation of a secondary structure within the SMN2 exon 7 pre-mRNA that serves as a putative substrate for negatively regulating factors, including hnRNPA1 and Sam68 [74–77]. In the SMN2 context, a new suite of molecular constituents are involved in exon 7 regulation now that the SF2/ASF site has been disrupted. In addition to the novel hnRNP-A1 site, exonic and intronic silencer elements are now able to exert an influence upon exon 7 splicing and promote exon 7 skipping including: the 3′ cluster or TSL at the 3′ end of exon 7 [78]; element 1 located upstream within intron 6 binds two factors, PTB and FUS-BP [79,80]; an additional SMN1/SMN2 non-polymorphic nucleotide difference within intron 7 mediates hnRNP–A1 binding [77,81]; ISE 17–1 located within intron 7 [81], and perhaps the most widely examined regulatory element, ISSN1, islocated near the 5′ splice site downstream of exon 7 [82,83].
One of the more important findings to come out of the molecular experimentation surrounding SMN exon 7 splicing was that SMN2 exon 7 is not irreversibly damaged. In fact, subtle mutations in the poly-pyrimidine tract upstream of exon 7 completely reverse the splicing ratio such that SMN2 produces exclusively full-length transcripts [63]. Similarly, mutations within exon 7 that modulate the inhibitor and enhancer binding sites can overcome the C/U-mediated splicing patterns [78,83–87], while overexpression of modified U1 snRNAs that specifically recognize the non-canonical splice site downstream of exon 7 largely correct the exon skipping phenotype [87]. Several reports have also demonstrated that by reducing splice site recognition at the exon 8 junction, the balance is shifted to the exon 7 splice site leading to an increased incorporation of SMN2 exon 7 [62,88–90]. Naturally occurring mutations also highlight the malleable nature of exon 7 splicing. For instance, discordant phenotypes that did not match the predicted disease severity based upon the SMN2 copy numbers revealed a novel mutation within SMN2 exon 7, 859G>C. This mutation was predicted to create a high affinity exonic splice enhancer for SF2/ASFand consistent with the in silico-based predications, the 859G>C mutation resulted in increased inclusion of SMN2 exon 7 – even in the presence of the 840C>T transition [91]. These important observations demonstrated that SMN2 exon 7 is not irreparably damaged and provided a biological foundation for strategies to modulate SMN2 exon 7 pre-mRNA splicing as a potential therapeutic for SMA.
Interestingly, a feedback mechanism may exist that contributes to the motor neuron specific sensitivity observed in SMN deficient contexts [92,93]. In in vitro assays, SMN reduction increased SMN2 exon 7 inclusion, suggesting that a reduction in SMN was further exacerbated by SMN-induced aberrant splicing of SMN2 exon 7 [92,93]. Consistent with this, RT-PCR analysis of SMN transcripts isolated from laser captured SMA motor neurons from SMA mice demonstrated that SMN2 exon 7 splicing was significantly more impacted by reduced SMN than non-motor neuron tissue [93].
SMN induction: modes of action for SMN therapeutic strategies
Gene replacement: viral vectors
Since SMA is monogenic and the SMN cDNA is relatively small, SMA is well suited for a viral-based replacement of SMN. Early work with pseudotyped lentivirus vectors and purported retrograde transport was capable of modest extensions in survival in severe SMA mice following intramuscular injections in SMA mice [94]. However, several hurdles still existed including achieving in vivo tropism for the appropriate tissues and being able to deliver a sufficiently high titer to the CNS. A breakthrough came when self-complementary adeno-associated virus vectors were used to delivery SMN to SMA mice. Intravenous (iv.) delivery of scAAV9–SMN at P1 resulted in a dramatic extension in survival and a significant rescue of the SMA phenotype [95–100]. Importantly, motor neuron transduction with the scAAV9 vector was high and an extension in survival, albeit reduced to an average of 22 days, could still be achieved if the vector was delivered at P5 [97]. Motor neuron transduction dropped dramatically at the later delivery time point, suggesting that the blood–brain barrier had blocked entry into the CNS or that disease progression has advanced to a stage that is no longer correctable [97]. In a separate report, delivery of AAV8–SMN or scAAV8–SMN via intracerebroventricular (icv.) injection also significantly extended survival; however, instead of the 250 plus days of survival observed in the scAAV9–SMN treated animals, these mice lived on average approximately 150 days [98]. Several possible explanations could account for the differences in life span for scAAV8 and scAAV9 treated animals. Recent work from several laboratories has demonstrated that severe SMA mice present a multi-organ system pathology including cardiac and vascular tissue and the pancreas [101–105]. scAAV9 has an exceptionally broad tropism, while scAAV8 tropism is more restricted and poorly transduces cardiac tissue [106]. This requirement for SMN within the CNS and the periphery may explain why high motor neuron expression of SMN following scAAV8–SMN results in a decreased degree of rescue compared to scAAV9–SMN delivery. In a head-to-head comparison of icv. versus iv. administration of scAAV9–SMN, icv. administration was shown to produce a greater degree of rescue in SMA [100,107]. Similar conclusions were also drawn regarding icv. administration in an even more severe model of disease that typically dies at approximately P5 [107]. While the extension in survival did not achieve the 150–250 days seen with the less severe model, this work demonstrated that delivery of the scA AV9-SMN vector can significantly extend survival even in severely symptomatic SMA animals.
Results using various mouse models indicated that CNS uptake following an iv. administration scAAV9–SMN was robust and provided the injection was performed almost immediately after birth, however, CNS penetration dropped off dramatically if the injection was given one week later. In contrast, adult cat and non-human primate studies have shown that scAAV9 can still enter the CNS following a single IV injection and, more importantly, efficient motor neuron uptake can still be achieved [108–110]. In terms of translating iv. delivery to SMA patients, current vector production technologies cannot accommodate a large scale clinical trial for older children or adults. However, sufficient virus could be generated for a clinical trial that utilized a more focused CNS-specific delivery paradigm. While neutralizing antibodies are clearly an issue that could hinder the advancement of this type of strategy [110], a SMA clinical trial is likely within the next 1–2 years.
Targeting SMN2: promoter activation
All SMA patients retain one or more copies of SMN2 and since SMN2 has the potential to encode fully functional SMN protein, this copy gene has been an invaluable therapeutic target. While total expression levels between SMN1 and SMN2 are similar, there has been significant interest in further stimulating the basal level of SMN2 transcription: even though SMN2 pre-mRNAs are preferentially producing SMNΔ7, a global increase in transcription would boost full-length and SMNΔ7 mRNA. While early reports suggested overexpression of SMNΔ7 protein functioned in a dominant-negative manner, the generation of the SMNΔ7 mouse demonstrated that SMNΔ7 was neither toxic nor a dominant-negative [22]. These animals are homozygous null for murine Smn, possess two human SMN2 genes and contain an additional transgene that overexpresses the cDNA for the SMNΔ7 isoform [22]. The addition of the SMNΔ7 cDNA extends survival to approximately 14 days, as compared with approximately 5 days for the Smn−/−; SMN2 SMA mice. This conclusively demonstrated that the SMNΔ7 product was not detrimental and the upregulating the SMN2 promoter could be a viable means of elevating SMN protein.
Multiple layers of regulation exist to tightly control eukaryotic gene expression. A global mechanism to regulate gene expression is through the compaction and relaxation of DNA accomplished in part by histone acetylases (HATs) which acetylate lysine residues found on histones, a primary building block for chromatin. Acetylation relaxes the DNA allowing for transcription while the activity of histone deacetylases (HDACs) promotes DNA chromatin compression and gene repression. Pharmacological manipulation of this system can be accomplished with a class of compounds referred to as HDAC inhibitors (HDACi) [111,112]. Within the context of SMA, repurposed compounds have shown promise in SMA reporter assays, SMA cells and in SMA mice. Sodium butyrate [113], valproic acid (VPA) [114–117], phenylbutyrate [118], trichostatin A (TSA) [119,120], LBH589 [121], and suberoylanilide hydroxamic acid (SAHA) [122,123] have been shown to increase SMN protein levels from the human SMN2 gene and in some instances from the murine Smn gene. Translating cell-based success into a phenotypic improvement in SMA mice has been challenging, in part, due to the extreme severity of the SMA models. However, TSA and SAHA were able to provide significant extensions in survival as well as lessening overall disease severity in SMA mice [119,120,123]. LBH589 elevated SMN protein by stimulating exon 7 inclusion and total SMN2 promoter expression, as well as increasing hTra2-β1 levels [121]. TSA plus a rigorous regimen of supportive and dietary care was further capable of extending survival by ~170% [120]. In addition to their SMN-inducing activity, many HDAC is appear to confer some degree of general neuroprotection independent of SMN, potentially through the suppression of atrogene pathways [124].
Perhaps one of the greatest challenges going forward for these types of compounds relates to specificity, or more to the point, the lack of specificity. In all likelihood, SMA patients will need to be on medication permanently and the pharmacological induction of a tangible percentage of the genome is likely to lead to off-target affects and toxicity. A recent inducible SMA mouse model of disease further suggests that elevated SMN levels will be required into adulthood as well [125]. While the investigations into the molecular regulation behind the SMN promoter structure(s) has provided some insight [126], additional work is needed to better understand the regulatory environment as well as to develop more specific HDACi that could be used for long-term administration. Since VPA and phenybutyrate are US FDA-approved compounds, it was possible to move these compounds into clinical trials for SMA type II and III patients, however, relatively little effect was observed [127–129]. Similar to the results seen with VPA and phenybutyrate, which performed well in cell-based assays, hydroxy urea was shown to increase SMN full-length RNA levels in SMA primary lymphoblast cultures but when analyzed in a placebo-controlled, double-blind trial, HU did not alter the SMA phenotype or SMN expression [130,131].
Prolactin (PRL), a blood–brain barrier permeable compound, has recently been shown to increase SMN expression dramatically in SMA cellular models and within the CNS of SMA mice [132]. While a significant extension in survival was observed in prolactin treated animals (~70%), this extension was not as robust as the SMN levels within the CNS might have predicted. One possible interpretation of these results is that while the receptor for PRL is widely expressed throughout the CNS, it is not widely present within the periphery. The relatively early mortality of PRL-treated animals did not appear to be due to NMJ-associated defects, but perhaps related to peripheral organ defects. PRL acts through the STAT5 pathway and was initially suggested as a potential target due to the functional overlap of three small molecules that increased SMN2 expression: sodium valproate, TSA and aclarubicin. STAT5 activation in SMA-like mouse embryonic fibroblasts and SMN2-expressing NSC34 cells increased SMN while a constitutively activated STAT5 mutant increased SMN in SMA patient lymphocytes [133]. Going forward, delivery of the FDA-approved recombinant PRL will likely be examined in SMA patients. Its activity and the absence of significant non-CNS activity will be of particular interest in the future.
During the first high-throughput screen for SMA-specific compounds, two parallel screens were run in an attempt to capture compounds with two distinct modes-of-action: SMN2 promoter activation and SMN2 exon 7 splicing. The SMN2 promoter assay identified a quinazoline structure that exhibited cell-based activity and led to a medicinal chemistry program that produced several compounds with drug-like properties [134,135]. The lead compound, 2,4-diaminoquinazoline, was shown to function through a distinct mechanism: inhibition of DcpS [136], which is an enzyme involved in 5′ cap-mediated degradation of mRNAs. Oral administration of the lead compound (RG3039) resulted in a dose-dependent increase of SMN in SMA mice and extended survival by ~20–30% [137]. A Phase I study performed by Repligen Corporation in 32 healthy individuals analyzing dosing, safety and tolerance has recently concluded and indicated a good safety profile for RG3039.
Targeting SMN2: modulating exon 7 inclusion with nucleic acid-based therapeutics
While early work demonstrated that exon 7 splicing could be modulated by the over-expression of transacting factors or the genetic removal of splicing inhibitors surrounding exon 7, there was little belief that these insights could translate into potential therapeutic strategies. However, this work laid the foundation for a new wave of research that leveraged the power of antisense oligonucleotides (ASOs) (Figure 3). ASOs are relatively short stretches of nucleic acid that recognize a target sequence with a high degree of specificity. In most instances, the molecular backbone has been chemically modified to reduce nuclease sensitivity, thereby extending the half-life for ASO compounds. While ASOs have often been used to knock-down expression of target mRNAs through siRNA pathways, ASOs within the context of SMA are designed as splice site-switching ASOs as a means to alter SMN2 exon 7 pre-mRNA splicing. The most straightforward strategy is to target an inhibitor of SMN2 exon 7 inclusion, such as the intronic splice silencer (ISS) elements surrounding exon 7, including ISS-N1 and E1 (Figure 3). ISS-N1 has been targeted by ASOs composed of several different backbone chemistries and of varying lengths [79,138–141], however, two compounds have demonstrated the greatest degree of efficacy in SMA mouse models. ISIS Pharmaceuticals has developed a backbone technology that utilizes a 2′-O-2-methyoxyethyl-modified back-bone in their ASO-10–27 (ISIS SMNRx). In an asymptomatic SMN2-transgenic model, ASO-10–27 fully reversed the SMN2 splicing pattern when administered via an icv. injection [142], whereas subsequent work in an SMA mouse model resulted in a significant extension in survival from approximately 14 days to approximately 26 days following a single icv. injection [143]. Somewhat unexpectedly, systemic administration of the same ASO resulted in nearly a 25-fold increase in survival, demonstrating the importance of peripheral tissues in severe SMA mouse models [144]. SMNRx has entered an open label Phase I clinical study that is designed to examine dosing following a single intrathecal injection.
Figure 3. Nucleic acid-based strategies to modulate SMN2 splicing.

(A)Antisense oligonucleotides that target E1, ISS-N1 and the intron 7/exon 8 juncture have been shown to stimulate exon 7 inclusion. (B) Bifunctional RNAs and similar derivatives targeting E1, exon 7, ISS-N1 stimulate exon 7 inclusion by recruiting SR proteins (green), whereas bifunctional RNAs targeting the intron 7/exon 8 junction recruit hnRNP proteins as a means to favor exon 7 inclusion. (C) Trans-splicing RNAs re-direct SMN2 splicing from the cis-SMN2 splicing to a ‘corrective’ SMN1 exon 7 that is supplied via vector.
An ASO with a different chemical backbone called Morpholinos has been used to target ISSN1 with similar success [145]. Using a slightly longer ASO, which spanned the sequences −10 to −29 (relative to the 3′ end of exon 7), a single high-concentration dose administered via icv. injection at P0 conferred a high degree of protection from SMA and extended survival to an average of 112 days. In contrast to the ISIS SMNRx ASO, no significant additional survival benefit was gained when ICV delivery was augmented with peripheral administration. It has become clear that while the murine blood–brain barrier in early neonatal pups allows compounds and ASOs to pass this will likely not be the case for larger animals and human clinical trials. However, without knowing the therapeutic time window or the pharmacokinetics of each compound, it is not possible to directly compare the two delivery paradigms or compounds. Therefore, it will be particularly interesting to determine whether a CNS-directed strategy versus a CNS/peripheral strategy will prevail in larger animals and in clinical trials. To this end, a swine model of SMA is under development and will be exceptionally useful as a means to examine the dosing and delivery strategies for novel molecular entities such as ASO and viral vectors [146,147].
Additional nucleic acid-based targets and strategies have been developed that redirect SMN2 splicing, including ASOs targeting element 1(E1) or the intron 7/exon 8 junction; bifunctional RNAs; peptide–nucleic acid ESSENCE compounds; and trans-splicing RNAs [62,71,88–90,141,148–154]. The bifunctional RNAs are comprised of two domains: an antisense domain and a separate region that serves as a recruiting and binding substrate for splicing factors, such as hTra2-β1 or SF2/ASF. Bifunctional RNAs and ESSENCE compounds have shown enhanced activation of SMN2 exon 7 splicing mediated by the recruiting platform over and above the activity conferred by the antisense domain, however, there is not a dramatic extension of survival using these molecules that is likely attributed to chemistry or stability differences. Similarly, trans splicing RNAs, which redirect SMN2 splicing from the endogenous pre-mRNA molecule to a vector-derived RNA in trans via a site-specific antisense domain, have shown promise in cell culture and modest activity in a severe mouse model of SMA.
Small molecules targeting SMN2 splicing
The novel molecular entities, including the nucleic acid-based therapeutics, have elicited a considerable amount of excitement based upon the successful studies in SMA animal models, however, more traditional small molecules have been identified that modulate SMN2 exon 7 splicing. One of the first compounds to be examined in SMA models was aclarubicin that demonstrated activity in cell-based assays [155]. Salbutamol has also been shown to increase the relative ratio of full-length: Δ7 SMN transcript in cell-based models [156,157]. As salbutamol is an approved compound, clinical trials were initiated based upon the positive SMN induction, demonstrating a high degree of tolerability yet only modest improvement in motor function [156,158].
In a cell-free screen designed to identify compounds that increased SMN2 exon 7 splicing, a tetracycline derivative, PTK-SMA1, was identified that increased in vitro SMN2 splicing as well as increasing exon 7 inclusion in mild SMA mouse model following intraperitoneal or iv. administration [159]. The beta-lactam antibiotic ceftriaxone has been evaluated in SMA mice and was shown to modestly, but significantly extend survival and decrease the severity of disease [160]. RNA transcripts were not examined in this report although total SMN protein levels were increased slightly. Ceftriaxone also exhibits a general in vitro and in vivo neuroprotective activity as it upregulates glutamate transporter expression, and in the G93A SOD1 amyotrophic lateral sclerosis mouse model it extends survival modestly and delays onset of disease [161].
In large part, the high-throughput screening vectors that have been utilized captured either SMN2 promoter activity or SMN2 exon 7 splicing. Recently, a new reporter was developed that incorporated the SMN2 promoter, the SMN cDNA-spanning exons 1–6 and the genomic cassette comprised exons 6–8. In this system, multiple SMN-inducing mechanisms could be screened simultaneously, such as promoter activation, exon 7 splicing, or RNA stability [162]. A series of novel compounds has been identified that appear to function through different mechanistic pathways including increasing SMN2 exon 7 inclusion. SMN exon 7 inclusion was increased not just in the reporter system but from the endogenous SMN2 gene within SMA patient fibroblasts [163]. Further confirmation of these new compounds is required to demonstrate in vivo activity and in disease relevant tissues.
SMN stabilization
Based upon an image-based cell screen for compounds that elevated SMN levels [164], GSK-3 inhibitors were shown to be a druggable target as inhibition of this pathway increased the intracellular pool of SMN by stabilizing the protein [165]. A potent inhibitor of GSK-3 called BIP-135 was able to extend survival modestly from 12.8 to 14.7 days in a severe SMA mouse model [165]. Recent work has also focused upon the proteasome as a means to stabilize the low intracellular levels of SMN and to some extent, the exon-skipped SMNΔ7 protein [166]. Bortezomib is a relatively specific proteosomal inhibitor as it selectively blocks chymotrypsin cleavage and has been used in the clinic. Bortezomib was shown to increase SMN and while it failed to significantly extend survival in SMA mice, there was a mild synergistic effect when Bortezomib was combined with TSA [167].
Aminoglycosides have been shown to stabilize SMN protein presumably by inducing a translational read-through of the SMNΔ7 protein [168]. The extension of the C-terminus either by translational read-through or by the synthetic addition of random amino acids stretches of varying lengths has demonstrated that extension of the C-terminus confers a greater degree of stability to the SMNΔ7 protein [168–171]. Consistent with these results, several FDA-approved compounds were shown to increase SMN levels in SMA fibroblasts and induced pluripotent stem cell-derived neuronal cultures [170–174]. A novel aminoglycoside, TC007, was delivered via ICV injections and was capable of extending survival from 12.6 to 16.0 days and decreasing disease severity in severe SMA mice [172]. Current FDA-approved compounds have several undesirable side effects, including ototoxicity and nephrotoxicity and it is currently unclear whether this approach will suffice as a stand alone therapy or it could be incorporated into a multidrug regimen.
SMN-independent therapeutic strategies
One of the more promising SMN-independent pathways identified is based upon the delivery of a compound that inhibits ROCK [58]. This compound, Y-27632, alters actin dynamics and without elevating Smn levels, dramatically extends survival in a relatively severe model of SMA called Smn2B/− [58]. One of the hallmarks of SMA involves significant neuromuscular junction pathology (Figure 4) [175–178]. Interestingly, the cellular phenotype including motor neuron and muscle pathology was significantly improved, however, the animals failed to gain weight above their untreated littermates. Similarly, another ROCK inhibitor, fasudil, which is FDA approved, significantly extended survival beyond 300 days and largely rescued the NMJ and muscle phenotypes in Smn2B−/− mice [57]. Not only does this work provide a potential new therapeutic direction for SMA, but it may also shed light upon the biology behind the disease as SMN’s role in actin dynamics may provide clues to the motor neuron sensitivity observed in SMA.
Figure 4. Spinal muscular atrophy pathology manifests in select neuromuscular junctions.
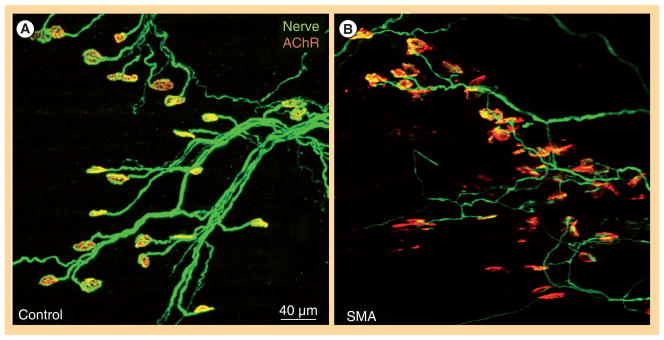
(A) Serratus posterior inferior muscles of control and (B) YFP-SMNΔ7 (right) mice at post-natal day 14 were immunostained for nerve terminals with anti-synaptophysin antibody (in green) and motor end plates with α-bungarotoxin (in red).
Unpublished images were generously provided by KKY Ling and CP Ko (Section of Neurobiology, University of Southern California, Los Angeles, CA, USA).
Other approaches have produced mixed or modest results in SMA mice, such as inhibition of the myostatin pathway or overexpressing IGF-1 [179–183]. One of the likely complications to SMN-independent strategies is that the most commonly used mouse model, the SMNΔ7 mouse, represents a very severe form of SMA. Clearly, any compound that exhibits activity and efficacy in this model would become immediately worthy of further analysis, however, it is equally important to not immediately disregard a compound that appears to function effectively in many parameters such as increased motor function or decreased muscle pathology yet fails to extend survival in the SMNΔ7 mouse. The recent development of less severe models may lead to the identification of compounds and molecules that function through different pathways that could still yield considerable benefit to SMA patients.
Future perspective
For several decades, SMA research has been pushing forward to the point of generating SMA-specific compounds. A significant grassroots movement for SMA research pioneered by Families of SMA, FightSMA, Muscular Dystrophy Association and the SMA Foundation has stimulated governmental funding including the SMA Project and the Network for Excellence in Neuroscience Clinical Trials. The strong research portfolio has also led to broad interest from pharmaceutical companies including: Isis Pharmaceuticals, Genzyme Corporation, Roche, Repligen Corporation, Paratek, Trophos, Biogen Idec and Novartis. Currently, Repligen Coproration, Trophos and Isis Pharmaceuticals have initiated clinical trials for SMA.
It will be important going forward to recall that clinical trials are not merely a confirmation of the preclinical work, but a completely independent series of experiments. Carefully designed clinical trials will be essential to moving compounds towards regulatory approval. The identification of the appropriate patient populations that can most clearly confirm the efficacy of a specific compound and its mode of action will be a complex process. It is possible that an effective compound for type I patients may not be the most effective compound for type III patients. A combinatorial approach may provide a SMN increase that addresses the
Executive summary.
Spinal muscular atrophy: disease & clinical manifestations
SMA is an autosomal recessive neurodegenerative disease that is the leading cause of infantile death worldwide.
SMA is caused by the loss of motor neurons within the anterior horn of the spinal cord.
SMN1 is the disease-determining gene, however, SMN2 is a critical disease modifier and is an excellent target for a variety of therapeutic strategies.
SMN is ubiquitously expressed and it is still unknown why decreased levels of SMN result in the motor neuron-specific defects associated with SMA.
Therapeutic strategies
-
Gene replacement
The SMN cDNA is relatively compact and able to easily fit within the confines of the scAAV genome and allows for robust expression in motor neurons in wildtype and SMA mice. Intravenous and intracerebroventricular delivery demonstrates that scAAV9 lead to a dramatic extension in survival and a general correction of disease symptoms. Currently, the early stages of a clinical trial have initiated and a Phase I study may initiate within 1–2 years.
-
SMN2 promoter activation
Suberoylanilide hydroxamic acid and trichostatin A have shown promise in preclinical models, while valproic acid and phenylbuturate have been examined in clinical trials with modest success. Additional non-histone deacetylase inhibitors include prolacatin which significantly elevates SMN in the CNS and increases SMA mouse survival by ~40%.
-
Modulating exon 7 inclusion with nucleic acid-based therapeutics
Antisense oligonucleotides directed against negatively regulating splice signals within SMN2 pre-mRNA have shown excellent activity in vivo. ISS-N1-targeting antisense oligonucleotides significantly extend survival of SMA mice and can effectively penetrate disease-relevant tissues. A Phase I study is underway for Isis Pharmaceutical’s SMNRx compound.
-
SMN stabilization
The low level of full-length SMN protein can be stabilized by treatment with proteasome inhibitors, including bortezomib, leading to an increased pool of intracellular SMN. Alternatively, compounds that induce a translational read-through event on SMNΔ7 transcripts can increase SMN levels through a distinct post-transcriptional mechanism.
-
SMN-independent strategies
Compounds such as Y-27632 and fasudil that modulate actin dynamics have been shown to significantly extend survival of SMA mice without altering the pathologically low levels of SMN protein. Neuroprotectants and skeletal muscle enhancement also present opportunities in SMA that are continuing to be explored, including the ongoing trial of olesoxmine, a compound produced by Trophos.
-
SMN1 and SMN2
SMA is caused by the homozygous loss of the ubiquitously expressed SMN1 gene.
A nearly identical SMN1-copy gene that produces low levels of full-length SMN and high levels of the truncated SMNΔ7 isoform. The full-length protein is identical to that produced by SMN1, therefore, many therapeutic strategies involve modulating SMN2 expression.
-
scAAV9–SMN
An adeno-associated virus vector with the serotype-9 capsid expressing the SMN cDNA. This vector and several similar variations have been shown to dramatically rescue the SMA phenotype in SMA mice.
-
Exon 7
In the full-length SMN protein, exon 7 is the final coding exon, however, the SMNΔ7 isoform lacks exon 7, and therefore incorporates four amino acids from exon 8. SMNΔ7 is highly unstable and dysfunctional compared to full-length SMN.
-
Antisense oligonucleotide
A short stretch of single-stranded nucleic acid that is designed to bind a target sequence with high affinity. Modification of the backbone chemistry can confer nuclease resistance, thereby dramatically extending the half-life of the antisense oligonucleotide.
-
Splice-site switching
Unlike a siRNA that is designed to knock-down a specific mRNA target, splice-site switching antisense oligonucleotides are designed to alter the pre-mRNA processing of specific transcripts. Within the SMA context, a primary goal is to competitively disable splicing repressor elements that block exon 7 inclusion.
Future perspective
Rarely has the first clinical trial become the gold standard for any disease entity. At this point in time, SMA lacks a validated benchmark for therapeutic efficacy and a workable definition of a clinically meaningful end point. This is not for a lack of effort as countless researchers and families have labored for years to bring the field so far.
We are entering uncharted waters but this is clearly the moment that the SMA field has been waiting and hoping to witness.
Will all patients respond similarly to a specific therapeutic? Probably not. Will delivery issues complicate therapeutic analysis? Perhaps. Will the evolving natural history and supportive care complicate an already broad clinical spectrum? Most certainly, yes.
Key Terms
- Spinal muscular atrophy
Pediatric neurodegenerative disease that is a leading genetic cause of infantile death worldwide
- SMN1
SMA-determining gene. Mutations or deletions of this gene give rise to spinal muscular atrophy
- Alternative splicing
Gene regulation process in which exons are joined together in a manner other than a linear progression. In SMN2, the majority of transcripts lack exon 7
- Adeno-associated virus
Dependo-parvovirus that is frequently a vector of choice for gene therapy based upon its broad tropism, low immunogenicity, and their ability to infect quiescent cells
- Antisense oligonucleotide
Short stretch of nucleic acid that is complementary to a specific sequence. By blocking regulatory elements, antisense oligonucleotides can alter pre-mRNA splicing patterns
Footnotes
Financial & competing interests disclosure
This work was supported by grants from FightSMA and the Gwendolyn Strong Foundation (MAL) and the NIH (MAL; CLL) (R21NS078299; R56NS041584). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;1(8174):919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- 2.Zerres K, Wirth B, Rudnik-Schoneborn S. Spinal muscular atrophy – clinical and genetic correlations. Neuromuscul Disord. 1997;7(3):202–207. doi: 10.1016/s0960-8966(97)00459-8. [DOI] [PubMed] [Google Scholar]
- 3.Chong JX, Oktay AA, Dai Z, Swoboda KJ, Prior TW, Ober C. A common spinal muscular atrophy deletion mutation is present on a single founder haplotype in the US Hutterites. Eur J Hum Genet. 2011;19(10):1045–1051. doi: 10.1038/ejhg.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewelt A, Newcomb TM, Swoboda KJ. New therapeutic approaches to spinal muscular atrophy. Curr Neurol Neurosci Rep. 2012;12(1):42–53. doi: 10.1007/s11910-011-0240-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munsat TL, Davies KE. International SMA consortium meeting (26–28 June 1992, Bonn, Germany) Neuromuscul Disord. 1992;2(5–6):423–428. doi: 10.1016/s0960-8966(06)80015-5. [DOI] [PubMed] [Google Scholar]
- 6.Meldrum C, Scott C, Swoboda KJ. Spinal muscular atrophy genetic counseling access and genetic knowledge: parents’ perspectives. J Child Neurol. 2007;22(8):1019–1026. doi: 10.1177/0883073807305672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zerres K, Rudnik-Schoneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. 1995;52(5):518–523. doi: 10.1001/archneur.1995.00540290108025. [DOI] [PubMed] [Google Scholar]
- 8.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 9.Munsat TL, Skerry L, Korf B, et al. Phenotypic heterogeneity of spinal muscular atrophy mapping to chromosome 5q11.2–13.3 (SMA 5q) Neurology. 1990;40(12):1831–1836. doi: 10.1212/wnl.40.12.1831. [DOI] [PubMed] [Google Scholar]
- 10.Brzustowicz LM, Lehner T, Castilla LH, et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2–13.3. Nature. 1990;344(6266):540–541. doi: 10.1038/344540a0. [DOI] [PubMed] [Google Scholar]
- 11.Melki J, Abdelhak S, Sheth P, et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature. 1990;344(6268):767–768. doi: 10.1038/344767a0. [DOI] [PubMed] [Google Scholar]
- 12.Roy N, Mahadevan MS, Mclean M, et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell. 1995;80(1):167–178. doi: 10.1016/0092-8674(95)90461-1. [DOI] [PubMed] [Google Scholar]
- 13.Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Human Mutation. 2000;15(3):228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 14.Burglen L, Lefebvre S, Clermont O, et al. Structure and organization of the human survival motor neurone (SMN) gene. Genomics. 1996;32(3):479–482. doi: 10.1006/geno.1996.0147. [DOI] [PubMed] [Google Scholar]
- 15.Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 16.Novelli G, Calza L, Amicucci P, et al. Expression study of survival motor neuron gene in human fetal tissues. Biochem Mol Med. 1997;61(1):102–106. doi: 10.1006/bmme.1997.2590. [DOI] [PubMed] [Google Scholar]
- 17.Gennarelli M, Lucarelli M, Capon F, et al. Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem Biophys Res Commun. 1995;213(1):342–348. doi: 10.1006/bbrc.1995.2135. [DOI] [PubMed] [Google Scholar]
- 18.Hahnen E, Schonling J, Rudnik-Schoneborn S, Zerres K, Wirth B. Hybrid survival motor neuron genes in patients with autosomal recessive spinal muscular atrophy: new insights into molecular mechanisms responsible for the disease. Am J Hum Genet. 1996;59(5):1057–1065. [PMC free article] [PubMed] [Google Scholar]
- 19.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96(11):6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48(6):885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Monani UR, Sendtner M, Coovert DD, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9(3):333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 22.Le TT, Pham LT, Butchbach ME, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14(6):845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 23.Campbell L, Potter A, Ignatius J, Dubowitz V, Davies K. Genomic variation and gene conversion in spinal muscular atrophy: implications for disease process and clinical phenotype. Am J Hum Genet. 1997;61(1):40–50. doi: 10.1086/513886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mcandrew PE, Parsons DW, Simard LR, et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997;60(6):1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wirth B, Herz M, Wetter A, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet. 1999;64(5):1340–1356. doi: 10.1086/302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mailman MD, Heinz JW, Papp AC, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genetics in Medicine. 2002;4(1):20–26. doi: 10.1097/00125817-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parano E, Pavone L, Falsaperla R, Trifiletti R, Wang C. Molecular basis of phenotypic molecular basis of this disease and an alternative compound could focus upon creating the appropriate cellular environment that fosters motor neuron protection and survival. SMA is a truly devastating disease and while the natural histories have changed significantly due in large part to herculean supportive care efforts, the future on a new wave of rationally designed SMA therapeutics is about to unfold and it is clear that there is a growing sense of promise heterogeneity in siblings with spinal muscular atrophy. Ann Neurol. 1996;40(2):247–251. doi: 10.1002/ana.410400219. [DOI] [PubMed] [Google Scholar]
- 29.Petit F, Cuisset JM, Rouaix-Emery N, et al. Insights into genotype-phenotype correlations in spinal muscular atrophy: a retrospective study of 103 patients. Muscle Nerve. 2011;43(1):26–30. doi: 10.1002/mus.21832. [DOI] [PubMed] [Google Scholar]
- 30.Jedrzejowska M, Borkowska J, Zimowski J, et al. Unaffected patients with a homozygous absence of the SMN1 gene. Eur J Hum Genet. 2008;16(8):930–934. doi: 10.1038/ejhg.2008.41. [DOI] [PubMed] [Google Scholar]
- 31.Prior TW. Spinal muscular atrophy: a time for screening. Curr Opin Ped. 2010;22(6):696–702. doi: 10.1097/MOP.0b013e32833f3046. [DOI] [PubMed] [Google Scholar]
- 32.Prior TW. Spinal muscular atrophy: newborn and carrier screening. Obstet Gynecol Clin North Am. 2010;37(1):23–36. doi: 10.1016/j.ogc.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 33.Gabanella F, Carissimi C, Usiello A, Pellizzoni L. The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum Mol Genet. 2005;14(23):3629–3642. doi: 10.1093/hmg/ddi390. [DOI] [PubMed] [Google Scholar]
- 34.Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA. 1997;94(18):9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Q, Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996;15(14):3555–3565. [PMC free article] [PubMed] [Google Scholar]
- 36.Coovert DD, Le TT, Mcandrew PE, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6(8):1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 37.Pellizzoni L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Reports. 2007;8(4):340–345. doi: 10.1038/sj.embor.7400941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10(8):597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z, Lotti F, Dittmar K, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133(4):585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baumer D, Lee S, Nicholson G, et al. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genetics. 2009;5(12):e1000773. doi: 10.1371/journal.pgen.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel AA, Steitz JA. Splicing double: insights from the second spliceosome. Nat Rev Mol Cell Biol. 2003;4(12):960–970. doi: 10.1038/nrm1259. [DOI] [PubMed] [Google Scholar]
- 42.Will CL, Schneider C, Macmillan AM, et al. A novel U2 and U11/U12 snRNP protein that associates with the pre-mRNA branch site. EMBO J. 2001;20(16):4536–4546. doi: 10.1093/emboj/20.16.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gabanella F, Butchbach ME, Saieva L, Carissimi C, Burghes AH, Pellizzoni L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE. 2007;2(9):e921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boulisfane N, Choleza M, Rage F, Neel H, Soret J, Bordonne R. Impaired minor tri-snRNP assembly generates differential splicing defects of U12-type introns in lymphoblasts derived from a type I SMA patient. Hum Mol Genet. 2011;20(4):641–648. doi: 10.1093/hmg/ddq508. [DOI] [PubMed] [Google Scholar]
- 45.Wu Q, Krainer AR. Splicing of a divergent subclass of AT-AC introns requires the major spliceosomal snRNAs. RNA. 1997;3(6):586–601. [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Q, Krainer AR. AT-AC pre-mRNA splicing mechanisms and conservation of minor introns in voltage-gated ion channel genes. Mol Cell Biol. 1999;19(5):3225–3236. doi: 10.1128/mcb.19.5.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossoll W, Bassell GJ. Spinal muscular atrophy and a model for survival of motor neuron protein function in axonal ribonucleoprotein complexes. Results Probl Cell Differ. 2009;48:289–326. doi: 10.1007/400_2009_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rossoll W, Jablonka S, Andreassi C, et al. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163(4):801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rossoll W, Kroning AK, Ohndorf UM, Steegborn C, Jablonka S, Sendtner M. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum Mol Genet. 2002;11(1):93–105. doi: 10.1093/hmg/11.1.93. [DOI] [PubMed] [Google Scholar]
- 50.Jablonka S, Wiese S, Sendtner M. Axonal defects in mouse models of motoneuron disease. J Neurobiol. 2004;58(2):272–286. doi: 10.1002/neu.10313. [DOI] [PubMed] [Google Scholar]
- 51.Fallini C, Zhang H, Su Y, et al. The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci. 2011;31(10):3914–3925. doi: 10.1523/JNEUROSCI.3631-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akten B, Kye MJ, Hao Le T, et al. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci USA. 2011;108(25):10337–10342. doi: 10.1073/pnas.1104928108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hubers L, Valderrama-Carvajal H, Laframboise J, Timbers J, Sanchez G, Cote J. HuD interacts with survival motor neuron protein and can rescue spinal muscular atrophy-like neuronal defects. Hum Mol Genet. 2011;20(3):553–579. doi: 10.1093/hmg/ddq500. [DOI] [PubMed] [Google Scholar]
- 54.Glinka M, Herrmann T, Funk N, et al. The heterogeneous nuclear ribonucleoprotein-R is necessary for axonal beta-actin mRNA translocation in spinal motor neurons. Hum Mol Genet. 2010;19(10):1951–1966. doi: 10.1093/hmg/ddq073. [DOI] [PubMed] [Google Scholar]
- 55.Peter CJ, Evans M, Thayanithy V, et al. The COPI vesicle complex binds and moves with survival motor neuron within axons. Hum Mol Genet. 2011;20(9):1701–1711. doi: 10.1093/hmg/ddr046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheever TR, Olson EA, Ervasti JM. Axonal regeneration and neuronal function are preserved in motor neurons lacking ss-actin in vivo. PLoS ONE. 2011;6(3):e17768. doi: 10.1371/journal.pone.0017768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bowerman M, Murray LM, Boyer JG, Anderson CL, Kothary R. Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Medicine. 2012;10:24. doi: 10.1186/1741-7015-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bowerman M, Beauvais A, Anderson CL, Kothary R. Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum Mol Genet. 2010;19(8):1468–1478. doi: 10.1093/hmg/ddq021. [DOI] [PubMed] [Google Scholar]
- 59.Oprea GE, Krober S, Mcwhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320(5875):524–527. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coady TH, Lorson CL. SMN in spinal muscular atrophy and snRNP biogenesis. Wiley Interdiscip Rev RNA. 2011;2(4):546–564. doi: 10.1002/wrna.76. [DOI] [PubMed] [Google Scholar]
- 61.Cheever TR, Li B, Ervasti JM. Restricted morphological and behavioral abnormalities following ablation of beta-actin in the brain. PLoS ONE. 2012;7(3):e32970. doi: 10.1371/journal.pone.0032970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lim SR, Hertel KJ. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3′ splice site pairing. J Biol Chem. 2001;276(48):45476–45483. doi: 10.1074/jbc.M107632200. [DOI] [PubMed] [Google Scholar]
- 63.Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9(2):259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- 64.Singh RN. Evolving concepts on human SMN pre-mRNA splicing. RNA biology. 2007;4(1):7–10. doi: 10.4161/rna.4.1.4535. [DOI] [PubMed] [Google Scholar]
- 65.Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2) Proc Natl Acad Sci USA. 2000;97(17):9618–9623. doi: 10.1073/pnas.160181697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen HH, Chang JG, Lu RM, Peng TY, Tarn WY. The RNA binding protein hnRNP Q modulates the utilization of exon 7 in the survival motor neuron 2 (SMN2) gene. Mol Cell Biol. 2008;28(22):6929–6938. doi: 10.1128/MCB.01332-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Young PJ, Didonato CJ, Hu D, Kothary R, Androphy EJ, Lorson CL. SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2 beta 1. Hum Mol Genet. 2002;11(5):577–587. doi: 10.1093/hmg/11.5.577. [DOI] [PubMed] [Google Scholar]
- 68.Hofmann Y, Wirth B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-beta1. Hum Mol Genet. 2002;11(17):2037–2049. doi: 10.1093/hmg/11.17.2037. [DOI] [PubMed] [Google Scholar]
- 69.Bose JK, Wang IF, Hung L, Tarn WY, Shen CK. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem. 2008;283(43):28852–28859. doi: 10.1074/jbc.M805376200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30(4):377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 71.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10(2):120–125. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 72.Singh NN, Seo J, Ottesen EW, Shishimorova M, Bhattacharya D, Singh RN. TIA1 prevents skipping of a critical exon associated with spinal muscular atrophy. Mol Cell Biol. 2011;31(5):935–954. doi: 10.1128/MCB.00945-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bebee TW, Gladman JT, Chandler DS. Splicing regulation of the survival motor neuron genes and implications for treatment of spinal muscular atrophy. Front Biosci. 2010;15:1191–1204. doi: 10.2741/3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pedrotti S, Bielli P, Paronetto MP, et al. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 2010;29(7):1235–1247. doi: 10.1038/emboj.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34(4):460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 76.Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16(24):3149–3159. doi: 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 77.Kashima T, Rao N, Manley JL. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci USA. 2007;104(9):3426–3431. doi: 10.1073/pnas.0700343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Singh NN, Androphy EJ, Singh RN. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA. 2004;10(8):1291–1305. doi: 10.1261/rna.7580704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet. 2009;18(9):1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miyajima H, Miyaso H, Okumura M, Kurisu J, Imaizumi K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J Biol Chem. 2002;277(26):23271–23277. doi: 10.1074/jbc.M200851200. [DOI] [PubMed] [Google Scholar]
- 81.Gladman JT, Chandler DS. Intron 7 conserved sequence elements regulate the splicing of the SMN genes. Hum Genet. 2009;126(6):833–841. doi: 10.1007/s00439-009-0733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82(4):834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26(4):1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cartegni L, Hastings ML, Calarco JA, De Stanchina E, Krainer AR. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet. 2006;78(1):63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baughan T, Shababi M, Coady TH, Dickson AM, Tullis GE, Lorson CL. Stimulating full-length SMN2 expression by delivering bifunctional RNAs via a viral vector. Mol Ther. 2006;14(1):54–62. doi: 10.1016/j.ymthe.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 86.Singh NN, Androphy EJ, Singh RN. An extended inhibitory context causes skipping of exon 7 of SMN2 in spinal muscular atrophy. Biochem Biophys Res Commun. 2004;315(2):381–388. doi: 10.1016/j.bbrc.2004.01.067. [DOI] [PubMed] [Google Scholar]
- 87.Fernandez Alanis E, Pinotti M, Dal Mas A, et al. An exon-specific U1 small nuclear RNA (snRNA) strategy to correct splicing defects. Hum Mol Genet. 2012;21(11):2389–2398. doi: 10.1093/hmg/dds045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Geib T, Hertel KJ. Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS ONE. 2009;4(12):e8204. doi: 10.1371/journal.pone.0008204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Madocsai C, Lim SR, Geib T, Lam BJ, Hertel KJ. Correction of SMN2 pre-mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005;12(6):1013–1022. doi: 10.1016/j.ymthe.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 90.Dickson A, Osman E, Lorson C. A negatively-acting bifunctional RNA increases survival motor neuron in vitro and in vivo. @Hum Gene Ther. 2008 doi: 10.1089/hum.2008.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85(3):408–413. doi: 10.1016/j.ajhg.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jodelka FM, Ebert AD, Duelli DM, Hastings ML. A feedback loop regulates splicing of the spinal muscular atrophy-modifying gene, SMN2. Hum Mol Genet. 2010;19(24):4906–4917. doi: 10.1093/hmg/ddq425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ruggiu M, Mcgovern VL, Lotti F, et al. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol. 2012;32(1):126–138. doi: 10.1128/MCB.06077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Azzouz M, Le T, Ralph GS, et al. Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J Clin Invest. 2004;114(12):1726–1731. doi: 10.1172/JCI22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dominguez E, Marais T, Chatauret N, et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2011;20(4):681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 96.Valori CF, Ning K, Wyles M, et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. 2010;2(35):35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- 97.Foust KD, Wang X, Mcgovern VL, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28(3):271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98.Passini MA, Bu J, Roskelley EM, et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. 2010;120(4):1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Glascock JJ, Osman EY, Wetz MJ, Krogman MM, Shababi M, Lorson CL. Decreasing disease severity in symptomatic, Smn(−/−); SMN2(+/+), spinal muscular atrophy mice following scAAV9-SMN delivery. Hum Gene Ther. 2012;23(3):330–335. doi: 10.1089/hum.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Glascock JJ, Shababi M, Wetz MJ, Krogman MM, Lorson CL. Direct central nervous system delivery provides enhanced protection following vector mediated gene replacement in a severe model of spinal muscular atrophy. Biochem Biophys Res Commun. 2012;417(1):376–381. doi: 10.1016/j.bbrc.2011.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Somers E, Stencel Z, Wishart TM, Gillingwater TH, Parson SH. Density, calibre and ramification of muscle capillaries are altered in a mouse model of severe spinal muscular atrophy. Neuromuscul Disord. 2012;22(5):435–442. doi: 10.1016/j.nmd.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 102.Bevan AK, Hutchinson KR, Foust KD, et al. Early heart failure in the SMN(Delta)7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19(20):3895–3905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Heier CR, Satta R, Lutz C, Didonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet. 2010;19(20):3906–3918. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shababi M, Habibi J, Ma L, Glascock JJ, Sowers JR, Lorson CL. Partial restoration of cardio-vascular defects in a rescued severe model of spinal muscular atrophy. J Mol Cell Cardiol. 2012;52(5):1074–1082. doi: 10.1016/j.yjmcc.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19(20):4059–4071. doi: 10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- 106.Inagaki K, Fuess S, Storm TA, et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14(1):45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Glascock J, Osman EY, Wetz MJ, Krogman MM, Shababi M, Lorson C. Decreasing disease severity in symptomatic spinal muscular atrophy mice following scAAV9-SMN delivery. Hum Gene Ther. 2011;23(3):330–335. doi: 10.1089/hum.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Duque S, Joussemet B, Riviere C, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17(7):1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Samaranch L, Salegio EA, San Sebastian W, et al. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum Gene Ther. 2012;23(4):382–389. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther. 2012;20(4):699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 112.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 113.Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA. 2001;98(17):9808–9813. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brichta L, Hofmann Y, Hahnen E, et al. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12(19):2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 115.Brichta L, Holker I, Haug K, Klockgether T, Wirth B. In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann Neurol. 2006;59(6):970–975. doi: 10.1002/ana.20836. [DOI] [PubMed] [Google Scholar]
- 116.Piepers S, Cobben JM, Sodaar P, et al. Quantification of SMN protein in leucocytes from spinal muscular atrophy patients: effects of treatment with valproic acid. J Neurol Neurosurg Psych. 2011;82(8):850–852. doi: 10.1136/jnnp.2009.200253. [DOI] [PubMed] [Google Scholar]
- 117.Van Bergeijk J, Haastert K, Grothe C, Claus P. Valproic acid promotes neurite outgrowth in PC12 cells independent from regulation of the survival of motoneuron protein. Chem Biol Drug Design. 2006;67(3):244–247. doi: 10.1111/j.1747-0285.2006.00369.x. [DOI] [PubMed] [Google Scholar]
- 118.Andreassi C, Angelozzi C, Tiziano FD, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12(1):59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- 119.Avila AM, Burnett BG, Taye AA, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117(3):659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Narver HL, Kong L, Burnett BG, et al. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann Neurol. 2008;64(4):465–470. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Garbes L, Riessland M, Holker I, et al. LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum Mol Genet. 2009;18(19):3645–3658. doi: 10.1093/hmg/ddp313. [DOI] [PubMed] [Google Scholar]
- 122.Hahnen E, Eyupoglu IY, Brichta L, et al. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J Neurochem. 2006;98(1):193–202. doi: 10.1111/j.1471-4159.2006.03868.x. [DOI] [PubMed] [Google Scholar]
- 123.Riessland M, Ackermann B, Forster A, et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010;19(8):1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- 124.Bricceno KV, Sampognaro PJ, Van Meerbeke JP, Sumner CJ, Fischbeck KH, Burnett BG. Histone deacetylase inhibition suppresses myogenin-dependent atrogene activation in spinal muscular atrophy mice. Hum Mol Genet. 2012;20(20):4448–4459. doi: 10.1093/hmg/dds286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Le TT, Mcgovern VL, Alwine IE, et al. Temporal requirement for high SMN expression in SMA mice. Hum Mol Genet. 2011;20(18):3578–3591. doi: 10.1093/hmg/ddr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kernochan LE, Russo ML, Woodling NS, et al. The role of histone acetylation in SMN gene expression. Hum Mol Genet. 2005;14(9):1171–1182. doi: 10.1093/hmg/ddi130. [DOI] [PubMed] [Google Scholar]
- 127.Mercuri E, Bertini E, Messina S, et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. 2007;68(1):51–55. doi: 10.1212/01.wnl.0000249142.82285.d6. [DOI] [PubMed] [Google Scholar]
- 128.Swoboda KJ, Scott CB, Reyna SP, et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS ONE. 2009;4(5):e5268. doi: 10.1371/journal.pone.0005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kissel JT, Scott CB, Reyna SP, et al. SMA Carnival Trial Part II: a prospective, single-armed trial of L-carnitine and valproic acid in ambulatory children with spinal muscular atrophy. PLoS ONE. 2011;6(7):e21296. doi: 10.1371/journal.pone.0021296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liang WC, Yuo CY, Chang JG, et al. The effect of hydroxyurea in spinal muscular atrophy cells and patients. J Neurol Sci. 2008;268(1–2):87–94. doi: 10.1016/j.jns.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 131.Chen TH, Chang JG, Yang YH, et al. Randomized, double-blind, placebo-controlled trial of hydroxyurea in spinal muscular atrophy. Neurology. 2010;75(24):2190–2197. doi: 10.1212/WNL.0b013e3182020332. [DOI] [PubMed] [Google Scholar]
- 132.Farooq F, Molina FA, Hadwen J, et al. Prolactin increases SMN expression and survival in a mouse model of severe spinal muscular atrophy via the STAT5 pathway. J Clin Invest. 2011;121(8):3042–3050. doi: 10.1172/JCI46276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ting CH, Lin CW, Wen SL, Hsieh-Li HM, Li H. Stat5 constitutive activation rescues defects in spinal muscular atrophy. Hum Mol Genet. 2007;16(5):499–514. doi: 10.1093/hmg/ddl482. [DOI] [PubMed] [Google Scholar]
- 134.Jarecki J, Chen X, Bernardino A, et al. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum Mol Genet. 2005;14(14):2003–2018. doi: 10.1093/hmg/ddi205. [DOI] [PubMed] [Google Scholar]
- 135.Thurmond J, Butchbach ME, Palomo M, et al. Synthesis and biological evaluation of novel 2,4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. J Med Chem. 2008;51(3):449–469. doi: 10.1021/jm061475p. [DOI] [PubMed] [Google Scholar]
- 136.Singh J, Salcius M, Liu SW, et al. DcpS as a therapeutic target for spinal muscular atrophy. ACS Chem Biol. 2008;3(11):711–722. doi: 10.1021/cb800120t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Butchbach ME, Singh J, Thornorsteinsdottir M, et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet. 2010;19(3):454–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Singh NN, Hollinger K, Bhattacharya D, Singh RN. An antisense microwalk reveals critical role of an intronic position linked to a unique long-distance interaction in pre-mRNA splicing. RNA. 2010;16(6):1167–1181. doi: 10.1261/rna.2154310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Williams JH, Schray RC, Patterson CA, Ayitey SO, Tallent MK, Lutz GJ. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J Neurosci. 2009;29(24):7633–7638. doi: 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Singh NN, Shishimorova M, Cao LC, Gangwani L, Singh RN. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009;6(3):341–350. doi: 10.4161/rna.6.3.8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Osman EY, Yen PF, Lorson CL. Bifunctional RNAs Targeting the Intronic Splicing Silencer N1 Increase SMN Levels and Reduce Disease Severity in an Animal Model of Spinal Muscular Atrophy. Mol Ther. 2012;20(1):119–126. doi: 10.1038/mt.2011.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Develop. 2010;24(15):1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Passini MA, Bu J, Richards AM, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3(72):72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478(7367):123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Porensky PN, Mitrpant C, Mcgovern VL, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21(7):1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lorson MA, Spate LD, Prather RS, Lorson CL. Identification and characterization of the porcine (Sus scrofa) survival motor neuron (SMN1) gene: an animal model for therapeutic studies. Dev Dyn. 2008;237(8):2268–2278. doi: 10.1002/dvdy.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lorson MA, Spate LD, Samuel MS, et al. Disruption of the survival motor neuron (SMN) gene in pigs using ssDNA. Transgenic Res. 2011;20(6):1293–1304. doi: 10.1007/s11248-011-9496-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Marquis J, Meyer K, Angehrn L, Kampfer SS, Rothen-Rutishauser B, Schumperli D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol Ther. 2007;15(8):1479–1486. doi: 10.1038/sj.mt.6300200. [DOI] [PubMed] [Google Scholar]
- 149.Meyer K, Marquis J, Trub J, et al. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum Mol Genet. 2009;18(3):546–555. doi: 10.1093/hmg/ddn382. [DOI] [PubMed] [Google Scholar]
- 150.Coady TH, Baughan TD, Shababi M, Passini MA, Lorson CL. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS ONE. 2008;3(10):e3468. doi: 10.1371/journal.pone.0003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Coady TH, Lorson CL. Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J Neurosci. 2010;30(1):126–130. doi: 10.1523/JNEUROSCI.4489-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Coady TH, Shababi M, Tullis GE, Lorson CL. Restoration of SMN function: delivery of a trans-splicing RNA re-directs SMN2 pre-mRNA splicing. Mol Ther. 2007;15(8):1471–1478. doi: 10.1038/sj.mt.6300222. [DOI] [PubMed] [Google Scholar]
- 153.Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci USA. 2003;100(7):4114–4119. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shababi M, Lorson CL. Optimization of SMN trans-splicing through the analysis of SMN introns. J Mol Neurosci. 2012;46(3):459–469. doi: 10.1007/s12031-011-9614-3. [DOI] [PubMed] [Google Scholar]
- 155.Andreassi C, Jarecki J, Zhou J, et al. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum Mol Genet. 2001;10(24):2841–2849. doi: 10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- 156.Angelozzi C, Borgo F, Tiziano FD, Martella A, Neri G, Brahe C. Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells. J Med Genet. 2008;45(1):29–31. doi: 10.1136/jmg.2007.051177. [DOI] [PubMed] [Google Scholar]
- 157.Tiziano FD, Lomastro R, Pinto AM, et al. Salbutamol increases survival motor neuron (SMN) transcript levels in leucocytes of spinal muscular atrophy (SMA) patients: relevance for clinical trial design. J Med Genet. 2010;47(12):856–858. doi: 10.1136/jmg.2010.080366. [DOI] [PubMed] [Google Scholar]
- 158.Pane M, Staccioli S, Messina S, et al. Daily salbutamol in young patients with SMA type II. Neuromuscul Disord. 2008;18(7):536–540. doi: 10.1016/j.nmd.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 159.Hastings ML, Berniac J, Liu YH, et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci Transl Med. 2009;1(5):5ra12. doi: 10.1126/scitranslmed.3000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nizzardo M, Nardini M, Ronchi D, et al. Beta-lactam antibiotic offers neuroprotection in a spinal muscular atrophy model by multiple mechanisms. Exp Neurol. 2011;229(2):214–225. doi: 10.1016/j.expneurol.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 161.Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 162.Xiao J, Marugan JJ, Zheng W, et al. Discovery, synthesis, and biological evaluation of novel SMN protein modulators. J Med Chem. 2011;54(18):6215–6233. doi: 10.1021/jm200497t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cherry JJ, Evans MC, Ni J, Cuny GD, Glicksman MA, Androphy EJ. Identification of novel compounds that increase SMN protein levels using an improved SMN2 reporter cell assay. J Biomol Screening. 2012;17(4):481–495. doi: 10.1177/1087057111431605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Makhortova NR, Hayhurst M, Cerqueira A, et al. A screen for regulators of survival of motor neuron protein levels. Nat Chem Biol. 2011;7(8):544–552. doi: 10.1038/nchembio.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen PC, Gaisina IN, El-Khodor BF, et al. Identification of a Maleimide-based glycogen synthase kinase-3 (GSK-3) inhibitor, BIP-135, that prolongs the median survival time of Delta7 SMA KO Mouse model of spinal muscular atrophy. ACS Chem Neurosci. 2012;3(1):5–11. doi: 10.1021/cn200085z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Burnett BG, Munoz E, Tandon A, Kwon DY, Sumner CJ, Fischbeck KH. Regulation of SMN protein stability. Mol Cell Biol. 2009;29(5):1107–1115. doi: 10.1128/MCB.01262-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kwon DY, Motley WW, Fischbeck KH, Burnett BG. Increasing expression and decreasing degradation of SMN ameliorate the spinal muscular atrophy phenotype in mice. Hum Mol Genet. 2011;20(18):3667–3677. doi: 10.1093/hmg/ddr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mattis VB, Bowerman M, Kothary R, Lorson CL. A SMNDelta7 read-through product confers functionality to the SMNDelta7 protein. Neurosci Lett. 2008;442(1):54–58. doi: 10.1016/j.neulet.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hua Y, Zhou J. Modulation of SMN nuclear foci and cytoplasmic localization by its C-terminus. Cell Mol Life Sci. 2004;61(19–20):2658–2663. doi: 10.1007/s00018-004-4300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Heier CR, Didonato CJ. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Hum Mol Genet. 2009;18(7):1310–1322. doi: 10.1093/hmg/ddp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wolstencroft EC, Mattis V, Bajer AA, Young PJ, Lorson CL. A non-sequence-specific requirement for SMN protein activity: the role of aminoglycosides in inducing elevated SMN protein levels. Hum Mol Genet. 2005;14(9):1199–1210. doi: 10.1093/hmg/ddi131. [DOI] [PubMed] [Google Scholar]
- 172.Mattis VB, Ebert AD, Fosso MY, Chang CW, Lorson CL. Delivery of a read-through inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Hum Mol Genet. 2009;18(20):3906–3913. doi: 10.1093/hmg/ddp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mattis VB, Fosso MY, Chang CW, Lorson CL. Subcutaneous administration of TC007 reduces disease severity in an animal model of SMA. BMC Neurosci. 2009;10:142. doi: 10.1186/1471-2202-10-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mattis VB, Rai R, Wang J, Chang CW, Coady T, Lorson CL. Novel aminoglycosides increase SMN levels in spinal muscular atrophy fibroblasts. Hum Genet. 2006;120(4):589–601. doi: 10.1007/s00439-006-0245-7. [DOI] [PubMed] [Google Scholar]
- 175.Cifuentes-Diaz C, Nicole S, Velasco ME, et al. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet. 2002;11(12):1439–1447. doi: 10.1093/hmg/11.12.1439. [DOI] [PubMed] [Google Scholar]
- 176.Kariya S, Park GH, Maeno-Hikichi Y, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17(16):2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ling KK, Gibbs RM, Feng Z, Ko CP. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2012;21(1):185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17(7):949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 179.Rose FF, Jr, Mattis VB, Rindt H, Lorson CL. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2009;18(6):997–1005. doi: 10.1093/hmg/ddn426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Shababi M, Glascock J, Lorson CL. Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum Gene Ther. 2010;22(2):121–125. doi: 10.1089/hum.2010.114. [DOI] [PubMed] [Google Scholar]
- 181.Tsai LK, Chen YC, Cheng WC, et al. IGF-1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol Dis. 2012;45(1):272–279. doi: 10.1016/j.nbd.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 182.Bosch-Marce M, Wee CD, Martinez TL, et al. Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum Mol Genet. 2011;20(9):1844–1853. doi: 10.1093/hmg/ddr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sumner CJ, Wee CD, Warsing LC, et al. Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum Mol Genet. 2009;18(17):3145–3152. doi: 10.1093/hmg/ddp253. [DOI] [PMC free article] [PubMed] [Google Scholar]