

Abstract
Background
African trypanosomes are capable of both pyrimidine biosynthesis and salvage of preformed pyrimidines from the host, but it is unknown whether either process is essential to the parasite.
Methodology/Principal Findings
Pyrimidine requirements for growth were investigated using strictly pyrimidine-free media, with or without single added pyrimidine sources. Growth rates of wild-type bloodstream form Trypanosoma brucei brucei were unchanged in pyrimidine-free medium. The essentiality of the de novo pyrimidine biosynthesis pathway was studied by knocking out the PYR6-5 locus that produces a fusion product of orotate phosphoribosyltransferase (OPRT) and Orotidine Monophosphate Decarboxylase (OMPDCase). The pyrimidine auxotroph was dependent on a suitable extracellular pyrimidine source. Pyrimidine starvation was rapidly lethal and non-reversible, causing incomplete DNA content in new cells. The phenotype could be rescued by addition of uracil; supplementation with uridine, 2′deoxyuridine, and cytidine allowed a diminished growth rate and density. PYR6-5−/− trypanosomes were more sensitive to pyrimidine antimetabolites and displayed increased uracil transport rates and uridine phosphorylase activity. Pyrimidine auxotrophs were able to infect mice although the infection developed much more slowly than infection with the parental, prototrophic trypanosome line.
Conclusions/Significance
Pyrimidine salvage was not an essential function for bloodstream T. b. brucei. However, trypanosomes lacking de novo pyrimidine biosynthesis are completely dependent on an extracellular pyrimidine source, strongly preferring uracil, and display reduced infectivity. As T. brucei are able to salvage sufficient pyrimidines from the host environment, the pyrimidine biosynthesis pathway is not a viable drug target, although any interruption of pyrimidine supply was lethal.
Introduction
Human African Trypanosomiasis (HAT, or sleeping sickness) is caused by infection with the protozoan parasites Trypanosoma brucei gambiense and T. b. rhodesiense in West Africa and in East and Southern Africa, respectively. In addition the subspecies T. b. brucei and the other non-human infective species T. vivax and T. congolense cause the veterinary condition African Animal Trypanosomiasis (AAT, or nagana) in livestock in much of sub-Saharan Africa. Both diseases continue to have profound health and economic implications in poor and isolated populations of the region. This problem is exacerbated by the inadequacies of the existing drugs, especially their toxicity, and a parenteral route of administration [1], and by high levels of treatment failure that reach about 30% in some areas [2]. The drug of choice for late stage HAT, eflornithine, is currently administered in the form of nifurtimox and eflornithine combination therapy (NECT) and is not suitable for T. b. rhodesiense infection, which still has to be treated by suramin or melarsoprol, for Stage I (periphery) or Stage II (central nervous system) disease, respectively; both have severe adverse effects on patients [1], [2]. The use of NECT lowers cost and toxicity but may not halt the spread of eflornithine resistance indefinitely [3], [4]. The quest for new drugs led to the study of nucleotide salvage and biosynthesis in protozoa, and initially focused on inhibitors of purine metabolism, as pathogenic protozoan parasites (but not free-living protists) have lost the de novo purine biosynthesis pathways [5], [6]. However, in many protozoa, including kinetoplastid parasites, redundancy of purine transporters [7], [8], [9] and interconversion pathways [10], [11], [12] makes therapy based on purine metabolism inhibitors extremely difficult to achieve.
In contrast, most parasitic protozoa are fully capable of synthesizing the pyrimidine ring de novo [10] and yet are also capable of salvaging pyrimidine nucleosides and/or nucleobases [13], [14], [15], [16]. Exceptions are Plasmodium spp, which are incapable of pyrimidine salvage [17], and the amitochondriate protozoa Trichomonas vaginalis, Tritrichomonas foetus and Giardia spp, which lack the de novo biosynthesis pathway [18], [19]. While possession of both the biosynthesis and salvage routes would appear to make pyrimidine metabolism an unattractive drug target, it has not been established whether either pyrimidine biosynthesis or salvage is essential in African trypanosomes. Moreover, the salvage and biosynthesis pathways actually share most of the pyrimidine metabolizing enzymes, many of which have now been shown to be essential because (in contrast to purine metabolism) there is little or no redundancy in the pathways. For example, dihydrofolate reductase - thymidylate synthase (DHFR-TS) is essential in trypanosomes and its knockout can only be rescued by high levels of thymidine [20], and CTP synthetase is essential as T. b. brucei are unable to incorporate extracellular cytosine or cytidine in their nucleic acids [21]. Furthermore, T. b. brucei deoxyuridine 5′-triphosphate nucleotidohydrolase (dUTPase) was recently shown to be essential [22] and it is clear that several other enzymes of the same pathways may equally be good drug targets.
However, it is as yet unclear whether either the uptake of extracellular pyrimidines or the de novo biosynthesis of the first pyrimidine nucleotide, UMP, is essential in kinetoplastid parasites. We have previously shown that in procyclic T. b. brucei pyrimidines are mainly taken up through the TbU1 uracil transporter [23], [16] and recently completed a study of pyrimidine transport activities in bloodstream form T. b. brucei showing the presence of only one high affinity uracil transporter, TbU3, and almost no uptake of other pyrimidines at physiological levels [24]. A previous study, by Arakaki et al [25] showed that RNAi disruption of one of the biosynthesis enzymes, dihydroorotate dehydrogenase, led to impaired growth which could be compensated for by pyrimidine uptake. The rescue by extracellular uracil, however, was not observed in the presence of the TbU3 inhibitor 5-fluorouracil [25]. In the present study we simulated complete inhibition of pyrimidine salvage by in vitro growth in pyrimidine-free medium and inhibition of de novo biosynthesis through the construction of a genetic deletion mutant lacking the final step of the pyrimidine biosynthesis pathway, which in trypanosomes is a fusion of the two enzymes Orotidine Monophosphate Decarboxylase (PYR6, OMPDCase) and orotate phosphoribosyltransferase (PYR5, OPRT) [26], [27]. The PYR6-5−/− trypanosomes were characterized in vitro and in vivo. While they were completely non-viable in the absence of pyrimidines in vitro, they were able to grow on low levels of pyrimidines, similar as reported for Leishmania donovani promastigotes [28]. The activity of the TbU3 transporter, and expression of Uridine Phosphorylase were both significantly increased when PYR6-5−/− trypanosomes were shifted to pyrimidine-free conditions. However, the observation that these parasites were able to establish an infection in mice showed that pyrimidine biosynthesis is not essential in vivo, with pyrimidine salvage from the blood sufficient for T. brucei viability and growth.
Materials and Methods
Ethics statement
The maintenance and care of experimental animals complied with the appropriate legislation; the UK Animals (Scientific Procedures) Act, 1986, and with the national and University of Glasgow maintenance and care guidelines. All procedures were carried out by trained, registered and licensed animal workers. Care of animals was done by professional staff in the designated University of Glasgow facility under supervision of qualified Veterinarians. Mice infected with trypanosomes were humanely euthanized before becoming seriously ill from the infection. Approval for these experiments was explicitly granted by the UK Home Office, project licence PPL 60/5760 and personal licence PIL60/2328.
Culture of trypanosomes
Bloodstream forms of T. b. brucei strain 427 were routinely cultured in HMI-9 medium [29] obtained from Invitrogen, supplemented with 10% Heat Inactivated Fetal Bovine Serum Gold (FBS; PAA Laboratories) in culture flasks, at 37°C, in a 5% CO2 atmosphere. Where indicated, trypanosomes were grown in a pyrimidine-free medium that was identical to the standard HMI-9/FBS, except that it did not contain thymidine (or any other pyrimidines) and that the serum was first thoroughly dialysed (12–14 kDa cut-off) against phosphate-buffered saline pH 7.4); this medium is referred to as HMI-9-tmd whereas the standard medium is simply referred to as HMI-9. The dialysis details and the exact composition of the HMI-9 and HMI-9-tmd media are given in the Supplementary materials.
Transport of [3H]-uracil and [3H]-uridine
Transport assays were performed exactly as described [30], [31]. Briefly, trypanosomes were washed into the appropriate assay buffer (AB; 33 mM HEPES, 98 mM NaCl, 4.6 mM KCl, 0.55 mM CaCl2, 0.07 mM MgSO4, 5.8 mM NaH2PO4, 0.3 mM MgCl2, 23 mM NaHCO3, 14 mM glucose, pH 7.3) to a final concentration of 108 cells ml−1. 100 µl cell suspension was incubated with either [5,6-3H]-uracil (Perkin Elmer, 40.3 Ci/mmol) or [5,6-3H]-uridine (American Radiolabeled Chemicals Inc, 30 Ci/mmol) at concentrations indicated in the results section, in the presence or absence of unlabeled substrate or other competitive inhibitors. The incubation was stopped after a predetermined interval using 1 ml of an ice-cold 1-mM solution of unlabeled substrate (uracil or uridine) and immediate centrifugation through oil (13,000×g) for 1 min. The resulting cell pellet was transferred to a scintillation tube and radioactivity was determined by liquid scintillation counting. The results were plotted to appropriate equations for linear or non-linear regression using the Prism 5 software package (GraphPad) after correction for non-specific association of radiolabel with the pellet, as described [30].
Generation of auxotrophic T. brucei bloodstream forms
The plasmid pLHTL-PYR6-5 [27] was generously donated by Professor George Cross of Rockefeller University, New York, NY. This construct contains a hygromycin resistance cassette (hygromycin B phosphotransferase) and a negative selection marker, Herpes simplex Thymidine Kinase (HSVTK) open reading frame between loxP domains [32] and is targeted to the PYR6-5 locus by flanking sequences of 496 bp immediately downstream of the target locus and of 365 bp commencing 134 bp upstream of the ORF.
Bloodstream forms of T. brucei s427 were cultivated to a density of ∼1–2×107 cells ml−1 and washed into Human T-Cell Solution for transfection with the LHTL-PYR6-5 cassette (liberated by digestion with PvuII) using an Amaxa Nucleofactor electroporator exactly as described [27], creating a PYR6-5+/− strain. Transformants were grown and cloned out in standard HMI-9 containing hygromycin (2 µg ml−1) and loss of the second PYR6-5 allele was induced by exposure of the clonal lines to 100 µM 5-fluoroorotic acid (5FOA; Sigma), resulting in a PYR6-5−/− strain, exactly as described [27], which was cloned by limiting dilution. PYR6-5 single and double knockout clones were confirmed by PCR. DNA was extracted from WT s427, and from the single and double PYR6-5 knockout strains using a DNeasy Blood and Tissue Kit (Qiagen). Primers were designed to amplify an 870 bp part of the PYR6-5 gene. PCR was performed on 200 ng of the isolated DNA using forward (5′ GTTCTCGAGTGCAAGCGGAT) and reverse (5′CACAATGCGGTCAAACTGCA) primers annealing at 56°C and extension at 72°C for 60 s. A Southern blot was also performed to confirm knockouts, using restricted digest of 10 µg DNA and blotting performed as described [33], using DNA probes specific for the PYR6-5 and hygromycin B phosphotransferase genes. The PYR6-5 probe was generated using the primers and conditions given above for the PCR confirmation, whilst the hygromycin probe was generated by a PCR using forward (5′ATGAAAAAGCCTGAACTCAC) and reverse (5′ACTCTATTCCTTTGCCCTCG) primers annealing at 55°C and extension at 72°C for 60 s.
Growth analysis
Growth of WT and PYR6-5−/− strains was assessed in standard HMI-9 and in HMI-9-tmd supplemented with specific pyrimidines as indicated in the text. Cells were seeded at 1×105 cells ml−1 and for this purpose grown in 12-well plates, with each condition set up in 2 wells; incubation was at 37°C and 5% CO2. Cells were counted every 12 or 24 h. The experiment was performed independently on three separate occasions.
Alamar blue drug sensitivity assays
Sensitivities of PYR6-5−/− and s427-WT cells to 5-fluorouracil, 5-fluoroorotic acid, 5-fluorouridine, 5-fluoro-2′deoxyuridine and 5-fluoro-2′deoxycytidine (all Sigma) were determined using the Alamar Blue assay exactly as described by [34], [35], using a FLUOstar Optima (BMG Labtech, Durham, NC); λexc was 544 nm and λem was 620 nm. Seeding density was 105 per well (final volume 200 µl) in doubling dilutions of test compounds and plates were incubated under standard conditions for 48 h, after which the blue, non-fluorescent indicator dye Alamar Blue (resazurin sodium salt; Sigma) was added and the plates were incubated for a further 24 h. Pentamidine was used as positive control throughout, with drug-free incubations as negative control (4 per plate). All drugs were doubly diluted over 23 wells with a starting concentration of test compound of 5 mM for pyrimidines and of 100 µM for pentamidine.
Flow cytometry
The DNA content of bloodstream trypanosomes was determined exactly as described [36]. Briefly, trypanosome samples were fixed o/n in methanol:PBS (7∶3, v/v), treated with RNase and stained with propidium iodide (both Sigma) and analysed with a FACSCalibur (Benton Dickinson) using the FL2-area detector.
Quantitative PCR of Uridine Phosphorylase
RNA isolated from WT s427 and PYR6-5−/− cells was quantified using a Nanodrop (Thermo Scientific); 2 µg of RNA was diluted in RNase-free water to a total volume of 25 µl. Complementary DNA (cDNA) was produced using a ReverseTranscriptase (RT) kit (Primerdesign, UK). cDNA for each sample was diluted 1∶10 and then used for Real Time-PCR. Amplification of cDNA was performed in a 7500 Real Time PCR System (Applied Biosystems). The dissociation curve was used to ensure the amplification of only one product; samples without RT or cDNA were used as controls. The constitutively expressed gene GPI8 [37] was used as endogenous control, with primer sequences 5′- TCTGAACCCGCGCACTTC and 5′-CCACTCACGGACTGCGTTT. For uridine phosphorylase (UP), the ΔΔCT method was used for relative quantification (RQ) using WT cells in HMI-9 as a calibrator or internal control. Data was analyzed using Applied Biosystems 7500 SDS Real-Time PCR systems software. Primers used for the amplification of UP were 5′-TTTGACCCCTCCACCATGA and 5′-GATTCAGCAGGTGAGCCACAA. The entire experiment was performed on three independent occasions, starting from cell culture and RNA isolation.
Infectivity in mice
Six-weeks-old female ICR (CD-1) Swiss outbred mice (Harlan) were divided into 3 groups of six mice each. Mice were injected intraperitoneally with 105 bloodstream forms of Trypanosoma brucei brucei strains .427 WT, Pyr6-5+/− and Pyr6-5−/− in 200 µl of HMI-9 medium supplemented with 10% FBS. To quantify parasitaemia, 1 µl of blood was daily harvested by tail venepuncture of each infected mice and appropriately diluted in red blood cell lysis buffer (Sigma). 10 µl of the diluted cells was examined under a light microscope using a haemocytometer and parasitaemia was expressed as number of parasites per ml blood.
Results
Generation and confirmation of pyrimidine auxotrophic T. brucei
A schematic representation of the generation of a PYR6-5−/− strain is shown in Figure 1A. Plasmids with the positive selection marker hygromycin phosphotransferase (HYG) and the negative selection marker Herpes simplex virus thymidine kinase (HSVTK) (generous donation from George Cross, Rockefeller University, New York) were used. Bloodstream form T. b. brucei s427 (1×106 cells ml−1) were transformed with the loxP-HYG-HSVTK-loxP cassette using an Amaxa Nucleofector. The transformants were grown in selective medium containing 4.5 µg ml−1 hygromycin (Sigma) and were cloned using limiting dilution, creating a heterozygote PYR6-5+/− strain. Viable clones with the desired insert were subjected to increasing drug pressure with 5-fluoroorotic acid (5-FOA) leading to loss of the second PYR6-5 gene (loss of heterozygosity) at 100 µM (Fig. 1B). Loss of heterozygosity (LOH) and the generated homozygous PYR6-5−/− were further confirmed using Southern blot (Fig. 1C).
Figure 1. Generation of Pyr6-5 Knockouts.

(A) Schematic representation of the generation of Orotidine Monophosphate Decarboxylase (OMPDCase) knockout. The first step is replacement of one PYR6-5 allele with a construct containing both positive selection marker hygromycin phosphotransferase (HYG) and negative marker Herpes simplex virus thymidine kinase (HSVTK) between 34-bp loxP elements. The second step creates the homozygous PYR6-5 −/− through drug pressure with 5-fluoroorotic acid (5 FOA), causing loss of heterozygosity. (B) PCR analysis of the PYR6-5 gene, generating an 870 bp amplicon as described in the Materials and Methods section confirmed the absence of the gene in Pyr6-5 −/− (lane 1) and its continued presence in Pyr6-5 +/− (lane 2). Lane 3 is the control with WT s427 DNA. (C) Southern blot confirming knockout strategy, using probes for the PYR6-5 locus and for the HYG-HSVTK cassette. Lane 1, Pyr6-5 −/−, lane 2, Pyr6-5 +/−, Lane 3, WT s427. Band ‘a’ is PYR6-5, band ‘b’ is HYG-HSVTK.
Growth of pyrimidine auxotrophs on different pyrimidine sources
Under standard culture conditions there was no clear growth phenotype associated with loss of the PYR6-5 locus, as growth of the knockout cells in standard HMI-9 was similar to that of WT s427 trypanosomes in HMI-9-tmd supplemented with 10% dialysed FBS (Fig. S1). PYR6-5−/− cells were grown either in standard HMI-9 or in HMI-9-tmd, which does not contain any pyrimidines but does contain 1 mM hypoxanthine as a purine source (Table S1) and is supplemented with FBS that was extensively dialysed to remove small molecules such as nucleosides. As expected, PYR6-5−/− cells were unable to grow in this semi-defined medium without pyrimidines, and the trypanosome population rapidly declined after 24 h (Fig. 2A,B). In contrast, a shift to purine-free conditions only caused growth arrest after approximately 48 h (Fig. 2B), consistent with previous observations in procyclic T. brucei [38]. Evidently, any interruption in pyrimidine supply rapidly makes trypanosomes non-viable and we investigated how quickly the damage becomes irreversible (Fig. 2A), by adding back 100 µM uracil at various times after passage of PYR6-5−/− to HMI-9-tmd. Cells grew to the same density as in standard HMI-9 when uracil was added immediately after passage (0 h control) but adding the uracil after as little as 12 h resulted in irreversible growth arrest and the eventual death of the parasite population. From 24 h, the addition of uracil was almost redundant, with the cell population rapidly declining as in continuously pyrimidine-free conditions (Fig. 2A).
Figure 2. Growth of bloodstream form T. b. brucei in media with various purine and pyrimidine content.
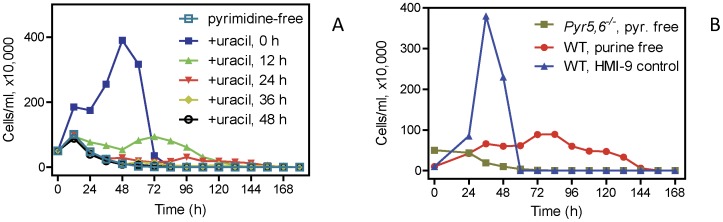
A. Pyr5-6−/− trypanosomes were transferred from HMI-9 to HMI-9-tmd (pyrimidine-free, □) medium to which subsequently uracil was added to a final concentration of 100 µM at the indicated time after seeding the culture. Samples were taken every 12 h and cell densities determined using a haemocytometer. In cultures with conditions that allowed fast growth, the trypanosome population declined after 36–48 h due to overgrowth. Cell population in the ‘0 h’ group declined after 60 h due to over-growth and exhaustion of the medium. B. Comparison of purine-free and pyrimidine conditions. WT s427 cells were passaged from mid-log cultures (grown in standard HMI-9 into fresh cultures with the same medium (control,▴) or the same medium without hypoxanthine and supplemented with dialysed serum (purine free, •). Pyrimidine-auxotrophic T. b. brucei (PYR6-5−/−) were transferred from standard HMI-9 into HMI-9-tmd (pyrimidine-free, ▪) medium. Cell population in the ‘WT, HMI-9 control’ group declined sharply after 26–48 h due to over-growth and exhaustion of the media.
We next tested the ability of these cells to grow on 100 µM or 1 mM (Fig. 3) of each natural pyrimidine nucleoside or nucleobase. At the lower concentration, uracil supported near-normal growth but at 1 mM appeared to have become somewhat growth inhibitory and growth was less pronounced, possibly because the resulting excessive uracil influx could cause an imbalance between pyrimidine nucleotides and 2′deoxyribonucleotides, or between purine and pyrimidine nucleotides. Uridine also supported growth at 100 µM and even better at 1 mM, whereas 2′-deoxyuridine barely had any effect at all at 100 µM. Of the other pyrimidines, only cytidine had any effect on growth, and only at 1 mM.
Figure 3. Growth of pyrimidine auxotrophic T. b. brucei bloodstream forms on various pyrimidine sources.
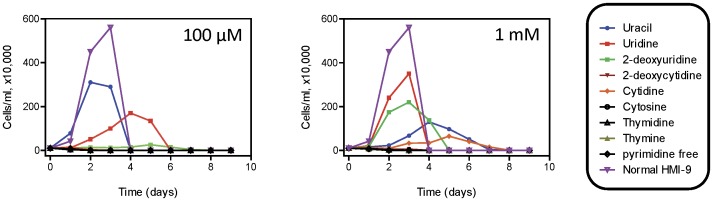
PYR6-5 −/− cell cultures were seeded at a density of 1×105 cells ml−1 and cultured at 37°C/5% CO2, either in normal HMI-9 or in a simplified pyrimidine version supplemented with dialysed FBS and the indicated pyrimidine source at (A) 100 µM or (B) 1 mM. Samples were taken every 24 h and cell densities determined using a haemocytometer, in duplicate. The experiment shown is representative of several similar experiments with essentially identical results. In cultures with conditions that allowed fast growth, the trypanosome population declined after 36–48 h due to overgrowth.
[3H]-Uracil uptake in pyrimidine auxotrophic Trypanosoma brucei
We determined uracil uptake rates in Pyr6-5 −/− and control s427 WT cells to assess whether uracil uptake capacity in bloodstream form T. b. brucei increased in the absence of pyrimidine biosynthesis. WT and Pyr6-5 −/− were grown in standard HMI-9 and uptake of 0.15 µM [3H]-uracil was measured in a timecourse over 120 s. The initial rate of uracil uptake was consistently higher in the Pyr6-5 −/− cells (Fig. 4A). The rates were 0.0109±0.0001 and 0.0241±0.0014 pmol(107 cells)−1s−1 for WT and Pyr6-5 −/− trypanosomes, respectively (n = 3; P<0.05, Student's T-test, unpaired). The increased uptake rate could not be attributed to the expression of an additional uracil transporter in the knockout strain not present in the WT cells, as Km values were identical in both strains (0.31±0.01 and 0.34±0.03 µM, respectively (n = 3)) and uracil transport was almost completely insensitive to uridine in both strains (Ki values >3 mM, n = 3; Fig. 4B). Indeed, uptake of 2.5 µM [3H]-uridine was almost undetectable in both strains (data not shown). However, the Vmax for uracil transport was significantly increased in Pyr6-5 −/− cells (0.14±0.01 versus 0.087±0.007 pmol(107 cells)−1s−1, respectively; n = 3, P = 0.012) (Fig. 4C), consistent with the increased initial rate seen in the time course, and probably reflecting a higher number of the uracil transporter in the plasma membrane rather than the expression of a different or additional transport protein.
Figure 4. Uracil transport by T. b. brucei bloodstream forms.

(A) Timecourse of 0.15 µM [3H]-uracil uptake by WT s427 and by PYR6-5 −/− cells, in the presence or absence of 1 mM unlabelled uracil, as indicated. Dashed lines represent linear regression over the first 50 s, yielding correlation coefficients of 0.97 and 0.92 for the knockout and WT strains, respectively. In the presence of excess unlabelled uracil uptake was not significantly different from zero (F-test) for both strains. The experiment shown is representative of three identical experiments with highly similar outcomes and shows average and SE of triplicate determinations. In the presence of 1 mM uridine, the lines for WT and PYR6-5 −/− were superimposed. (B) Uptake of [3H]-uracil by PYR6-5 −/− trypanosomes was measured over 30 s in the presence or absence of unlabelled uracil (○) or uridine (▪) at the indicated concentrations. The data was plotted to a sigmoid curve with variable slope which in the case of uridine inhibition was set at zero for its minimum. The data are the average and SE of triplicate determinations and the experiment shown is representative of several independent experiments with essentially identical outcomes. (C) Michaelis-Menten saturation plots for uracil uptake of [3H]-uracil by WT (○) or PYR6-5 −/− (▪) T. b. brucei bloodstream forms. The data represents the average and SE of three identical experiments, each performed in triplicate.
Sensitivity of pyrimidine auxotrophic of trypanosomes to pyrimidine analogues
We tested whether pyrimidine auxotrophs were more sensitive to cytotoxic pyrimidine analogues and found that Pyr6-5 −/− cells are approximately one order of magnitude more sensitive to most analogues, including 5-fluorouracil and 5-fluoro-2′deoxyuridine (Table 1). The only exception was 5-fluoroorotic acid, whose action is dependent on OPRT and OMPDCase, and to which the Pyr6-5 −/− cells were completely impervious up to 5 mM although WT trypanosomes were sensitive to this compound with an EC50 of 13.2±1.2 µM. Interestingly, the Pyr6-5 −/− strain was also sensitized to 5-fluorouridine whereas the WT cells were not sensitive to this compound up to the limit tested (5 mM).
Table 1. EC50 values for some pyrimidine analogues tested on WT s427 and PYR6-5 −/− bloodstream forms grown in standard HMI-9, using a standard protocol based on the fluorescent indicator dye Alamar Blue.
| WTs427 | PYR6-5 −/− | RF | P value | |||
| AVG ± SE(µM) | n | AVG ± SE(µM) | n | |||
| 5-Fluorouracil | 35.9±1.5 | 4 | 2.3±0.07 | 4 | 0.06 | <0.001 |
| 5-Fluoro-2′deoxyuridine | 4.6±0.5 | 3 | 0.77±0.10 | 3 | 0.16 | 0.002 |
| 5-Fluoro-2′deoxycytidine | 43.7±4.4 | 3 | 4.6±0.9 | 3 | 0.105 | <0.001 |
| 5-Fluorouridine | >5000 | 5 | 472±3 | 3 | <0.09 | <0.001 |
| 5-Fluoroorotic acid | 13.2±1.2 | 3 | >5000 | 4 | >380 | <0.001 |
RF, resistance factor, being the ratio of the EC50 values (µM) for knockout over WT strains. P value is based on an unpaired Students t-test.
Expression of Uridine Phosphorylase in pyrimidine auxotrophs
We observed that cultures of Pyr6-5 −/− cells appeared to be able to adapt to uridine as a sole pyrimidine source (data not shown). In order to utilize uridine for the synthesis of pyrimidine nucleotides they need to generate uracil from it, using uridine phosphorylase (UP) [39]. We thus inferred that upregulation of UP could be a possible adaptation to pyrimidine starvation and performed quantitative PCR to assess relative UP mRNA levels in WT and Pyr6-5 −/− cells grown in different media. As shown in Figure 5, UP expression was identical in WT cells grown in standard HMI-9 or in HMI-9-tmd supplemented with 100 µM uracil, but was significantly increased after 48 h growth on HMI-9-tmd supplemented with 1 mM uridine (P<0.001). In Pyr6-5 −/− cells cultured long-term in standard HMI-9 but transferred to HMI-9-tmd/uracil for 48 h UP expression levels were higher than for s427-WT under the same conditions (P<0.001) and the level was further increased for Pyr6-5 −/− cells grown 48 h in HMI-9-tmd/uridine (P<0.001). These data appear to indicate that T. b. brucei can adjust its UP expression levels to accommodate growth on uridine as its sole pyrimidine source, whether these cells are pyrimidine auxotrophs or prototrophs. We next investigated whether Pyr6-5 −/− strains can adapt when long-term cultured on uridine as sole pyrimidine source. We found that these cells do express significantly higher UP levels than s427-WT control cells grown in standard HMI-9 or on HMI-9-tmd/uracil (P<0.01), but revert quickly to control levels of expression when shifted to HMI-9-tmd/uracil (Fig. 5).
Figure 5. Comparative expression of uridine phosphorylase in wild-type and pyrimidine auxotrophic trypanosomes.

Expression of uridine phosphorylase (UP) was assessed by Real Time PCR in WT and PYR6-5 −/− strains grown under various conditions. The results are presented normalized to the control (group1) and are the average and SE of 8 replicates. 1. Control: WT grown in HMI-9; 2. WT grown 48 h in HMI-9-tmd+100 µM uracil; 3. WT grown 48 h in HMI-9-tmd+1 mM uridine; 4. PYR6-5−/− grown 48 h in HMI-9-tmd+100 µM uracil; 5. PYR6-5−/− grown 48 h in HMI-9-tmd+1 mM uridine; 6. PYR6-5−/−long-term adapted to growth on uridine, grown 48 h in HMI-9-tmd+100 µM uracil; 7. PYR6-5−/−long-term adapted to growth on uridine, grown 48 h in HMI-9-tmd+1 mM uridine. Data were analysed with a one-way ANOVA with Tukey's correction. Horizontal asterisks indicate significant differences from control; vertical asterisks indicate significant differences between individual bars as indicated.
The effect of pyrimidine starvation on DNA content and integrity of pyrimidine auxotrophic T. b. brucei
Pyrimidine auxotrophs die relatively rapidly in the absence of a salvageable pyrimidine source (uracil>uridine>2′deoxyuridine>cytidine; see figure 3), with death of the population progressing soon after 24 hours. To investigate the cause of the rapid cell death we examined DNA content of the Pyr6-5 −/− cells grown in HMI-9-tmd supplemented with 100 µM of various pyrimidines, using flow cytometry with the DNA-binding fluorophore propidium iodide; Pyr6-5 −/− cells grown in normal HMI-9 served as control. We found that DNA content in control cells presented a classical distribution of most cells in G1 phase (diploid), a small proportion in S-phase undergoing DNA synthesis and finally a percentage of the population in G2 phase (double set of chromosomes) (Fig. 6). This profile was stable over the 48 hours of the experiment, although the proportion in G2 phase increased somewhat over this period, probably reflecting the mid-log phase of growth of the population at the end of the experiment, compared to early log phase at the start (% in G2 was 4.9±2.8%, 11.6±3.1% and 13.9±1.9% at 24 h, 36 and 48 h, respectively; quantified using the ModFit software package). In sharp contrast, there was a rapid increase in cells displaying an incomplete complement of chromosomes in Pyr6-5 −/− cells grown in HMI-9-tmd, resulting both in cells with less fluorescence (i.e. less DNA) than should be associated with normal G1 phase cells, or cells with a DNA content between G1 and G2 phase (Fig. 6). This clearly indicates that the cells are attempting cell division ‘as normal’ but are unable to complete chromosome synthesis due to lack of pyrimidine nucleotides, leading to aberrant cells with incomplete and fragmented chromosomes that are ultimately non-viable. This phenomenon progressed rapidly and at 48 h few live cells could be detected. The cells that could be counted by the flow cytometer almost all contained incomplete and presumably fragmented DNA. Highly similar flow cytometry results were obtained when supplementing HMI-9-tmd with cytosine, thymine or thymidine, whereas supplementation with uracil or uridine produced profiles highly similar to the control (growth in standard HMI-9); addition of 2′deoxyuridine, cytidine or 2′deoxycytidine resulted in intermediate levels of DNA damage over 48 h (results not shown). In an effort to quantify the emergence of aberrant cells the flow cytometry profiles were analyzed with the ModFit software package which models the peak area. This was not successful for the 48-h time points because of the lack of viable cells and too extensive DNA damage, which did not allow reliable estimates of relevant peaks. However, some results for the 24-h and 36-h time points are shown in figure 7. The use of thymidine as sole pyrimidine source caused a highly significant increase in cells in G2 phase, possibly because of the anticipated imbalance between thymidine nucleotides and deoxycytidine nucleosides, which the cell cannot generate from thymidine. The peak classified as ‘DNA debris’ increased within 24 h of pyrimidine-free conditions and this was highly significant (P<0.01) after 36 h; the debris amount was also significantly increased by culturing on thymidine (Fig. 7). We conclude that any significant interruption of pyrimidine nucleotide availability leads to major defects in DNA synthesis.
Figure 6. Flow cytometry for DNA content in bloodstream form PYR6-5 −/− cells.

Pyrimidine auxotrophic trypanosomes were either incubated in standard HMI-9 or, in parallel, in pyrimidine-free HMI-9-tmd for up to 48 h, stained with propidium iodide and prepared for flow cytometric analysis. Whereas control cultures show a classical distribution of cells in G1, S and G2 phase, as well as a small percentage of cells with less than the normal diploid DNA content (debris, d), cells grown in pyrimidine-free medium showed a much higher percentage of cells with partial DNA content, and this increased dramatically between 24 and 48 h.
Figure 7. Quantitative analysis of DNA content in pyrimidine-starved trypanosomes.

The peak area of G1, G2 and debris of flow cytometric analysis of DNA content (Fig. 6) was determined using the ModFit software package after 24 or 36 of growth under various culturing conditions. Growth was in HMI-9 (control) or in HMI-9-tmd with or without the addition of 100 µM of one pyrimidine as indicated. The data are the average of 3–6 independent determinations and statistical analysis was performed using one-way ANOVA with Tukey's correction (Prism 5, GraphPad). *, P<0.05; **, P<0.01.
Infectivity of pyrimidine auxotrophic of T. b. brucei in mice
The observation (Fig. 3) that pyrimidine auxotrophic Pyr6-5 −/− cells grow in standard HMI-9, which contains only thymidine as a pyrimidine source, but cannot grow in thymidine-supplemented medium with dialyzed FBS strongly suggest that (1) T. b. brucei cannot use thymidine as its sole pyrimidine source and (2) it is able to salvage sufficient amounts of other pyrimidines from the non-dialyzed serum. It could thus be speculated that pyrimidine auxotrophic trypanosomes should be able to survive in vivo. To test this, we infected groups of 6 mice with a high inoculum of 105 trypanosomes of s427-WT, Pyr6-5 +/− or Pyr6-5 −/− strains and followed survival and parasitaemia for 15 days. Figure 8A shows that WT trypanosomes were the most virulent and killed all mice between 4 and 8 days. The single allele knockout strain Pyr6-5 +/− caused the death of four mice by day 5 but two of the animals survived until day 12. In contrast, all the animals inoculated with Pyr6-5 −/− survived until day 15, when the experiment was terminated. However, the auxotrophs were able to survive and to multiply in the host, as evidenced by the average parasitaemia, which reached similar levels as for the other strains albeit much more slowly (Fig. 8B). We conclude that T. brucei can salvage just enough uracil and/or uridine in vitro to maintain an infection.
Figure 8. Infectivity of pyrimidine auxotrophic T. b. brucei.

(A) Survival of mice in groups of 6, each inoculated with 105 bloodstream form trypanosomes of various clonal lines. (B) Parasitaemia of the same mice as depicted for survival in panel A. The average parasitaemia of the surviving mice is shown. Detection was by phase-contrast microscopy and detection limit was 1×104; where the infected was sub-patent, a value of 5000 cells ml−1 was inserted in order to arrive at a reasonable average. Both panels: ○, WT s427; ▪, PYR6-5 +/−; ▴, PYR6-5 −/−.
Discussion
Kinetoplastid parasites are able to salvage preformed pyrimidine nucleobases and/or nucleosides [6], [7], [15], [40], [41] as well as synthesise them de novo from glutamine and aspartate [42]. The two pathways converge at UMP, the end-product of the 6-step biosynthesis pathway as well as the nexus for salvaged cytidine, uridine, 2′dUrd, 2′dCtd and uracil, through the actions of cytidine deaminase, uridine phosphorylase and uracil phosphoribosyltransferase (UPRT). From UMP the cell can then make all pyrimidine ribonucleotides and 2′deoxyribonucleotides that it needs, through non-redundant pathways. Salvaged thymidine can be utilised as thymidine nucleotides but not to produce any other pyrimidine nucleotides ([24]; reviewed in 10 and 19) and both procyclic and bloodstream form trypanosomes take up thymidine very poorly [16], [24]. Therefore it is clear that pyrimidine metabolism in protozoa must be replete with good drug targets. Indeed, T. b. brucei DHFR-TS, CTP synthetase and dUTPase have all been shown already to be essential enzymes [20], [21], [22]. These enzymes are all in the pathways downstream from UMP and thus shared by the salvage route and the biosynthesis route.
What is less clear is whether either of the two biochemical pathways to obtain UMP in the first place might be essential and thus a potential drug target. In order to therapeutically target the salvage pathway to UMP it would be necessary to inhibit either the uptake of uracil, uridine, 2′deoxyuridine, cytidine and 2′deoxycytidine, or to inhibit UPRT. With regards to the former option it should be noted that cytidine and 2′deoxycytidine are incorporated very poorly into the T. b. brucei nucleotide pool [21], [24] and it would thus only be necessary to inhibit the carriers for uracil, 2′deoxyuridine and uridine, and we recently reported that all three are mediated by the same transporter, TbU3, in bloodstream forms [24]. However, we report here that WT trypanosomes (i.e. pyrimidine prototrophs) grow almost unimpeded in the absence of any pyrimidine source and must conclude that neither pyrimidine transporters nor UPRT are essential functions in bloodstream form T. b. brucei - consistent with a recent report that deletion of UPRT in L. donovani promastigotes created a pyrimidine prototrophic parasite with normal in vitro growth [43]. It can thus be concluded that pyrimidine salvage is not an essential function for trypanosomes.
Arakaki et al [25] previously investigated whether the de novo biosynthesis route to UMP was essential to T. b. brucei in vitro; they employed RNA-interference (RNAi) to reduce expression of T. brucei dihydroorotate dehydrogenase (DHODH). These authors reported that knockdown of this enzyme did not affect growth in standard HMI-9 but greatly reduced growth in pyrimidine-depleted medium using a commercial dialysed serum. Our own observations with a PYR6-5−/− strain are entirely consistent with Arakaki's report: the growth rate of pyrimidine auxotrophs is at most slightly affected in normal medium with non-dialysed serum. As shown in the Supplementary data, thymidine (∼83 µM) is the only pyrimidine added to that medium but since this nucleoside cannot be converted to uridine and cytidine nucleotides by T. b. brucei, it is redundant [24], [25] and clearly the serum provides sufficient pyrimidines for growth, consistent with the average uracil concentration of 0.17±0.05 µM in human plasma [44] and high affinity uptake of pyrimidines, particularly uracil, by T. brucei [23], [24]. We thus conclude that pyrimidine biosynthesis is not essential for in vitro growth, and the fact that even 10% FBS supplies sufficient pyrimidines, seems to indicate that it may not be essential for in vivo growth either.
This was tested by infecting mice with s427-WT, PYR6-5+/− and PYR6-5−/− trypanosomes. All three strains were able to maintain an infection and although the homozygous knockout strain was clearly less virulent, it unambiguously establishes that inhibition of the de novo pyrimidine biosynthesis is not a viable therapeutic strategy against African trypanosomes. These findings are very similar to those reported for promastigote L. donovani [28] but, in contrast to the authors of that report, we contend that the fact that disruption of pyrimidine biosynthesis can be compensated for by physiological levels of pyrimidines demonstrates that this pathway is not essential in kinetoplastids, and not a viable drug target. Indeed, the same authors very recently demonstrated that L. donovani that lack carbamoyl phosphate synthetase, and are thus pyrimidine auxotrophic, were able to establish a ‘robust’ infection in mice [43]. It is noteworthy, however, that Leishmania species are obligated intracellular parasites and that this manuscript is the first assessment of in vivo growth of an extracellular pyrimidine-auxotrophic protozoan. This is relevant as the intracellular and extracellular nucleoside and nucleobase levels are potentially very different, with the intracellular purines and pyrimidines overwhelmingly existing as nucleotides, which cannot be taken up directly by protozoan transporters [6]. In addition, a previous report on pyrimidine-auxotrophic Toxoplasma gondii, another obligate intracellular protozoan, showed that these parasites were completely avirulent even in immunocompromised mice [45] - a phenotype attributed to the lack of free pyrimidines within animal cells which also prevents growth of pyrimidine auxotrophic bacteria [46].
Inhibition of the pyrimidine biosynthesis pathway in T. b. brucei greatly sensitises the trypanosomes to cytotoxic pyrimidine analogues such as 5-fluorouracil, 5-fluoro-2′deoxyuridine, 5-fluorodeoxycytidine and 5-fluorouridine (Table 1). The enhanced effect of 5-fluorouracil was also noted by Arakaki et al [25], who attributed it to inhibition of uracil uptake. Whilst this analogue is indeed a competitive inhibitor of uracil transport in T. brucei, none of these fluorinated pyrimidines would sufficiently inhibit uracil uptake in bloodstream forms at the EC50 values given in Table 1, especially not 5F-2′deoxyuridine or 5F-2′deoxycytidine [24]. As an alternative explanation, we propose that these fluorinated pyrimidines enter the trypanosomes as prodrugs and subversive substrates for the pyrimidine salvage enzymes, are converted to nucleotides and incorporated into nucleic acids, as indeed is the case in mammalian cells [47] and as we have recently shown for 5-fluorouracil in T. b. brucei [24]. This incorporation is more efficient in the absence of a newly synthesised pool of pyrimidine metabolites that would compete with the halogenated analogues at the level of each enzyme as well as for RNA and/or DNA polymerases.
We thus conclude that an inhibitor of any one of the enzymes of the de novo pathway together with either an inhibitor of uracil/uridine uptake, or with a cytotoxic nucleoside analogue, would be a powerful and synergistic combination that would act on trypanosomes through both misincorporation of false nucleotides, and by causing pyrimidine starvation through inhibition of pyrimidine carriers, and we found that trypanosome populations die much more quickly from a lack of pyrimidines than from a lack of purines [38]. It can easily be speculated that kinetoplastid parasites, having evolved without the capacity to synthesise their own purines, must be relatively well-adapted to periods with relatively low purine availability, as demonstrated by the reversibility of purine starvation-induced growth arrest [38]. In contrast, they have not needed to develop a mechanism to cope with a prolonged dearth of pyrimidines, being able to make sufficient amounts themselves, and trypanosomes are therefore unable to recover from even short periods of pyrimidine starvation. We did observe, consistently, increased expression of uridine phosphorylase, and an increase in uracil uptake capacity (both about two-fold), in pyrimidine-starved trypanosomes but this hardly constitutes a major upregulation of the pyrimidine salvage pathway and it is at best uncertain whether this is a regulated, physiological response to low pyrimidine levels. Indeed, the lack of a regulated response to the insufficient level of pyrimidine nucleotides was manifest in the major defects in DNA synthesis after only 24 h, leading to fragmented and incomplete chromosomes.
In summary, we have shown that neither pyrimidine uptake or de novo biosynthesis is essential in African trypanosomes but that a drug combination targeting both systems would be a very powerful approach to novel therapeutic approaches against kinetoplastid parasites.
Supporting Information
Growth of PYR6-5 −/− T. b. brucei bloodstream forms in standard HMI-9 and s427-WT in HMI-9-tmd supplemented with 10% dialysed FBS. Seeding density was 1×105 cells ml−1 and cells were manually counted every 24 h. On day 3 cells were passaged to relevant fresh medium, again at 1×105 cells ml−1.
(TIF)
Composition of standard HMI-9 medium.
(DOCX)
(DOCX)
Acknowledgments
The authors would like to thank Prof. George A. M. Cross (Rockefeller University, New York, NY, USA) for his generous donation of the PYR6-5 replacement cassette. The authors thank Mr Abdulsalam Alkhaldi (University of Glasgow) for expert technical assistance.
Funding Statement
This work was supported by the Wellcome Trust. The Wellcome Trust Centre for Molecular Parasitology is supported by core funding from the Wellcome Trust [085349], and DNAT is the recipient of a Wellcome Trust scholarship. The authors also gratefully acknowledge financial support from the Libyan Government (studentship to JAMA) and the Medical Research Council (contract number 84733). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Brun R, Blum J, Chappuis F, Burri C (2010) Human African trypanosomiasis. Lancet 375: 148–159. [DOI] [PubMed] [Google Scholar]
- 2. Brun R, Schumacher R, Schmid C, Kunz C, Burri C (2001) The phenomenon of treatment failure in Human African Trypanosomiasis. Trop Med Int Health 6: 906–914. [DOI] [PubMed] [Google Scholar]
- 3. Balasegaram M, Young H, Chappuis F, Priotto G, Raguenaud ME, et al. (2009) Effectiveness of melarsoprol and eflornithine as first-line regimens for gambiense sleeping sickness in nine Médecins Sans Frontières programmes. Trans R Soc Trop Med Hyg 103: 280–290. [DOI] [PubMed] [Google Scholar]
- 4. Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, et al. (2009) Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet 374: 56–64. [DOI] [PubMed] [Google Scholar]
- 5.Martin DW (1981) Metabolism of purine and pyrimidine nucleotides. In: Martin DW, Mayers PA, Rodwell VW, editors. Harper's Review of Biochemistry: Lange Medical Publications, California. pp. 331–348.
- 6. De Koning HP, Bridges DJ, Burchmore RJ (2005) Purine and pyrimidine transport in pathogenic protozoa: from biology to therapy. FEMS Microbiol Rev 29: 987–1020. [DOI] [PubMed] [Google Scholar]
- 7. Landfear SM, Ullman B, Carter NS, Sanchez MA (2004) Nucleoside and nucleobase transporters in parasitic protozoa. Eukaryot Cell 3: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Koning HP, Jarvis SM (1997) Purine nucleobase transport in bloodstream forms of Trypanosoma brucei brucei is mediated by two novel transporters. Mol Biochem Parasitol 89: 245–258. [DOI] [PubMed] [Google Scholar]
- 9. Al-Salabi MI, Wallace LJ, Lüscher A, Mäser P, Candlish D, et al. (2007) Molecular interactions underlying the unusually high adenosine affinity of a novel Trypanosoma brucei nucleoside transporter. Mol Pharmacol 71: 921–929. [DOI] [PubMed] [Google Scholar]
- 10.Berens RL, Krug EC, Marr JJ (1995) Purine and pyrimidine metabolism. In: Marr JJ, Muller M, editors. Biochemistry and Molecular biology of parasites. London: Academic Press. pp. 89–117.
- 11. Berg M, Kohl L, Van der Veken P, Joossens J, Al-Salabi MI, et al. (2010) Evaluation of nucleoside hydrolase inhibitors in the treatment of African trypanosomiasis. Antimicrob Agents Chemother 54: 1900–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berg M, Van der Veken P, Goeminne A, Haemers A, Augustyns K (2010) Inhibitors of the purine salvage pathway: a valuable approach for antiprotozoal chemotherapy? Curr Med Chem 17: 2456–2481. [DOI] [PubMed] [Google Scholar]
- 13. Aronow B, Kaur K, McCartan K, Ullman B (1987) Two high affinity nucleoside transporters in Leishmania donovani . Mol Biochem Parasitol 22: 29–37. [DOI] [PubMed] [Google Scholar]
- 14. De Koning HP, Al-Salabi MI, Cohen AM, Coombs GH, Wastling JM (2003) Identification and characterisation of high affinity nucleoside and nucleobase transporters in Toxoplasma gondii . Int J Parasitol 33: 821–831. [DOI] [PubMed] [Google Scholar]
- 15. Papageorgiou IG, Yakob L, Al Salabi MI, Diallinas G, Soteriadou K, et al. (2005) Identification of the first pyrimidine nucleobase transporter in Leishmania: similarities with the Trypanosoma brucei U1 transporter and antileishmanial activity of uracil analogues. Parasitology 130: 275–283. [DOI] [PubMed] [Google Scholar]
- 16. Gudin S, Quashie NB, Candlish D, Al-Salabi MI, Jarvis SM, et al. (2006) Trypanosoma brucei: a survey of pyrimidine transport activities. Exp Parasitol 114: 118–125. [DOI] [PubMed] [Google Scholar]
- 17. Van Dyke K, Tremblay GC, Lantz CH, Szustkiewicz C (1970) The source of purines and pyrimidines in Plasmodium berghei. Am J Trop Med Hyg 19: 202–208. [DOI] [PubMed] [Google Scholar]
- 18. Wang CC, Cheng HW (1984) Salvage of pyrimidine nucleosides by Trichomonas vaginalis . Mol Biochem Parasitol 10: 171–184. [DOI] [PubMed] [Google Scholar]
- 19. Hassan HF, Coombs GH (1988) Purine and pyrimidine metabolism in parasitic protozoa. FEMS Microbiol Rev 4: 47–83. [DOI] [PubMed] [Google Scholar]
- 20. Sienkiewicz N, Jarosławski S, Wyllie S, Fairlamb AH (2008) Chemical and genetic validation of dihydrofolate reductase–thymidylate synthase as a drug target in African trypanosomes. Mol Microbiol 69: 520–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hofer A, Steverding D, Chabes A, Brun R, Thelander L (2001) Trypanosoma brucei CTP synthetase: a target for the treatment of African sleeping sickness. Proc Natl Acad Sci USA 98: 6412–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Castillo-Acosta VM, Estévez AM, Vidal AE, Ruiz-Perez LM, González-Pacanowska D (2008) Depletion of dimeric all-α dUTPase induces DNA strand breaks and impairs cell cycle progression in Trypanosoma brucei . Int J Biochem Cell Biol 40: 2901–2913. [DOI] [PubMed] [Google Scholar]
- 23. De Koning HP, Jarvis SM (1998) A highly selective, high-affinity transporter for uracil in Trypanosoma brucei brucei: evidence for proton-dependent transport. Biochem Cell Biol 76: 853–858. [DOI] [PubMed] [Google Scholar]
- 24. Ali JA, Creek DJ, Burgess K, Allison HC, Field MC, et al. (2013) Pyrimidine salvage in Trypanosoma brucei bloodstream forms and the trypanocidal action of halogenated pyrimidines. Mol Pharmacol 2012 Nov 27. [Epub ahead of print] PMID: 23188714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arakaki TL, Buckner FS, Gillespie JR, Malmquist NA, Phillips MA, et al. (2008) Characterization of Trypanosoma brucei dihydroorotate dehydrogenase as a possible drug target; structural, kinetic and RNAi studies. Mol Microbiol 68: 37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao G, Nara T, Nakajima-Shimada J, Aoki T (1999) Novel organisation and sequences of five genes encoding all six enzymes for the de novo pyrimidine biosynthesis in Trypanosoma brucei . J Mol Biol 285: 149–161. [DOI] [PubMed] [Google Scholar]
- 27. Scahill MD, Pastar I, Cross GA (2008) CRE recombinase-based positive-negative selection systems for genetic manipulation in Trypanosoma brucei . Mol Biochem Parasitol 157: 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. French JB, Yates PA, Soysa DR, Boitz JM, Carter NS, et al. (2011) The Leishmania donovani UMP synthase is essential for promastigote viability and has an unusual tetrameric structure that exhibits substrate-controlled oligomerization. J Biol Chem 286: 20930–20941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirumi H, Hirumi K (1989) Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol 75: 985–989. [PubMed] [Google Scholar]
- 30. Wallace LJ, Candlish D, De Koning HP (2002) Different substrate recognition motifs of human and trypanosome nucleobase transporters. Selective uptake of purine antimetabolites. J Biol Chem 277: 26149–26156. [DOI] [PubMed] [Google Scholar]
- 31. Natto MJ, Wallace LJ, Candlish D, Al-Salabi MI, Coutts SE, et al. (2005) Trypanosoma brucei: expression of multiple purine transporters prevents the development of allopurinol resistance. Exp Parasitol 109: 80–86. [DOI] [PubMed] [Google Scholar]
- 32. Sternberg N, Hamilton D, Hoess R (1981) Bacteriophage P1 site-specific recombination. II. Recombination between loxP and the bacterial chromosome. J Mol Biol 150: 487–507. [DOI] [PubMed] [Google Scholar]
- 33. Martin KL, Smith TK (2006) Phosphatidylinositol synthesis is essential in bloodstream form Trypanosoma brucei . Biochem J 396: 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodenko B, Van der Burg AM, Wanner MJ, Kaiser M, Brun R, et al. (2007) 2,N6-Disubstituted adenosine analogues with antitrypanosomal and antimalarial activity. Synthesis, uptake studies and in vivo evaluation. Antimicrob Agents Chemother 51: 3796–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gould MK, Vu XL, Seebeck T, De Koning HP (2008) Propidium iodide-based methods for monitoring drug action in the kinetoplastidae: comparison with the Alamar Blue assay. Anal Biochem 382: 87–93. [DOI] [PubMed] [Google Scholar]
- 36. Ibrahim HMS, Al-Salabi MI, El Sabbagh N, Quashie NB, Alkhaldi AAM, et al. (2011) Symmetrical choline-derived dications display strong anti-kinetoplastid activity. J Antimicrob Chemother 66: 111–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lillico S, Field MC, Blundell P, Coombs GH, Mottram JC (2003) Essential roles for GPI-anchored proteins in African trypanosomes revealed using mutants deficient in GPI8. Mol Biol Cell 14: 1182–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Koning HP, Watson CJ, Sutcliffe L, Jarvis SM (2000) Differential regulation of nucleoside and nucleobase transport in Crithidia fasciculata and Trypanosoma brucei in response to purine stress. Mol Biochem Parasitol 106: 93–107. [DOI] [PubMed] [Google Scholar]
- 39. Larson ET, Mudeppa DG, Gillespie JR, Mueller N, Napuli AJ, et al. (2010) The crystal structure and activity of a putative trypanosomal nucleoside phosphorylase reveal it to be a homodimeric uridine phosphorylase. J Mol Biol 396: 1244–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bellofatto V (2007) Pyrimidine transport activities in trypanosomes. Trends Parasitol 23: 187–189. [DOI] [PubMed] [Google Scholar]
- 41. De Koning HP (2007) Pyrimidine transporters of protozoa – A class apart? Trends Parasitol 23: 190. [Google Scholar]
- 42. Hammond DJ, Gutteridge WE (1982) UMP synthesis in the kinetoplastida. Biochim Biophys Acta 718: 1–10. [DOI] [PubMed] [Google Scholar]
- 43. Wilson ZN, Gilroy CA, Boitz JM, Ullman B, Yates PA (2012) Genetic dissection of pyrimidine biosynthesis and salvage in Leishmania donovani . J Biol Chem 287: 12759–12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bi D, Anderson LW, Shapiro J, Shapiro A, Grem JL, et al. (2000) Measurement of plasma uracil using gas chromatography - mass spectrometry in normal individuals and in patients receiving inhibitors of dihydropyrimidine dehydrogenase. J Chromatogr B Biomed Sci Appl 738: 249–258. [DOI] [PubMed] [Google Scholar]
- 45. Fox BA, Bzik DJ (2002) De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii . Nature 415: 926–929. [DOI] [PubMed] [Google Scholar]
- 46. Fields PI, Swanson RV, Haidaris CG, Heffron F (1986) Mutants of Salmonella tyhimurium that cannot survive within the macrophage are avirulent. Proc Natl Acad Sci USA 83: 5189–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Longley DB, Harkin DP, Johnston PG (2003) 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3: 330–338. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Growth of PYR6-5 −/− T. b. brucei bloodstream forms in standard HMI-9 and s427-WT in HMI-9-tmd supplemented with 10% dialysed FBS. Seeding density was 1×105 cells ml−1 and cells were manually counted every 24 h. On day 3 cells were passaged to relevant fresh medium, again at 1×105 cells ml−1.
(TIF)
Composition of standard HMI-9 medium.
(DOCX)
(DOCX)