

Abstract
Chromosome region maintenance 1/exportin1/Exp1/Xpo1 (CRM1) is the major transport receptor for the export of proteins from the nucleus. It binds to nuclear export signals (NESs) that are rich in leucines and other hydrophobic amino acids. The prediction of NESs is difficult because of the extreme recognition flexibility of CRM1. Furthermore, proteins can be exported upon binding to an NES-containing adaptor protein. Here we present an approach for identifying targets of the CRM1-export pathway via quantitative mass spectrometry using stable isotope labeling with amino acids in cell culture. With this approach, we identified >100 proteins from HeLa cells that were depleted from cytosolic fractions and/or enriched in nuclear fractions in the presence of the selective CRM1-inhibitor leptomycin B. Novel and validated substrates are the polyubiquitin-binding protein sequestosome 1, the cancerous inhibitor of protein phosphatase 2A (PP2A), the guanine nucleotide-binding protein-like 3-like protein, the programmed cell death protein 2-like protein, and the cytosolic carboxypeptidase 1 (CCP1). We identified a functional NES in CCP1 that mediates direct binding to the export receptor CRM1. The method will be applicable to other nucleocytoplasmic transport pathways, as well as to the analysis of nucleocytoplasmic shuttling proteins under different growth conditions.
The transport of macromolecules across the nuclear envelope occurs through large proteinaceous structures called nuclear pore complexes (NPCs).1 NPCs are composed of ∼30 nucleoporins that occur in copy numbers of eight or multiples of eight, leading to a complex with a total size of ∼125 MDa in vertebrate cells (1, 2). Active nucleocytoplasmic transport of proteins is a signal- and energy-dependent process that is mostly mediated by transport receptors of the importin β-superfamily called karyopherins or importins/exportins (3, 4). These proteins interact not only with their cargos, but also with certain nucleoporins, facilitating the translocation of the transport complex across the NPC. For nuclear export, at least seven nuclear export receptors/exportins have been identified (3, 4). Chromosome region maintenance 1/exportin1/Exp1/Xpo1 (CRM1) is the most important export receptor for proteins in yeast and vertebrates, and it is also involved in the export of several RNA species (5). Very little is known about the interaction of CRM1 with nucleoporins. Binding to cargo molecules, in contrast, is very well described. Exported proteins typically carry a nuclear export signal (NES) that is enriched with leucines or other hydrophobic amino acids. Such leucine-rich NESs were first discovered in the HIV type 1 Rev protein (6) and the cAMP-dependent protein kinase inhibitor (7). The consensus sequence consists of four key hydrophobic amino acids (leucine, isoleucine, valine, and phenylalanine or methionine; denoted by Φ1–Φ4) following the sequence Φ1-(x)2–3-Φ2-(x)2–3-Φ3-(x)-Φ4, with x preferentially being a charged polar or small amino acid (for a review, see Ref. 8). A structural analysis of different NES peptides revealed a fifth hydrophobic amino acid in some substrates involved in CRM1 recognition, leading to a revised consensus sequence of Φ0-(x)-Φ1-(x)2–3-Φ2-(x)2–3-Φ3-(x)-Φ4 (9). In a very recent study, Chook and coworkers established a novel database, NESdb, for NES-containing proteins and analyzed the sequence requirements for proteins in that database in detail (10, 11). “Supraphysiological ” substrates with NESs that fulfill all criteria bind CRM1 with very high affinity and can outcompete other substrates (9, 12). Apart from linear sequences, CRM1 might recognize more complex export signals, such as in fatty acid binding protein 4, in which a functional NES is established only in the tertiary structure of the protein (13), or in snurportin 1 (SPN1), in which sequences outside of the NES proper contribute to CRM1 binding (14–16). This high level of complexity in the recognition sequence for the export receptor makes it very difficult to predict potential CRM1-dependent export cargos using bioinformatics tools. Nevertheless, >200 potential CRM substrates have been described so far (11, 17–19; see also NESbase 1.0 and NESdb).
The small GTP-binding protein Ran also plays an essential role in CRM1-mediated nuclear export, as it binds cooperatively to the export receptor, together with the NES cargo. As the affinity of many NES substrates for CRM1 is rather low, the formation of this trimeric transport complex seems to be a rate-limiting step in nuclear export (20). On the nuclear side of the NPC, a number of accessory factors such as RanBP3 (21, 22), Nup98 (23), and NLP1 (24) can further promote the formation of export complexes. Following export, RanBP1 and RanGAP initiate the disassembly of the export complex (for a review, see Ref. 3).
A powerful tool for the analysis of CRM1-mediated export is the fungal metabolite leptomycin B (LMB). LMB originally was discovered as an antifungal antibiotic in Streptomyces (25) and later turned out to be a specific and selective inhibitor of the CRM1-mediated nuclear export pathway (26, 27). It binds covalently to cysteine 528 in the NES-binding region of human CRM1 (28), preventing the formation of trimeric export complexes (for a review, see Ref. 5).
We used a quantitative MS-based approach (stable isotope labeling with amino acids in cell culture (SILAC)) to evaluate the nuclear export characteristics of proteins by measuring changes in their relative abundance in subcellular fractions after blocking the CRM1-mediated nuclear export with LMB. Using this approach, we identified known and novel CRM1-targets and characterized the NES of one cargo, cytosolic carboxypeptidase 1 (CCP1), in detail.
EXPERIMENTAL PROCEDURES
SILAC Media
Dulbecco's modified Eagle medium (DMEM), a high-glucose medium deficient in the amino acids arginine and lysine, was supplemented with 10% dialyzed fetal calf serum, penicillin-streptomycin (100 IU/ml and 100 μg/ml, respectively), and either unlabeled l-arginine · HCl and l-lysine · HCl (SILAC: R0K0, “light”) or l-arginine-U-13C6 · HCl (Cambridge Isotope Laboratories, Andover, MA) and l-lysine-U-13C6-15N2 · 2HCl (Cambridge Isotope Laboratories) (SILAC: R6K8, “heavy”) at concentrations of 50 μg/ml.
Cell Culture, Transfections, and LMB Treatment
HeLa P4 cells (29) were grown at 37 °C in a humidified incubator with a 5% CO2 atmosphere. Cells were adapted to the appropriate SILAC medium for at least five passages in order to achieve complete incorporation of the isotopically labeled amino acids. Cells were transfected with Rev(48–116)-GFP2-M9 plasmid, as a positive control for LMB treatment, using the calcium phosphate method (30). After 24 h, cells were treated with 10 nm LMB (Alexis Biochemicals, Lausen, Switzerland) for 3 h at 37 °C to block CRM1-dependent nuclear export and then subjected to subcellular fractionation. For the validation of novel CRM1 substrates, HeLa cells were transfected with appropriate plasmids using the calcium phosphate method (30), treated with LMB as described above, and then subjected to indirect immunofluorescence.
Subcellular Fractionation
Untreated light cells were combined with LMB-treated heavy cells (“forward experiment”), or LMB-treated light cells were combined with untreated heavy cells (“reverse experiment”), at a ratio of 1:1, and the cells were subjected to subcellular fractionation as previously described (31). Briefly, 8 × 106 HeLa cells were trypsinized, washed in cold medium, collected via centrifugation, resuspended in PBS, and centrifuged again at 100g at 4 °C. 10% of the cells were boiled directly in SDS sample buffer and centrifuged at 14,000g, and the supernatant was collected as total lysate (T). The remaining cells were resuspended in 400 μl of ice-cold buffer 1 (150 mm NaCl, 50 mm HEPES pH 7.4, 1% digitonin (1 μl/106 cells), and protease inhibitors), incubated at 4 °C for 10 min, and centrifuged at 2000g. The supernatant was collected as the cytosol-enriched fraction (C). The pellet was resuspended in 400 μl of ice-cold buffer 2 (150 mm NaCl, 50 mm HEPES pH 7.4, 1% Nonidet P-40, and protease inhibitors), incubated on ice for 30 min, and centrifuged at 7000g. The supernatant comprising extract of membrane-bound organelles such as the endoplasmic reticulum, Golgi, mitochondria, and some nuclear lumenal proteins (M) was removed. The pellet was resuspended in 400 μl of ice-cold buffer 3 (150 mm NaCl, 50 mm HEPES pH 7.4, 0.5% sodium deoxycholate, 0.1% SDS, benzonase (1 U/ml), and protease inhibitors), incubated at 4 °C for 1 h, and centrifuged at 7000g for 10 min. The supernatant comprising the extracted nuclear membranes and soluble nuclear proteins (N) was collected.
Plasmids
Plasmid codings for GFP-Sequestosome 1 (Dr. Terje Johansen, Tromsø, Norway), HA-DDX3 (Dr. Kuan-Teh Jeang, Bethesda, MD), YFP-CCP1 (Dr. Carsten Janke, Paris, France), FLAG-CIP2A (Dr. Jukka Westermarck, Turku, Finland), HA-GNL3L (Dr. Robert Tsai, Houston, TX), and GST-SPN1 (Dr. Achim Dickmanns, Göttingen, Germany) were received as kind gifts. The M9 core sequence of human hnRNPA1 was amplified using primers (5′ AATTCTTCAAACTTTGGGCCCATGAAAGGAGGAAACTTTGGAGGAAGGTCATCAGGACCATATTGAG and 5′ TCGACTCAATATGGTCCTGAGACCTTCCTCCAAAGTTTCCTCCTTTCATGGGCCCAAAGTTTGAAG) and cloned into the Rev(48–116)-GFP2-cNLS plasmid (32) to exchange the cNLS and to generate Rev(48–116)-GFP2-M9core (details upon request). The coding sequence for RanBP1 was amplified using primers (5′ TTTTTCCATGGCGGCCGCCAAGG and 5′ TTTTTGAATTCTCATTGCTTCTCCTCAGCATCCT) and inserted into the NcoI/EcoRI sites of the pEF-HA-plink vector (33) to generate HA-RanBP1. The coding sequence for Nmd3 was amplified from HeLa cell cDNA using appropriate primers and cloned via XhoI/EcoRI of digested pEGFP-C1 vector (Clontech) to obtain GFP-Nmd3. The coding sequence for PDCD2L was amplified from HeLa cell cDNA using a set of nested primers (sense: 5′ GCGTTTTCACCTGGTCGCCCG and 5′ TTTTAAGCTTGGATGGCGGCCGTTCTGAAGCCG; antisense: 5′ GGATGTAAACATTTGTTTTAA and 5′ TTTGAATTCCTACTTAAACAATAATTCATCTGG) and cloned in frame into the HindIII/EcoRI-digested pEGFP-C1 vector (Clontech) to obtain GFP-PDCD2L. For constructs coding for HA-CCP1 fl, HA-CCP1 ΔN50, HA-CCP1 ΔN100, HA-YFP, HA-YFP-CCP1 aa1–75, HA-YFP-CCP1 aa1–120, and GST-CCP1 aa1–120, fragments were amplified using the YFP-CCP1 plasmid as a template and appropriate primers and cloned via NotI/SalI into a modified EF-HA vector (33) or via SalI/NotI into pGEX-6P-1. HA-YFP-CCP1 aa1–120 L80/84A and HA-YFP-CCP1 aa1–120 L80/84/87A were prepared via site-directed mutagenesis using the HA-YFP-CCP1 aa1–120 as a template. Details of the construction of fragments and the mutants thereof can be obtained upon request. All constructs were verified via DNA sequencing.
Gel Electrophoresis, In-gel Tryptic Digestion of Proteins, and LC-MS/MS
Equal amounts of nuclear and cytosolic extracts were separated via one-dimensional SDS-PAGE (4%–12% NuPAGE Bis-Tris Gel, Invitrogen). Following Coomassie Brilliant Blue staining, each lane was cut into 23 equally sized pieces. In-gel digestion was performed as described elsewhere (34). Briefly, proteins were reduced with 10 mm DTT for 50 min, alkylated with 55 mm iodoacetamide for 20 min at 26 °C, and in-gel digested with trypsin (Roche Applied Science) overnight. First, extracted peptides were loaded into an in-house packed C18 trap column (1.5 cm, 360 μm outer diameter, 150 μm inner diameter, Reprosil-Pur 120 Å, 5 μm, C18-AQ, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) at a flow rate of 10 μl/min. Retained peptides were eluted and separated on an analytical C18 capillary column (15 cm, 360 μm outer diameter, 75 μm inner diameter, Reprosil-Pur 120 Å, 5 μm, C18-AQ, Dr. Maisch GmbH, Germany) at a flow rate of 300 μl/min with a gradient from 5% to 38% acetonitrile in 0.1% formic acid for 50 min using an Agilent 1100 nano-flow LC system (Agilent Technologies, Santa Clara, CA) coupled to an LTQ-Orbitrap Velos hybrid mass spectrometer (Thermo Electron, Bremen, Germany). The LTQ-Orbitrap Velos was operated in data-dependent mode and survey scans were acquired in the Orbitrap (m/z 350–1600) with a resolution of 30,000 at m/z 400 with a target value of 1 × 106. Up to 15 of the most intense ions with charges ≥ 2 from the survey scan were sequentially isolated for collision-induced dissociation with a normalized collision energy of 35. Dynamic exclusion was set to 60 s to avoid repeating the sequencing of peptides.
Data and Bioinformatics Analysis
Raw MS files from the LTQ-Orbitrap Velos were analyzed using MaxQuant software (version 1.0.13.13 unless otherwise stated) (35) and the Mascot search engine (version 2.3.2). Peak lists generated by Quant.exe were matched against the International Protein Index human protein database (version 3.86, containing 91,695 entries) supplemented with 179 common contaminants (e.g. keratins, serum albumin) and concatenated with the reverse sequences of all entries. Mascot search parameters were as follows: carbamidomethylation of cystein was set as a fixed modification, whereas oxidation of methionine and N-terminal protein acetylation were set as variable modifications; tryptic specificity with no proline restriction and up to two missed cleavages was used. The MS survey scan mass tolerance was 7 ppm and for MS/MS 0.5 Da. Only peptides with a minimal length of five amino acids were considered for identification. The false discovery rate was set to 1% at both the peptide and the protein level. Peptides with a posterior error probability of less than 0.05 were considered for identification and quantification. “Re-quantify ” was enabled, and “keep low scoring versions of identified peptides ” was disabled. Quantification of SILAC pairs was performed with a minimum ratio count of two by considering unique and razor peptides. To generate results with a high confidence interval, four biological replicates were performed, and each biological replicate was analyzed twice. To avoid false positives due to the experimental workflow, label-swap experiments were performed. Proteins behaving adversely in forward and reverse labeling experiments were excluded from the analysis. Significance B was used as a main criterion for data analysis. This prevents the arbitrary or empirical setting of fold-change cut-off values. For proteins with p values ≤ 0.01, changes were considered significant. Gene Ontology enrichment analysis was done using DAVID (36). Protein interaction network analysis was performed using interaction data from the STRING database (37), in which only high-confidence (score > 0.7) interactions were represented in the network. The online tools NetNES1.1 (18) and ELM (38) were used to predict putative NESs. For single peptide identifications, annotated spectra were obtained via MaxQuant (supplemental data).
Indirect Immunofluorescence and Confocal Microscopy
24 h after transfection, immunofluorescence staining was essentially performed as described previously (39) using Hoechst 33258 as a DNA stain. Cells were analyzed, and images were taken using an LSM 510-Meta confocal microscope and processed using AxioVision Rel. 4.8 LE and Adobe Photoshop 6.0. For quantification of fluorescence signal intensities, areas within the nuclear (N) and cytoplasmic (C) regions of individual cells were selected and the fluorescence intensity was measured using ImageJ software. Quantification was performed from at least three independent experiments, each with more than 20 cells with similar expression levels. The results were plotted as the fold-change of the intensity ratios (N/C) upon LMB treatment.
Protein Expression
His-CRM1 (40) and RanQ69L (41) were expressed as described and dialyzed against transport buffer (TPB) (20 mm HEPES-KOH, pH 7.3, 110 mm KOAc, 2 mm Mg(OAc)2, 1 mm EGTA, 2 mm DTT, and 1 μg/ml each of aprotinin, leupeptin, and pepstatin). The expression of GST-SPN1 in BL21 codon+ was induced with 0.5 mm isopropyl 1-thio- â-D-galactopyranoside at 16 °C overnight. Bacteria were lysed in buffer C (50 mm Tris pH 6.8, 300 mm NaCl, 1 mm MgCl2, 0.25 mm EDTA with protease inhibitors as above, and 1 mm DTT). Expression of GST-CCP1 aa1–120 in BL21-DE3 was induced with 0.1 mm isopropyl 1-thio- â-D-galactopyranoside at 16 °C overnight. Bacteria were lysed in buffer A (50 mm Tris pH 8, 250 mm NaCl, 2 mm MgCl2, 10% glycerol) containing 1% Triton X-100, 4 mm β-mercaptoethanol, 0.4 μg/ml lysozyme, and protease inhibitors. Insoluble components were pelleted via centrifugation at 100,000g for 45 min, and the supernatant was incubated with glutathione Sepharose beads (GE Healthcare) for 1.5 h at 4 °C. The beads were washed and bound proteins were eluted with 15 mm glutathione in the appropriate buffer and dialyzed against TPB containing 1 mm DTT and protease inhibitors. All proteins were frozen in liquid nitrogen and stored at −80 °C.
Binding Studies
For in vitro binding studies, 5 μg GST-fusion proteins were immobilized on glutathione Sepharose beads (GE Healthcare) that had been pre-incubated with 10 mg/ml BSA in buffer B (50 mm Tris pH 7.4, 200 mm NaCl, 1 mm MgCl2, 5% glycerol). The beads were washed and incubated with 5 μg of His-CRM1 alone or loaded with RanQ69L (GTP) in 300 μl of buffer B containing 2 mg/ml BSA. After 1.5 h at 4 °C, the beads were washed three times with buffer B. Bound proteins were eluted with SDS-sample buffer and subjected to SDS-PAGE followed by immunoblotting.
FACS Assay
His-CRM1 was labeled with Cy3 according to the instructions from the manufacturer (Cy3 Mono-Reactive Dye Pack, GE Healthcare). GST-fusion proteins were immobilized on glutathione Sepharose High Performance beads (GE Healthcare) (50 pmol of protein per 2.5 μl of beads) and washed twice with TPB containing 10 mg/ml BSA. TPB was added to the beads to obtain a 50% slurry. 5 μl of the slurry was incubated with 0.77 μg (6.64 pmol) of labeled CRM1 in the absence or presence of 2.5 μg (120 pmol) of Ran preloaded with GTP and 7 μl 4x assay buffer (500 mm NaCl, 40 mg/ml BSA, 1 mm DTT, 2% 1,6-hexanediole) in a final reaction volume of 28 μl. After the reaction had progressed at 4 °C for 60 min, the beads were washed once with TPB and subjected to flow cytometry using a FACSCanto II (BD Biosciences) and FACS Diva6.1.1 software. The median fluorescence of 2500 beads was measured (585/42 bandwidth and 556LP filter), and the results were plotted as arbitrary units.
Antibodies and Reagents
For the detection of HA-epitope-tagged proteins via immunofluorescence or immunoblotting, a monoclonal mouse anti-HA antibody (16B12, Covance, Emeryville, CA, USA) was used. Rabbit anti-SP1 antibody was obtained from Thermo Scientific and was used as a marker for soluble nuclear proteins according to the specifications of the company. Rabbit anti-tubulin and rabbit anti-GAPDH antibodies were obtained from Proteintech (Chicago, IL), mouse anti-lamin A/C from BD Biosciences, goat anti-GST from Amersham Biosciences, and mouse anti-penta-His from Qiagen (Hilden, Germany). For immunofluorescence, donkey anti-mouse Alexa-Fluor-488 (1:1000; Molecular Probes, Darmstadt, Germany) was used as secondary antibody. For immunoblotting, HRP-coupled donkey anti-mouse or donkey anti-rabbit IgG (Dianova, Hamburg, Germany) was used as a secondary antibody.
RESULTS
CRM1 is the major transport receptor for the export of proteins out of the nucleus, with more than 100 substrates described. We set out to devise a method to systematically search for CRM1-substrates that would be applicable for different cell lines under different growth conditions. The protocol involves subcellular fractionation and high-throughput quantitative proteomics analysis using SILAC. In the presence of the CRM1-inhibitor LMB, we expected (i) an accumulation of CRM1 substrates in the nuclear fraction and/or (ii) a depletion of substrates from the cytosolic fraction relative to a control experiment in the absence of LMB. The general workflow (depicted in Fig. 1A) involved the incubation of cells in culture with isotopically labeled amino acids prior to subcellular fractionation. As a control for a CRM1-dependent substrate, we transfected HeLa cells with a plasmid coding for the shuttling reporter protein Rev(48–116)-GFP2-M9. This protein contains the NES of the viral protein HIV-1-Rev (6) and the M9-NLS that is recognized by the import receptor transportin (42). In the absence of LMB, Rev(48–116)-GFP2-M9 mostly localized to the nucleus, but it was also detected in the cytoplasm to some extent. Upon the addition of LMB, nuclear export was inhibited and the protein was exclusively nuclear (Fig. 1B). GFP2, in contrast, was not affected by LMB. Transfected cells were grown in two different SILAC media containing either unlabeled l-arginine and l-lysine (SILAC: R0K0, light) or l-arginine-U-13C6 and l-lysine-U-13C6-15N2 (SILAC: R6K8, heavy). Cells were then treated with or without LMB for 3 h, mixed at a 1:1 ratio, and subjected to subcellular fractionation. Experiments were performed in the “forward” mode (heavy cells treated with LMB, as in Fig. 1A) and in the “reverse” mode (light cells treated with LMB; not depicted in Fig. 1A). Each independent label-swap experiment was performed twice (yielding a total of four biological replicates), and each biological replicate was analyzed twice (technical replicates) via MS. Fig. 1C demonstrates the quality of the fractionation. The cytoplasmic marker proteins α-tubulin and GAPDH were present predominantly in the cytosolic fraction and were absent from the nuclear fraction. The nuclear marker proteins lamin A/C and the transcription factor SP1, in contrast, were detected predominantly in the nuclear fraction and were largely absent from the cytosolic fraction. This successful fractionation was a prerequisite for the extensive analysis of LMB effects on the subcellular localization of proteins. In addition to subcellular fractions, total cell extracts were analyzed to assess the effect of LMB on the whole proteome and to rule out secondary effects when analyzing the fractions. Here, we performed one “forward” and one “reverse” experiment, with two technical replicates each.
Fig. 1.

A, experimental workflow. HeLa cells were grown in DMEM containing either light or heavy amino acids. An equal number of cells was mixed prior to subcellular fractionation. Soluble proteins from cytosolic and nuclear fractions were separated via SDS-PAGE. After in-gel digestion, extracted peptides were analyzed via LC-MS/MS, and raw data were processed using MaxQuant. B, HeLa cells were transfected with plasmids coding for (EGFP)2 or Rev(48–116)-GFP2-M9. After 24 h, cells were treated with or without 10 nm leptomycin B (LMB) for 3 h, fixed, and analyzed via fluorescence microscopy. C, subcellular fractions (T, total; C, cytosolic; N, nuclear) were analyzed via SDS-PAGE followed by immunoblotting to detect lamin A/C, the transcription factor SP1, GAPDH, and tubulin.
Almost complete incorporation (∼99%; supplemental Fig. S1A) of heavy amino acids was achieved after five passages of the cells. In order to assess the mass spectrometric quantification accuracy, untreated heavy and light cells were mixed and analyzed. Almost 95% of quantified proteins showed a logarithmic fold change of close to zero (supplemental Fig. S1B).
Effects of LMB on the Proteome
The treatment of cells with LMB could potentially lead to changes in protein abundances in addition to the expected changes in subcellular localization. Therefore, we first analyzed total cell extracts (i.e. omitting any subcellular fractionation) to monitor changes of the cellular proteome upon treatment with the CRM1 inhibitor. As expected for a highly selective drug, most cellular proteins (∼4300 proteins identified in total) were not affected by the treatment of cells with LMB (supplemental Fig. S2A; see supplemental Table S1 for a list of all identified proteins). Label-swap experiments were performed to avoid experimental bias, and “significance B ” values were analyzed. Differences were considered significant for p ≤ 0.01 (see “Experimental Procedures ” for details). Using these criteria, 70 proteins were found to be enriched in LMB-treated cells, and 58 proteins were depleted. Functional interaction mapping (STRING analysis) revealed two major clusters of proteins. Many of the depleted proteins were found to interact with each other (supplemental Fig. S2B, red circles). These proteins are structural components of the ribosome and/or involved in translation, as revealed by gene ontology analysis (data not shown). Enriched proteins, in contrast, interact during various steps of RNA processing (supplemental Fig. S2B, blue circles) and are also involved in RNA splicing according to the gene ontology analysis. Thus, inhibition of the CRM1 pathway for a short period of time affects the abundance of a rather select set of proteins and is not expected to have a big impact on the identification of potential CRM1 substrates. We therefore set out to analyze the proteome of the cytosolic and nuclear fractions to detect changes in protein quantities upon LMB treatment.
Identification of Potential CRM1 Substrates
When we analyzed subcellular fractions, a total of ∼3300 proteins (cytosolic fraction; Fig. 2A) or ∼3200 proteins (nuclear fraction; Fig. 2B) were identified via LC-MS/MS after SILAC labeling, with an overlap of ∼1900 proteins (see the Venn diagram in supplemental Fig. S3A). In both the cytosolic and the nuclear fractions, proteins showed a surprisingly tight distribution around logarithmic fold change values close to zero (i.e. 1:1 ratios of individual proteins; see Figs. 2A, 2B) upon LMB treatment. This indicates a rather small effect of LMB on the majority of the proteins and also shows the accuracy of our quantitative MS analysis. As a main filtering criterion, we used “significance B,” as calculated by MaxQuant, which has advantages over just considering the fold change: (i) it avoids setting any empirical cut-off values but shows a better statistical significance of outlier proteins, as these are identified from the bulk of the distribution by calculating the variances of all proteins; and (ii) it takes into account the fact that high-abundance proteins are more accurately quantified than low-abundance ones, so that small changes also can be considered as significant.
Fig. 2.

Inhibition of the CRM1 export pathway leads to changes in the subcellular localization of proteins. A, B, scatter plots of quantified proteins in cytosolic (A) or nuclear (B) fractions after LMB treatment of HeLa cells. Proteins are colored according to significance B; gray triangles signify p values > 0.01, blue < 0.01, yellow < 0.0001, and red < 1 × 10−11. C, Venn diagram showing the overlap between proteins identified in A and B. See Table I for details.
Applying this high level of stringency, we detected changes for 138 proteins that upon LMB treatment were either depleted from the cytosolic fraction (84 proteins) or accumulated in the nuclear fraction (59 proteins), or both (5 proteins; see Fig. 2C and Table I). In a first step to analyze our data, we compared these 138 proteins with those that were identified as a result of changes in their total cellular levels (see above and supplemental Fig. S3B). Only 8 out of 84 proteins that were depleted from the cytosolic fraction upon LMB treatment were also depleted from the total cell extract after treatment with the CRM1 inhibitor. Among these proteins were eukaryotic translation initiation factor 6 (eIF6) (43) and RNA-helicase DDX3 (44), two published CRM1 substrates. We analyzed another prominent hit in this category of proteins that were depleted from the cytosol as well as from the total cell extract (sequestosome 1; see below) and found that a GFP-tagged version of the protein clearly shifted toward the nucleus upon LMB treatment of the cells. One possible explanation for the depletion from the total cell extract is a reduction of the half-life of such proteins upon the inhibition of nuclear export. In our studies, however, we did not further address the issue of protein stability under different conditions. The remaining 76 proteins that were depleted from the cytosolic fraction upon LMB treatment were not affected in the total cell extract. Similarly, the 59 proteins that were found to be enriched in the nuclear fraction were not enriched in the total cell extract under LMB treatment (supplemental Fig. S3C), and none of the proteins depleted from the nuclear fraction were also depleted from the total cell extract (data not shown). We conclude from these results that the changes we observed in total cell extracts upon LMB treatment cannot account for most of the putative CRM1 substrates as found in the subcellular fractions.
Table I. List of proteins that were depleted from the cytosol and/or accumulated in the nucleus in HeLa cells upon LMB treatment as identified via LC-MS/MS.
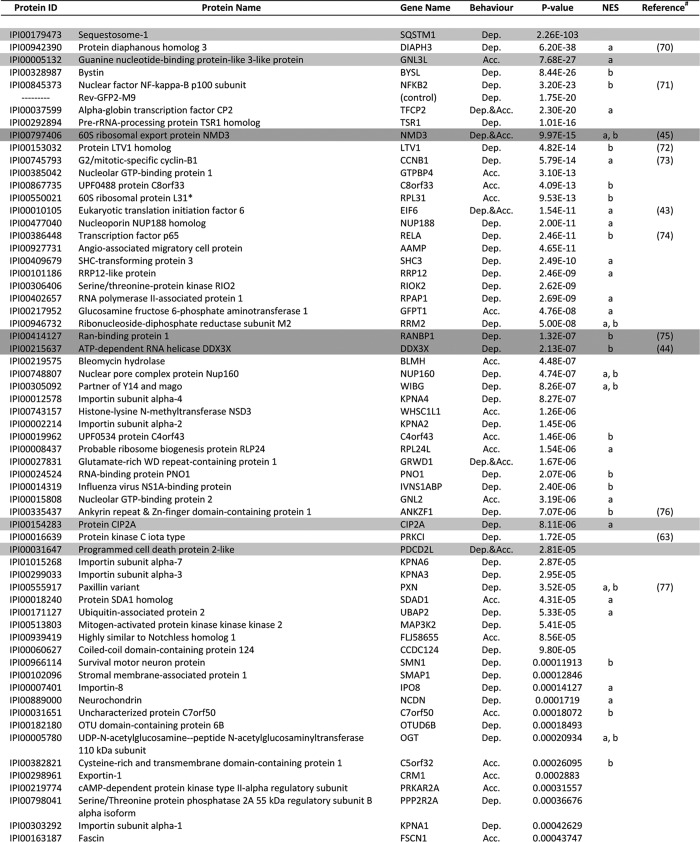
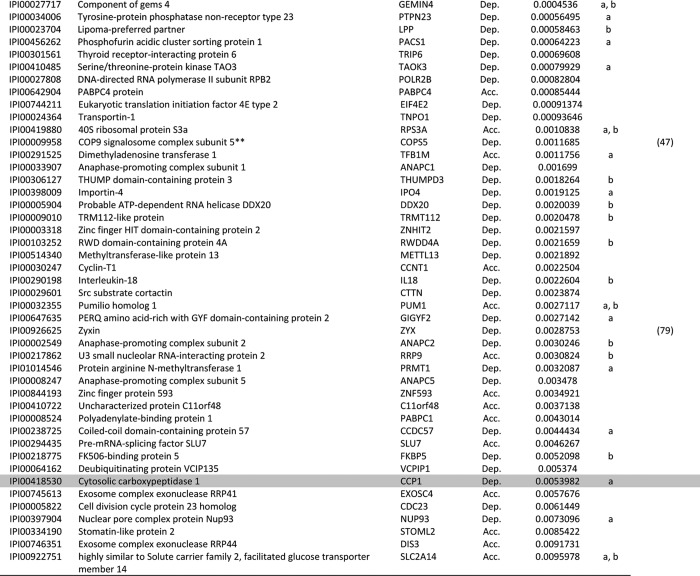
Above-listed proteins were extracted from supplementary Table S1 according to our filtering criteria. For a list of identified peptides, see supplementary Table S2.
Dep, depleted from the cytosol of HeLa cells in response to LMB treatment; Acc, accumulated in the nucleus of HeLa cells in response to LMB treatment.
For proteins that were both depleted from the cytoplasm and accumulated in the nucleus, the higher significance value is presented.
Protein sequences were analyzed using NES-prediction algorithms, and hits are marked as “a” or “b” (a, NetNES prediction algorithm; b, Eukaryotic Linear Motive Resource for Functional Sites in Proteins).
Proteins highlighted in dark gray are known CRM1 substrates validated in this study (see Figure. 3A), and proteins highlighted in light gray are novel CRM1 substrates validated in this study (see Fig. 3B).
a References indicate proteins previously identified as CRM1 targets. These proteins are also listed in databases such as NESbase or NESdb.
b One of the 60s ribosomal complex proteins identified (see Supplementary Table S3A).
c One of the COP9 signalosome complex proteins identified (see Supplementary Table S3B).
One might expect CRM1 substrates to be depleted from the cytosolic fraction and simultaneously enriched in the nuclear fraction upon LMB treatment. In our list of potential substrates, however, only five proteins showed this behavior (Fig. 2C). These are glutamate-rich WD repeat-containing protein 1, eIF6, alpha-globin transcription factor CP2, programmed cell death protein 2-like protein (PDCD2L), and the 60S ribosomal export protein NMD3. eIF6 and NMD3 have previously been identified as CRM1 substrates (43, 45). Other established CRM1 substrates that were depleted from the cytosolic fraction did not accumulate in the nucleus. One prominent example is our positive control, Rev(48–116)-GFP2-M9, which was identified with a high level of significance as depleted from the cytosolic fraction (see Table I).
Besides NMD3 and eIF6, a number of proteins from our list of CRM1 substrates have previously been identified as affected by LMB (see Table I). We used epitope-tagged versions of NMD3, RanBP1, and DDX3 to confirm some of these hits. Indeed, HA-RanBP1 and HA-DDX3 were predominantly cytoplasmic in control cells and shifted to the nucleus upon treatment with LMB (Fig. 3A). GFP-NMD3 was nuclear also in the absence of the CRM1 inhibitor, but a small shift toward the nucleus upon the addition of LMB was observed (Fig. 3A).
Fig. 3.

Validation of known (A) and novel (B) CRM1 cargos. HeLa cells were transfected with plasmids coding for GFP-NMD3, HA-RanBP1, HA-DDX3, GFP–sequestosome 1, HA-GNL3L, FLAG-CIP2A, GFP-PDCD2L, or YFP-CCP1, as indicated. After 24 h, cells were treated with or without 10 nm LMB for 3 h. Expressed proteins were detected via indirect immunofluorescence using anti-HA or anti-FLAG antibodies or directly via the GFP/YFP-tag. C, quantitative analysis of the LMB effect on reporter proteins as used in A and B. For comparison, EGFP2 and Rev(48–116)-GFP2-M9 were included in the analysis. Error bars indicate the standard deviation from the mean of at least three independent experiments.
Some of the proteins that we identified are known components of large cytoplasmic or nuclear multi-protein complexes. Prominent examples are several proteins of the 60S ribosomal subunit (supplemental Table S3A). This complex is known to be exported from the nucleus by CRM1, with NMD3 as an adapter protein (45), explaining the appearance of the individual proteins on our list of potential CRM1 substrates. Likewise, almost all components of the COP9 signalosome, a multi-subunit complex that functions as a de-neddylase and is involved in the control of protein stability (46), were identified in our screen (supplemental Table S3B). It has previously been reported that the treatment of cells with LMB results in an accumulation of Jab1/CSN5, one component of the COP9 signalosome, in the nucleus (47). In summary, our screen identified several known CRM1 substrates, validating our experimental approach.
Validation of Novel CRM1 Substrates
Next, we performed a thorough microscopic analysis of reporter constructs for a selected set of proteins from our list that had not previously been identified as CRM1 substrates. We chose proteins with varying levels of significance (i.e. from the top, from the middle, and from the bottom of Table I). GFP-, YFP-, HA-, or FLAG-tagged versions of these proteins were expressed, and cells were treated with or without LMB, followed by microscopic analysis. On our list of potential CRM1 substrates, sequestosome 1 (also called p62) exhibited the highest level of significance. Sequestosome 1 was initially identified as a protein that binds polyubiquitin chains (48) and has been implicated in the development of Parkinson's disease, Alzheimer's disease, and Paget's disease, among other diseases (49). It functions as an adapter protein with multiple binding partners (for a review, see Ref. 50), but a role in the nucleus has, to our knowledge, not yet been described. When expressed as a GFP-fusion protein, sequestosome 1 localized to the cytoplasm with a number of cytoplasmic speckles, as described previously. Upon the addition of LMB, the protein clearly shifted toward the nucleus (Fig. 3B).
Another candidate with a high level of significance was the guanine nucleotide-binding protein-like 3-like protein (GNL3L), which has been described as a nucleolar protein (51). An HA-tagged version of GNL3L was found to accumulate in the nucleus but was also detectable in the cytoplasm (Fig. 3B). After LMB treatment, HA-GNL3L was exclusively nuclear, suggesting inhibited export in the presence of the drug.
The cancerous inhibitor of PP2A (CIP2A) is an example of a protein with an intermediate level of significance (see Table I). CIP2A binds the transcription factor c-myc, inhibits the phosphatase activity of PP2A (52), and has been implicated in various cancers. Again, a YFP-tagged fusion protein was found in the cytoplasm of control cells and strongly accumulated in the nucleus of LMB treated cells (Fig. 3B).
PDCD2L, a poorly characterized protein that both accumulated in the nucleus and was depleted from the cytoplasm in our screen, showed a level of significance similar to that of CIP2A (Table I). We expressed GFP-PDCD2L in HeLa cells and clearly saw cytoplasmic localization in control cells and nuclear accumulation in LMB-treated cells (Fig. 3B).
A protein with a rather low level of significance is CCP1, also called Nna1 (53). It functions as a tubulin deglutamylase and is involved in certain types of neurodegeneration (54). Despite its low ranking in our list of potential CRM1 substrates (see Table I), a YFP-tagged version of CCP1 was clearly affected by LMB: in the absence of the CRM1 inhibitor, the protein was exclusively cytoplasmic, whereas it showed an equal distribution between the nucleus and the cytoplasm in the presence of the drug (Fig. 3B).
A quantitative analysis of the effect of LMB on the localization of previously known and newly identified CRM1 substrates is presented in Fig. 3C. Of note, sequestosome 1, the top-scoring protein on our list of CRM1 substrates (Table I), also showed the strongest effect in this analysis. We tested several other epitope-tagged proteins from our list (data not shown). Some, such as bystin, a protein with a high significance level that is known to interact with the 40S ribosomal subunit, were detected exclusively in the nucleus, with and without LMB treatment. Thus, no LMB effect could be detected. Notably, however, none of the epitope-tagged proteins tested showed exclusive cytoplasmic localization in the presence or absence of LMB, indicating a lack of false positives in our list. Together, these results clearly demonstrate that our experimental approach is well suited for the identification of proteins that change their subcellular localization upon inhibition of the CRM1-dependent nuclear export pathway.
Characterization of Novel CRM1 Substrates
What is the molecular basis for the accumulation of proteins in the nucleus and/or the depletion from the cytoplasm upon the treatment of cells with LMB? First and foremost, the affected protein might contain a bona fide NES and be a direct binding partner of CRM1, resulting in reduced export from the nucleus upon inhibition of the CRM1-mediated export pathway. Alternatively, the affected protein might be part of a larger complex that is a substrate for CRM1; in this case, the protein itself does not interact directly with CRM1 and probably lacks a functional NES. Two bioinformatic tools were used to identify potential NESs in our putative CRM1 substrates. The first was based on a dedicated prediction algorithm for leucine-rich NESs (18). The second was the Eukaryotic Linear Motive Resource for Functional Sites in Proteins (ELM). For the majority of our candidates, NESs were predicted by at least one of the programs, including CIP2A and CCP1 (see below and compare with Table I). Such proteins may well be direct binding partners of CRM1. It is well known, however, that many predicted NESs are non-functional, possibly because they are hidden in the three-dimensional structure of the protein. Other proteins, including sequestosome 1, did not yield a predicted NES in this analysis. Indeed, we were unable to detect significant binding of CRM1 to bacterially expressed sequestosome 1 (data not shown). Such proteins might either interact with CRM1 only upon a specific post-translational modification or use an adaptor protein for binding to the export receptor. We next set out to test CIP2A and CCP1 for direct CRM1 binding and to experimentally characterize predicted NESs. Analysis of CIP2A by ELM yielded three putative NESs (amino acids 414–430, 470–484, and 598–612). We obtained fusion constructs of CIP2A (GST-CIP2A 1–560 and GST-CIP2A 587–905, kind gifts from J. Okkeri and J. Westermarck) and performed CRM1-binding assays with these proteins. A specific interaction with the export receptor in a RanGTP-dependent manner, however, could not be detected (data not shown), suggesting that CIP2A, despite the predicted NESs, requires an adapter protein for CRM1 binding. For CCP1, ELM predicted an NES in the N-terminal region between amino acids 79 and 92 (DLQTTLNILSILVE). In the CCP1 sequence, E92 is followed by two additional hydrophobic amino acid residues (L93 and V94) that could be part of the NES. To address the functionality of this putative NES, we prepared N-terminal deletion constructs for CCP1 (Fig. 4A) and expressed them in HeLa cells. Although the full-length protein (HA-CCP1 fl) and HA-CCP1ΔN50 (deletion of N-terminal 50 amino acids) were largely excluded from the nucleus, deletion of the first 100 amino acids (HA-CCP1 Δ100) resulted in an equal distribution of the protein between the nucleus and the cytoplasm (Fig. 4A). This result suggested that CCP1 contained a functional NES in the region between amino acids 51 and 100. The addition of LMB to transfected cells resulted in a shift of HA-CCP1 fl and HA-CCP1ΔN50 toward the nucleus, as expected. Interestingly, LMB further increased the nuclear localization of HA-CCP1 Δ100, suggesting that the protein might contain a second, as yet unidentified NES.
Fig. 4.

Identification of a functional NES in CCP1. A, schematic representation of N- and C-terminal deletion constructs of CCP1 (left-hand panel). HeLa cells were transfected with plasmids coding for HA-CCP1 full-length, HA-CCP1 ΔN50, HA-CCP1 ΔN100, HA-CCP1 ΔC210, or HA-CCP1 ΔN100ΔC210. After 24 h, cells were treated with or without 10 nm LMB for 3 h. Expressed proteins were detected via indirect immunofluorescence using antibodies against the HA tag (right-hand panel). B, schematic representation of N-terminal fragments of CCP1, with or without mutations at the putative NES (left-hand panel). HeLa cells were transfected with plasmids coding for HA-YFP, HA-YFP-CCP1 aa 1–75, HA-YFP-CCP1 aa 1–120, HA-YFP-CCP1 aa 1–120 L80/84A, or HA-YFP-CCP1 aa 1–120 L80/84/87A. After 24 h, cells were treated with or without 10 nm LMB for 3 h. Expressed proteins were detected via fluorescence microscopy based on the YFP tag (right-hand panel).
ELM also predicted a nuclear localization signal between amino acids 996 and 1017 of CCP1. As a first step toward the analysis of this putative nuclear localization signal, we deleted the last 210 amino acids from the full-length protein, yielding HA-CCP1 ΔC210, and from the N-terminal deletion mutant, yielding HA-CCP1 ΔN100/ΔC210. The latter protein was excluded from nuclei in HeLa cells (Fig. 4A), suggesting that the last 210 amino acids of CCP1 indeed contain a nuclear localization signal. Further details of the nuclear import properties of CCP1 remain to be elucidated.
We next fused N-terminal fragments of CCP1 to YFP. YFP alone and YFP-CCP1 (1–75) were found predominantly in the nucleus of transfected cells, and LMB did not affect their localization (Fig. 4B). YFP-CCP1 (1–120), in contrast, was largely cytoplasmic in the absence of LMB and shifted toward the nucleus in the presence of the CRM1 inhibitor, demonstrating that the protein can be actively exported by CRM1. Finally, we introduced leucine to alanine mutations at hydrophobic positions of the putative NES of YFP-CCP1 (1–120). Clearly, the mutations resulted in a shift of the fusion protein from the cytoplasm toward the nucleus. Together, these results strongly suggest that the predicted NES (amino acids 79–94) in CCP1 is indeed functional and is required for export from the nucleus.
Next, we addressed the question of whether CCP1 interacts directly with CRM1. As the full-length protein could not be expressed in bacteria, we fused an N-terminal fragment containing the putative NES to GST (GST-CCP1 1–120) and used the purified protein for CRM1-binding studies. GST or GST-CCP1 1–120 was immobilized on beads and incubated with His-tagged CRM1 in the absence or presence of Ran pre-loaded with GTP. As shown in Fig. 5A, significant binding to GST-CCP1 1–120, but not to GST, could be detected upon the addition of RanGTP. Finally, we developed a novel flow-cytometry-based assay to evaluate the interaction of potential CRM1 substrates with the export receptor in a more quantitative manner. GST, GST-CCP1 (1–120), or GST-SPN1 was immobilized on glutathione beads and incubated with fluorescently labeled CRM1 in the absence or presence of RanGTP. After a short incubation, the beads were washed once and the association of CRM1 with individual beads was analyzed via flow cytometry. As shown in Fig. 5B, CRM1 did not bind significantly to GST. GST-SPN1, in contrast, showed a strong interaction that was further enhanced by the addition of RanGTP, consistent with previous observations (16). For GST-CCP1 (1–120), we observed a specific interaction, as the addition of RanGTP resulted in a strong increase of bound CRM1. This rapid and simple assay will be very helpful for semi-quantitative analyses of the interaction of CRM1 with potential export substrates.
Fig. 5.

CCP1 interacts directly with CRM1. A, GST or GST-CCP1 (aa 1–120) were immobilized on beads and incubated with His-CRM1 with or without RanQ69L-GTP. Bound proteins were analyzed via SDS-PAGE and immunoblotted using anti-His or anti-GST antibodies. B, quantitative analysis of substrate binding to CRM1. GST, GST-snurportin 1 (SPN1), or GST-CCP1 (aa 1–120) were immobilized on beads and incubated with fluorescently labeled CRM1 in the absence or presence of RanGTP. Fluorescence intensities of individual beads were analyzed by means of flow cytometry. Error bars indicate the standard deviation from the mean of at least three independent experiments. Note that original values were plotted, reflecting the strong interaction of the high-affinity substrate SPN1 with CRM1.
DISCUSSION
Effects of LMB on the Proteome
In this study, we analyzed the effects of the selective nuclear export inhibitor LMB on the subcellular localization of proteins using an MS-based high-throughput quantitative approach. SILAC has previously been used for a variety of quantification studies including post-translational modifications (55–57), protein turnover (58), and changes in the subcellular localization of proteins upon DNA damage (59).
Analysis of the proteome of total cell lysates showed that LMB is indeed very selective, affecting the abundance of less than 3% of all identified proteins. ∼50% of those proteins that were depleted from total cell extracts upon LMB treatment were structural components of ribosomes (supplemental Fig. S2B). Previously published work has shown that ribosomal subcomplexes are exported from the nucleus into the cytoplasm by CRM1, and we also identified many ribosomal proteins accumulating in the nucleus of LMB-treated cells (supplemental Table S3). These results suggest that inhibition of the nuclear export of ribosomal subunits promotes their turnover (e.g. by the proteasome). The accumulation of proteins in the nucleus accompanied with a higher turnover rate thus resulted in an overall decrease of ribosomal proteins identified in the total cell extract upon LMB treatment. On the other hand, ∼50% of proteins whose total cellular concentration increased upon LMB treatment are involved in RNA splicing/processing (supplemental Fig. S2B). For future characterization of LMB effects on the proteome, proteins will have to be analyzed with respect to gene expression levels and stability.
Known and Novel CRM Substrates
We identified 138 proteins that were depleted from the cytosolic fraction and/or accumulated in the nuclear fraction after LMB treatment of cells for 3 h. Importantly, one of these proteins was our positive control, Rev(48–116)-GFP2-M9. Our initial microscopic analysis revealed that this reporter protein was largely nuclear even in the absence of LMB (Fig. 1B) and showed only a weak cytoplasmic signal. Upon the addition of the drug, however, the weak cytoplasmic signal derived from Rev(48–116)-GFP2-M9 disappeared completely. Despite the low cytoplasmic levels relative to the nuclear levels, we were able to detect changes in the cytosolic fraction upon LMB treatment. This result demonstrates that our approach even allows the identification of shuttling CRM1 substrates that are largely nuclear under physiological conditions as a result of a dominant nuclear import pathway.
Approximately one-quarter of the 138 potential CRM1 substrates are components of either the 60S ribosomal subunit or the COP9 signalosome (supplemental Table S3), complexes known to be exported out of the nucleus via the CRM1-dependent transport pathway. Surprisingly, we identified only two components of the 40S ribosomal subunit (RPS3A and bystin) accumulated in the nucleus, although 40S-export (like 60S-export) was reported to be inhibited upon the depletion of CRM1 using siRNAs (60). Perhaps strong effects on 40S-export become apparent only after a longer exposure to LMB.
For ∼60 of the remaining proteins, putative NESs could be identified using one or two of the available search engines (Table I, “NES” column, a, b). Such searches, however, could lead to false positive results, as hydrophobic NES-like sequences could be buried in the three-dimensional structure of the protein and not be available for CRM1 binding. Also, NESs could be formed only upon proper folding of the protein, precluding their identification by analysis of the linear sequence.
For ∼15 of the identified proteins, CRM1-dependent export has been described previously in the literature and/or in cargo databases (see Table I and references cited therein). We confirmed the effect of LMB on the subcellular localization of three such proteins—NMD3, RanBP1, and DDX3—via microscopic analyses. Together, this demonstrates that we are able to identify proteins that shuttle between the nucleus and the cytoplasm in a CRM1-dependent manner with our SILAC-based approach.
In addition to those known substrates, we confirmed nuclear accumulation upon CRM1 inhibition for five proteins that had not been previously identified as CRM1 targets (see Table I; known and novel cargos that were validated in this study are highlighted in dark and light gray, respectively). A clear result was obtained for CCP1. Although this protein exhibited one of the highest p values (i.e. lowest significance) in our analysis (∼0.005), LMB induced a very clear shift of YFP-CCP1 toward the nucleus. Furthermore, we were able to identify a functional leucine-rich NES in the N-terminal region of the protein and showed that it directly interacts with CRM1 in a RanGTP-dependent manner. CCP1 had originally been named Nna1 (nervous system nuclear protein induced by axotomy (53)), referring to its partially nuclear localization. The biological significance of the CRM1-dependent nuclear export of CCP1/Nna1 remains to be investigated.
For four other candidates—sequestosome 1, CIP2A, GNL3L, and PDCD2L—we confirmed CRM1-dependenent nuclear export through the expression of tagged versions in HeLa cells. Sequestosome 1 and CIP2A probably interact with the export receptor in an indirect manner. Sequestosome 1 acts as a multidomain scaffold protein that sequesters other proteins. It interacts with atypical protein kinase C lambda/iota (61) and also regulates NFκB activation (62). The CRM1-dependent nuclear export of atypical protein kinase C lambda/iota (63) and IκBα (64), a protein involved in NFκB regulation, has been demonstrated previously. Similarly, CIP2A interacts directly with the transcription factor c-Myc and inhibits PP2A activity (52). Cytoplasmic localization of the PP2A subunit B56alpha depends on CRM1-mediated nuclear export (65). Alternatively, CRM1 binding could be regulated by post-translational modifications that are absent from the bacterially expressed proteins used for our interaction studies. During the course of this project, CRM1-dependent nuclear accumulation of CIP2A was also observed by Jukka Westermarck and coworkers.2 Again, the biological significance of the CRM1-dependent nuclear export of CIP2A and sequestosome 1 remains to be investigated. Likewise, the export characteristics of GNL3L and PDCD2L were not further analyzed in this study.
Some of the proteins listed in Table I could certainly result from secondary effects of LMB. Examples are the import factors importin alpha, importin 4, importin 8, and transportin. Also, CRM1 itself accumulated in the nucleus of LMB-treated cells. Under physiological conditions, these proteins exit the nucleus in a complex with RanGTP. Inhibition of the CRM1 pathway and the accumulation of RanBP1 in the nucleus thus also might affect the shuttling of other transport factors in an indirect manner. Importantly, none of the proteins that we analyzed via fluorescence microscopy turned out to be a false positive, demonstrating the quality of our screen.
However, we expect false negatives, as our collection of potential CRM1 substrates is certainly not a comprehensive list of all possible cellular targets of the export receptor. Several factors could account for the fact that some established substrates were not identified in our screen. (i) The protein might simply not be expressed in our HeLa cells, or it might be of very low abundance. (ii) Certain CRM1 substrates might yield few or no ionizable tryptic peptides. (iii) The protein might be exported only upon a certain stimulus. (iv) A shuttling protein could be confined to the nucleus because of nuclear import's being highly dominant over nuclear export. In that case, a complete blockade of export by CRM1 would not lead to changes in concentrations in the cytosol or the nucleus. (v) Our protocol for subcellular fractionation might be inappropriate for certain proteins. Very small proteins, for example, could rapidly exit the nucleus via passive diffusion during cell lysis. As a result, they would not be found to accumulate in the nuclear fraction upon LMB treatment. Also, very large or very hydrophobic proteins could co-fractionate with nuclear proteins, even though they are in fact cytoplasmic proteins. Hence, bona fide CRM1 substrates would end up in the nuclear fraction, irrespective of the LMB treatment. Finally, proteins could be insoluble during the last extraction step. To circumvent these issues, different methods for subcellular fractionations would have to be compared.
Despite these shortcomings, our MS-based approach proved to be a valid and powerful method for the quantitative analysis of nucleocytoplasmic transport. Importantly, all of the CRM1 candidates that were tested could be validated through fluorescence microscopy, demonstrating the quality of our list of potential substrates. This approach can be used to analyze different cell lines under different growth conditions, leading to a more comprehensive list of CRM1 substrates. It will also allow the discovery of substrates with export signals that only occur in the three-dimensional structure of a protein and cannot be identified with the current bioinformatic tools. Recently, CRM1 inhibitors were suggested as therapeutic drugs for the treatment of various forms of cancer (66, 67). A list of CRM1 targets thus might help to enhance our understanding of cancer progression. Furthermore, other nucleocytoplasmic transport pathways could be investigated in a similar manner. For many nuclear import or export receptors, only a limited number of physiological substrates are known. Very recently, a SILAC-based approach was used to identify substrates of the import receptor transportin (68). Cells could be treated with or without siRNAs against transport receptors, and subcellular fractions could be analyzed via mass spectrometry. In a similar approach, nucleoporins could be analyzed, as some of them are known to specifically affect the localization of a subset of cellular proteins (69).
Supplementary Material
Acknowledgments
We thank Drs. Terje Johansen (Tromsø, Norway), Kuan-Teh Jeang (Bethesda, MD), Carsten Janke (Paris, France), Jukka Westermarck (Turku, Finland), Robert Tsai (Houston, TX) and Achim Dickmanns (Göttingen, Germany) for very helpful reagents; Uwe Plessmann (Göttingen, Germany) for technical support; Miroslav Nikolov (Göttingen, Germany) for discussions; and Anne Clancy (Göttingen, Germany) for critically reading the manuscript.
Footnotes
* This project was supported by a grant from the DFG to R.K. (KE 660/5-2) and by funding from the International Max Planck Research School (IMPRS) in Molecular Biology awarded to S.K.
 This article contains supplemental material.
This article contains supplemental material.
2 J. Westermarck and J. Okkeri, personal communication.
1 The abbreviations used are:
- CCP1
- cytosolic carboxypeptidase 1
- CIP2A
- cancerous inhibitor of protein phosphatase 2A
- CRM1
- chromosome region maintenance 1/exportin1/Exp1/Xpo1
- eIF6
- eukaryotic translation initiation factor 6
- GNL3L
- guanine nucleotide-binding protein-like 3-like protein
- GTP
- guanosine-5′-triphosphate
- LMB
- leptomycin B
- NES
- nuclear export signal
- NPC
- nuclear pore complex
- PDCD2L
- programmed cell death protein 2-like protein
- PP2A
- protein phosphatase 2A
- SILAC
- stable isotope labeling with amino acids in cell culture
- SPN1
- snurportin 1
- TPB
- transport buffer.
REFERENCES
- 1. Fernandez-Martinez J., Rout M. P. (2012) A jumbo problem: mapping the structure and functions of the nuclear pore complex. Curr. Opin. Cell Biol. 24, 92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Onischenko E., Weis K. (2011) Nuclear pore complex—a coat specifically tailored for the nuclear envelope. Curr. Opin. Cell Biol. 23, 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fried H., Kutay U. (2003) Nucleocytoplasmic transport: taking an inventory. Cell. Mol. Life Sci. 60, 1659–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wente S. R., Rout M. P. (2010) The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010, 10.1101/cshperspect.a000562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hutten S., Kehlenbach R. H. (2007) CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol. 17, 193–201 [DOI] [PubMed] [Google Scholar]
- 6. Fischer U., Huber J., Boelens W. C., Mattaj I. W., Lührmann R. (1995) The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 82, 475–483 [DOI] [PubMed] [Google Scholar]
- 7. Wen W., Meinkoth J. L., Tsien R. Y., Taylor S. S. (1995) Identification of a signal for rapid export of proteins from the nucleus. Cell 82, 463–473 [DOI] [PubMed] [Google Scholar]
- 8. Kutay U., Güttinger S. (2005) Leucine-rich nuclear-export signals: born to be weak. Trends Cell Biol. 15, 121–124 [DOI] [PubMed] [Google Scholar]
- 9. Güttler T., Madl T., Neumann P., Deichsel D., Corsini L., Monecke T., Ficner R., Sattler M., Görlich D. (2010) NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat. Struct. Mol. Biol. 17, 1367–1376 [DOI] [PubMed] [Google Scholar]
- 10. Xu D., Farmer A., Collett G., Grishin N. V., Chook Y. M. (2012) Sequence and structural analyses of nuclear export signals in the NESdb database. Mol. Biol. Cell 23, 3677–3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu D., Grishin N. V., Chook Y. M. (2012) NESdb: a database of NES-containing CRM1 cargos. Mol. Biol. Cell 23, 3673–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Engelsma D., Bernad R., Calafat J., Fornerod M. (2004) Supraphysiological nuclear export signals bind CRM1 independently of RanGTP and arrest at Nup358. EMBO J. 23, 3643–3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ayers S. D., Nedrow K. L., Gillilan R. E., Noy N. (2007) Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPARgamma by FABP4. Biochemistry 46, 6744–6752 [DOI] [PubMed] [Google Scholar]
- 14. Dong X., Biswas A., Suel K. E., Jackson L. K., Martinez R., Gu H., Chook Y. M. (2009) Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature 458, 1136–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Monecke T., Güttler T., Neumann P., Dickmanns A., Görlich D., Ficner R. (2009) Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science 324, 1087–1091 [DOI] [PubMed] [Google Scholar]
- 16. Paraskeva E., Izaurralde E., Bischoff F. R., Huber J., Kutay U., Hartmann E., Lührmann R., Görlich D. (1999) CRM1-mediated recycling of snurportin 1 to the cytoplasm. J. Cell Biol. 145, 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fu S. C., Imai K., Horton P. (2011) Prediction of leucine-rich nuclear export signal containing proteins with NESsential. Nucleic Acids Res. 39, e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. la Cour T., Kiemer L., Molgaard A., Gupta R., Skriver K., Brunak S. (2004) Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 17, 527–536 [DOI] [PubMed] [Google Scholar]
- 19. la Cour T., Gupta R., Rapacki K., Skriver K., Poulsen F. M., Brunak S. (2003) NESbase version 1.0: a database of nuclear export signals. Nucleic Acids Res. 31, 393–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kehlenbach R. H., Assheuer R., Kehlenbach A., Becker J., Gerace L. (2001) Stimulation of nuclear export and inhibition of nuclear import by a Ran mutant deficient in binding to Ran-binding protein 1. J. Biol. Chem. 276, 14524–14531 [DOI] [PubMed] [Google Scholar]
- 21. Mueller L., Cordes V. C., Bischoff F. R., Ponstingl H. (1998) Human RanBP3, a group of nuclear RanGTP binding proteins. FEBS Lett. 427, 330–336 [DOI] [PubMed] [Google Scholar]
- 22. Nemergut M. E., Lindsay M. E., Brownawell A. M., Macara I. G. (2002) Ran-binding protein 3 links Crm1 to the Ran guanine nucleotide exchange factor. J. Biol. Chem. 277, 17385–17388 [DOI] [PubMed] [Google Scholar]
- 23. Oka M., Asally M., Yasuda Y., Ogawa Y., Tachibana T., Yoneda Y. (2010) The mobile FG nucleoporin Nup98 is a cofactor for Crm1-dependent protein export. Mol. Biol. Cell 21, 1885–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waldmann I., Spillner C., Kehlenbach R. H. (2012) The nucleoporin-like protein NLP1 (hCG1) promotes CRM1-dependent nuclear protein export. J. Cell Sci. 125, 144–154 [DOI] [PubMed] [Google Scholar]
- 25. Hamamoto T., Gunji S., Tsuji H., Beppu T. (1983) Leptomycins A and B, new antifungal antibiotics. I. Taxonomy of the producing strain and their fermentation, purification and characterization. J. Antibiot. (Tokyo) 36, 639–645 [DOI] [PubMed] [Google Scholar]
- 26. Fornerod M., Ohno M., Yoshida M., Mattaj I. W. (1997) CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90, 1051–1060 [DOI] [PubMed] [Google Scholar]
- 27. Wolff B., Sanglier J. J., Wang Y. (1997) Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 4, 139–147 [DOI] [PubMed] [Google Scholar]
- 28. Kudo N., Matsumori N., Taoka H., Fujiwara D., Schreiner E. P., Wolff B., Yoshida M., Horinouchi S. (1999) Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. U.S.A. 96, 9112–9117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Charneau P., Mirambeau G., Roux P., Paulous S., Buc H., Clavel F. (1994) HIV-1 reverse transcription. A termination step at the center of the genome. J. Mol. Biol. 241, 651–662 [DOI] [PubMed] [Google Scholar]
- 30. Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. (1994) Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley-Interscience, New York [Google Scholar]
- 31. Holden P., Horton W. A. (2009) Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes 2, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hutten S., Flotho A., Melchior F., Kehlenbach R. H. (2008) The Nup358-RanGAP complex is required for efficient importin alpha/beta-dependent nuclear import. Mol. Biol. Cell 19, 2300–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gasteier J. E., Madrid R., Krautkramer E., Schroder S., Muranyi W., Benichou S., Fackler O. T. (2003) Activation of the Rac-binding partner FHOD1 induces actin stress fibers via a ROCK-dependent mechanism. J. Biol. Chem. 278, 38902–38912 [DOI] [PubMed] [Google Scholar]
- 34. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 35. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 36. Huang D. W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 37. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., von Mering C. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dinkel H., Michael S., Weatheritt R. J., Davey N. E., Van Roey K., Altenberg B., Toedt G., Uyar B., Seiler M., Budd A., Jodicke L., Dammert M. A., Schroeter C., Hammer M., Schmidt T., Jehl P., McGuigan C., Dymecka M., Chica C., Luck K., Via A., Chatr-Aryamontri A., Haslam N., Grebnev G., Edwards R. J., Steinmetz M. O., Meiselbach H., Diella F., Gibson T. J. (2012) ELM—the database of eukaryotic linear motifs. Nucleic Acids Res. 40, D242–D251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hutten S., Kehlenbach R. H. (2006) Nup214 is required for CRM1-dependent nuclear protein export in vivo. Mol. Cell. Biol. 26, 6772–6785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guan T., Kehlenbach R. H., Schirmer E. C., Kehlenbach A., Fan F., Clurman B. E., Arnheim N., Gerace L. (2000) Nup50, a nucleoplasmically oriented nucleoporin with a role in nuclear protein export. Mol. Cell. Biol. 20, 5619–5630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Melchior F., Sweet D. J., Gerace L. (1995) Analysis of Ran/TC4 function in nuclear protein import. Methods Enzymol. 257, 279–291 [DOI] [PubMed] [Google Scholar]
- 42. Pollard V. W., Michael W. M., Nakielny S., Siomi M. C., Wang F., Dreyfuss G. (1996) A novel receptor-mediated nuclear protein import pathway. Cell 86, 985–994 [DOI] [PubMed] [Google Scholar]
- 43. Biswas A., Mukherjee S., Das S., Shields D., Chow C. W., Maitra U. (2011) Opposing action of casein kinase 1 and calcineurin in nucleo-cytoplasmic shuttling of mammalian translation initiation factor eIF6. J. Biol. Chem. 286, 3129–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yedavalli V. S., Neuveut C., Chi Y. H., Kleiman L., Jeang K. T. (2004) Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 119, 381–392 [DOI] [PubMed] [Google Scholar]
- 45. Ho J. H., Kallstrom G., Johnson A. W. (2000) Nmd3p is a Crm1p-dependent adapter protein for nuclear export of the large ribosomal subunit. J. Cell Biol. 151, 1057–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kato J. Y., Yoneda-Kato N. (2009) Mammalian COP9 signalosome. Genes Cells 14, 1209–1225 [DOI] [PubMed] [Google Scholar]
- 47. Tomoda K., Kubota Y., Arata Y., Mori S., Maeda M., Tanaka T., Yoshida M., Yoneda-Kato N., Kato J. Y. (2002) The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J. Biol. Chem. 277, 2302–2310 [DOI] [PubMed] [Google Scholar]
- 48. Shin J. (1998) P62 and the sequestosome, a novel mechanism for protein metabolism. Arch. Pharm. Res. 21, 629–633 [DOI] [PubMed] [Google Scholar]
- 49. Geetha T., Vishwaprakash N., Sycheva M., Babu J. R. (2012) Sequestosome 1/p62: across diseases. Biomarkers 17, 99–103 [DOI] [PubMed] [Google Scholar]
- 50. Moscat J., Diaz-Meco M. T., Wooten M. W. (2007) Signal integration and diversification through the p62 scaffold protein. Trends Biochem. Sci. 32, 95–100 [DOI] [PubMed] [Google Scholar]
- 51. Meng L., Zhu Q., Tsai R. Y. (2007) Nucleolar trafficking of nucleostemin family proteins: common versus protein-specific mechanisms. Mol. Cell. Biol. 27, 8670–8682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Junttila M. R., Puustinen P., Niemela M., Ahola R., Arnold H., Bottzauw T., Ala-aho R., Nielsen C., Ivaska J., Taya Y., Lu S. L., Lin S., Chan E. K., Wang X. J., Grenman R., Kast J., Kallunki T., Sears R., Kahari V. M., Westermarck J. (2007) CIP2A inhibits PP2A in human malignancies. Cell 130, 51–62 [DOI] [PubMed] [Google Scholar]
- 53. Harris A., Morgan J. I., Pecot M., Soumare A., Osborne A., Soares H. D. (2000) Regenerating motor neurons express Nna1, a novel ATP/GTP-binding protein related to zinc carboxypeptidases. Mol. Cell. Neurosci. 16, 578–596 [DOI] [PubMed] [Google Scholar]
- 54. Rogowski K., van Dijk J., Magiera M. M., Bosc C., Deloulme J. C., Bosson A., Peris L., Gold N. D., Lacroix B., Grau M. B., Bec N., Larroque C., Desagher S., Holzer M., Andrieux A., Moutin M. J., Janke C. (2010) A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 143, 564–578 [DOI] [PubMed] [Google Scholar]
- 55. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 56. Golebiowski F., Matic I., Tatham M. H., Cole C., Yin Y., Nakamura A., Cox J., Barton G. J., Mann M., Hay R. T. (2009) System-wide changes to SUMO modifications in response to heat shock. Sci. Signal 2, ra24. [DOI] [PubMed] [Google Scholar]
- 57. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal 3, ra3. [DOI] [PubMed] [Google Scholar]
- 58. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 59. Boisvert F. M., Ahmad Y., Gierlinski M., Charriere F., Lamont D., Scott M., Barton G., Lamond A. I. (2012) A quantitative spatial proteomics analysis of proteome turnover in human cells. Mol. Cell. Proteomics 11, M111.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wild T., Horvath P., Wyler E., Widmann B., Badertscher L., Zemp I., Kozak K., Csucs G., Lund E., Kutay U. (2010) A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS 8, e1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sanchez P., De Carcer G., Sandoval I. V., Moscat J., Diaz-Meco M. T. (1998) Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol. Cell. Biol. 18, 3069–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wooten M. W., Geetha T., Seibenhener M. L., Babu J. R., Diaz-Meco M. T., Moscat J. (2005) The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J. Biol. Chem. 280, 35625–35629 [DOI] [PubMed] [Google Scholar]
- 63. Perander M., Bjorkoy G., Johansen T. (2001) Nuclear import and export signals enable rapid nucleocytoplasmic shuttling of the atypical protein kinase C lambda. J. Biol. Chem. 276, 13015–13024 [DOI] [PubMed] [Google Scholar]
- 64. Johnson C., Van Antwerp D., Hope T. J. (1999) An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J. 18, 6682–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Flegg C. P., Sharma M., Medina-Palazon C., Jamieson C., Galea M., Brocardo M. G., Mills K., Henderson B. R. (2010) Nuclear export and centrosome targeting of the protein phosphatase 2A subunit B56alpha: role of B56alpha in nuclear export of the catalytic subunit. J. Biol. Chem. 285, 18144–18154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Inoue H., Kauffman M., Shacham S., Landesman Y., Yang J., Evans C. P., Weiss R. H. (2012) CRM1 blockade by selective inhibitors of nuclear export (SINE) attenuates kidney cancer growth. J Urol. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ranganathan P., Yu X., Na C., Santhanam R., Shacham S., Kauffman M., Walker A., Klisovic R., Blum W., Caligiuri M., Croce C. M., Marcucci G., Garzon R. (2012) Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 120, 1765–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kimura M., Kose S., Okumura N., Imai K., Furuta M., Sakiyama N., Tomii K., Horton P., Takao T., Imamoto N. (2012) Identification of cargo proteins specific for the nucleocytoplasmic transport carrier transportin by combination of an in vitro transport system and SILAC-based quantitative proteomics. Mol. Cell. Proteomics (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wälde S., Thakar K., Hutten S., Spillner C., Nath A., Rothbauer U., Wiemann S., Kehlenbach R. H. (2012) The nucleoporin Nup358/RanBP2 promotes nuclear import in a cargo- and transport receptor-specific manner. Traffic 13, 218–233 [DOI] [PubMed] [Google Scholar]
- 70. Miki T., Okawa K., Sekimoto T., Yoneda Y., Watanabe S., Ishizaki T., Narumiya S. (2009) mDia2 shuttles between the nucleus and the cytoplasm through the importin-{alpha}/{beta}- and CRM1-mediated nuclear transport mechanism. J. Biol. Chem. 284, 5753–5762 [DOI] [PubMed] [Google Scholar]
- 71. Huang T. T., Kudo N., Yoshida M., Miyamoto S. (2000) A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc. Natl. Acad. Sci. U.S.A. 97, 1014–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Seiser R. M., Sundberg A. E., Wollam B. J., Zobel-Thropp P., Baldwin K., Spector M. D., Lycan D. E. (2006) Ltv1 is required for efficient nuclear export of the ribosomal small subunit in Saccharomyces cerevisiae. Genetics 174, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Toyoshima F., Moriguchi T., Wada A., Fukuda M., Nishida E. (1998) Nuclear export of cyclin B1 and its possible role in the DNA damage-induced G2 checkpoint. EMBO J. 17, 2728–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Harhaj E. W., Sun S. C. (1999) Regulation of RelA subcellular localization by a putative nuclear export signal and p50. Mol. Cell. Biol. 19, 7088–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Richards S. A., Lounsbury K. M., Carey K. L., Macara I. G. (1996) A nuclear export signal is essential for the cytosolic localization of the Ran binding protein, RanBP1. J. Cell Biol. 134, 1157–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Samuels A. L., Klinken S. P., Ingley E. (2009) Liar, a novel Lyn-binding nuclear/cytoplasmic shuttling protein that influences erythropoietin-induced differentiation. Blood 113, 3845–3856 [DOI] [PubMed] [Google Scholar]
- 77. Dong J. M., Lau L. S., Ng Y. W., Lim L., Manser E. (2009) Paxillin nuclear-cytoplasmic localization is regulated by phosphorylation of the LD4 motif: evidence that nuclear paxillin promotes cell proliferation. Biochem. J. 418, 173–184 [DOI] [PubMed] [Google Scholar]
- 78. Petit M. M., Fradelizi J., Golsteyn R. M., Ayoubi T. A., Menichi B., Louvard D., Van de Ven W. J., Friederich E. (2000) LPP, an actin cytoskeleton protein related to zyxin, harbors a nuclear export signal and transcriptional activation capacity. Mol. Biol. Cell 11, 117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nix D. A., Fradelizi J., Bockholt S., Menichi B., Louvard D., Friederich E., Beckerle M. C. (2001) Targeting of zyxin to sites of actin membrane interaction and to the nucleus. J. Biol. Chem. 276, 34759–34767 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.