

Abstract
Enzymes can be post-translationally modified, leading to isoforms with different properties. The phenotypic consequences of the quantitative variability of isoforms have never been studied. We used quantitative proteomics to dissect the relationships between the abundances of the enzymes and isoforms of alcoholic fermentation, metabolic traits, and growth-related traits in Saccharomyces cerevisiae. Although the enzymatic pool allocated to the fermentation proteome was constant over the culture media and the strains considered, there was variation in abundance of individual enzymes and sometimes much more of their isoforms, which suggests the existence of selective constraints on total protein abundance and trade-offs between isoforms. Variations in abundance of some isoforms were significantly associated to metabolic traits and growth-related traits. In particular, cell size and maximum population size were highly correlated to the degree of N-terminal acetylation of the alcohol dehydrogenase. The fermentation proteome was found to be shaped by human selection, through the differential targeting of a few isoforms for each food-processing origin of strains. These results highlight the importance of post-translational modifications in the diversity of metabolic and life-history traits.
The key problem in understanding genotype-phenotype relationships is the complexity arising from multiple levels of cellular functioning. Among them, metabolic networks involve series of interconnected chemical reactions catalyzed by enzymes, allowing the transformation of input substrates into intermediate or final metabolites. These networks play an essential role in an organism's growth, reproduction, and ability to maintain cell integrity and to respond to environmental changes (1, 2). The metabolic fluxes, as well as the metabolite concentrations, are governed by the activity of the enzymes, which depends on three types of factors: kinetic parameters, enzyme abundance, and activation state of the enzyme. The kinetic parameters are determined by the sequence and the three-dimensional structure of the protein (3). The abundance of the enzymes is the result of numerous molecular processes taking place from the transcriptional to the translational level, including epigenetic modifications of the DNA and chromatin (4), transcriptional regulation by transcription factors (5), mRNA capping and splicing and small RNA regulation (6), protein turnover (7), etc. The enzyme activation state is primarily because of post-translational modifications of the native protein, themselves highly regulated (8, 9). Other mechanisms involved in enzyme activity are protein-protein interactions and allosteric regulation, such mechanisms being sometimes mediated through post-translational modifications (10, 11). The resulting isoforms can display differences in activity, affinity for partners (protein or effectors), and stability (12). The most studied modification is the reversible activation and inactivation of enzymes by phosphorylation (11, 13, 14), but other modifications are documented, such as acetylation that alters enzymatic activity and stability (15, 16) or fatty-acid modifications affecting cellular localization (17).
Thus, there are multiple levels to modulate metabolic phenotypes, and the identification of the most effective ones has been the subject of much interest (18–26). Recent data suggest that upstream levels of regulation have moderate control over metabolic changes. For example, genes involved in redox regulation in Arabidopsis thaliana have quite stable expression whereas the corresponding fluxes and metabolite contents display marked genetic variations (27). Similarly, various studies on bacteria have shown that transcription is not sufficient to explain the variation of metabolic fluxes or phenotypes in Escherichia coli (28), Bacillus subtilis (29), Corynebacterium glutamicum (30), Synechocystis sp (31), or Mycoplasma pneumonia (19). Systems biology studies, including transcriptomic and proteomic approaches, have suggested that the transcriptome alone does not provide a reliable indication of flux distribution in metabolic networks in yeast (32–34). By contrast, manipulating post-translational processes may have marked consequences on metabolic flux. For example in plants, abolition of the post-translational regulation of just one enzyme (a nitrate reductase) is sufficient to increase the corresponding flux of nitric oxide (35). In Bacillus subtilis, the phosphorylation state of one protein (Crh, a phosphocarrier protein) controls the flux through the methylglyoxal pathway, which is an alternative route of glycolysis in bacteria (36). In Salmonella enterica, lysine acetylation was shown to coordinate central metabolic pathways such as carbon use (37) or glycolysis and the TCA cycle in human (15). Thus, phosphorylation, acetylation, and other post-translational modifications emerge as major regulators of central metabolic pathways, yet are largely underestimated because of the lack of reliable approaches for systematic analyses (38). Their genetic and plastic variability together with their effects on the phenotype remain to be studied.
The present work focuses on the genetic and plastic variability of enzyme and isoform abundances in yeast, and on the possible consequences of this variability on metabolic and “life-history” traits, i.e. traits characterizing the lifespan of the organism such as growth or survival. Quantitative proteomics based on two-dimensional electrophoresis (2-DE)1 is well adapted for this purpose, because the different isoforms of a protein often have different electrophoretic mobility, resulting in distinguishable spots. We applied quantitative proteomics to Saccharomyces cerevisiae alcoholic fermentation (AF), a central metabolic pathway exploited for millennia in three important human food-processes: beer and wine production (39–41), and bread leavening (42). The yeast AF enzymes are well-known and most of them have been identified on 2-DE maps (43–47). In a previous work, we showed that life-history traits (carrying capacity and cell size) and metabolic phenotypes (maximum CO2 flux, ethanol, acetate, and glycerol content) displayed large variation, with medium effects usually higher than the strain effects (48). On the other hand, trade-offs were found between metabolic and life-history traits (49). A recent work showed that the expression variation of a few genes involved in the upper part of glycolysis could drive changes in life-history strategies (50), indicating that life-history traits might be under the control of some metabolic enzymes.
All these observations prompted us to investigate the possible control of metabolic and life-history traits by a large panel of AF enzymes tested under various conditions. Our experimental design included nine food-processing strains grown in triplicate in three different synthetic media mimicking the dough/wort/grape must found in bakery, brewery, and enology, to: (1) quantify thoroughly the abundances of 18 AF enzymes and their isoforms in a sample of 27 medium x strain combinations; (2) compare the genetic and plastic variability of the enzymes and their isoforms; (3) search which enzymes or isoforms, if any, are related to CO2 flux, AF metabolite concentrations, and life-history traits, and thus may exert control over metabolism and life-history strategy. Our results highlight the preponderant role of post-translational modifications in the variation of metabolic phenotypes and life-history traits.
EXPERIMENTAL PROCEDURES
A detailed Materials and Methods section is available as Supporting Information.
Biological Material and Synthetic Fermentative Media
Nine S. cerevisiae strains were used (supplemental Table S1), from enology (E1 to E4), brewery (B1 and B2), and distillery origins (D1 to D3). All strains were grown in triplicates in three synthetic fermentative media differing by their amount of sugar, nitrogen, pH, osmotic pressure, and anaerobic growth factors to reflect main changes of fermentation medium between brewery (BREM), bakery (BAM), and winery (WIM) contexts (supplemental Table S2).
Metabolic and Life-history Traits
For each of the 81 fermentations (nine strains × three media × three repetitions), we measured the following metabolic and life-history traits: CO2 specific flux (the CO2 production rate per cell, g/h/cell), ethanol production (% vol/cell), acetic acid concentration (g/cell), glycerol concentration (g/cell), biomass (gcell), carrying capacity (K or maximum population size in cells/ml), and cell size (μm [diameter]).
Quantitative Proteomics
One sample per fermentation (81 fermentations) was harvested at comparable physiological stage (maximal CO2 production rate before nutriment starvation). One 2-DE gel per sample was run and stained with colloidal-blue, which offers a linear relationship between spot quantification and protein abundance (47) and thus allows accurate comparison of spot abundance between and within 2-DE gels. Spots of interest were quantified using Progenesis software (Nonlinear Dynamics, Newcastle, UK) and identified using mass spectrometry (MS). Almost all enzymes involved in glycolysis and ethanol pathways were identified, or at least the major and most abundant isozymes in case of paralogous genes.
Statistical Analyses
The variation of each isoform or enzyme abundance (in the latter case the isoforms of the enzyme were summed) was investigated through a mixed ANOVA model:
 |
where Z is the variable, medium is the medium effect (i = 1, 2, 3), strain is the strain effect (j = 1, …, 9), block is the random block effect (effect of each weekly experimental repetition, k = 1, …, 11), position is the random position effect (bioreactor position, l = 1, …, 15), batch is the random 2-DE batch effect (m = 1, …, 6), medium * strain is the interaction effect between medium and strain factors, and ε is the residual error. For further analyses (hierarchical clustering, PCA, LDA, regression analysis, etc.), we used the mean abundances predicted by the ANOVA model, that is, corrected for the random effects (block, position, and batch effects). The final data set is available as supplementary Data set S4. Hierarchical clustering was made using R (Ward's clustering method and Euclidean distances). Proteomic-trait relationships were explored using multiple linear regression to find enzymes and isoforms whose abundance was significantly related to metabolic and life-history traits. The impact of human domestication was investigated using linear discriminant analysis (LDA) to discriminate beer, distillery, and wine strains using R. Discriminant isoforms were identified through stepwise variable selection and through the calculation of the « ability to separate » (AS) criterion.
RESULTS
To explore the extent of phenotypic diversity of enzymes abundance in alcoholic fermentation pathway, we chose nine food-processing strains of S. cerevisiae (supplemental Table S1) from different food origins, and we performed anaerobic alcoholic fermentations in triplicate using three different synthetic media (supplemental Table S2) that mimicked the dough/wort/grape must found in bakery, brewery, and enology (48). For each of the 81 fermentations (9 strains × 3 media × 3 repetitions), cell samples for proteomics assays were harvested during the fermentations when the CO2 production rate per cell (the flux) was close to its maximum, so that the cells displayed comparable physiological stage. Using quantitative proteomics, we identified and quantified the relative abundance of 15 enzymes of glycolysis and ethanol pathways, one enzyme of acetate pathway and two enzymes of glycerol pathway (Fig. 1). Those 18 enzymes were representative of the alcoholic fermentation metabolic process and will be thereafter called the fermentation proteome. For most enzymes, several spots, corresponding to different post-translational forms (isoforms) were identified (Fig. 1), allowing subsequent analyses both at the enzyme level (sum of all isoforms for each enzyme) and at the post-translational modification level (individual isoforms). The few suspected allelic variants identified by 2-DE (shifting trains of spots, Fig. 1B) were confirmed by gene sequence (supplementary Information Data set S1). In these last cases, we compared isoforms having the same position within the train of spots (acidic, basic and intermediary isoforms) rather than co-located spots (see Materials and Methods in Supplementary Information). The mean coefficient of variation between biological triplicates for isoforms was 18.4%, which is low enough to accurately detect small abundance variations. Proteomic data were released in the PROTICdb database, a web-based application designed for large-scale proteomic programs to store and query data related to protein separation by 2-DE and protein identification by MS (http://moulon.inra.fr/protic/adaptalevure. See Supplementary Information for details).
Fig. 1.

Linking glycolysis, ethanol, glycerol, and acetate pathways to 2-DE proteomics. Plain boxes: identified enzymes. Striped boxes: unidentified enzymes corresponding to minor or low-abundant isozymes. Stars indicate specific spots corresponding to allelic variants (see B). A, 2-DE: Localization of the different enzymes within the master gel (co-electrophoresis of all samples). MW and pI stand for Molecular Weight (kDa) and isoelectric point, respectively. B, Allelic variants: five allelic variants were identified for Pfk1p, Pgk1p, Tdh3p, Eno1p, and Eno2p on the basis of the electrophoretic mobility of the corresponding spots, and were confirmed by protein sequence (supplementary Information Dataset S1). The detail of enzymes and metabolites abbreviations is Pgi, Phosphoglucoisomerase; Pfk, Phosphofructokinase; Fba, Fructose-biphosphatase aldolase; Tpi, Triose-phosphate isomerase; Tdh, Triose-phosphate dehydrogenase; Pgk, 3-Phosphoglycerate kinase; Gpm, Glycerate phosphomutase; Eno, Enolase; Pyk, Pyruvate kinase; Pdc, Pyruvate decarboxylase; Adh, Alcohol dehydrogenase; Gpd, Glycerol-3-phosphate dehydrogenase; Hor, Hyperosmolarity-responsive (dl-glycerol-3-phosphatase); Rhr, Related to HOR2 (dl-glycerol-3-phosphatase); Ald, Aldehyde dehydrogenase; Glucose-6P, glucose-6-phosphate; F6P, fructose-6-phosphate; FBP, Fructose-1,6-biphosphate; DHAP, dihydroxyacetone phosphate; G3P, Glycerol-3-phosphate; GA3P, glyceraldehyde-3-phosphate; BPG, glycerate-1,3-biphosphate; 3PG, glycerate-3-phosphate; 2PG, glycerate-2-phosphate; PEP, phosphoenol-pyruvate.
The Fermentation Proteome is Constrained
We first analyzed the different sources of variation for protein abundance at different levels: At the whole fermentation proteome level (sum of enzymes of glycolysis, ethanol, acetate, and glycerol pathways), at the enzymes level (sum of isoforms for each enzyme), and at the post-translational level using individual isoforms (Table I). Considered globally, the sum of the abundance of the enzymes involved in the fermentation proteome (42 isoforms) represents on average 32.87 ± 1.89% of the total analyzed proteome (2265 ± 209 spots depending on the 2-DE gel). Variance analysis (ANOVA) revealed that such fermentation pool displayed no medium, no strain, and no medium × strain interaction effects, indicating that the enzymatic pool allocated to glycolysis, ethanol, acetate, and glycerol pathways is invariant whatever the medium and strain considered. Within the fermentation proteome, the abundance from one enzyme to another (sum of all isoforms for each enzyme) varied greatly (supplemental Fig. S1), with highly abundant proteins (Tdh2p, Tdh3p, Eno2p, Fba1p) and poorly represented enzymes (PfK1p, Ald6p, Pgi1p, Hor2p, Rhr2p). However, although abundance had important variation within enzymes, among strains, the proportion allocated to each enzyme appeared to be globally conserved (Fig. 2). Indeed, the mean coefficients of variation of the 18 enzymes (CV = 0.26) and 42 isoforms (CV = 0.35) were significantly lower than the mean coefficient of variation of the 688 other common spots (non-AF proteins) on the 2-DE gels (CV = 1.24, Kolmogorov-Smirnov test, p value = 2.48 × 10−10 and 2.89 × 10−15, respectively). However, although the abundance of AF enzymes appeared more constrained than the whole proteome, significant variations were found, in particular for the enzymes of the last part of glycolysis (except Tdh3p and Gpm1p), as well as for the enzymes of ethanol, acetate, and glycerol pathways (Table I). A significant strain effect was found for most enzymes (13/18), which accounted for 21% to 68% of total variation (Table I). The medium effect was significant for only 6/18 enzymes and accounted for much less of the total variation (between 4 and 28%, Table I). The medium × strain interaction effect was significant for 2/18 enzymes, and accounted for 15% to 16% of total variation of the enzyme. Finally only 5/18 enzymes exhibited no strain or medium effect, and the average abundance of Pgi1p, Fba1p, and Tpi1p, corresponding to the first part of glycolysis, was similar in all the 27 medium × strain combinations. Therefore, we found a significant variation for enzyme abundance, which was better explained by genetic differences between strains than by plastic changes in response to variations of the culture medium.
Table I. Results of the ANOVAs: sums of squares for abundance of enzymes and their isoforms involved in glycolysis, ethanol, acetate, and glycerol pathways. For some enzymes/isoforms. data transformation was necessary to obtain normally-distributed residues: log transformation for the fermentation proteome, Pgi1p-1660, Pfk2p-4832, Fba1p-4759, Tdh1p-2775, Tdh2p-4872, Tdh2p-4740, Tdh3p-all isoforms, Tdh3p-acidic, Tdh3p-intermediary, Tdh3p-basic, Pgk1p-basic, Gpm1p-all isoforms, Gpm1p-3333, Gpm1p-3345, Gpm1p-3313, Eno1p-all isoforms, Eno2p-all isoforms, Eno2p-intermediary, Eno2p-acidic, Pyk1p-1587, Pdc1p-1606, Adh1p-all isoforms, Adh1p-4799, Ald6p-1565; inverse transformation for Fba1p-all isoforms, Fba1p-4758, Eno1p-acidic, Pyk1p-all isoforms, Pyk1p-1630, Pyk1p-1310, Pdc1p-4854, Adh1p-4808; and finally square root transformation for Tdh1p-2824 and Eno1p-intermediary. Variance was calculated across the 27 strain x medium combinations.
| Metabolic pathway | Enzymes | Isoforms reference | % of total sum of squares (df)a |
Variance | |||
|---|---|---|---|---|---|---|---|
| medium (2) | strain (8) | medium × strain (16) | residual (52) | ||||
| Fermentation proteome | all AF enzymes | all isoforms | 3.55 | 10.84 | 28.86 | 56.74 | 6.21E-02 |
| Glycolysis | Pgi1p | 1660 | 0.33 | 19.9 | 17.9 | 61.87 | 1.86E-05 |
| Pfk1p | all isoforms | 2.2 | 34.9*** | 27.62 | 35.29 | 1.37E-05 | |
| intermediary | 2.57 | 43.99*** | 24.85 | 28.59 | 3.85E-06 | ||
| basic | 1.96 | 31.2** | 19.09 | 47.75 | 1.81E-07 | ||
| acidic | 5.12 | 27.45** | 28.87 | 38.57 | 2.02E-06 | ||
| Pfk2p | 4832 | 0.82 | 35.26** | 27.79 | 36.13 | 1.94E-06 | |
| Fba1p | all isoforms | 2.18 | 9.57 | 24 | 64.25 | 5.69E-04 | |
| 4759 | 2.14 | 21.07 | 26.3 | 50.5 | 5.23E-05 | ||
| 4758 | 0 | 8.3 | 31.08 | 60.62 | 4.87E-04 | ||
| 2414 | 9.22 | 17.38 | 16.95 | 56.45 | 9.35E-05 | ||
| Tpi1p | 3406 | 1.71 | 11.57 | 19.81 | 66.9 | 9.08E-05 | |
| Tdh1p | all isoforms | 18.73** | 21.9** | 20.82 | 38.55 | 3.31E-03 | |
| 2775 | 13.79** | 19.81* | 27.79 | 38.61 | 2.58E-04 | ||
| 2757 | 14.46* | 20.09* | 19.27 | 46.18 | 9.84E-04 | ||
| 2729 | 12* | 21.56** | 28.25 | 38.19 | 5.41E-05 | ||
| 2824 | 10.55 | 29.07** | 10.8 | 49.58 | 1.02E-04 | ||
| Tdh2p | all isoforms | 1.77 | 37.89*** | 24.82 | 35.52 | 1.71E-03 | |
| 4732 | 2.56 | 50.81*** | 11.24 | 35.4 | 7.68E-05 | ||
| 4872 | 4.21* | 77.63*** | 5.2 | 12.96 | 9.01E-05 | ||
| 4740 | 0.49 | 56.38*** | 17.4 | 25.73 | 1.73E-03 | ||
| Tdh3p | all isoforms | 1.3 | 14.91 | 33.52 | 50.27 | 1.63E-03 | |
| basic | 1.51 | 17.71 | 22.79 | 57.99 | 5.16E-05 | ||
| intermediary | 1.22 | 12.24 | 34.52 | 52.02 | 1.13E-03 | ||
| acidic | 1.28 | 79.49*** | 5.79 | 13.44 | 3.11E-04 | ||
| Pgk1p | all isoforms | 8.49* | 48.63*** | 9.48 | 33.39 | 2.68E-03 | |
| basic | 20.61*** | 34.57*** | 13.09 | 31.74 | 2.70E-05 | ||
| intermediary | 5.58 | 42.82*** | 11.97 | 39.63 | 1.61E-03 | ||
| acidic | 7.67* | 53.39*** | 5.53 | 33.41 | 9.13E-05 | ||
| Gpm1p | all isoforms | 9.22 | 6.03 | 23.08 | 61.67 | 2.98E-04 | |
| 3333 | 9.71 | 2.83 | 23.73 | 63.72 | 1.98E-04 | ||
| 3345 | 5.47 | 6.35 | 17.42 | 70.75 | 1.39E-05 | ||
| 3313 | 2.89 | 24.26** | 32.97 | 39.88 | 3.74E-05 | ||
| Eno1p | all isoforms | 27.55*** | 26.87*** | 18.24 | 27.34 | 2.31E-03 | |
| basic | 27.34*** | 27.65*** | 15.64 | 29.37 | 6.54E-05 | ||
| intermediary | 21.82*** | 26.52** | 15.69 | 35.96 | 1.30E-03 | ||
| acidic | 16.07** | 31.99*** | 15.23 | 36.71 | 1.02E-04 | ||
| Eno2p | all isoforms | 3.39 | 27.88** | 20.81 | 47.92 | 1.88E-03 | |
| basic | 4.18 | 32.77*** | 21.16 | 41.9 | 5.08E-04 | ||
| intermediary | 1.52 | 22.85* | 32.17 | 43.46 | 1.38E-03 | ||
| acidic | 1.9 | 15.67 | 14.51 | 67.92 | 6.40E-05 | ||
| Pyk1p | all isoforms | 1.7 | 41.85*** | 19.56 | 36.89 | 2.99E-03 | |
| 1587 | 0.79 | 21.51 | 22.41 | 55.29 | 8.72E-05 | ||
| 1310 | 1.1 | 36.36*** | 22.03 | 40.51 | 1.46E-03 | ||
| 1630 | 2.74 | 42.2*** | 14.38 | 40.68 | 1.55E-04 | ||
| Ethanol | Pdc1p | all isoforms | 7.69 | 20.88* | 24.64 | 46.79 | 1.08E-03 |
| 4854 | 0.65 | 55.53*** | 11.52 | 32.3 | 9.72E-04 | ||
| 1589 | 2.28 | 66.54*** | 11.99 | 19.19 | 1.16E-03 | ||
| 1605 | 6.71 | 39.02*** | 9.49 | 44.78 | 1.15E-04 | ||
| 1606 | 3.64 | 29.71* | 10.56 | 56.1 | 1.65E-05 | ||
| Adh1p | all isoforms | 0.94 | 33.43*** | 23.38 | 42.24 | 7.22E-04 | |
| 4799 | 1.78 | 49.82*** | 17.3 | 31.1 | 1.90E-04 | ||
| 4808 | 0.92 | 27.33** | 28.75 | 43 | 2.45E-04 | ||
| Glycerol | Hor2p | 3129 | 6.76** | 63.72*** | 14.62* | 14.9 | 3.75E-05 |
| Rhr2p | 3003 | 4.08** | 68.49*** | 15.92** | 11.52 | 1.95E-04 | |
| Acetate | Ald6p | 1565 | 23.67 | 18.32 | 19.39 | 38.61 | 8.96E-06 |
a df: degree of freedom. Significance is indicated as follow: * significant at 5%; ** significant at 1%; *** significant at 0.1% (Benjamini-Hochberg correction for multiple testing).
Fig. 2.
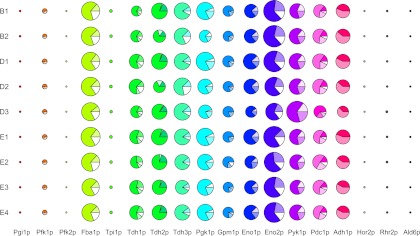
Distribution of the fermentation proteome within nine food-processing strains. B1 and B2, brewery strains; D1 to D3, distillery strains; E1 to E4, enology strains. The 18 enzymes involved in glycolysis, ethanol, glycerol and acetate pathways are illustrated by pies whose size is proportional to the mean enzyme abundance over the three media. Within each pie the different isoforms (if any) are represented by pie's slice of different colors.
The Isoforms of a Given Enzyme Display Different Patterns of Variation
The different isoforms of the fermentation enzymes were also analyzed individually (Table I). The isoforms of a given enzyme generally displayed different abundance patterns that were hidden when the analysis was performed at the enzyme level (sum of the isoforms). For example, three spots were identified for Gpm1p. Summing all isoforms, Gpm1 displayed neither medium nor strain effects, whereas spot 3313 displayed strain effect (higher relative abundance for E2 and D1 strains). For most enzymes, isoforms vary in different ways with respect to genetic and environmental factors (Table I). In addition, for some enzymes, the abundance variation of the different isoforms compensate, in part, for each other. For example, the global variance of Pdc1p abundance was twice lower than the sum of the variances of the four individual Pdc1p isoforms (1.08 × 10−3 versus 2.26 × 10−3). This showed that the different Pdc1p isoforms had negative covariance. Indeed, the abundance of three Pdc1p isoforms (1605, 1606, and 1589) was significantly lower for strain D3 whereas the abundance of the remaining Pdc1p isoform (spot 4854) was significantly higher, compensating in part for the variation of the others. Isoform compensation was found for four enzymes (Fba1p, Tdh2p, Eno2p, Pdc1p) that displayed less variation when considering the variance of the sum of the isoforms rather than the sum of the variance of the isoforms (Table I). Isoforms compensation was visible on Fig. 2: the total enzyme abundances (illustrated by pie sizes) varied weakly between strains whereas the distribution of the different isoforms of the enzymes (illustrated by pie's slices) was more variable for Tdh2p, Pdc1p, Fba1p, and Eno2p. Overall, analyses of variance showed that the different isoforms of a given enzyme can respond differently to environmental and genetic changes, and in some cases can compensate each other.
Global Patterns of Isoform Abundance Variation Reflect the Strain Genetic Diversity
To represent global patterns of protein abundance variation, we performed a hierarchical clustering of all 27 medium × strain combinations on the basis of individual mean isoform abundance over replicates (Fig. 3A). The resulting dendrogram showed a clustering according to the strains rather than the culture media and was close to the one obtained from genetic data (Fig. 3B). The hierarchical clustering was thus congruent with the analyses of variance of the isoforms and suggested that the variations in isoform abundance of the fermentation proteome are mainly genetically determined and displays limited plastic variation.
Fig. 3.

Hierarchical clustering using proteomic and genetic data. A, Proteomic relationships between nine food-processing strains in three fermentative media using the fermentation proteome data. B, Genetic relationships between nine food-processing strains using eight microsatellites. The robustness of the nodes was assessed through multiscale bootstrap resampling.
Because yeast strains used in different food processes may have experienced independent human domestication (51–53), we searched for enzymes and/or isoforms that could be involved in differentiating the strains according to their food origin (beer, distillery, and wine). We ran a LDA on the basis of isoform relative abundance with the food origin of the strains as grouping factor (Fig. 4A). The a posteriori probability to infer correctly the food origin of a strain was 0.96, indicating that it was possible to find a linear combination of isoforms that almost perfectly separated the samples according to the food origin of the strains, whatever the culture medium. A stepwise variable selection was then performed to determine which isoforms allowed such food origin discrimination and the ability to separate (AS) criterion was calculated (Fig. 4B). The isoform with the highest AS was Pdc1p-4854 that accounted for 32.56% of food origin discrimination, indicating that human domestication differentially targeted this isoform, directly or indirectly. Indeed, Pdc1p-4854 was significantly more abundant in distillery strains than in beer and wine strains (Fig. 4C). The acidic isoform of Tdh3p also accounted for 15.39% of food origin discrimination and separated wine strains from both distillery and beer strains. The unique isoform of Ald6–1565, one isoform of Tdh1p (2729), and one isoform of Pgk1p (intermediary) were associated with 7.51%, 7.13%, and 6.54% of food-origin discrimination, respectively. The other isoforms had lower ability to separate food origins (<5%). Notice that only few isoforms of the same enzyme appeared among the most discriminant factors in the LDA: one isoform out of the four of Pdc1p, one isoform out the three of Tdh3p, Tdh1p, and Pgk1p. This result indicates that the fermentation proteome was significantly shaped by human domestication, through the differential targeting of some isoforms of a few enzymes involved in fermentation.
Fig. 4.
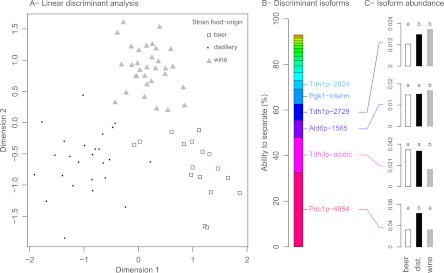
Discrimination of the food origin of the strains on the basis of the fermentation proteome. A, Linear discriminant analysis of the food origins of the strains (beer, distillery, and wine strains) on the basis of the isoform relative abundances. B, Values of the “Ability to Separate” (AS) criterion for the most significant isoforms. The other isoforms have an AS below 4%. C, Isoform mean relative abundance for each food origin. Means with different letters differ significantly (Duncan's multiple comparison, p < 0.05).
The CO2 Specific Flux is Related to Variation in Abundance of Specific Isoforms of Different Enzymes
The net outcome of glycolysis and alcoholic fermentation is the production of ethanol and ATP from glucose, along with CO2 release. Stepwise multiple regression analyses were performed to determine which enzymes or isoforms of the fermentation proteome (if any) were related to CO2 specific flux (i.e., the CO2 production rate per cell). For enzyme-flux regression analysis, the best model (i.e., lowering AIK criterion) accounted for 44.49% of the variation of the CO2 specific flux. For the isoform-flux regression analysis, the best model accounted for 79.50% of the variation of the CO2 specific flux (Table II), suggesting that the efficiency of the fermentation process is more related to the abundance of specific forms of different enzymes than to global enzyme abundances. Therefore we considered individual isoforms for further functional analysis. Eighteen isoforms were found to be significantly associated with the variation of the CO2 specific flux (Fig. 5A). Among them, the Pdc1p-1589 isoform accounted for 12.47% of the CO2 flux variation, which was the largest part of variation explained by a single isoform. However, its abundance was negatively correlated to the flux, which suggests that it may correspond to an inactive or poorly active form of Pdc1p. In addition, two isoforms of Tdh1p (spots 2757 and 2729) accounted for 10.07% and 8.50% of the flux variation, respectively, and both were positively correlated to the flux. An isoform of the alcohol dehydrogenase, Adh1p (spot 4799) was also found negatively correlated to the flux and accounted for 7.57% of its variation. Finally, an enzyme involved in the glycerol pathway, Hor2p (spot 3129), was found negatively related to the CO2 flux and accounted for 5.02% of the flux variation, whereas the isoforms of the other enzymes accounted for less than 5% of the flux variation. Thus, CO2 flux variation was associated with the variation in abundance of specific isoforms rather than by the variation in abundance of all isoforms belonging to a peculiar enzyme. Moreover, each isoform accounted for a limited proportion of the flux variation, suggesting that the control of CO2 flux is distributed among the different isoforms.
Table II. Percentage of variation accounted for by the multiple regression model for whole enzymes and for individual isoforms for seven metabolic or life-history traits.
| Metabolic of life-history traits | Whole enzymes | Individual isoforms |
|---|---|---|
| Flux | 44.49% | 79.50% |
| Ethanol per cell | 46.14% | 60.23% |
| Glycerol per cell | 49.96% | 69.05% |
| Acetate per cell | 46.53% | 68.00% |
| Biomass per cell | 34.22% | 56.58% |
| K (carrying capacity) | 67.28% | 87.60% |
| Cell size | 60.92% | 88.39% |
Fig. 5.
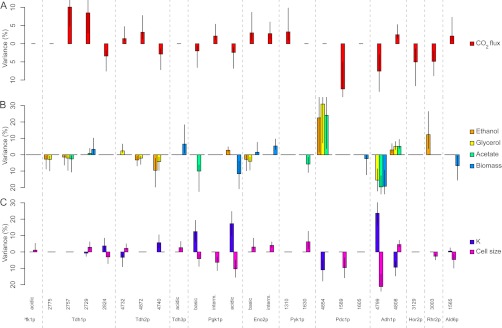
Relationship between seven metabolic and life-history traits and individual isoforms of the enzymes involved in alcoholic fermentation. The best model accounting for trait variation was established using stepwise multiple regression (see equations in supplemental Information - text file). All the isoforms included in the model are presented. A, Percentage of variance of the CO2 specific flux per cell accounted for by each isoform. B, Percentage of variance accounted for by each isoform for the concentration of ethanol, glycerol, and acetate per cell, and for biomass per cell. C, Percentage of variance accounted for by each isoform for two life-history traits: K (carrying capacity) and cell size.
The CO2 Specific Flux is Related to Phosphorylation and N-Terminal Acetylation of Some Enzymes
Additional experiments were run with a high-resolution mass spectrometer (QExactive, Thermo Scientific) to identify the underlying post-translational modification(s) differentiating the isoforms of the three enzymes (Pdc1p, Tdh1p, and Adh1p) that accounted for the highest parts of variation of CO2 specific flux. The mass spectrometry (MS) data were used to search specifically for phosphorylation and N-terminal acetylation, which are very common post-translational modifications in yeast and were previously described for Pdc1p, Tdh1p, and Adh1p (http://www.ibgc.u-bordeaux2.fr/YPM/, http://www.phosphogrid.org).
For the four isoforms of Pdc1p, we were unable to identify the causal post-translational modifications, due either to the absence of phosphorylation or N-terminal acetylation for this enzyme or to accessibility problems of the modified peptides. For Tdh1p-2824, we identified a phosphorylated serine (position 201) that discriminated Tdh1p-2824 and Tdhp-2757 (which may correspond to the native protein). Protein phosphorylation induces an acidic shift, which is congruent with isoform position on gel (Fig. 1). The post-translational modifications associated with the two other isoforms of Tdh1p (2729 and 2775) were not identified, but multiple combinations of post-translational modifications are possible, rendering hazardous their identification by mass spectrometry.
For Adh1p, we identified an N-terminal acetylation (after methionine excision) harbored by Adh1p-4808, whereas Adh1p-4799, the other isoform, probably corresponded to the native protein. N-terminal acetylation also induces an acidic shift, in accordance with Adh1p isoform location on 2-DE gels. Hence, from a functional viewpoint, our mass spectrometry analyses indicated that CO2 flux variation was mainly negatively associated with the variation in abundance of an unknown post-translational modified form of Pdc1p, the phosphorylation status of Tdh1p and the N-terminal acetylation status of Adh1p.
Final Metabolite Concentrations are Related To Some Specific Isoforms of Different Enzymes
During alcoholic fermentation, most of the consumed glucose (89.08%) is used to produce ethanol. The remaining glucose is used for biomass production (5.96%), glycerol (3.86%) and acetate (0.61%) synthesis (48). Other minor by-products, such as carbohydrate storage, represent less than 5‰ of initial glucose content. However, the AF product concentration at the end of the fermentation process varies greatly depending on the medium and the strain for a given fermentation volume (48). To determine whether some enzymes or isoforms could be related to the final concentration of AF products and production of biomass, stepwise multiple regression and bootstrap resampling were applied for ethanol, acetate, glycerol, and biomass production per cell. With no exception, individual isoforms accounted for a larger part of variation of each of the four AF products than did the whole enzymes (Table II).
Nine isoforms were found significantly related to ethanol production per cell (Fig. 5B), the first one being Pdc1p-4854 that accounted for 22.52% of its variation with a positive correlation between the abundance of the isoform and ethanol production. Two other isoforms, Rhr2p-3003 and Tdh2p-4740, accounted for a substantial part of the variation (12.29% and 9.55%, positive and negative correlation respectively). For acetate production (Fig. 5B), the fittest model included seven isoforms, two of them accounting for most of the variation: Pdc1p-4854 (24.08%, positive correlation) and Adh1p-4799 (19.66%, negative correlation). Variation in glycerol production was also mostly associated with the variation in abundance of these two isoforms (30.93% variation, positive correlation for Pdc1p-4854 and 15.54% variation, negative correlation for Adh1p-4799), whereas it was not associated with the variation of Hor2p and Rhr2p isozymes, that are directly involved in the glycerol pathway (Fig. 5B). Finally, regarding biomass production per cell, the fittest regression model contained eight isoforms of which the first ones, Adh1p-4799, Pgk1p-acidic, and Ald6p-1565, were negatively correlated to biomass and accounted for 19.31%, 11.59%, and 6.67% of variation respectively. Thus, the fate of glucose during AF seems to be mostly related to some specific isoforms of a few enzymes: increased level of Pdc1p-4854 was associated with an increase of ethanol, glycerol, and acetate production, whereas increased level of the nonacetylated form of Adh1p (Adh1p-4799) was associated with a decrease of biomass formation and to a lesser extent of acetate and glycerol concentrations. For Pdc1p, the isoform associated with ethanol, glycerol, and acetate production (Pdc1p-4854) differs from the one associated with CO2 specific flux variation (Pdc1p-1589) but belong to the same enzymes.
“Ant” and “Grasshopper” Strategies are Mainly Determined by the Degree of Acetylation of Adh1p
In previous works, two life-history traits, cell size and maximum population size (K, the carrying capacity) were found to be related to the glycolytic flux and to define a range of life-history strategies distributed between two extreme behaviors, metaphorically designated as ants and grasshoppers (49, 50, 54). Similarly to what was done for metabolic traits, stepwise multiple regression and bootstrap resampling on isoform abundances were applied to cell size and K, the carrying capacity (Fig. 5C). The two isoforms of alcohol dehydrogenase, Adh1p, accounted for a large part of variation of these two life-history traits. Variation in abundance of the non-N-terminal acetylated isoform (Adh1p-4799) was found to be positively associated with K and negatively associated to cell size (23.59% and 21.16% of variation respectively), whereas it was the opposite for the acetylated isoform of Adh1p (spot 4808) (9.38% and 4.55% for K and cell size, negative and positive correlation respectively). Some isoforms of Pgk1p and Pdc1p were also significantly related to K and cell size: Pgk1p-acidic, Pgk1pbasic, and Pdc1p-4854 accounted for 17.29%, 12.46%, and 10.92% of K variation, whereas Pgk1p-acidic and Pdc1p-1589 accounted for 10.33% and 9.58% of cell size variation. Interestingly, this analysis allowed us to explore the metabolic bases for the correlation between K and cell size. From the isoforms that were retained by the multiple regression, eight (Tdh1p-2729, Tdh1p-2824, Tdh2p-4732, Pgk1p-basic, Pgk1p-acidic, Adh1p-4799, Adh1p-4808, and Ald6p-1565) were positively correlated with one trait, and negatively correlated with the other, which is consistent with the negative correlation previously observed between the two life-history traits. The other isoforms were specific for either K or cell size. In addition, we found a spectacular specialization of the isoforms of Adh1p, with isoform-4799 correlated with high K and low cell size whereas isoform-4808 was correlated to low K and high cell size (Fig. 5C). Although these two isoforms accounted for around 26–33% of the variation for cell size and K by linear regression, it raised up to 52–65% when considering the degree of acetylation of Adh1p (Adh1p-4808/sum of both Adh1p isoform). As shown Fig. 6, the percentage of acetylated Adh1p is highly positively correlated to K (ρ = 0.77, p < 10−15) and highly negatively correlated to cell size (ρ = –0.75, p < 10−15). This shows that ant and grasshopper strategies are mainly associated with the degree of N-terminal acetylation of the alcohol dehydrogenase.
Fig. 6.
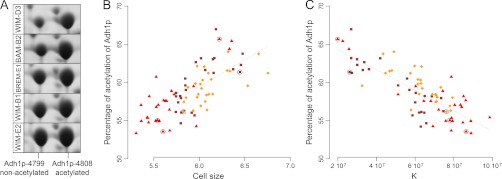
Relationships between Adh1p isoform ratio and two life-history traits, cell size, and carrying capacity. A, 2-DE gel portions of the two isoforms of Adh1p ordered in decreasing Adh1p isoform ratio (spot 4799/spot 4808). B1 and B2, brewery strains; D3, distillery strains; E1 to E4, enology strains. BAM, BREM, and WIM: bakery, brewery, and winery media. B, Relationship between cell size (diameter, μm) and Adh1p isoforms ratio (spot 4799/spot 4808). A significant negative correlation was found (ρ = −0.74, p < 10−15). C, Relationship between K (cells per ml) and Adh1p isoform ratio (spot 4799/spot 4808). A significant positive correlation was found (ρ = 0.77, p < 10−15). The points corresponding to the 2-DE gel portions (A) are black-circled.
Using Natural Variation for Metabolic Engineering
Although multiple regression analyses gave a good indication of the correlation existing between metabolic or life-history traits and isoform abundances, they cannot predict whether the change of abundance of a single isoform will have a large impact on the traits. Indeed, an isoform may be significantly correlated to a trait but the slope may be low (a variation in abundance has little effect on the flux) and/or the range of variation in abundance of the isoform may be restricted. To determine the extent to which the different isoforms could affect maximum CO2 flux during alcoholic fermentation, we used the equation of the multiple regression model previously established. For each strain, we used the mean abundance of their isoforms observed in all three media (supplementary Information Data set S2), except for one isoform the concentration of which varied over its natural range of variation in the nine strains. This allowed us to predict how the CO2 specific flux changes when the abundance of one isoform changes (bar lengths and positions, Fig. 7). For instance, the natural range of variation of Ald6p-1565 was associated with a very low CO2 flux variation, whereas the response of the flux upon Pdc1p-1589 variation was large. The magnitude of these predicted variations was not strain-dependent (the same equation of multiple regression was used), but there were clear differences between strains regarding the max-min flux profiles over the isoforms (bar positions on the y axis in Fig. 7). In D3 strain, which had the highest observed flux (3.04 × 10−11 g/h/cell), but the lowest abundance of Pdc1p-1589 (remember that the correlation between the flux and Pdc1p-1589 is negative), changing Pdc1p-1589 abundance was associated with a flux decrease. But for the eight remaining strains, Pdc1p-1589 variation was mostly associated with a strong increase of the flux, even exceeding D3's (best) flux. For example, strain B1 had a mean flux of 2.17 × 10−11 g/h/cell. The abundance decrease of Pdc1p-1589 isoform could be associated to a virtual flux increase of 54% (3.35 × 10−11 g/h/cell) compared with the measured B1's flux. For B2 the virtual flux was even more increased: B2 strain had a mean flux of 2.88 × 10−11 g/h/cell that could be increased up to 4.27 × 10−11 g/h/cell along with the virtual abundance decrease of Pdc1p-1589. This result elects Pdc1p-1589 as a relevant molecular target for further flux improvement for most strains. In a lesser extent Tdh2p-4740 could also be targeted to increase flux in all strains but B1 and D2, and Rhr2p-3003 in D3 and E2.
Fig. 7.
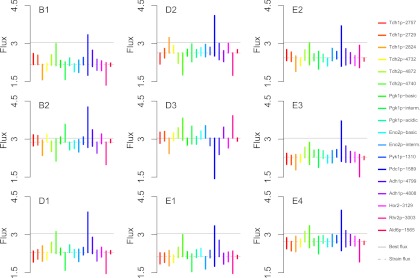
Predicted response of CO2 flux to individual isoform variation. To predict the response of CO2 flux to individual isoform variation, we used the equation of the multiple regression accounting for CO2 flux from isoform abundance. For each strain, the abundance of all isoforms but one was fixed equal to their mean over the three media, and the last isoform varied over the range of natural variation among the nine strains. D1 to D3, distillery strains; E1 to E4, enology strains. BAM, BREM, and WIM: bakery, brewery, and winery media. The CO2 specific flux is expressed in 10−11 g h−1 cell−1. The “best flux” is indicated by the gray horizontal lines and the “strain flux” by the dotted line.
DISCUSSION
Global Constraints on the Fermentation Proteome
We reported here a comprehensive study of the sources of variation for the yeast fermentation proteome during alcoholic fermentation, and its relationship with metabolic and life-history trait variation. We compared strains from different food origins grown in different fermentation media, and chose the peak CO2 production rate as the reference physiological stage. At this stage the enzymatic pool allocated to the fermentation proteome, which represents one third of the total proteome, appeared to be constant over the media and strains considered. Previous work has suggested that enzyme concentrations cannot increase indefinitely and are probably bounded because of cellular constraints in space and energy (55), avoiding macromolecular crowding (56) and lowering the energetic cost associated with enzyme transcription, translation, and maintenance under limited resources (57, 58). Although our data demonstrated the existence of such a constraint at the level of the whole fermentation proteome, we have also shown that the AF enzymes have reduced variance compared with non-AF proteins, highlighting the existence of evolutionary constraints. Such constraints may be related to the existence of macromolecular complexes associating enzymes belonging to the same metabolic pathway. These so-called metabolons (59) were described for glycolytic enzymes in several organisms ranging from plants (60, 61), to human (62, 63) and yeast (64). Metabolon allows passing (channeling) the intermediary metabolites from one enzyme to the consecutive one within a given metabolic pathway, forming a metabolite tunnel. Substrate channeling is assumed to increase the efficiency and velocity of metabolic pathways, relative to the performances of a set of independent enzymes, but also to prevent the release of unstable intermediates (65). In addition, protein associations may protect the metabolic pathways in stressful environments (66). Indeed, we can hypothesize that the abundance of metabolon enzymes is closely regulated, which is consistent with the strong evolutionary constraint observed here for AF enzymes.
Variation and Trade-offs Between Isoform Abundances
For most of the AF enzymes studied here, several spots were identified in the 2-DE gels. Those isoforms could be unambiguously attributed to post-translational modifications: (1) all the isoforms were detected in all nine strains. Within a given strain, different isoforms corresponding to a given enzyme encoded by a given gene necessarily arose through post-translational modifications; (2) in case of proteins encoded by paralogous genes (Tdh1/Tdh2/Tdh3, Eno1/Eno2, etc.) mass spectrometry specific peptides allowing the clear discrimination of the paralogs were identified (supplementary Information Data set S3); (3) in the only five cases where we observed a shift of electrophoretic mobility of one enzyme for a particular strain (Fig. 1B), all the spots corresponding to the enzyme shifted, indicating a change in an amino acid, confirmed from gene sequences; (4) we were able to identify the post-translational modifications for some isoforms. Adh1p-4808 was identified as the acetylated form of Adh1p, whereas Adh1p corresponded to the native protein. Tdh1p-2824 displayed a phosphorylated residue (serine 201), whereas Tdh1p-2757 corresponded to the nonphosphorylated isoform. The post-translational modifications differentiating the other isoforms could not be identified.
Our data allowed us to measure the magnitude of the abundance variation of the isoforms of each enzyme, and to compare them to the variation observed at the whole enzyme level represented by the sum of isoform abundances. Many enzymes had isoforms that displayed different ranges of genetic and environmental variation, indicating isoform specialization. For some enzymes, most of the variation for protein abundances was observed at the isoform level, and the abundances of the different isoforms of a given enzyme were negatively correlated between strains, resulting in a lower genetic variation at the enzyme level. Moreover, variation of isoform abundances seemed to have clear functional consequences. Some specific isoforms—rather than all the isoforms of some enzymes—were associated to the variation of CO2 flux, AF products, cell size, or carrying capacity (K). A few isoforms were also associated to differences between the food origins of the strains. The relationships between isoforms and phenotypic traits can be interpreted in three ways: (1) the isoforms of the enzymes control the phenotype, (2) the phenotype regulates back isoforms abundance, (3) trade-offs related to different allocations of the same resources lead to correlations between isoforms and traits. As post-translational modifications of enzymes are largely involved in the modulation of catalytic activity, ranging from inactivation to full activation, in protein-protein interactions or the regulation of enzymatic turnover (12), we interpret our results as the consequence of isoform control on phenotypic traits. To our knowledge, this is the first time that post-translational modifications are shown to be associated with traits related to fermentation metabolism, suggesting that “fine-tuning” of yeast AF is sustained at the post-translational level.
Sharing-out the Control of Flux
In metabolic control analysis (MCA), the flux control coefficient is a dimensionless measure of how much a flux varies in response to an infinitesimal change in the rate of a particular reaction (67, 68). Provided the flux-enzyme (or isoform) relationship is concave hyperbolic, flux control coefficients can also be estimated from changes in enzyme activities (69). By extension, the proportion of variance of CO2 flux accounted for by the abundance variation of an individual isoform can be regarded as a proxy of the flux control exerted by the isoform. Although the relationship between flux control coefficient and the proportion of accounted for variance is complex (70), an enzyme that accounts for a significant part of the flux variance has necessarily a nonnull control coefficient. In our work, 18 isoforms were found to be related to the CO2 flux, each isoform accounting for a limited proportion of flux variation (maximum 12.34%), in agreement with MCA that predicts flux control to be split over several enzymes rather than one. The CO2 flux control was mainly distributed among an unknown post-translational modification of Pdc1p, the phosphorylation status of Tdh1p and the degree of N-terminal acetylation of Adh1p. Some isoforms of Pfk1p and Pfk2p exhibited strong genetic variation but no association with the CO2 flux, which suggests that those enzymes have no control of the CO2 flux. Conversely, our experimental set-up did not allow us to say anything about the control of the flux for isoforms that showed no genetic or environmental variations like Pgi1p and Tpi1p. Our results can be compared with previous works that specifically under- or overexpressed some enzymes of the fermentation proteome. From a MCA perspective, allosteric enzymes Pfkp and Pyk1p are unlikely to exert a high control on the glycolytic flux (71). This was confirmed experimentally for Pfkp (72–74) which is consistent with our own findings. However, Pyk1p was found to exert a significant level of control over both the rate and direction of carbon flux in yeast during growth on glucose (73), as well as in Lactococcus lactis (75). Accordingly, the Pyk1p-1310 isoform was found significantly associated with CO2 flux in our study. Our results also show some discrepancies with what had been previously observed. For instance Schaaff et al. (74) reported that the yeast glycolytic flux remained unaffected by the overexpression of hexokinase, Pfk1p, Pgk1p, Pyk1p, Pdc1p, Gpm1p, or Adh1p, whereas we found a strong association between the CO2 flux and some isoforms of Pgk1p, Pyk1p, Pcd1p, and Adh1p. Similarly, over and underexpression of Pgi1p and Fba1p were shown to be associated with changes in glucose consumption rate, cell size, and the carrying capacity K (50), whereas we found no genetic and no environmental variation for isoform abundance of those enzymes. However, those results may not be contradictory, keeping in mind that the variations that we observed between strains are the result of evolutionary processes that occurred during yeast domestication, whereas Schaaf et al. (74) and Wang et al. (50) had analyzed expression mutants. Indeed, in the MCA perspective, the enzyme selection coefficients for changing the flux are proportional to the flux control coefficients (76). Enzymes having a strong control on the flux are expected to be the primary targets of selection and show less genetic variation within populations. In Drosophila, a survey of within and between species polymorphism of 17 enzymes pointed the glucose-6P branchpoint as a specific target of selection (77). It is often assumed that the first steps in a metabolic pathway are exerting strong control over flux (78). This prediction was verified for the pathway of anthocyanin biosynthesis by comparing the rate of evolution of enzyme genes along the pathway in three plant species: Rausher et al. (79) showed that upstream enzymes of the pathway were much less variable than downstream enzymes. In yeast, previous works suggested that specific selective pressures shaped the first part of the glycolysis: Pgi1p, Fba1p, and Tpi1p have been conserved as single copy even after independent whole-genome duplication, meaning that one duplicated copy has been lost (80–82). Our results show less genetic variation between strains for the abundance of enzymes of the upper part of the glycolytic pathway, and more variation for the abundance of downstream enzymes, together with strong associations with CO2 flux as well with metabolic or life-history traits. We propose to interpret these features as evidence for unequal sharing-out of the flux control, with a higher control exerted by the upstream enzymes leading to higher evolutionary constraints. Because of those constraints, human selection for modulating the CO2 flux or other traits related to food processing has been only possible through small changes in the abundance of less constrained enzymes, downstream in glycolysis or belonging to the glycerol, acetate, or ethanol pathways, resulting in a higher genetic diversity observed today for those enzymes.
Human Domestication Shaped the Fermentation Proteome
Using linear discriminant analysis and subsequent studies, we showed that the fermentation proteome was significantly shaped by human domestication, through the differential targeting of a few isoforms. The distillery strains were separated from beer and wine strains on the basis of Pdc1p-4854, an isoform associated to the main AF metabolites (ethanol, acetate, glycerol concentration per cell). Wine and beer strains have been specifically selected to lower acetate production that is responsible for a well-known off-flavor, the vinegar taste (83), whereas this feature is less important for distilling yeasts. This could explain why winemakers and brewers' selection significantly lowered this isoform. In addition, winemaking selection appeared to have specially targeted the acidic isoform of Tdh3p. In a recent work, Jimenez-Marti and coworkers suggested that Tdh3p might account for strain adaptation to enological conditions (84). This could explain why isoforms of this enzyme had been specially targeted for wine strains. Industrial yeast improvement strategies could build on this result of empirical human domestication and target the regulation of the post-translational forms of the others enzymes involved in metabolic control such as Pdc1p or Adh1p.
Pdc1p and Adh1p: The Last, but not the Least, Steps of Alcoholic Fermentation
Deciphering the relationships between the fermentation proteome and metabolism revealed the implication of several isoforms belonging to different enzymes. In particular, isoforms of two proteins, Pdc1p and Adh1p, were associated to alcoholic fermentation: Pdc1p-1589 isoform was found to be related to the CO2 flux, whereas Pdc1p-4854 accounted for an important part (20–32%) of the final ethanol, glycerol, and acetate concentrations. Importantly, this last isoform's abundance was low, and considering the four identified Pdc1p's spots as a whole would have hidden Pdc1p-4854 variation and forbidden the identification of Pdc1p as an essential enzyme controlling AF metabolites production in addition to its role in CO2 flux control. Previous work described the presence of N-terminally acetylated and nonacetylated isoforms of Pdc1p in yeast (85), and close comparison of our 2-DE gels with the yeast proteome map (http://www.ibgc.u-bordeaux2.fr/YPM/) suggests that Pdc1p-4854 may correspond to the nonacetylated isoform whereas Pdc1p-1589 and Pdc1p-1605 may be acetylated forms. Pdc1p N-terminal acetylation is achieved through the excision of the initial methionine and subsequent addition of an acetyl group. Our mass spectrometry analyses allowed us to identify the native (nonacetylated) peptide for Pdc1p-4854 (MSEITLGK), whereas this peptide was not identified for other Pdc1p isoforms. We hypothesized that our mass spectrometry analyses failed to detect the acetylated peptide (*SEITLGK) because of its short length (peptides with length inferior to eight amino acids usually have low yet not-significant p values (>0.05) and are thus missed by the analysis).
Besides Pdc1p, Adh1p's isoforms were found to be significantly related to different metabolic and life-history traits: the nonacetylated isoform of Adh1p (4799) was associated with a significant part of the variation of glycerol, acetate, and biomass formation per cell (15–20%), and was also related to two fitness traits (21–24%), the carrying capacity (K) and cell size, that define ant and grasshopper life-history strategies. In particular, the degree of acetylation of Adh1p was strongly related to these traits, highlighting the functional importance of N-terminal acetylation. In yeast, the N-terminal acetyltransferase responsible for Adh1p acetylation is NatA (85). NatA is composed of two main subunits (Nat1p and Ard1p), whose mutants are related to the disappearance of acetylated isoforms of several enzymes, including Adh1p and Pdc1p (85). N-terminal acetylation is one of the most common protein modifications in eukaryotes, as 85% of the proteins have an acetylated isoform (86), but it displays several specificities. Unlike other post-translational modifications, N-terminal acetylation is irreversible and occurs during protein synthesis after about 50 amino acid residues have emerged from the ribosome (87). Thus, N-terminal acetylation is sometimes designed as a cotranslational rather than post-translational modification. The biological significance of N-terminal acetylation is still unclear, because no global trend regarding the functional consequences of N-terminal acetylation emerges. For some proteins, N-terminal acetylation may act as a degradation signal (88), whereas for others N-terminal acetylation may protect from proteolytic degradation and subsequently increases their half-life (89). N-terminal acetylation was also shown to be involved in protein sorting and addressing to cellular organelles (90) or to membrane (91). Indeed, although Adh1p is frequently described as a cytoplasmic protein, it is also associated with the plasma membrane (92) like many other fermentation enzymes (Pgi1p, Tpi1p, Eno1p, Eno2p, Tdh1p, Tdh2p, Tdh3p, Pgk1p, Pyk1p). In addition, these enzymes display protein-protein interactions in yeast (93) and also in Human (94), suggesting they may form a large metabolon complex whose plasma membrane localization (92) may be useful for the rapid processing of the glucose entering the cell. This is particularly true for larger cells in which independent cytosolic proteins have less chance to be close together and form an enzyme-to-enzyme channeling of glycolytic intermediates. Moreover, in human erythrocytes, glycolytic enzymes are organized into complexes on the membrane (63) via N-terminal residues of some proteins (95) and phosphorylation (63), highlighting the importance of post-translational modifications in metabolon efficiency. We speculate that a few fermentation enzymes could act as a “plasma membrane anchor” of fermentation metabolon, such as acetylated Adh1p. This would be in agreement with the fact that larger cells show a higher degree of N-terminal acetylation of Adh1p. It could also explain why the nonacetylated isoform of Adh1p is negatively correlated to the flux, in contrast to the acetylated one.
In any case, although the real impact of N-terminal acetylation is unknown for Adh1p, we showed a clear correlation between the degree of acetylation of Adh1p and both cell size and carrying capacity, as well as between the nonacetylated isoform and CO2 flux/glycerol/acetate/biomass production. Recently, some works have shown that lysine acetylation was involved in the control of central metabolic pathways in both prokaryotes and eukaryotes (15, 37), but to our knowledge, this is the first time that N-terminal acetylation of an enzyme is shown to be unambiguously associated with both metabolic and life-history traits.
In conclusion, using Saccharomyces cerevisiae alcoholic fermentation as a model, we have highlighted the importance of post-translational modifications such as phosphorylation and N-terminal acetylation in metabolic control. Isoforms were also shown to govern other key fitness traits related to cell growth, showing their importance from the functional and evolutionary viewpoints and underlining the usefulness of large-scale approaches at the post-translational level.
Supplementary Material
Acknowledgments
We thank Michel Rigoulet, Anne Devin, Michel Zivy, and Olivier Martin for their comments that helped improving the manuscript.
Footnotes
* This work was supported by the ANR program “blanc” Adaptalevure NT05-4_45721 and the ANR program “ALIA” HeterosYeast ANR-08-ALIA-9.
 This article contains supplemental Information, Data sets S1 to S4, Tables S1 and S2 and Fig. S1.
This article contains supplemental Information, Data sets S1 to S4, Tables S1 and S2 and Fig. S1.
Author contributions: P.M., M.B., M.A., C.D., D.V. and D.S. designed research; W.A., P.M. M.B. and A.B. performed research; T.B., D.C. and B.V conducted mass spectrometry analyses, O.L. and T.B. released proteomic data through PROTICdb, W.A., P.M., M.B., M.A., T.S., C.D., D.V. and D.S. analyzed data and wrote the paper.
Conflict of Interest: The authors declare that they have no conflict of interest.
1 The abbreviations used are:
- 2-DE
- two-dimensional electrophoresis
- BREM
- brewery medium
- BAM
- bakery medium
- WIM
- winery medium
- LDA
- linear discriminant analysis
- AF
- alcoholic fermentation
- MS
- mass spectrometry
- MCA
- metabolic control analysis.
REFERENCES
- 1. Skovran E., Crowther G. J., Guo X., Yang S., Lidstrom M. E. (2010) A systems biology approach uncovers cellular strategies used by Methylobacterium extorquens AM1 during the switch from multi- to single-carbon growth. PLoS One 5, e14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sokolova I. M., Pörtner H. O. (2003) Metabolic plasticity and critical temperatures for aerobic scope in a eurythermal marine invertebrate (Littorina saxatilis, Gastropoda: Littorinidae) from different latitudes. J. Exp. Biol. 206, 195–207 [DOI] [PubMed] [Google Scholar]
- 3. Stein M., Gabdoulline R. R., Wade R. C. (2008) Calculating enzyme kinetic parameters from protein structures. Biochem. Soc. Trans. 36, 51–54 [DOI] [PubMed] [Google Scholar]
- 4. Shilatifard A. (2006) Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem. 75, 243–269 [DOI] [PubMed] [Google Scholar]
- 5. Latchman D. S. (1997) Transcription factors: an overview. Int. J. Biochem. Cell Biol. 29, 1305–1312 [DOI] [PubMed] [Google Scholar]
- 6. Proudfoot N. J., Furger A., Dye M. J. (2002) Integrating mRNA processing with transcription. Cell 108, 501–512 [DOI] [PubMed] [Google Scholar]
- 7. Hinkson I. V., Elias J. E. (2011) The dynamic state of protein turnover: It's about time. Trends Cell Biol. 21, 293–303 [DOI] [PubMed] [Google Scholar]
- 8. Molina M., Cid V. J., Martin H. (2010) Fine regulation of Saccharomyces cerevisiae MAPK pathways by post-translational modifications. Yeast 27, 503–511 [DOI] [PubMed] [Google Scholar]
- 9. Doble B. W., Woodgett J. R. (2003) GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nussinov R., Tsai C. J., Xin F., Radivojac P. (2012) Allosteric post-translational modification codes. Trends Biochem. Sci. 37, 447–455 [DOI] [PubMed] [Google Scholar]
- 11. Oliveira A. P., Sauer U. (2012) The importance of post-translational modifications in regulating Saccharomyces cerevisiae metabolism. FEMS Yeast Res. 12, 104–117 [DOI] [PubMed] [Google Scholar]
- 12. Mann M., Jensen O. N. (2003) Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261 [DOI] [PubMed] [Google Scholar]
- 13. Cohen P. (2000) The regulation of protein function by multisite phosphorylation–a 25 year update. Trends Biochem. Sci. 25, 596–601 [DOI] [PubMed] [Google Scholar]
- 14. Rubin C. S., Rosen O. M. (1975) Protein phosphorylation. Annu. Rev. Biochem. 44, 831–887 [DOI] [PubMed] [Google Scholar]
- 15. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guan K. L., Xiong Y. (2011) Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci. 36, 108–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nadolski M. J., Linder M. E. (2007) Protein lipidation. FEBS J. 274, 5202–5210 [DOI] [PubMed] [Google Scholar]
- 18. Yamada T., Bork P. (2009) Evolution of biomolecular networks: lessons from metabolic and protein interactions. Nat. Rev. Mol. Cell Biol. 10, 791–803 [DOI] [PubMed] [Google Scholar]
- 19. Heinemann M., Sauer U. (2010) Systems biology of microbial metabolism. Curr. Opin. Microbiol. 13, 337–343 [DOI] [PubMed] [Google Scholar]
- 20. Kotte O., Zaugg J. B., Heinemann M. (2010) Bacterial adaptation through distributed sensing of metabolic fluxes. Mol. Syst. Biol. 6, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kohlstedt M., Becker J., Wittmann C. (2010) Metabolic fluxes and beyond-systems biology understanding and engineering of microbial metabolism. Appl. Microbiol. Biotechnol. 88, 1065–1075 [DOI] [PubMed] [Google Scholar]
- 22. Fraenkel D. G. (2003) The top genes: on the distance from transcript to function in yeast glycolysis. Curr. Opin. Microbiol. 6, 198–201 [DOI] [PubMed] [Google Scholar]
- 23. Fiehn O., Weckwerth W. (2003) Deciphering metabolic networks. Eur. J. Biochem. 270, 579–588 [DOI] [PubMed] [Google Scholar]
- 24. Wiechert W., Schweissgut O., Takanaga H., Frommer W. B. (2007) Fluxomics: mass spectrometry versus quantitative imaging. Curr. Opin. Plant Biol. 10, 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nielsen J. (2011) Transcriptional control of metabolic fluxes. Mol. Syst. Biol. 7, 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haverkorn van Rijsewijk B. R., Nanchen A., Nallet S., Kleijn R. J., Sauer U. (2011) Large-scale (13)C-flux analysis reveals distinct transcriptional control of respiratory and fermentative metabolism in Escherichia coli. Mol. Syst. Biol. 7, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kolbe A., Oliver S. N., Fernie A. R., Stitt M., van Dongen J. T., Geigenberger P. (2006) Combined transcript and metabolite profiling of Arabidopsis leaves reveals fundamental effects of the thiol-disulfide status on plant metabolism. Plant Physiol. 141, 412–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hua Q., Joyce A. R., Palsson B. Ø., Fong S. S. (2007) Metabolic characterization of Escherichia coli strains adapted to growth on lactate. Appl. Environ. Microbiol. 73, 4639–4647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schilling O., Frick O., Herzberg C., Ehrenreich A., Heinzle E., Wittmann C., Stülke J. (2007) Transcriptional and metabolic responses of Bacillus subtilis to the availability of organic acids: transcription regulation is important but not sufficient to account for metabolic adaptation. Appl. Environ. Microbiol. 73, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krömer J. O., Sorgenfrei O., Klopprogge K., Heinzle E., Wittmann C. (2004) In-depth profiling of lysine-producing Corynebacterium glutamicum by combined analysis of the transcriptome, metabolome, and fluxome. J. Bacteriol. 186, 1769–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang C., Hua Q., Shimizu K. (2002) Integration of the information from gene expression and metabolic fluxes for the analysis of the regulatory mechanisms in Synechocystis. Appl. Microbiol. Biotechnol. 58, 813–822 [DOI] [PubMed] [Google Scholar]
- 32. Daran-Lapujade P., Jansen M. L., Daran J. M., van Gulik W., de Winde J. H., Pronk J. T. (2004) Role of transcriptional regulation in controlling fluxes in central carbon metabolism of Saccharomyces cerevisiae. A chemostat culture study. J. Biol. Chem. 279, 9125–9138 [DOI] [PubMed] [Google Scholar]
- 33. Daran-Lapujade P., Rossell S., van Gulik W. M., Luttik M. A., de Groot M. J., Slijper M., Heck A. J., Daran J. M., de Winde J. H., Westerhoff H. V., Pronk J. T., Bakker B. M. (2007) The fluxes through glycolytic enzymes in Saccharomyces cerevisiae are predominantly regulated at posttranscriptional levels. Proc. Natl. Acad. Sci. U.S.A. 104, 15753–15758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Groot M. J., Daran-Lapujade P., van Breukelen B., Knijnenburg T. A., de Hulster E. A., Reinders M. J., Pronk J. T., Heck A. J., Slijper M. (2007) Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals post-transcriptional regulation of key cellular processes. Microbiology 153, 3864–3878 [DOI] [PubMed] [Google Scholar]
- 35. Lillo C., Meyer C., Lea U. S., Provan F., Oltedal S. (2004) Mechanism and importance of post-translational regulation of nitrate reductase. J. Exp. Bot. 55, 1275–1282 [DOI] [PubMed] [Google Scholar]
- 36. Landmann J. J., Busse R. A., Latz J. H., Singh K. D., Stülke J., Gorke B. (2011) Crh, the paralogue of the phosphocarrier protein HPr, controls the methylglyoxal bypass of glycolysis in Bacillus subtilis. Mol. Microbiol. 82, 770–787 [DOI] [PubMed] [Google Scholar]
- 37. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P. (2010) Acetylation of Metabolic Enzymes Coordinates Carbon Source Utilization and Metabolic Flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gerosa L., Sauer U. (2011) Regulation and control of metabolic fluxes in microbes. Curr. Opin. Biotechnol. 22, 566–575 [DOI] [PubMed] [Google Scholar]
- 39. Cavalieri D., McGovern P. E., Hartl D. L., Mortimer R., Polsinelli M. (2003) Evidence for S. cerevisiae fermentation in ancient wine. J. Mol. Evol. 57, S226–232 [DOI] [PubMed] [Google Scholar]
- 40. Meussdoerffer F. G. (2009) A comprehensive history of beer brewing. In: Eβlinger H. M., ed. Handbook of Brewing: Processes, Technology, Markets, pp. 1–42, WILEY-VCH, Weinheim [Google Scholar]
- 41. Pretorius I. S. (2000) Tailoring wine yeast for the new millennium: novel approaches to the ancient art of winemaking. Yeast 16, 675–729 [DOI] [PubMed] [Google Scholar]
- 42. Samuel D. (1996) Investigation of ancient egyptian baking and brewing methods by correlative microscopy. Science 273, 488–490 [DOI] [PubMed] [Google Scholar]
- 43. Kobi D., Zugmeyer S., Potier S., Jaquet-Gutfreund L. (2004) Two-dimensional protein map of an “ale”-brewing yeast strain: proteome dynamics during fermentation. FEMS Yeast Res. 5, 213–230 [DOI] [PubMed] [Google Scholar]
- 44. Garrels J. I., McLaughlin C. S., Warner J. R., Futcher B., Latter G. I., Kobayashi R., Schwender B., Volpe T., Anderson D. S., Mesquita-Fuentes R., Payne W. E. (1997) Proteome studies of Saccharomyces cerevisiae: identification and characterization of abundant proteins. Electrophoresis 18, 1347–1360 [DOI] [PubMed] [Google Scholar]
- 45. Perrot M., Sagliocco F., Mini T., Monribot C., Schneider U., Shevchenko A., Mann M., Jenö P., Boucherie H. (1999) Two-dimensional gel protein database of Saccharomyces cerevisiae (update 1999). Electrophoresis 20, 2280–2298 [DOI] [PubMed] [Google Scholar]
- 46. Shevchenko A., Jensen O. N., Podtelejnikov A. V., Sagliocco F., Wilm M., Vorm O., Mortensen P., Shevchenko A., Boucherie H., Mann M. (1996) Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. U.S.A. 93, 14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fiévet J., Dillmann C., Lagniel G., Davanture M., Negroni L., Labarre J., de Vienne D. (2004) Assessing factors for reliable quantitative proteomics based on two-dimensional gel electrophoresis. Proteomics 4, 1939–1949 [DOI] [PubMed] [Google Scholar]
- 48. Albertin W., Marullo P., Aigle M., Dillmann C., de Vienne D., Bely M., Sicard D. (2011) Population size drives the industrial yeast alcoholic fermentation and is under genetic control. Appl. Environ. Microbiol. 77, 2772–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Spor A., Wang S., Dillmann C., de Vienne D., Sicard D. (2008) “Ant” and “grasshopper” life-history strategies in Saccharomyces cerevisiae. PLoS ONE 3, e1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang S., Spor A., Nidelet T., Montalent P., Dillmann C., de Vienne D., Sicard D. (2011) Switch between life history strategies due to changes in glycolytic enzyme gene dosage in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 77, 452–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fay J. C., Benavides J. A. (2005) Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genet. 1, 66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liti G., Carter D. M., Moses A. M., Warringer J., Parts L., James S. A., Davey R. P., Roberts I. N., Burt A., Koufopanou V., Tsai I. J., Bergman C. M., Bensasson D., O'Kelly M. J., van Oudenaarden A., Barton D. B., Bailes E., Nguyen A. N., Jones M., Quail M. A., Goodhead I., Sims S., Smith F., Blomberg A., Durbin R., Louis E. J. (2009) Population genomics of domestic and wild yeasts. Nature 458, 337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sicard D., Legras J. L. (2011) Bread, beer and wine: Yeast domestication in the Saccharomyces sensu stricto complex. C. R. Biol. 334, 229–236 [DOI] [PubMed] [Google Scholar]
- 54. Spor A., Nidelet T., Simon J., Bourgais A., de Vienne D., Sicard D. (2009) Niche-driven evolution of metabolic and life-history strategies in natural and domesticated populations of Saccharomyces cerevisiae. BMC Evol. Biol. 9, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lion S., Gabriel F., Bost B., Fiévet J., Dillmann C., de Vienne D. (2004) An extension to the metabolic control theory taking into account correlations between enzyme concentrations. Eur. J. Biochem. 271, 4375–4391 [DOI] [PubMed] [Google Scholar]
- 56. Ellis R. J. (2001) Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26, 597–604 [DOI] [PubMed] [Google Scholar]
- 57. Koehn R. K. (1991) The cost of enzyme synthesis in the genetics of energy balance and physiological performance. Biol. J. Linnean Soc. 44, 231–247 [Google Scholar]
- 58. Vilaprinyo E., Alves R., Sorribas A. (2010) Minimization of biosynthetic costs in adaptive gene expression responses of yeast to environmental changes. PLoS Comput. Biol. 6, e1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Srere P. A. (1985) The metabolon. Trends Biochem. Sci. 10, 109–110 [Google Scholar]
- 60. Graham J. W., Williams T. C., Morgan M., Fernie A. R., Ratcliffe R. G., Sweetlove L. J. (2007) Glycolytic enzymes associate dynamically with mitochondria in response to respiratory demand and support substrate channeling. Plant Cell 19, 3723–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Williams T. C., Sweetlove L. J., Ratcliffe R. G. (2011) Capturing Metabolite Channeling in Metabolic Flux Phenotypes. Plant Physiol. 157, 981–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mazurek S., Hugo F., Failing K., Eigenbrodt E. (1996) Studies on associations of glycolytic and glutaminolytic enzymes in MCF-7 cells: role of P36. J. Cell. Physiol. 167, 238–250 [DOI] [PubMed] [Google Scholar]
- 63. Campanella M. E., Chu H., Low P. S. (2005) Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc. Natl. Acad. Sci. U.S.A. 102, 2402–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brandina I., Graham J., Lemaitre-Guillier C., Entelis N., Krasheninnikov I., Sweetlove L., Tarassov I., Martin R. P. (2006) Enolase takes part in a macromolecular complex associated to mitochondria in yeast. Biochim. Biophys. Acta 1757, 1217–1228 [DOI] [PubMed] [Google Scholar]
- 65. Huang X., Holden H. M., Raushel F. M. (2001) Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem. 70, 149–180 [DOI] [PubMed] [Google Scholar]
- 66. Araiza-Olivera D., Sampedro J. G., Mújica A., Pena A., Uribe-Carvajal S. (2010) The association of glycolytic enzymes from yeast confers resistance against inhibition by trehalose. FEMS Yeast Res. 10, 282–289 [DOI] [PubMed] [Google Scholar]
- 67. Heinrich R., Rapoport T. A. (1974) A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 42, 89–95 [DOI] [PubMed] [Google Scholar]
- 68. Kacser H., Burns J. A. (1973) The control of flux. Symp. Soc. Exp. Biol. 27, 65–104 [PubMed] [Google Scholar]
- 69. Small J. R., Kacser H. (1993) Responses of metabolic systems to large changes in enzyme activities and effectors. 1. The linear treatment of unbranched chains. Eur. J. Biochem. 213, 613–624 [DOI] [PubMed] [Google Scholar]
- 70. Bost B., Dillmann C., de Vienne D. (1999) Fluxes and metabolic pools as model traits for quantitative genetics. I. The L-shaped distribution of gene effects. Genetics 153, 2001–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fell D. A. (1997) Understanding the control of metabolism, Portland Press, London [Google Scholar]
- 72. Davies S. E., Brindle K. M. (1992) Effects of overexpression of phosphofructokinase on glycolysis in the yeast Saccharomyces cerevisiae. Biochemistry 31, 4729–4735 [DOI] [PubMed] [Google Scholar]
- 73. Pearce A. K., Crimmins K., Groussac E., Hewlins M. J., Dickinson J. R., Francois J., Booth I. R., Brown A. J. (2001) Pyruvate kinase (Pyk1) levels influence both the rate and direction of carbon flux in yeast under fermentative conditions. Microbiology 147, 391–401 [DOI] [PubMed] [Google Scholar]
- 74. Schaaff I., Heinisch J., Zimmermann F. K. (1989) Overproduction of glycolytic enzymes in yeast. Yeast 5, 285–290 [DOI] [PubMed] [Google Scholar]
- 75. Koebmann B. J., Andersen H. W., Solem C., Jensen P. R. (2002) Experimental determination of control of glycolysis in Lactococcus lactis. Antonie Van Leeuwenhoek 82, 237–248 [PubMed] [Google Scholar]
- 76. Gout J. F., Duret L., Kahn D. (2009) Differential retention of metabolic genes following whole-genome duplication. Mol. Biol. Evol. 26, 1067–1072 [DOI] [PubMed] [Google Scholar]
- 77. Flowers J. M., Sezgin E., Kumagai S., Duvernell D. D., Matzkin L. M., Schmidt P. S., Eanes W. F. (2007) Adaptive evolution of metabolic pathways in Drosophila. Mol. Biol. Evol. 24, 1347–1354 [DOI] [PubMed] [Google Scholar]
- 78. Okumoto S., Takanaga H., Frommer W. B. (2008) Quantitative imaging for discovery and assembly of the metabo-regulome. New Phytol. 180, 271–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rausher M. D., Miller R. E., Tiffin P. (1999) Patterns of evolutionary rate variation among genes of the anthocyanin biosynthetic pathway. Mol. Biol. Evol. 16, 266–274 [DOI] [PubMed] [Google Scholar]
- 80. Conant G. C., Wolfe K. H. (2007) Increased glycolytic flux as an outcome of whole-genome duplication in yeast. Mol. Syst. Biol. 3, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Marland E., Prachumwat A., Maltsev N., Gu Z., Li W. H. (2004) Higher gene duplicabilities for metabolic proteins than for nonmetabolic proteins in yeast and E. coli. J Mol Evol 59, 806–814 [DOI] [PubMed] [Google Scholar]
- 82. Seoighe C. (1999) Gene Order Evolution and Genomic Analyses of the Model Eukaryote, Saccharomyces cerevisiae, and Other Yeast Species. University of Dublin [Google Scholar]
- 83. Bely M., Stoeckle P., Masneuf-Pomarède I., Dubourdieu D. (2008) Impact of mixed Torulaspora delbrueckii-Saccharomyces cerevisiae culture on high-sugar fermentation. Int. J. Food Microbiol. 122, 312–320 [DOI] [PubMed] [Google Scholar]
- 84. Jiménez-Marti E., Aranda A., Mendes-Ferreira A., Mendes-Faia A., del Olmo M. L. (2007) The nature of the nitrogen source added to nitrogen depleted vinifications conducted by a Saccharomyces cerevisiae strain in synthetic must affects gene expression and the levels of several volatile compounds. Antonie Van Leeuwenhoek 92, 61–75 [DOI] [PubMed] [Google Scholar]
- 85. Polevoda B., Norbeck J., Takakura H., Blomberg A., Sherman F. (1999) Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J. 18, 6155–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Polevoda B., Sherman F. (2000) Nα-terminal acetylation of eukaryotic proteins. J. Biol. Chem. 275, 36479–36482 [DOI] [PubMed] [Google Scholar]
- 87. Pestana A., Pitot H. C. (1975) Acetylation of nascent polypeptide chains on rat liver polyribosomes in vivo and in vitro. Biochemistry 14, 1404–1412 [DOI] [PubMed] [Google Scholar]
- 88. Hwang C.-S., Shemorry A., Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hershko A., Heller H., Eytan E., Kaklij G., Rose I. A. (1984) Role of the alpha-amino group of protein in ubiquitin-mediated protein breakdown. Proc. Natl. Acad. Sci. U.S.A. 81, 7021–7025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Forte G. M., Pool M. R., Stirling C. J. (2011) N-Terminal Acetylation Inhibits Protein Targeting to the Endoplasmic Reticulum. PLoS Biol 9, e1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jackson C. L. (2004) N-terminal acetylation targets GTPases to membranes. Nat. Cell Biol. 6, 379–380 [DOI] [PubMed] [Google Scholar]
- 92. Delom F., Szponarski W., Sommerer N., Boyer J. C., Bruneau J. M., Rossignol M., Gibrat R. (2006) The plasma membrane proteome of Saccharomyces cerevisiae and its response to the antifungal calcofluor. Proteomics 6, 3029–3039 [DOI] [PubMed] [Google Scholar]
- 93. Collins S. R., Kemmeren P., Zhao X. C., Greenblatt J. F., Spencer F., Holstege F. C., Weissman J. S., Krogan N. J. (2007) Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol. Cell. Proteomics 6, 439–450 [DOI] [PubMed] [Google Scholar]
- 94. Havugimana P. C., Hart G. T., Nepusz T., Yang H., Turinsky A. L., Li Z., Wang P. I., Boutz D. R., Fong V., Phanse S., Babu M., Craig S. A., Hu P., Wan C., Vlasblom J., Dar V. U., Bezginov A., Clark G. W., Wu G. C., Wodak S. J., Tillier E. R., Paccanaro A., Marcotte E. M., Emili A. (2012) A census of human soluble protein complexes. Cell 150, 1068–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chu H., Low P. S. (2006) Mapping of glycolytic enzyme-binding sites on human erythrocyte band 3. Biochem. J. 400, 143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.