

Abstract
Proteolysis is a critical post-translational modification for regulation of cellular processes. Our lab has previously developed a technique for specifically labeling unmodified protein N termini, the α-aminome, using the engineered enzyme, subtiligase. Here we present a database, called the DegraBase (http://wellslab.ucsf.edu/degrabase/), which compiles 8090 unique N termini from 3206 proteins directly identified in subtiligase-based positive enrichment mass spectrometry experiments in healthy and apoptotic human cell lines. We include both previously published and unpublished data in our analysis, resulting in a total of 2144 unique α-amines identified in healthy cells, and 6990 in cells undergoing apoptosis. The N termini derive from three general categories of proteolysis with respect to cleavage location and functional role: translational N-terminal methionine processing (∼10% of total proteolysis), sites close to the translational N terminus that likely represent removal of transit or signal peptides (∼25% of total), and finally, other endoproteolytic cuts (∼65% of total). Induction of apoptosis causes relatively little change in the first two proteolytic categories, but dramatic changes are seen in endoproteolysis. For example, we observed 1706 putative apoptotic caspase cuts, more than double the total annotated sites in the CASBAH and MEROPS databases. In the endoproteolysis category, there are a total of nearly 3000 noncaspase nontryptic cleavages that are not currently reported in the MEROPS database. These studies significantly increase the annotation for all categories of proteolysis in human cells and allow public access for investigators to explore interesting proteolytic events in healthy and apoptotic human cells.
Annotation of the human α-aminome, the full set of unmodified protein N termini, can provide a wealth of information regarding protein turnover, protein trafficking, and protease activity (1). The vast majority of protein N termini in eukaryotic cells are cotranslationally blocked by acetylation through the action of N-acetyl transferases (2). Free α-amines occur on some proteins that are never N-terminally acetylated, and can also be regenerated by signal or transit peptide removal during protein trafficking, and endo- or exoproteolysis during protein maturation and signaling. Thus, there has been considerable effort to develop unbiased proteomic methods to characterize the α-aminome in healthy and diseased states (3–8).
We have developed a positive enrichment method in which the α-amines of intracellular (8) or extracellular proteins (9) can be specifically and directly tagged and captured, without pretreatment or protection, using subtiligase, an engineered peptide ligase (Fig. 1A) (10, 11). Following purification, tryptic digestion, and LC-MS/MS, the protein sequence and exact site of proteolysis are readily identified. We have applied this approach to study proteolysis by caspases, cysteine-class aspartyl specific proteases, during cellular apoptosis (8, 12–14), and inflammatory response (15). These studies, in a variety of cell types and apoptotic inducers, have revealed much about the targets, substrate recognition, timing, logic, and evolution of caspase cleavage events. These efforts have generated a huge amount of data that requires systematic compilation, organization, and normalization so that it can be shared and queried easily by all investigators and compared with other databases describing proteolytic events (16–18).
Fig. 1.

Experimental schema, database design, and database summary. (A), For all experiments, human cells were grown under standard conditions, either with or without treatment with apoptosis inducing agents. Cells are lysed and proteins biotinylated on their free α-amines using subtiligase, followed by purification and identification by LC-MS/MS. N termini identifications from every experiment were entered into the database to create the untreated and apoptotic datasets, and a subset apoptotic caspase-cleaved dataset for apoptotic N termini following aspartic acid cleavage. (B), The DegraBase database is structured around four main tables linking the experimental data to the MS identifications and external database information at both the N terminus and protein level (for more details see Supplemental File S1). (C), Summary statistics of the DegraBase for all experiments in the DegraBase and for both the untreated and apoptotic datasets (more details in Supplemental Table S1A). The blue box highlights the apoptotic caspase-cleaved dataset within the apoptotic dataset.
Here we present the results of both previously published and new experiments that detect α-amines in both untreated and apoptotic human cells. These studies reveal new translational N-terminal processing, signal and transit peptide removal, and other proteolytic events associated with normal protein maturation and function in healthy cells. Comparing these data to the apoptotic dataset reveals that the greatest changes in apoptosis are caused by endoproteolysis, owing to the induction of caspases as well as other proteases. We find a total of 1706 putative caspase sites in nearly 1300 different human proteins. We further find an additional 2900 noncaspase, nontryptic, nontransit, and nonsignal peptide cleavage sites in 1415 proteins.
In addition to the analyses described here, we provide a publically available database, the DegraBase, that is dynamic, expandable, searchable, and readily accessible (http://wellslab.ucsf.edu/degrabase/). With this database, investigators can query all 8090 unique α-amines detected with high confidence from 26,043 peptide observations in both previously published (8, 12, 13) and new subtiligase α-aminome labeling experiments. The DegraBase substantially expands annotated intracellular proteolytic events in healthy and apoptotic cells.
EXPERIMENTAL PROCEDURES
Cell Cultures
Jurkat, THP-1, DB, RPMI 8226, MM1-S and U266 human cell lines were acquired from the American Type Culture Collection (ATCC, Manassas, VA) and were cultured under the recommended conditions. When cells reached a density of 1 × 106 cells/ml, an apoptotic inducer (doxorubicin, etoposide, bortezomib, FasL, CD95, staurosporine, or TRAIL) was added from 1000x stock (for individual experimental details, see supplemental Table 1A). Cell viability and caspase activity were monitored by CellTiter-Glo, Caspase-Glo (Promega, Madison, WI) and Ac-DEVD-AFC activity assays. Cells were harvested by centrifugation after 0–40 h, washed with phosphate buffered saline solution, pelleted, and stored at –80°C. For untreated experiments, healthy cells cultured under the same conditions were harvested without any inducer added.
N-terminal Labeling
The lysis and N-terminal labeling were performed as described previously (8, 12, 13, 15). For experiments not previously published, the following protocol was used. Cells were lysed in a bicine buffer with triton or SDS containing the protease inhibitors EDTA, PMSF, E-64, z-VAD-fmk, and AEBSF. Proteins were reduced with 2 mm tris(2-carboxyethyl) phosphine hydrochloride at 90°C for 15 min and alkylated once cooled with 4 mm iodoacetamide in dark for 1 h, then quenched with 10 mm dithiothreitol. Labeling was performed with 1 mm of a biotinylated synthesized peptide ester called TEVest and 1 μm subtiligase for at least an hour at room temperature (11, 19). There were four different TEVest peptide esters used to facilitate the identification of the labeled products; they were identical except for the small tag left after processing to aid in mass spectrometry recognition: serine-tyrosine (SY), glycine-tyrosine (GY), phenylalanine (Phe), or 2-aminobutyric acid (Abu). The biotinylated proteins were separated by gel filtration or precipitation and captured on NeutrAvidin agarose beads (Pierce, Rockford, Illinois, USA). The samples were digested with sequence grade modified trypsin (Promega, Madison, WI) before or after capture. After capture, the labeled peptides were released with recombinant TEV protease and collected. Samples were desalted by chromatography with C18 ZipTip Pipette Tips (Millipore, Billerica, MA) or C18 high-performance liquid chromatography (HPLC) (Waters, Milford, MA). Further offline strong cation exchange fractionation was performed on some samples. For further individual experimental details, see supplemental Table S1A.
Liquid Chromatograph Tandem Mass Spectrometry (LC-MS/MS) and Peptide Identification
For all experiments, samples were separated by reverse phase HPLC coupled to a mass spectrometer: QSTAR Pulsar, QSTAR XL, QSTAR Elite (Applied Biosystems, Foster City, CA), LTQ-Orbitrap XL or QExactive (Thermo Fisher Scientific, San Jose, CA). Spectra were converted into peak lists for database searching using the mascot dll in Analyst for QSTAR instruments or using an in-house script based on the Raw_Extract script in Xcalibur v2.4 (Thermo Fisher Scientific). Peptide identification was performed using Protein Prospector version 5.10.0 (20). Search parameter mass allowances were tailored for each instrument: 100 ppm precursor and 0.15 Da fragment for QSTAR instruments, 20 ppm precursor and 0.6 Da fragment for LQT-Orbitrap XL, and 20 ppm precursor and 0.8 Da for QExactive. All searches were performed with constant modification of the peptide N terminus with the appropriate TEVest tag, variable modifications of carbamidomethylation of cysteines and oxidation of methionine, and allowing for up to three missed tryptic cleavages. All datasets were searched assuming tryptic specificity at the peptide C terminus, but no cleavage specificity at the N terminus. All fractions (including re-analysis of previously published data) were searched against the human SwissProt library release 2012_03 (20,255 entries) to provide consistent accession number annotations for all data. Maximum expectation value scores for protein and peptide of 0.02 were employed as acceptance criteria. Searches against a decoy library of random and reversed protein sequences revealed an average false discovery rate (FDR) across all datasets of 0.55%.
Data Analysis
The DegraBase framework was created using FileMakerPro version 9.0, and houses three types of data: the sample, peptide and N terminus/protein tables (Fig. 1B). Experimental parameters are entered by investigators, mass spectrometry data are imported from files created by Protein Prospector, and protein- and cleavage site-specific annotation data are imported from a number of external databases including UniProtKB (21), the CASBAH (16), and MEROPS (17). Full documentation, including FileMakerPro scripts for data analysis and Perl scripts for processing of UniProtKB data before input, is available as supplemental File S1. The DegraBase also exists as an HTML-based website (http://wellslab.ucsf.edu/degrabase/) to allow for more accessible searching.
Abundance data were taken from PaxDB version 2.1 using the integrated dataset called “Weighted average of 'H. sapiens PeptideAtlas Build May 2010′(weighting 50%), 'H. sapiens PeptideAtlas Build March 2009′,(weighting 50%)” available from the downloads tab at www.pax-db.org (22). Sequence logos were made using iceLogo with the whole human SwissProt library as background (23). All logo images were made with the percent difference scoring system, except when stated as “Filled Logos” representing amino acid frequency, not information content. Significance was determined by chi-square analyses. Data for methionine processing, mitochondrial transit peptide removal and signal peptide removal were compared with SwissProt library release 2012_03. Mitochondrial localization was determined based on the MitoCarta database (24).
Gene Ontology (GO)1 term enrichment was determined using the GO::TermFinder software (25). A list of unique proteins for each dataset was created and uploaded to the database and tested for enrichment against the human SwissProt background using all evidence codes except ND (No biological Data available) and IEA (Inferred from Electronic Annotation. Enriched terms were defined using a corrected p value cutoff of less than 0.01. To compare terms between datasets, a pairwise chi-square test was performed using the Benjamini-Hochberg multiple testing correction procedure.
RESULTS
The DegraBase
Given the massive amount of data generated from multiple experiments under different conditions, it was necessary to create a simple and normalized database. The DegraBase is a relational database built to house our α-aminomics data (Fig. 1B). It is available in three formats (see supplemental Information): a FileMakerPro file (supplemental File S2), an excel document containing worksheets for each of the major tables (supplemental File S3), and a web interface (http://wellslab.ucsf.edu/degrabase/) where users may search by substrate name or accession number. Full documentation of the database is available in Supplemental File S1.
Data and Datasets
The current DegraBase contains a total of 26043 independent peptide identifications from 44 different proteomic labeling experiments (11 untreated and 33 apoptotic) (Fig. 1C and supplemental Table 1A). There are a total of 8090 unique N terminus identifications from 3206 proteins. We subdivided our data into three sets: (1) untreated, (2) apoptotic, and (3) apoptotic caspase-cleaved. In a separate study using our labeling method, we have seen that there is cell line- and drug-specific variability in the data, but most differences show up in detected abundance of cleavage product over time rather than the presence or absence (reported here) of the specific identified N termini (13). Therefore, we were comfortable pooling our multiple apoptotic experiments together to compare all proteins detected in all untreated cells tested versus those undergoing apoptosis.
The untreated dataset contains all observations from the 11 experiments performed in five different cell lines (supplemental Table S1B). This dataset has 3732 identified N termini corresponding to 2144 unique N terminus start sites from 1239 proteins. The apoptotic dataset consists of all observations from the 33 experiments using seven different chemotherapeutic inducers in five cell lines (supplemental Table S1C). This generated a total of 22311 independent peptide identifications, corresponding to 6990 unique N terminus sites from 3020 different proteins. This reflects the dramatic activation of caspases following the induction of apoptosis of our samples, also observed with caspase activity and cell death assays (data not shown). We defined the third dataset, the apoptotic caspase-cleaved dataset, as a subset of the apoptotic dataset that includes all apoptotic aspartic acid-cleaved N termini (supplemental Table S1D). This dataset includes 1706 unique N termini from 1268 proteins, and in combination with our previous studies, MEROPS and CASBAH, increases the number of published human caspase-cleavage events to over 2200. The apoptotic dataset contains 1706 aspartate cleaved peptides compared with the 140 seen in the untreated dataset, reflecting a dramatic induction of caspase activity.
To estimate to what degree the α-aminome MS data are biased by protein abundance in cells, we compared the datasets to PaxDB (22), a database that provides an independent estimate of relative protein abundance based on MS spectral counting data. All three α-aminome datasets cover more than six orders of magnitude of ppm (supplemental Fig. S1A–S1C). Only for the small set of low abundance proteins did our α-aminome identification tail off, which presumably reflects the limits of detection of the methodology. There is a slight enrichment for higher abundance proteins overall (supplemental Fig. S1D).
At the protein level, there is a large overlap between the untreated and apoptotic datasets; 1053 of the 1239 proteins (85%) from untreated cells were also found in the apoptotic dataset (Fig. 2A). In contrast, we observed a smaller overlap between datasets when considering the particular N termini within each protein (Fig. 2B); only 1328 of the 2144 untreated N termini (62%) were labeled under apoptotic conditions. There is a small set of 361 proteins, but only 129 N termini, that overlap between the untreated and apoptotic caspase-cleaved datasets. The presence of caspase-cleaved products in healthy cells likely reflects low levels of apoptosis that occurs in any healthy cell population, and make up a very small portion of the total untreated set. The protein overlap may represent apoptotic caspase substrates that also undergo endoproteolysis in healthy cells by noncaspases. Interestingly, many of the proteolytic substrates in healthy cells are cleaved at different positions upon induction of apoptosis.
Fig. 2.

Dataset overlap. Venn diagrams show a larger overlap of unique protein (A) than peptide (B) identifications between untreated, apoptotic and apoptotic caspase-cleaved datasets. The apoptotic caspase-cleaved dataset is defined as a subset of the apoptotic dataset, and is therefore contained wholly within the apoptotic set.
To compare the functional properties of the different datasets, we performed Gene Ontology (GO) term enrichment using GO::TermFinder (supplemental Table S2) (25). We looked at the terms unique to each dataset to identify specific process, function or component annotations related to healthy or dying cellular states. The untreated dataset was enriched in terms related to homeostatic functions like metabolic and biosynthetic processes (specifically related to ribosomal, coenzyme, amino acids and fatty acids, NADH dehydrogenase, and isomerase functions), the mitochondrial proton-transporting ATP synthase complex, and organelle envelope lumen (prominently related to the endoplasmic reticulum). In the apoptotic set, we compared the significant terms from caspase substrates to the noncaspase apoptotic terms, and to the terms unique to the apoptotic set only. The caspase substrates are enriched in the regulation of transcription, and there were many terms related to cell morphogenesis, specifically chromosome and microtubule structure, which are known to change and break down during apoptosis. The noncaspase apoptotic enriched terms in process, function and component ontologies relate to chromatin assembly (especially DNA binding, vesicle coating, and targeting), signal transduction involved in DNA damage and cell cycle checkpoints, and nucleotide catabolic processes. We also saw enrichment in the non-caspase apoptotic set for proteins associated with terms for proteolysis and cell death.
We next analyzed the precise sequences surrounding the N termini identified in each dataset. We used iceLogo (23) to visualize the sequence specificity for cleavage events for each dataset using the human SwissProt database to establish background amino acid frequencies (Fig. 3). The cleavage sites are presented in the standard Schechter-Berger form, with the scissile bond between the P1 residue and the P1′ residue (26). All three logos show a strong preference for small amino acids (glycine, serine, or alanine) at the P1′ position, but significant differences at the P1 position. In healthy cells, there is enrichment for cleavage sites following lysine, arginine, and methionine. The methionine cleavages mainly represent N-terminal methionine processing. The large number of cuts following basic residues is consistent with a high activity of trypsin-like enzymes in both healthy and apoptotic cells. In apoptotic cells this tryptic-like activity is overshadowed by the large number of caspase cleavages following aspartic acid residues. The apoptotic caspase-cleaved dataset shows a degenerate specificity with moderate enrichment for aspartic acid-glutamic acid-valine in the P4-P2 positions, matching the classic “DEVD” substrate preference for executioner caspases-3 and -7, the signature proteases of apoptosis (17).
Fig. 3.

iceLogo diagrams show amino acid frequencies. The untreated (A) and apoptotic (B) datasets show distinct patterns of amino acid frequency for the eight positions surrounding the labeled α-amine (P4-P4′) from all unique N termini. Enrichment (above the line) or depletion (below the line) of amino acid frequency is determined using the human SwissProt library release 2012_03 as background. The apoptotic caspase-cleaved iceLogo (C) represents all cleavages following aspartic acid (P1 = D) in the apoptotic dataset.
Three Categories of Proteolysis: Translational N Terminus Processing, Signal/Transit Peptide Removal, and Endoproteolysis
In the global analysis presented above, we have discussed all the proteolytic events in healthy and apoptotic cells without distinguishing between the different kinds of proteolytic processing known to occur in cells. We now look more closely at three important areas of proteolytic processing: (1) processing around the methionine at the translational N terminus (N termini labeled at residues 1 and 2), (2) cleavage of possible secretory or transit peptides during organelle trafficking (labeled at residues 3–65), and (3) other endoproteolytic events (labeled at residues 66+) (Fig. 4). We chose to define possible signal or transit peptides within residues 3–65 based on patterns from previously published datasets (27, 28) and from our own data (see below). Subdividing each dataset into these different groups, we see that the majority of cuts results from endoproteolysis (55–80%), then putative signal or transit peptide removal (20–35%), and finally processing around the initiator methionine (<10%).
Fig. 4.

There are three distinct groups of proteolytic processing within the data: (i) processing around the methionine at the translational N terminus (N termini observed at residues 1 and 2), (ii) cleavage of possible secretory or transit peptides during organelle trafficking (observed residues 3–65), and (iii) other endoproteolytic events (considered as cuts after residue 65). The untreated and apoptotic datasets had similar levels of translational N terminus labeling (∼10%), but differed for the latter categories, with the apoptotic datasets having more cleavage events past residue 65. The apoptotic caspase-cleaved set is shifted even more toward endoproteolytic cleavages than the apoptotic set.
Initiator Methionine Processing
Eukaryotic proteins are typically cotranslationally acetylated, rendering the translational N terminus inaccessible to the subtiligase labeling technique (29, 30). Recent work suggests that this acetylation is largely irreversible (31). However, there are some proteins, both with and without initiator methionine removal, that do not undergo cotranslational acetylation; these have translational N termini that are accessible to labeling. In the full database (both untreated and apoptotic samples), we observed labeling of the initiator methionine in 154 proteins (12% of identified proteins) (supplemental Table S3A), and labeling of the second residue, indicating methionine removal, in 198 proteins (15% of identified proteins) (supplemental Table S3B). It is noteworthy that the majority of these translational N termini are not yet annotated in SwissProt (supplemental Fig. S2).
The sequence logo for proteins in the untreated dataset retaining the initiator methionine suggests enrichment for a hydrophobic amino acid at the second residue (Fig. 5A), whereas proteins with the initiator methionine removed had enrichment for small amino acids at the second residue (Fig. 5B). This is largely consistent with previous studies showing that methionine removal is most efficient for proteins with small amino acids following the initiator methionine (32). Almost identical patterns were seen for the proteins in the apoptotic set (Figs. 5C–5D). Out of 182 methionine processing events seen in healthy cells, only 35 were not found in the apoptotic set, suggesting there is little change in the translational N-terminal processing events during apoptosis.
Fig. 5.

Initiator methionine processing. The iceLogos for the untreated (A, B) and apoptotic (C, D) sets are very similar for processing around the methionine at the translational N terminus. Enrichment or depletion of amino acid frequency is determined using human SwissProt library release 2012_03 as background. (A) and (C) represent retention (but not acetylation) of the initiator methionine (Met[1]); (B) and (D) represent removal of the initiator methionine without acetylation of the second residue (Xaa[2]). Two proteins labeled at residue 1 but annotated in UniProt as not containing an initiator methionine (Ig lambda chain V-IV region HII (P01717, serine) and 40S ribosomal protein S30 (P62861, lysine)) were removed from the datasets for the iceLogo creation.
Mitochondrial Transit Peptide Removal
Proteins expressed from nuclear genes but destined for the mitochondria generally contain positively charged N-terminal regions that direct them to the mitochondrial import machinery (33). Once inside the mitochondria, these mitochondrial transit peptides (mTPs) are removed by the mitochondrial processing peptidase, and in some cases the truncated proteins are further processed by other proteases that remove one or a few additional residues (27). Although the mitochondrial proteome has been well characterized, data on the precise location of mTP cleavage sites remains minimal. Moreover, sequence specificities of the proteases involved are only partially understood (24, 28, 34). Considering only the untreated dataset to avoid possible apoptosis-induced cleavages, we identified roughly 250 labeled N termini from the ∼1000 human SwissProt proteins that are in MitoCarta, a highly curated database of mitochondrial proteins (24).
The distribution of N terminus placement found in mitochondrial proteins is quite different from that of nonmitochondrial proteins. We see a significant spike in the range of position 10 to position 65, roughly the location of most known mTP cleavage sites (Fig. 6A) (27). We therefore focused our examination on the 171 N termini seen in this range in MitoCarta proteins. For the purpose of these analyses, in cases where one protein had more than one N terminus in this region, we chose the site closest to the translational N terminus. We found that 58 had no mTP cleavage site annotated in SwissProt, and 67 had an annotated site different from the one we observed (supplemental Fig. S3A and supplemental Table S3C). Additionally, 24 peptide removal start sites are within one residue of their SwissProt annotations. This may reflect the secondary proteolysis known to occur in the mTP removal pathway (27), and may be a part of a mitochondrial version of the N-end rule (35). It is notable that 46 out of the 67 cases (69%) where our data disagrees with SwissProt annotation involve cleavage sites that SwissProt describes as “By Similarity,” “Potential,” or “Probable,” indicating a lack of strong evidence for these cuts. In contrast, in only 17 out of the 46 cases (37%) where our data agree with SwissProt does the annotation contain this qualifying language (supplemental Table S3C). We generated an iceLogo for the 16 residues before and four residues after the 171 mTP cleavage sites (Fig. 6B). As expected based on previous studies, the iceLogo shows enrichment for arginine and de-enrichment for acidic residues throughout the transit peptide. The strongest arginine signal is in the P2 and P3 positions, as expected from previous studies (28) and is the most enriched signal in our untreated position 10–65 logo (Fig. 6C).
Fig. 6.

Signal/Transit peptide removal sites. (A) Annotated mitochondrial proteins were greatly enriched for labeling between residues 10–65 compared with all other proteins in the untreated dataset, reflecting labeling at mitochondrial transit peptide removal sites. (B–D), iceLogos for subsets of the set of cleavages at positions 3 to 65 in the untreated dataset: (B), all unique N termini from proteins thought to be mitochondrial; (C), all N termini in the untreated dataset labeled between 10–65; and (D), all unique N termini thought to contain signal peptides.
Signal Peptide Removal
Proteins destined for the secretory pathway are subjected to proteolytic removal of signal peptides near the N terminus (36). As with mTPs, we looked for these cleavages only in the untreated dataset to avoid any apoptosis-specific events. In this case, we focused on the 63 proteins that were annotated in SwissProt as having a signal peptide removal site between positions 10 and 65 (supplemental Table S3D). Most of these were ER or Golgi resident proteins; our technique does not efficiently capture secreted proteins after they have dissociated from the cell. In 30 of these proteins, the cleavage site we observed matched the one annotated in SwissProt (supplemental Fig. S3B). In this case, the qualifiers “By Similarity” and “Potential” were used by SwissProt to describe at least 60% of their signal peptide removal site annotations both in the set that agrees with our data, and the set that does not. We cannot be sure what accounts for the differences between our data and SwissProt, but we hope that these data will prove useful to researchers who work in this area. As with the mTP sites, we generated an iceLogo for positions P16 to P4′ relative to the cleavage site (Fig. 6D). In this case, we saw a substantial enrichment for leucine residues in the P16-P6 positions, which is consistent with the known requirement for hydrophobicity in the signal peptide to facilitate interaction with the ER membrane (36, 37).
Endoproteolysis Before and After Apoptosis
We next consider the endoproteolytic events that occur after residue 65, which represent the majority of our identified N termini (Fig. 4). Although many of the cleavages that occur before or at residue 65 are endoproteolytic, we chose to focus on the 66+ set to reduce the contamination of this set with possible signal or transit peptide removal sites. Considering the apoptotic set cleaved after residue 65, 28% of them occur with an aspartic acid residue at the P1 position, suggesting a caspase cleavage event (supplemental Fig. S4). To visualize noncaspase protease activity, we removed all cleavage sites with aspartic acid at the P1 position and then used iceLogo to generate filled logos (where letter height represents amino acid frequency) for the P1 and P1′ positions in the untreated and apoptotic datasets (Fig. 7). Both logos show the predominance of basic amino acids (arginine and lysine) at P1 and small amino acids (glycine, serine, or alanine) at P1′. However, there is an overall decrease in the fraction of cleavages following arginine or lysine in the apoptotic dataset. This likely reflects the induction of other noncaspase and nontryptic proteases during apoptosis that are different from proteases in healthy cells.
Fig. 7.

Filled logos for endoproteolysis occurring at residue 66 or above for the untreated (A) and apoptotic (B) datasets with all aspartic acid cleavages removed (9% of the untreated and 28% of the apoptotic N termini). The size of each letter represents its relative frequency within the dataset. Distributions are very similar for the P1′ position, but show differences at the P1 position.
Many human proteins undergo degradation by the proteasome, however we do not expect to see very many proteasome products using our subtiligase method because the majority are quickly degraded into their amino acid components (38). Those that remain intact (for example, in order to be displayed on an MHC class I complex) are mostly less than 10 amino acids long; the fractionation steps required to separate the small biotin label from the larger labeled protein fragments in the subtiligase N-terminal enrichment technique causes significant loss of short peptides. Furthermore, our mass spectrometry data searches only considered peptides with a tryptic site on the C terminus, and only a subset of proteasome peptide products would fit this criteria.
The N-End Rule Before and After Apoptosis
The P1′ position of a cleavage site is important for the half-life of the resulting protein fragment, as determined by the Arg/N-end rule pathway (35). Many proteases, including caspases (39), prefer the small amino acids glycine, serine, or alanine in the P1′ pocket; in fact, these three residues make up 32% of the P1′ residues of the ∼56,000 protease cleavages (of both native and synthetic substrates) described in the substrate section of the MEROPS database. However, small amino acids are not a requirement, as we saw all twenty possible amino acids in the P1′ position in the untreated, apoptotic and apoptotic caspase-cleaved datasets (Table I). The untreated dataset had a greater proportion of cleavages with charged amino acids (lysine, arginine, aspartic acid, and glutamic acid) at the P1′ position than apoptotic or apoptotic caspase-cleaved datasets, whereas the apoptotic and apoptotic caspase-cleaved datasets had more cleavages with serine and glycine in the P1′ position.
Table I. P1′ amino acid frequency and Arg/N-End Rule Pathway half-lives.
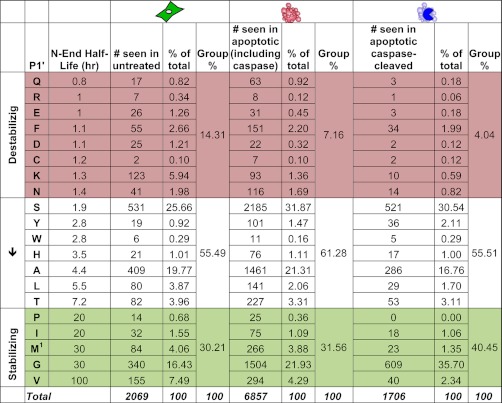
a Initiator methionines removed.
The Arg/N-end rule pathway degrades proteins and proteolysis products through ubiquitination and targeting to the proteasome. Different N-terminal amino acids may be stabilizing or destabilizing, and thus affect the half-life of the protein (35). To investigate the potential for a biological effect of the proteolysis products, we analyzed the data with respect to the theoretical half-lives for each P1′ amino acid (40). We grouped the amino acids into stabilizing P1′ amino acids (half-life greater than 20 h), destabilizing (half-life less than 1.5 h), and intermediate (half-live between 1.5 and 20 h) (Table I). For all three datasets, the majority (53–60%) of N termini were found in the intermediate group. However, there is a clear difference in the pattern of stabilizing and destabilizing cuts depending on cell condition. Almost 14% of all untreated cleavage events occurred before destabilizing amino acids. In contrast, only 7% of apoptotic cleavages and 4% of apoptotic caspase cleavages occurred before destabilizing amino acids, with most shifting into the intermediate range of predicted half-lives. In general, untreated proteolysis events were more destabilizing and had shorter theoretical half-lives than apoptotic events, and caspase cleavages leave particularly stable and longer lasting predicted N termini.
DISCUSSION
The subtiligase N-terminal labeling method yields high selectivity for α-amines with accurate peptide and protein identifications and very low false discovery rate (< 1%). Overall, we observed 3206 proteins, corresponding to almost 16% of the entire SwissProt human proteome and covering over 6 logs in protein abundance. We see internally consistent labeling between the datasets. For example, the same mitochondrial transit sites and initiator methionine sites were labeled in 35–40 out of 44 untreated and apoptotic experiments. These cleavage events are independent of apoptosis, and therefore show the consistency in our labeling and detection method. Additionally, we believe that little bias originates because of subtiligase specificity. For example, all 20 amino acids were labeled at P1′, including proline and valine, which are known to be slow substrates for subtiligase in vitro (10).
We are confident that the distinctions between healthy and apoptotic datasets reflect the biology of the human cell lines, as the apoptotic samples showed decreased cell viability and increased caspase activity (as measured both in cell culture and in observed caspase-cleaved N termini relative to the untreated samples). In fact, 129 of the 140 aspartic-cleaved N termini identified in healthy cells were also in the 1706 apoptotic N terminus set, and likely reflect a small population of apoptotic cells within the healthy cell population. Comparing our apoptotic protein dataset to other published datasets shows that we capture most known apoptosis-related proteins. We have labeled more than 60% of the proteins listed in the ApoptoProteomics database (41), an apoptotic proteomics database. Additionally, 75 of our caspase substrates overlap with the literature-curated CASBAH database (16), and 79 overlap with the caspase-3, -6, or -7 substrates listed in MEROPS (17) (in both of these comparisons, we excluded >200 entries present in these databases but derived from the original subtiligase study (8)). These 1706 apoptotic caspase-cleaved sites, in combination with MEROPS, the CASBAH, and other work from our laboratory (8, 12, 13), bring the total known human caspase cleavage sites to more than 2200. Importantly, the DegraBase contains a larger number of new noncaspase proteolytic events that have yet to be assigned to a specific protease. Surprisingly, there are only 45 sites in the 2900 noncaspase, nontryptic (not cleaved after lysine or arginine) endoproteolytic events (residue 66+) that are present in the MEROPS database (release 9.6), and only 10 of these sites are annotated as “physiological” cleavages in MEROPS (supplemental Table S4). Interestingly, there is little exoproteolysis of these intracellular proteins, whereas laddering produced by sequential exoproteolysis was observed in 24% of all proteins identified in a subtiligase-based study of human serum (9).
It is probable that only a subset of the total identified apoptotic proteolytic substrates needs to be cleaved to complete apoptosis. For example, there are 1706 putative caspase cleavages, but 784 was the largest number of sites labeled in any single cellular experiment. Additionally, about 50% of all apoptotic N termini identified were only seen in one experiment. This may reflect the diversity in drug induction and cell types chosen as well as the expected stochasticity in mass spectrometry and our labeling technology. Some of the apoptotic labeling patterns may also be caused by induced polyspecific proteases, like the caspases, with large and diverse sets of possible substrates. Only 110 sites (87 caspase-cleaved) from 109 proteins were seen consistently in at least 10 apoptotic experiments and only one or zero untreated experiments (supplemental Table S5). Interestingly, these common cleavages have a wide kinetic range. Some are shown to be cleaved by up to three different apoptotic caspases (12), and many have homologs in mouse and fly that are also known caspase substrates (14). These apoptosis-enriched sites may represent important apoptotic nodes, whereas other cleavage sites may be unique to the experimental conditions, or possibly bystander cleavages.
Remarkably, the protease actors in healthy and apoptotic cells appear to target an overlapping set of substrates, but not always at the same cleavage sites (Fig. 2). This could allow for different regulation of these targets depending on the cellular conditions. An important difference between the untreated and apoptotic datasets is the theoretical half-lives of the newly created N termini. We found the neo-N termini created by caspase cleavages have a higher proportion of stabilizing N-terminal amino acids than those in the untreated and apoptotic datasets (Table I). A similar conclusion that apoptotic fragments tended to persist during apoptosis was reached using the PROTOMAP method (42). Stable apoptotic cleavage products, in particular protein fragments of caspase substrates, may function in a different manner from the parent protein. We realize that these half-life values are largely dependent on the assay method and may not be representative of specific in vivo half-lives of a given protein fragment. However, Piatkov et al. have recently showed the greater extent that caspases and the Arg/N-End Rule pathway interact: proapoptotic protein fragments contain evolutionarily conserved destabilizing N-terminal amino acids, targeting them for quick degradation by the proteasome in healthy cells; caspases cleave and inactivate members of the proteasome pathway, preventing peptide degradation (43). Indeed, we do see caspase cleavages in UBR4 and UBR5, which function as the Arg/N-End Rule pathway E3 Ubiquitin ligases.
During apoptosis, caspase cleavages may result in loss-of-function, gain-of-function, or no functional effect on the substrate. As many caspase cleavage events occur between protein domains (8), many substrates have the potential for gain of function events in which a catalytic domain is relocated or an inhibitor removed. Several such cleavages have been thoroughly studied in kinases, as reviewed by Kurokawa and Kornbluth (44). In a preliminary search through our database, we see enrichment for caspase cleavages between annotated domains. For example, in the kinase family, 52 of the 57 caspase-derived N termini occurred between domains, compared with 51 of 76 noncaspase apoptotic N termini and 15 of 26 untreated N termini (45, 46) (supplemental Table S6). This is consistent with a recent study by Dix et al., that demonstrated crosstalk between caspases and kinases, where phosphorylation can direct caspase cleavages on kinases that may lead to a change in kinase activity (47).
In sum, we provide an unbiased and global annotation of the human cellular α-aminome. The data are consolidated into a searchable database, the DegraBase, revealing a large amount homeostatic and apoptotic proteolysis in cells. To our knowledge, these untreated and apoptotic datasets are the most extensive published to date using a single methodology. We confidently identified many new sites related to protein processing, including initiator methionine retention or removal, and the specific cleavage locations for signal or transit peptide removal during protein trafficking. Additionally, our dataset shows the abundance of healthy homeostatic and non-caspase apoptotic endoproteolytic events that occur in cells. We hope that our colleagues across many areas of biology will find the DegraBase to be a useful resource for further understanding and characterization of proteolytic events in cells.
Supplementary Material
Acknowledgments
We thank David Wildes, Alan Barber, Jason Porter, Gemma Holliday, and Aenoch Lyn for their helpful discussions. We thank David Maltby, Jonathan Trinidad, Al Burlingame, and the rest of the UCSF Mass Spectrometry Facility, plus Iman Mohtashemi and Yan Chen of Thermo Scientific, for mass spectrometry assistance.
Footnotes
* This project was supported by a grant from the UCSF Stephen and Nancy Grand Multiple Myeloma Translational Initiative. It was supported by the National Science Foundation GRFP (E.D.C.), the University of California Systemwide Biotechnology Research & Education Program GREAT Training Grant #2007- 20 (E.D.C.), National Institutes of Health Training Grant T32 GM007175 (J.E.S.), and Banting Postdoctoral Fellowships funded by the Government of Canada (O.J.). Additional support from National Institutes of Health R01 GM081051, R01 GM097316, and RO1 CA154802 (J.A.W.), The Rogers Family Foundation (J.A.W.), and an award from the Sandler Program in Biological Sciences (J.A.W.). Mass spectrometry was performed at the Bio-Organic Biomedical Mass Spectrometry Resource at UCSF, which is supported by grants from the National Center for Research Resources (5P41RR001614) and the National Institute of General Medical Sciences (8P41GM103481 and 1S10RR015804) from the National Institutes of Health.
 This article contains supplemental Figs S1 to S4, Files S1 to S3, and Tables S1 to S6.
This article contains supplemental Figs S1 to S4, Files S1 to S3, and Tables S1 to S6.
1 The abbreviations used are:
- GO
- gene ontology
- LC-MS/MS
- liquid chromatograph tandem mass spectrometry
- mTP
- mitochondrial transit peptide.
REFERENCES
- 1. Arnesen T. (2011) Towards a functional understanding of protein N-terminal acetylation. PLoS Biol. 9, e1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Starheim K. K., Gevaert K., Arnesen T. (2012) Protein N-terminal acetyltransferases: when the start matters. Trends Biochem. Sci. 37, 152–161 [DOI] [PubMed] [Google Scholar]
- 3. van den Berg B. H., Tholey A. (2012) Mass spectrometry-based proteomics strategies for protease cleavage site identification. Proteomics 12, 516–529 [DOI] [PubMed] [Google Scholar]
- 4. Staes A., Impens F., Van Damme P., Ruttens B., Goethals M., Demol H., Timmerman E., Vandekerckhove J., Gevaert K. (2011) Selecting protein N-terminal peptides by combined fractional diagonal chromatography. Nat. Protoc. 6, 1130–1141 [DOI] [PubMed] [Google Scholar]
- 5. Impens F., Colaert N., Helsens K., Plasman K., Van Damme P., Vandekerckhove J., Gevaert K. (2010) MS-driven protease substrate degradomics. Proteomics 10, 1284–1296 [DOI] [PubMed] [Google Scholar]
- 6. auf dem Keller U., Schilling O. (2010) Proteomic techniques and activity-based probes for the system-wide study of proteolysis. Biochimie 92, 1705–1714 [DOI] [PubMed] [Google Scholar]
- 7. Drag M., Bogyo M., Ellman J. A., Salvesen G. S. (2010) Aminopeptidase fingerprints, an integrated approach for identification of good substrates and optimal inhibitors. J. Biol. Chem. 285, 3310–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wildes D., Wells J. A. (2010) Sampling the N-terminal proteome of human blood. Proc. Natl. Acad. Sci. U.S.A. 107, 4561–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang T. K., Jackson D. Y., Burnier J. P., Wells J. A. (1994) Subtiligase: a tool for semisynthesis of proteins. Proc. Natl. Acad. Sci. U.S.A. 91, 12544–12548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jackson D. Y., Burnier J., Quan C., Stanley M., Tom J., Wells J. A. (1994) A designed peptide ligase for total synthesis of ribonuclease A with unnatural catalytic residues. Science 266, 243–247 [DOI] [PubMed] [Google Scholar]
- 12. Agard N. J., Mahrus S., Trinidad J. C., Lynn A., Burlingame A. L., Wells J. A. (2012) Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proc. Natl. Acad. Sci. U.S.A. 109, 1913–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shimbo K., Hsu G. W., Nguyen H., Mahrus S., Trinidad J. C., Burlingame A. L., Wells J. A. (2012) Quantitative profiling of caspase-cleaved substrates reveals different drug-induced and cell-type patterns in apoptosis. Proc. Natl. Acad. Sci. U.S.A. 109, 12432–12437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crawford E. D., Seaman J. E., Barber A. E., 2nd, David D. C., Babbitt P. C., Burlingame A. L., Wells J. A. (2012) Conservation of caspase substrates across metazoans suggests hierarchical importance of signaling pathways over specific targets and cleavage site motifs in apoptosis. Cell Death Differ. 19, 2040–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agard N. J., Maltby D., Wells J. A. (2010) Inflammatory stimuli regulate caspase substrate profiles. Mol. Cell Proteomics 9, 880–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luthi A. U., Martin S. J. (2007) The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 14, 641–650 [DOI] [PubMed] [Google Scholar]
- 17. Rawlings N. D., Barrett A. J., Bateman A. (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lange P. F., Huesgen P. F., Overall C. M. (2012) TopFIND 2.0–linking protein termini with proteolytic processing and modifications altering protein function. Nucleic Acids Res. 40, D351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshihara H. A., Mahrus S., Wells J. A. (2008) Tags for labeling protein N-termini with subtiligase for proteomics. Bioorg. Med. Chem. Lett. 18, 6000–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chalkley R. J., Baker P. R., Medzihradszky K. F., Lynn A. J., Burlingame A. L. (2008) In-depth analysis of tandem mass spectrometry data from disparate instrument types. Mol. Cell Proteomics 7, 2386–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang M., Weiss M., Simonovic M., Haertinger G., Schrimpf S. P., Hengartner M. O., von Mering C. (2012) PaxDb, a Database of Protein Abundance Averages Across All Three Domains of Life. Mol. Cell Proteomics 11, 492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 24. Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S. E., Walford G. A., Sugiana C., Boneh A., Chen W. K., Hill D. E., Vidal M., Evans J. G., Thorburn D. R., Carr S. A., Mootha V. K. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boyle E. I., Weng S., Gollub J., Jin H., Botstein D., Cherry J. M., Sherlock G. (2004) GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20, 3710–3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schechter I., Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 27. Vögtle F. N., Wortelkamp S., Zahedi R. P., Becker D., Leidhold C., Gevaert K., Kellermann J., Voos W., Sickmann A., Pfanner N., Meisinger C. (2009) Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell 139, 428–439 [DOI] [PubMed] [Google Scholar]
- 28. Emanuelsson O., Brunak S., von Heijne G., Nielsen H. (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2, 953–971 [DOI] [PubMed] [Google Scholar]
- 29. Brown J. L., Roberts W. K. (1976) Evidence that approximately eighty per cent of the soluble proteins from Ehrlich ascites cells are Nalpha-acetylated. J. Biol. Chem. 251, 1009–1014 [PubMed] [Google Scholar]
- 30. Van Damme P., Arnesen T., Gevaert K. (2011) Protein alpha-N-acetylation studied by N-terminomics. Febs J. 278, 3822–3834 [DOI] [PubMed] [Google Scholar]
- 31. Hwang C. S., Shemorry A., Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bradshaw R. A., Brickey W. W., Walker K. W. (1998) N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem. Sci. 23, 263–267 [DOI] [PubMed] [Google Scholar]
- 33. Neupert W., Herrmann J. M. (2007) Translocation of proteins into mitochondria. Annu. Rev. Biochem. 76, 723–749 [DOI] [PubMed] [Google Scholar]
- 34. Taylor A. B., Smith B. S., Kitada S., Kojima K., Miyaura H., Otwinowski Z., Ito A., Deisenhofer J. (2001) Crystal structures of mitochondrial processing peptidase reveal the mode for specific cleavage of import signal sequences. Structure 9, 615–625 [DOI] [PubMed] [Google Scholar]
- 35. Varshavsky A. (2011) The N-end rule pathway and regulation by proteolysis. Protein Sci. 20, 1298–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rapoport T. A. (1992) Transport of proteins across the endoplasmic reticulum membrane. Science 258, 931–936 [DOI] [PubMed] [Google Scholar]
- 37. Imai K., Nakai K. (2010) Prediction of subcellular locations of proteins: where to proceed? Proteomics 10, 3970–3983 [DOI] [PubMed] [Google Scholar]
- 38. Kisselev A. F., Akopian T. N., Woo K. M., Goldberg A. L. (1999) The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J. Biol. Chem. 274, 3363–3371 [DOI] [PubMed] [Google Scholar]
- 39. Stennicke H. R., Renatus M., Meldal M., Salvesen G. S. (2000) Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem. J. 350, 563–568 [PMC free article] [PubMed] [Google Scholar]
- 40. Gonda D. K., Bachmair A., Wünning I., Tobias J. W., Lane W. S., Varshavsky A. (1989) Universality and structure of the N-end rule. J. Biol. Chem. 264, 16700–16712 [PubMed] [Google Scholar]
- 41. Arntzen M. Ø., Thiede B. (2012) ApoptoProteomics, an integrated database for analysis of proteomics data obtained from apoptotic cells. Mol. Cell Proteomics 11, M111.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dix M. M., Simon G. M., Cravatt B. F. (2008) Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell 134, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Piatkov K. I., Brower C. S., Varshavsky A. (2012) The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc. Natl. Acad. Sci. U.S.A. 109, E1839–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kurokawa M., Kornbluth S. (2009) Caspases and kinases in a death grip. Cell 138, 838–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 46. Miranda-Saavedra D., Barton G. J. (2007) Classification and functional annotation of eukaryotic protein kinases. Proteins 68, 893–914 [DOI] [PubMed] [Google Scholar]
- 47. Dix M. M., Simon G. M., Wang C., Okerberg E., Patricelli M. P., Cravatt B. F. (2012) Functional Interplay between Caspase Cleavage and Phosphorylation Sculpts the Apoptotic Proteome. Cell 150, 426–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.