

Abstract
Many studies of molecular microbial ecology rely on the characterization of microbial communities by PCR amplification, cloning, sequencing, and phylogenetic analysis of genes encoding rRNAs or functional marker enzymes. However, if the established clone libraries are dominated by one or a few sequence types, the cloned diversity is difficult to analyze by random clone sequencing. Here we present a novel approach to deplete unwanted sequence types from complex nucleic acid mixtures prior to cloning and downstream analyses. It employs catalytically active oligonucleotides containing locked nucleic acids (LNAzymes) for the specific cleavage of selected RNA targets. When combined with in vitro transcription and reverse transcriptase PCR, this LNAzyme-based technique can be used with DNA or RNA extracts from microbial communities. The simultaneous application of more than one specific LNAzyme allows the concurrent depletion of different sequence types from the same nucleic acid preparation. This new method was evaluated with defined mixtures of cloned 16S rRNA genes and then used to identify accompanying bacteria in an enrichment culture dominated by the nitrite oxidizer “Candidatus Nitrospira defluvii.” In silico analysis revealed that the majority of publicly deposited rRNA-targeted oligonucleotide probes may be used as specific LNAzymes with no or only minor sequence modifications. This efficient and cost-effective approach will greatly facilitate tasks such as the identification of microbial symbionts in nucleic acid preparations dominated by plastid or mitochondrial rRNA genes from eukaryotic hosts, the detection of contaminants in microbial cultures, and the analysis of rare organisms in microbial communities of highly uneven composition.
INTRODUCTION
The “rRNA approach” (1–5), i.e., the PCR amplification, cloning, sequencing, and phylogenetic analysis of rRNA genes, has been fundamental for our current understanding of natural microbial diversity. By revealing a huge number of as-yet-uncultured microbes and novel phylogenetic lineages that have been overlooked by traditional cultivation-based methods of microbiology (reviewed in references 6, 7, 8, and 9), this approach has substantially influenced our perception of microbial communities in the environment. Moreover, variations of the same principle using suitable functional marker genes have become the gold standard for diversity analyses of selected microbial guilds (e.g., see references 10, 11, 12, 13, and 14).
In all of these approaches, a highly uneven abundance of the target organisms in the sample, different target gene copy numbers per genome (15), mismatches between primers and some templates, and other PCR biases can lead to the dominance of a few sequence types in the resulting clone libraries. In such cases, an encompassing diversity census of the microbial consortium to be analyzed would require extensive redundant DNA sequencing to retrieve sequences that occur less frequently in the clone library. Rare sequences in a library can be more easily retrieved by the separation of PCR amplicons using denaturing gradient gel electrophoresis (DGGE) prior to cloning and sequencing or by the prescreening of clone libraries by restriction fragment length polymorphism (RFLP) or other fingerprinting methods (16, 17). However, these and related methods can be time-consuming and have their own limitations, such as problems in distinguishing organisms that are represented by highly similar or identical RFLP patterns or by comigrating bands in DGGE gels.
Other approaches to reduce the workload for microbial community analyses prevent the PCR amplification of selected (usually dominant) templates in DNA extracts made from environmental samples (18, 19). Here we present a novel method to specifically remove such unwanted targets that can be applied to either RNA or DNA extracts. It employs catalytically active, locked nucleic acid (LNA)-containing oligonucleotides (“LNAzymes”) that hybridize to their target RNA and cleave it at a specific site. This approach is based on the discovery of Breaker and Joyce (20), who used in vitro selection to isolate the first catalytically active molecule that was completely composed of deoxyribonucleotides (a “DNAzyme”) and that was able to cleave RNA in the presence of Pb2+ ions. In 1997, Santoro and Joyce (21) isolated two DNAzymes capable of cleaving RNA at specific sites under simulated in vivo conditions. Since then, two DNAzyme sequence motifs, referred to as “8-17” and “10-23,” have been used in many applications in molecular biology (22, 23). DNAzymes with the motif 10-23, named after the 23rd clone of the 10th round of in vitro selection (21), consist of a 15-nucleotide catalytic domain that is flanked by two substrate-binding arms (Fig. 1). The catalytic domain usually is not amenable to any sequence changes without losing its activity. However, the sequences of the substrate-binding arms, which usually consist of 7 to 11 nucleotides, can be freely modified. DNAzymes of type 10-23 cut the phosphodiester bond in RNA at a purine-pyrimidine (R-Y) junction (Fig. 1A), with the efficiency of cleavage following the scheme AU ≈ GU > GC ≫ AC (24). If the substrate-binding arms contain LNA or 2-O-methyl RNA nucleotides, the efficiency is significantly improved, especially for cutting RNA molecules that form secondary structures. Locked nucleic acids contain at least one nucleotide whose sugar moiety is modified and has a bicyclic structure, which “locks” the conformation of the sugar (25). Due to the higher binding affinity of LNA nucleotides, LNAzymes can better resolve and hybridize to complex RNA structures. This leads to an improved efficiency of cleavage without compromising the specificity for a particular target (26–28). Interestingly, DNAzymes and LNAzymes are highly sensitive to base mismatches to the substrate RNA in one of the substrate-binding arms. Just one strong mismatch close to the cleavage site is sufficient to dramatically reduce the cleavage efficiency (29, 30) (Fig. 1B).
Fig 1.

Mechanism of LNAzyme-mediated depletion of specific RNA templates. (A) Binding of the LNAzyme to its target due to hybridization of the substrate-binding arms up- and downstream of the cleavage site, followed by specific cleavage of the target and the resulting failure of RT-PCR amplification. (B) Base mismatches between the substrate-binding arms of the LNAzyme and a nontarget RNA template prevent the cleavage even if hybridization occurs. Nontarget templates, which are identical in sequence to the substrate-binding arms but do not contain any RY (purine-pyrimidine) cleavage site in the binding region of the LNAzyme, are also not cleaved. In either case, the nontarget template remains intact for subsequent RT-PCR amplification and downstream analyses or cloning. This figure is partly based on illustrations of DNAzymes by Santoro and Joyce (29), Cairns et al. (24), and Suenaga et al. (30).
To date, DNAzymes and LNAzymes have seldom been used in molecular microbial ecology (30). Our approach to deplete unwanted sequence types relies on LNAzymes whose substrate-binding arms are sequence complementary to specific target sites on the RNA molecules to be cleaved. We evaluated the specificity and performance of this method by using defined mixtures of 16S rRNA genes, and subsequently, we demonstrated its usefulness for identifying rare members of a microbial community that is dominated by one organism.
MATERIALS AND METHODS
Outline of the protocol for selective template removal.
As LNAzymes cleave RNA exclusively, they can be applied directly to RNA preparations from environmental samples for the specific removal of unwanted RNA molecules (Fig. 2). However, by implementing additional steps in the workflow, LNAzymes can also be used with DNA preparations, which more commonly serve as templates for the PCR amplification of rRNA genes in the “rRNA approach.” In this case, the forward (sense) PCR primer must contain a T3 or T7 RNA polymerase promoter at its 5′ end. The obtained PCR amplicons subsequently serve as templates for in vitro transcription (IVT) to RNA. The resulting mixture of RNA molecules is then subjected to the specific cleavage of unwanted targets by LNAzymes. The remaining intact RNAs are converted back to DNA by reverse transcriptase PCR (RT-PCR), whereas the cleaved RNAs do not serve as templates for RT-PCR (Fig. 1 and 2). Thus, the obtained DNA is depleted in those targets that were cleaved by the LNAzymes. If a forward primer with a T3 or T7 RNA polymerase promoter is also used in the RT-PCR step, the procedure can be repeated for further depletion of the unwanted targets (Fig. 2).
Fig 2.
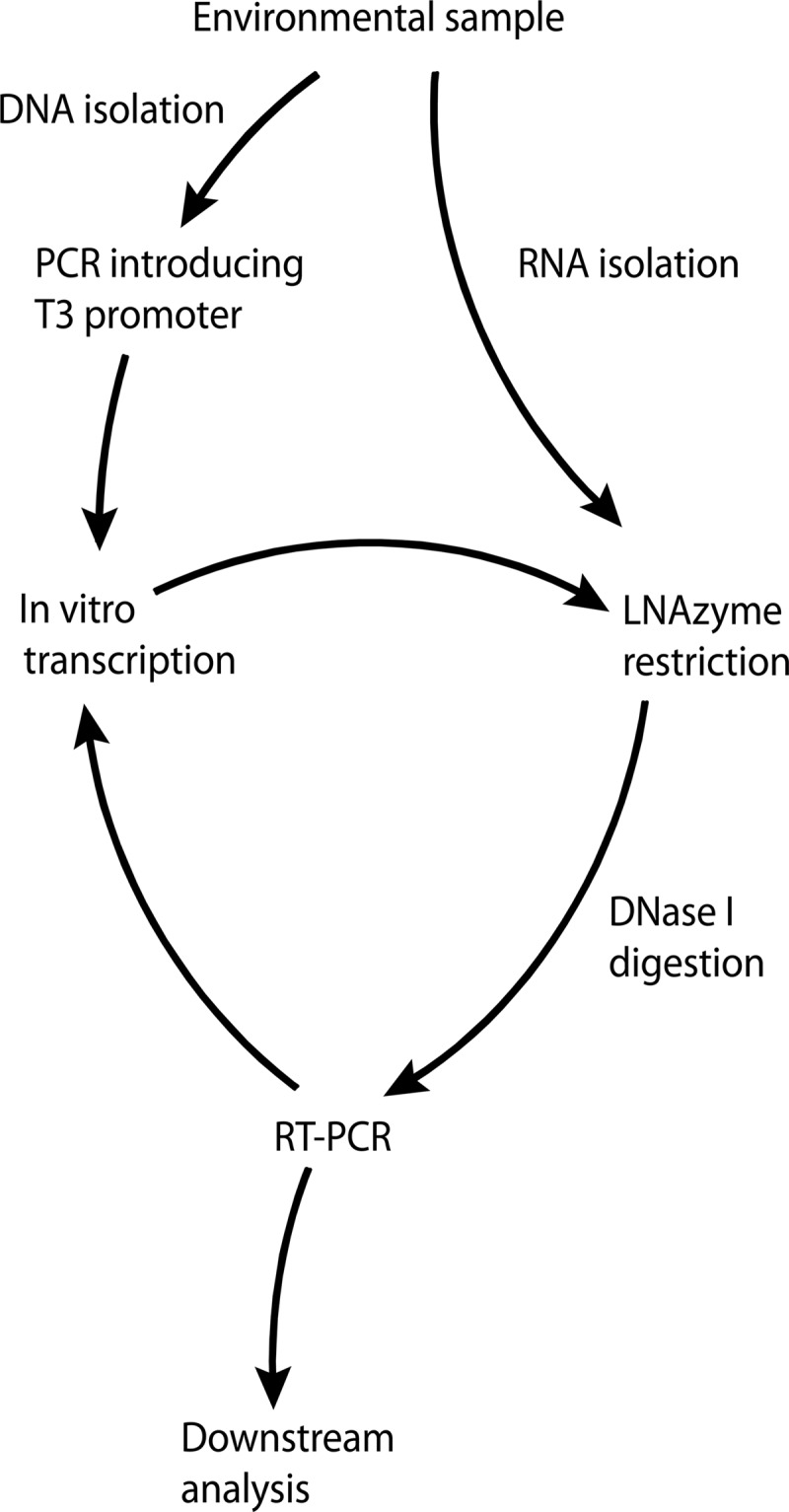
Schematic illustration of the workflow for the specific depletion of unwanted templates using LNAzymes. The procedure starts with the extraction of DNA or RNA from an environmental sample and results in a final PCR product, which is suitable for cloning or analyses by fingerprinting techniques such as T-RFLP.
Nucleic acid extraction.
The nitrite-oxidizing enrichment culture of “Candidatus Nitrospira defluvii” was maintained as described previously (31). An aliquot of 50 ml of the culture was centrifuged at 6,000 × g at room temperature for 4 min, and the pellet was resuspended in 1 ml of TRIzol (Invitrogen, Carlsbad, CA). Activated sludge samples were collected from a nitrifying sequencing batch reactor at the municipal wastewater treatment plant of Ingolstadt, Germany. Sludge flocs were allowed to settle, liquid supernatant was removed, and the concentrated sludge was frozen in liquid N2. Aliquots were stored at −80°C and, before use, were resuspended in 1 ml of TRIzol. The suspensions (“Ca. Nitrospira defluvii” enrichment culture or activated sludge) were then transferred into Lysing Matrix A tubes (MP Biomedicals, Solon, OH), and cells were disrupted in a bead beater (Bio 101, Vista, CA) set at 6.5 m s−1 for 45 s. RNA and DNA were extracted according to recommendations provided with TRIzol. As a gelatinous matrix was coextracted with the RNA, 0.25 ml of a salt solution (2 M NaCl, 0.2 M LiCl) per ml of TRIzol was added to the aqueous phase prior to precipitation with 2-propanol to obtain pure RNA. Any DNA present in the RNA preparation was removed by DNase I (Sigma-Aldrich, St. Louis, MO) digestion in a modified DNase I buffer (20 mM Tris-HCl [pH 8.0], 2 mM MgCl2, 0.2 mM CaCl2) for 45 min at 37°C. Following digestion, the RNA was extracted again with TRIzol. DNA extracted with TRIzol was further purified by using the AllPrep DNA/RNA minikit (Qiagen, Hilden, Germany).
PCR and RT-PCR.
All PCRs were performed according to the following scheme: DNA was denatured at 94°C for 2 min, followed by 25 cycles of denaturation at 94°C for 30 s, primer annealing at 54°C for 45 s, and elongation at 72°C for 90 s. Cycling was completed with a final elongation step at 72°C for 10 min. The reaction mixtures contained 1× enzyme buffer, 2 mM MgCl2, 0.2 mM deoxynucleoside triphosphates (dNTPs), 0.5 μM each primer, 0.75 U of Taq polymerase, 0.05 mg ml−1 bovine serum albumin (all from Fermentas, St. Leon-Rot, Germany), and 2 μl of template DNA in a final volume of 25 μl. For RT-PCR, the Access One Step RT-PCR kit (Promega, Madison, WI) was used. Briefly, cDNA was synthesized at 45°C for 45 min, followed by denaturation at 94°C for 2 min and by 25 cycles of denaturation at 94°C for 30 s, primer annealing at 54°C for 45 s, and elongation at 68°C for 2 min. Cycling was finished with a final elongation step at 68°C for 7 min. The absence of DNA in all RNA templates was confirmed by PCR without reverse transcription. All primers used in this study for PCR and RT-PCR are listed in Table S1 in the supplemental material. Primers were obtained from Thermo Fisher (Thermo Fisher Scientific, Ulm, Germany). For PCRs and for RT-PCR, the nucleic acid templates were diluted 1:50 in ultrapure water in order to avoid PCR inhibition by highly concentrated template or inhibitory compounds.
In vitro transcription.
PCR-amplified 16S rRNA genes containing a 5′ T3 promoter, for in vitro transcription by T3 RNA polymerase, were obtained by PCR with primers T3-8F and 907R. These amplicons were analyzed by agarose gel electrophoresis, and bands of the correct size were excised from the gels and purified with the QiaQuick gel extraction kit (Qiagen). The reaction mixture for in vitro transcription contained 1× T3 polymerase transcription buffer (Fermentas), 10 mM dithiothreitol (DTT) (Carl Roth, Karlsruhe, Germany), 1 U of T3 polymerase (Fermentas), 0.5 mM nucleoside triphosphates (NTPs) (Hoffmann-La Roche, Basel, Switzerland), and 100 to 200 ng of purified PCR product. The final volume was 20 μl. The mixture was incubated at 37°C for 4 h.
LNAzyme design and digestion of target rRNAs.
Already existing 16S rRNA-targeted probes for fluorescence in situ hybridization (FISH) were used as a starting point for the design of specific LNAzymes, which was accomplished by using the software package ARB (32). In particular, the ARB “Probe Match” tool was applied to find optimal LNAzyme cleavage positions in the target rRNAs. For this purpose, the FISH probe sequences were entered and then elongated and/or shortened at their 5′ or 3′ ends to shift the probe-binding sites along the target rRNA sequence until a suitable cleavage (AU or GU) site was reached. The candidate LNAzymes obtained from this procedure were checked for unwanted base mismatches to the target bacteria and for required mismatches to nontarget organisms as well as for self-complementary regions that could reduce cleavage efficiency due to hairpin formation or LNAzyme dimerization.
In this study, we designed three 16S rRNA-targeted LNAzymes specific for ammonia-oxidizing bacteria (AOB) or nitrite-oxidizing bacteria (NOB) (see Table S1 in the supplemental material). All three LNAzymes contain the 10-23 motif. LNAzyme Nso215 covers the AOB Nitrosomonas oligotropha, N. europaea, N. ureae, N. halophila, N. cryotolerans, Nitrosomonas sp. Nm143 and Nm84, Nitrosococcus (Nitrosomonas) mobilis, and most environmental sequences related to these cultured strains in the SILVA, release 102, full-length, nonredundant database. The 16S rRNA sequences of other nitrosomonads have base mismatches to the substrate-binding arms that most likely decrease the cleavage efficiency of this LNAzyme. The target region of Nso215 on the 16S rRNA overlaps the target site of probe Nmo218 that is commonly used for the FISH detection of N. oligotropha-related AOB (33). The cleavage site of Nso215 is GU, and the substrate-binding arms, determining the specificity of the LNAzyme, contain four LNA nucleotides (see Table S1 and Fig. S1 in the supplemental material). The LNA nucleotides were placed at the mismatch positions to most nontarget organisms that had a similar target sequence in the SILVA, release 102, database (34), because LNAs provide better discrimination against single-base mismatches (25). The LNAzymes Ntspa668 and Ntspa665, both targeting NOB of the genus Nitrospira, were derived from the Nitrospira-specific FISH probe Ntspa662 (35). This probe was elongated in the 5′ direction along the target 16S rRNA, resulting in a suitable AU cleavage site being flanked by the substrate-binding arms (see Table S1 and Fig. S1 in the supplemental material). Both LNAzymes cover all known sublineages of the genus Nitrospira (35–37), except for some members of marine lineage IV. The substrate-binding arms of Ntspa665 are longer (15 nucleotides up- and downstream of the cleavage site, instead of 12 and 10 nucleotides in Ntspa668). In addition, Ntspa665 contains only cytosine LNA nucleotides, which do not form base pairs with each other and thus greatly reduce the likelihood of intramolecular strong secondary structure formation. In both LNAzymes, the four LNA nucleotides were placed at positions of frequent mismatches to nontarget organisms.
The LNAzymes were obtained from Exiqon (Vedbaek, Denmark). We observed that both highly concentrated stocks as well as working solutions of LNAzymes partly lose their activity after storage at −20°C for several months. This problem was avoided by storing them in concentrated aliquots at −80°C. The LNAzyme digestion buffer contained 100 mM NaCl, 50 mM RNase-free Tris (pH 8.0) (Ambion, Austin, TX), 10 mM LiCl, 1 μM LNAzyme, and 5 μl of an in vitro transcription reaction mixture containing the transcribed RNA (see above). The final volume of the reaction mixture was 60 μl. All reagents except Tris were treated with 0.1% (vol/vol) diethylpyrocarbonate (DEPC) to inactivate nucleases. Previous studies found that Mg2+ or other divalent cations are required for DNAzyme activity (21, 29). Good LNAzyme efficiency was obtained even though Mg2+ was not added to the LNAzyme digestion buffer in an effort to preserve the integrity of RNA at elevated temperatures (see Results). However, we cannot exclude that the buffer contained trace amounts of divalent cations, because no chelating agent was added.
The efficiency of LNAzyme-mediated cleavage was improved by repeated heating and cooling of the reaction mixture (38). The mixture was incubated first for 90 s at 90°C for RNA denaturation, then for 30 s at 60°C to allow for annealing of LNAzymes to their target RNA molecules, and finally for 45 min at 37°C for the actual cleavage. These steps were repeated three times, resulting in four identical cycles of denaturation, annealing, and cleavage. PCR products (DNA), which had served as templates in the previous in vitro transcription step and which had been carried over into the LNAzyme reaction mixture, were then digested with DNase I for 45 min at 37°C. This digestion step also removed residual oligonucleotides (PCR primers) and the LNAzymes. Subsequently, 6 μl of 50 mM EDTA was added, and DNase I was inactivated by heating the mixture to 70°C for 10 min. After this step, the noncleaved RNA was ready for use as the template for RT-PCR.
T-RFLP.
For terminal restriction fragment length polymorphism (T-RFLP) analysis, primer 8F labeled with 6-carboxyfluorescein (6-FAM) was used for PCR or RT-PCRs. Amplicons of the correct size were excised from agarose gels and purified by using the QiaQuick gel extraction kit (Qiagen), and approximately 100 ng of purified amplicons was digested for 3 h at 37°C by using the restriction endonuclease MspI (Fermentas). The digestion products were analyzed by using an ABI 3130xl genetic analyzer (Applied Biosystems, Foster City, CA) and PeakScanner 1.0 software (Applied Biosystems). Real peaks were distinguished from noise by using methods described previously by Abdo et al. (39) and by using the Perl and “R” scripts provided on their website (http://www.ibest.uidaho.edu/tools/trflp_stats/index.php). The effects of the LNAzyme treatments were tested for significance by one-way analysis of variance (ANOVA) including Tukey's honestly significant difference (HSD) post hoc test, which was performed by using the PASW Statistics 17 software package (IBM SPSS, Armonk, NY).
Cloning, sequencing, and phylogenetic analysis of 16S rRNA genes.
One clone library was established from the “Ca. Nitrospira defluvii” enrichment using DNA as the initial template, by applying LNAzyme Ntspa668 (see Table S1 in the supplemental material) after PCR and IVT and by performing RT-PCR with primer pair 8F/907R (Fig. 2). Two clone libraries were constructed by using RNA as the initial template, which was treated with LNAzyme Ntspa665 and the helper probes (see Table S1 in the supplemental material). For one of these “RNA-based” libraries, primer pair 8F/907R was used for RT-PCR, whereas primer pair 8F/1492R was applied for the other library, which thus contained almost full-length 16S rRNA genes. For comparison, a fourth library was made by using DNA extracted from the enrichment culture as the template for PCR with primer pair 8F/907R and without any LNAzyme treatment.
PCR products of the correct size were excised from agarose gels, purified with the QiaQuick gel extraction kit (Qiagen), and cloned by using the TOPO TA cloning system (Invitrogen). Cloned genes were subjected to RFLP analysis with the restriction endonuclease MspI (Fermentas). Based on the obtained band patterns in agarose gels, Good's coverage of the clone libraries was calculated as
where n1 is the number of RFLP patterns that occurred only once and N is the total number of clones analyzed. The Simpson diversity index was calculated as follows
where R is the number of different RFLP profiles observed and ni is the number of clones with a particular RFLP pattern. A subset of the clones was selected for sequencing based upon the RFLP patterns. Sanger sequencing was performed by using BigDye Terminator cycle sequencing kit v3.1 and an ABI 3130xl genetic analyzer (Applied Biosystems), according to the manufacturer's instructions. The obtained 16S rRNA gene sequences were aligned with SINA Webaligner (http://www.arb-silva.de/aligner) and further analyzed by using ARB software (32) with the SSU Ref 108 SILVA NR sequence database (34). Automatically generated sequence alignments were manually refined. The phylogenetic affiliation of each sequence was determined by adding the sequences to the tree “tree_SSURef_1200_99_slv_108,” which is included in the database, by using the ARB parsimony function and the “pos_var_ssuref:bacteria” sequence conservation filter that is also included in the database. Prior to adding the new sequences, all sequences with a Pintail (40) score below 80 were removed from the tree. Putative chimeric sequences were identified by independent phylogenetic analyses of the first 513 5′ base positions, the central 513 base positions, and the last 513 3′ base positions of the sequences. Sequences that showed an unstable phylogenetic affiliation in these tests were further checked for anomalies with Pintail software (40).
Fluorescence in situ hybridization.
Bacteria in the “Ca. Nitrospira defluvii” enrichment culture were visualized by FISH with rRNA-targeted oligonucleotide probes (see Table S1 in the supplemental material), as described previously (41). Fluorescence-labeled oligonucleotide probes were obtained from Thermo Fisher. Microscopic observation and documentation were performed with an LSM 510 confocal laser scanning microscope and the provided software (Zeiss, Oberkochen, Germany). The relative biovolume of accompanying bacteria in the enrichment culture (i.e., cells that were labeled by the EUB338 probe mix but not by probe Ntspa1431) was quantified by FISH and digital image analysis (42) and by using daime software (43).
Nucleotide sequence accession numbers.
The sequences obtained in this study have been deposited in the GenBank database under accession no. JF449905 to JF449947. The sequences of the 16S rRNA genes in Table 1 have been deposited in the GenBank database under accession no. JQ068103 to JQ068106.
Table 1.
Concentration ratios of plasmid vectors carrying the respective partial 16S rRNA genes in the artificial mixtures that were used to evaluate the LNAzymes and the protocol developed in this study
| Mixture | Concn ratio |
|||
|---|---|---|---|---|
| Nitrosomonas sp. | Nitrospira sp. | Chloroflexi sp. | Comamonas sp. | |
| A | 100 | 15 | 10 | 1 |
| B | 15 | 100 | 1 | 10 |
| C | 100 | 1 | 1 | 1 |
| D | 200 | 100 | 1 | 1 |
RESULTS
Evaluation of the protocol and the LNAzymes.
The newly designed LNAzymes and our protocol for the specific removal of unwanted template nucleic acids (Fig. 2) were tested with artificial mixtures containing template DNAs from target and nontarget organisms. For this purpose, we used four cloned partial 16S rRNA genes that had been retrieved from a nitrifying activated sludge and belonged to bacteria related to the Nitrosomonas oligotropha lineage (44), Comamonas aquatica, the phylum Chloroflexi, and Nitrospira lineage I (35), respectively. The plasmid vectors, which contained these cloned genes, were mixed in defined ratios (Table 1). The mixed 16S rRNA genes were then PCR amplified by using primers 8F (with a 5′ T3 promoter) and 907R (see Table S1 in the supplemental material). Subsequently, RNA was obtained by IVT of the mixed PCR amplicons. Further following the scheme in Fig. 2, this in vitro-synthesized RNA was incubated with the LNAzymes and was then converted back to cDNA and amplified by RT-PCR with primers 8F and 907R. Finally, T-RFLP analysis was used to determine the composition of the resulting PCR product mixture and to assess how efficiently the respective targets had been depleted by the LNAzyme approach.
LNAzyme-mediated RNA cleavage led to the depletion of specific targets in four different mixtures of the 16S rRNA genes (Fig. 3). LNAzyme Nso215 cleaved the partial 16S rRNA derived from the Nitrosomonas-related clone with high efficiency, and a single iteration of the protocol was sufficient to drastically reduce the abundance of this target (Fig. 3A and C). A second iteration of the protocol resulted in an almost complete elimination of the Nitrosomonas-related 16S rRNA gene from mixtures that originally contained large relative amounts of this target (Fig. 3A and C and Table 1). Concomitantly, the relative abundances of the other three 16S rRNA types increased in the mixtures (Fig. 3A and C), with the sole exception being the Comamonas-related 16S rRNA in mixture “A” (Table 1), whose relative abundance remained constant throughout the experiment (Fig. 3A). In contrast, the relative abundance of this 16S rRNA type increased in the experiment with mixture “C” (Table 1 and Fig. 3C), where the same LNAzyme, Nso215, was used to deplete Nitrosomonas-related 16S rRNA.
Fig 3.

Evaluation of the LNAzyme cleavage protocol with artificial mixtures of cloned partial 16S rRNA genes (Table 1). The bars show the relative abundances of the rRNA genes in the mixtures before and after one or two iterations of the LNAzyme-mediated depletion of the Nitrosomonas and/or Nitrospira rRNA. Data for control experiments without added LNAzymes are also shown (the second iteration without LNAzyme was performed after a first iteration with LNAzyme). The relative abundances were determined by T-RFLP analysis. Only the relevant T-RFLP peaks are shown; for the complete T-RFLP profiles, please refer to Fig. S2 in the supplemental material. (A) Depletion of the Nitrosomonas-related 16S rRNA gene by LNAzyme Nso215 using 16S rRNA gene plasmid mixture “A” (Table 1). (B) Depletion of the Nitrospira-related 16S rRNA gene by LNAzyme Ntspa668 using plasmid mixture “B” (Table 1). (C) Like panel A but with a different initial abundance of the Nitrosomonas-related and other 16S rRNA genes in the mixture (mixture “C” in Table 1). (D) Simultaneous depletion of the Nitrosomonas- and the Nitrospira-related 16S rRNA genes by LNAzymes Nso215 and Ntspa668 (mixture “D” in Table 1). Error bars depict standard deviations from three replicate experiments. One asterisk above the bars indicates that a single iteration of target depletion was sufficient to obtain a significant (P < 0.05) difference compared to the untreated plasmid mixture. Two asterisks mark cases in which two iterations of target depletion were needed to achieve a significant difference.
The Nitrospira-specific LNAzyme Ntspa668 was also successful at strongly depleting Nitrospira-related 16S rRNA (Fig. 3B). However, in this case, the difference between the first and second iterations of the protocol was more pronounced than with LNAzyme Nso215 (Fig. 3B). Finally, we tested whether two different LNAzymes can be combined in the same experiment for depleting different targets. For this purpose, the LNAzymes Nso215 and Ntspa668 were used simultaneously with an artificial mixture containing large relative amounts of Nitrosomonas- and Nitrospira-related 16S rRNA genes but very small relative amounts of the Comamonas- and Chloroflexi-related rRNA genes. Both LNAzymes cleaved their respective targets with efficiencies that were similar to those observed for single LNAzyme experiments (Fig. 3D). It is important to note that the apparently weak decrease of the Nitrospira-like 16S rRNA levels during the first iteration (Fig. 3D) did not result from a low efficiency of LNAzyme Ntspa668. The very pronounced depletion of the Nitrosomonas-like 16S rRNA (Fig. 3D) must have increased the relative amounts of the other 16S rRNA types in the mixture, including Nitrospira. This relative increase in the amount of the Nitrospira-like 16S rRNA was neutralized by Ntspa668. After two iterations, the activity of this LNAzyme led to the strong depletion of the Nitrospira-like 16S rRNA, which was not observed in the control experiment without Ntspa668 (Fig. 3D).
In preliminary experiments, the obtained T-RFLP profiles contained numerous peaks apparently belonging to short fragments, which were inconsistent with the known positions of restriction endonuclease cleavage sites in the 16S rRNA gene sequences. The number of those peaks increased after each iteration of the LNAzyme cleavage protocol (data not shown). These artifacts were eliminated by agarose gel electrophoresis of the PCR products and excision of the correct bands prior to IVT or T-RFLP analysis (see also Materials and Methods).
Identification of accompanying bacteria in a Nitrospira enrichment culture.
After the new LNAzymes and the cleavage protocol had been tested with artificial template mixtures, the method was used to identify accompanying bacteria (“contaminants”) in an enrichment culture of the nitrite-oxidizing bacterium “Candidatus Nitrospira defluvii” (31). This enrichment culture consisted mainly of “Ca. Nitrospira defluvii” and had been used for sequencing of the genome of this nitrifier by environmental genomics (45) but still contained minor amounts of bacteria that do not belong to the genus Nitrospira according to FISH with rRNA-targeted probes (31). The presence of contaminants in the Nitrospira culture, which was used for this study, was confirmed by quantitative FISH, revealing that 13% of the bacterial community in this enrichment culture did not hybridize to any Nitrospira-specific FISH probe but was detected by the Bacteria-specific EUB338 probe mix.
DNA and RNA were extracted and purified from aliquots of the “Ca. Nitrospira defluvii” enrichment culture. Subsequently, the 16S rRNA of Nitrospira was depleted by using LNAzymes and the protocol (Fig. 2), with either DNA or RNA as the initial template. When the procedure started with DNA, most of the Nitrospira target was already removed after the first iteration using LNAzyme Ntspa668 (Fig. 4A). This led to higher relative abundances of other 16S rRNA types, which was evident from the increasing areas of some peaks in the T-RFLP profile (Fig. 4A).
Fig 4.

Depletion of 16S rRNA of “Ca. Nitrospira defluvii” from DNA (A) or native RNA (B) that was extracted from the “Ca. Nitrospira defluvii” enrichment culture. The bars show the relative abundances of the 16S rRNA genes of “Ca. Nitrospira defluvii” and of other bacteria, as determined by T-RFLP analysis, before and after one or two iterations of the LNAzyme-mediated depletion. Data for control experiments without added LNAzymes are also shown (the second iteration without LNAzyme was performed after a first iteration with LNAzyme). Only selected T-RFLP peaks are shown; for the complete T-RFLP profiles, please refer to Fig. S3 in the supplemental material. Applied LNAzymes were Ntspa668 (A) and Ntspa668 or Ntspa665 (B) (which also shows the effects of additional helper probes [HP] on the efficiency of target depletion). The decrease of the peaks at positions 487 and 490 (A) in the second iteration probably reflects artifacts of PCR or IVT. Error bars depict standard deviations from three replicate experiments. Asterisks above bars indicate that a significant (P < 0.05) difference compared to the untreated DNA or RNA was obtained. Where this was achieved in the experiment with DNA (A), a single iteration of target depletion was sufficient. In panel B, the results of the significance test are shown for the treatment with helper probes and LNAzyme Ntspa665.
Interestingly, LNAzyme Ntspa668 cleaved native 16S rRNA of Nitrospira with poor efficiency (Fig. 4B). This result was in contrast to the good performance of this LNAzyme when cleaving RNA that had been produced by IVT of a DNA template (Fig. 3B and 4A). Therefore, the second Nitrospira-specific LNAzyme (Ntspa665) was designed with longer substrate-binding arms and different LNA nucleotides (see above). Application of Ntspa665 to the native RNA resulted in a significantly stronger relative decrease in the level of the Nitrospira target than achieved by Ntspa668 (Fig. 4B). As we assumed that secondary structures of the native 16S rRNA hampered LNAzyme-mediated cleavage, we combined the LNAzymes with helper probes (46) that were newly developed and targeted Nitrospira lineages I and II, Ntspa630help and Ntspa700help (see Table S1 in the supplemental material). These helper probes were supposed to bind to the Nitrospira 16S rRNA adjacent to the target sites of the LNAzymes, thereby resolving secondary structures and facilitating hybridization of the LNAzymes to their target. Indeed, the helper probes improved the depletion of native Nitrospira 16S rRNA by both LNAzymes Ntspa668 and Ntspa665 (Fig. 4B). As observed when DNA was used as an initial template (Fig. 4A), the relative abundances of other 16S rRNA types increased as the abundance of Nitrospira 16S rRNA decreased (Fig. 4B). However, this shift was less pronounced, most likely due to the lower efficiency of both Nitrospira-specific LNAzymes when cleaving native 16S rRNA, even in the presence of helper probes.
To identify the accompanying bacteria in the enrichment culture, 16S rRNA genes were cloned after the depletion of the Nitrospira 16S rRNA. Between 40 and 50 randomly selected clones from each library were subjected to RFLP analysis to determine the proportion of cloned Nitrospira 16S rRNA genes. As expected, their frequency was much lower in all libraries produced from LNAzyme-treated templates than in the library that was made from untreated template DNA (Table 2). Analysis of the 31 obtained 16S rRNA gene sequences (after LNAzyme treatment) revealed a surprising diversity of accompanying bacteria in the “Ca. Nitrospira defluvii” enrichment culture, comprising representatives of seven bacterial phyla (see Table S2 in the supplemental material).
Table 2.
Frequencies of the 16S rRNA genes of “Ca. Nitrospira defluvii” and other bacteria in clone libraries established with and without preceding treatment by LNAzymese
| Treatmenta | No. of distinct RFLP patterns (no. of singletons)d | No. of clones assigned to “Ca. Nitrospira defluvii” | No. of clones assigned to other bacteria | Frequency of “Ca. Nitrospira defluvii” (%) | Good's coverage (%) | Simpson diversity index |
|---|---|---|---|---|---|---|
| Untreated DNA 907R | 4 (2) | 40 | 11 | 78.4 | 96.1 | 0.36 |
| DNA 907Rb | 12 (5) | 3 | 39 | 7.1 | 88.1 | 0.8 |
| RNA 907Rc | 14 (9) | 9 | 35 | 20.5 | 79.6 | 0.76 |
| RNA 1492Rc | 14 (9) | 1 | 45 | 2.2 | 80.4 | 0.84 |
DNA, DNA was used as the initial template; RNA, RNA was used as the initial template. 907R and 1492R are the reverse primers used for 16S rRNA gene-specific PCR.
Treatment by LNAzyme Ntspa668 prior to cloning.
Treatment by LNAzyme Ntspa665 prior to cloning.
Singletons are RFLP patterns that occurred only once.
Assignments of cloned genes to “Ca. Nitrospira defluvii” or other bacteria were based on RFLP band patterns, exploiting the fact that “Ca. Nitrospira defluvii” has a characteristic RFLP pattern after digestion of its 16S rRNA gene amplicon with the endonuclease MspI (this was confirmed by sequencing of selected clones showing this RFLP pattern).
DISCUSSION
Rank-abundance analyses have shown that complex microbial communities can be dominated by relatively few and highly abundant community members, whereas the bulk of diversity (richness) is “hidden” in a large number of rare taxa (47). This uneven structure is mirrored in the composition of cloned rRNA gene libraries established from samples of these communities. Similarly, rRNA gene libraries made from associations between microorganisms and eukaryotes are often dominated by clones carrying plastid or mitochondrial rRNA genes that were targeted by the applied Bacteria-specific PCR primers (e.g., see reference 48). To facilitate the detection of less frequent sequence types, a few methods have been developed for the specific depletion of unwanted (dominant) targets before or during PCR amplification (18, 19, 49). A common motif for these approaches is the use of nucleic acid oligomers, which must specifically hybridize to DNA templates that should not be amplified during PCR. The optimal hybridization conditions must be determined for each oligomer to avoid unspecific binding to nontarget DNA and inefficient hybridization to the target. The parallel elimination of more than one unwanted target would be complicated if the required stringent hybridization conditions prohibit the simultaneous application of different target-specific oligomers.
In this study, we developed an alternative approach that employs catalytically active LNAzymes (Fig. 1). The LNAzymes must also hybridize to their respective target, but the cleaving efficiency highly depends on perfect base complementarity close to the cleaving site. Hence, as demonstrated by previous fundamental work on the properties of DNAzymes and LNAzymes, even unspecific hybridization to a nontarget template (which has mismatches) would not lead to significant cleavage (24, 29, 30, 50). By including in vitro transcription, we demonstrated that the RNA-cleaving LNAzymes can also be used if DNA is the initial template.
The experiments with artificial mixtures of four different 16S rRNA genes confirmed that the newly designed LNAzymes Nso215 and Ntspa668 cleaved their respective targets efficiently and with high specificity. The depletion of their targets led to a relative increase of the initially less frequent sequence types (Fig. 3). The absence of such an increase in two cases (Comamonas [Fig. 3A] and Nitrosomonas [Fig. 3B]) may be due to biases of IVT or one of the amplification steps in the protocol. The increase of the Comamonas-like template in another experiment with the same LNAzyme, Nso215 (Fig. 3C), suggests that such biases can be stochastic and are unlikely to hamper the approach in general. In addition, we noticed that the T-RFLP profiles obtained for the untreated plasmid mixtures did not correctly reflect the initial composition of these mixtures (Fig. 3 and Table 1). This result was expected because T-RFLP profiles often do not represent the quantitative structure of microbial communities because of biases in PCR, enzymatic digestion, and capillary electrophoresis (see reference 51 and references therein). However, relative changes in the composition of the same microbial community are accurately detected by T-RFLP analysis (51). Thus, this method is suitable for monitoring the shifts in relative abundance caused by the LNAzyme-mediated depletion of selected targets. We also noticed that even without the use of any LNAzyme, the IVT and RT-PCR steps can slightly modify the relative abundances of the templates (Fig. 3). Altogether, these observations show that our protocol may not be able to detect all rare sequence types in a complex mixture and that it, like other approaches using nonquantitative IVT or PCR (52), would not be suitable for a quantitative census of the sequence types in a mixture of different templates.
The Nitrospira-specific LNAzyme Ntspa668 was less efficient than the Nitrosomonas-targeting LNAzyme Nso215 (Fig. 3A, B, and D). Aside from base mismatches, factors reported to affect the efficiency of DNAzymes and LNAzymes include secondary structures of complex RNA substrates (27, 53) and the thermodynamic stability of the enzyme-substrate complex (29). Although less efficient, the Nitrospira-like 16S rRNA gene was almost completely depleted after a second iteration of the cleavage protocol with Ntspa668 (Fig. 3B and D). Thus, repeating the procedure (Fig. 2) can overcome difficulties with less efficient LNAzymes and problematic target sites. The efficiency can easily be monitored over successive iterations by a fingerprinting technique such as T-RFLP. The experiment with both LNAzymes Nso215 and Ntspa668 (Fig. 3D) confirmed that two LNAzymes can be combined in the same workflow step and under the same conditions to simultaneously deplete different unwanted targets. Future experiments should test whether more LNAzymes can be used together for the depletion of multiple targets from complex mixtures.
Enrichment cultures of an organism that still contain accompanying bacteria (“contaminants”) are prime examples of highly uneven communities. Here we used LNAzymes to identify contaminants in the enrichment of “Ca. Nitrospira defluvii” (31), which still contained small amounts of other bacteria. For this purpose, the entire approach was started with either DNA or native RNA as the initial template in order to compare the results and identify possible difficulties with the method. Additional helper probes were needed to facilitate the LNAzyme-mediated cleavage of native 16S rRNA from “Ca. Nitrospira defluvii” (Fig. 4B), whereas no helper probes were required with DNA as the initial template (Fig. 4A). Thus, target site accessibility influences the efficiency of LNAzymes, at least when they cleave native rRNA. Although a pronounced relative increase was not achieved for all rare 16S rRNA sequence types in the enrichment culture (Fig. 4), the selection of non-Nitrospira clones required little effort after the LNAzyme treatments. All gene libraries were reasonably well covered by the analyzed sets of clones, but analyses of similar clone numbers retrieved a much higher level of diversity of 16S rRNA types from the libraries that were established after the use of LNAzymes (Table 2). Nevertheless, the detected phylotypes were not evenly distributed among the clones selected from these three libraries (see Table S2 in the supplemental material). This might be caused by nonexhaustive sequencing of non-Nitrospira clones from each library or by other factors, such as an unequal coverage of the organisms by the different PCR primers applied. The frequencies of the different sequence types in the “RNA-based” libraries may also be affected by secondary structures of the template rRNA interfering with RT-PCR or by the cellular ribosome content. The LNAzyme-based approach led to the discovery of a surprisingly high level of diversity of cocultured bacteria that are possibly involved in biological interactions with Nitrospira (discussed in the supplemental material).
In general, similar criteria must be considered for the design of rRNA-targeted DNAzymes (or LNAzymes) and of rRNA-targeted oligonucleotide probes that are used for hybridization techniques such as FISH. The sites containing mismatches to nontarget organisms should be located near the center of the binding region to achieve an optimal specificity of the probe or LNAzyme. Second, LNAzymes and probes should cover all or at least most members of their phylogenetic target groups. Moreover, the secondary structure of the rRNA is likely to influence the binding of LNAzymes to their target site, as it affects the hybridization efficiency of FISH probes (54). Good target sites for FISH (54, 55) could also be suitable for LNAzymes, although this might not always apply, because FISH targets fixed intact ribosomes, whereas the LNAzymes in our protocol bind to isolated rRNA. Therefore, we suggest starting the design of new rRNA-targeted LNAzymes by screening a resource, such as probeBase (56), for existing FISH probes that are specific for the respective target organisms and have been tested in previous studies. The next step is to identify a suitable LNAzyme cleavage site in the probe sequence or in adjacent regions of the target rRNA. If a cleavage site is found, the probe, if necessary, can be shifted and/or extended in the 5′ and 3′ directions in silico along the rRNA. Either substrate-binding arm must be long enough for efficient hybridization and cleavage (29). In preliminary experiments using native 23S rRNA of Escherichia coli and an LNAzyme based on the FISH probe Gam42a, we observed good cleavage efficiency with substrate-binding arms of 8 and 9 nucleotides, respectively (data not shown). The LNAzymes used here to target nitrifiers had arms of 12 and 10 nucleotides (Ntspa668), 15 and 15 nucleotides (Ntspa655), and 15 and 12 nucleotides (Nso215). Finally, the specificity and coverage of the resulting LNAzyme should be evaluated by tools such as the “Probe Match” function of ARB software (32), probeCheck (57), or TestProbe (http://www.arb-silva.de/search/testprobe).
We evaluated all probes deposited in probeBase until 9 March 2010 to determine how suitable they would be for LNAzyme design. Among these 2,479 probes, 2,270 target an rRNA region that contains a potential RU cleavage site, which results in the highest cutting efficiency (24). No less than 807 probes would lead, without any sequence modifications, to LNAzymes with two substrate-binding arms of at least 8 nucleotides (see Fig. S4 in the supplemental material). An additional 703 probes would lead to LNAzymes with one substrate-binding arm of at least 6 nucleotides and the other arm of at least 8 nucleotides (see Fig. S4 in the supplemental material). Thus, only slight modifications (elongation of one arm by up to 2 nucleotides) might be needed to design an efficient LNAzyme based on these probes. In total, 61% of all probes in probeBase could be converted to efficient LNAzymes with few or no sequence changes, mostly preserving the already evaluated coverage of these probes and, due to the LNA nucleotides, possibly even increasing their specificity. However, the different cleavage efficiencies of Ntspa668 and Ntspa665 (see above) show that particular LNAzymes and/or target sites may require additional optimization and that tests to evaluate newly designed LNAzymes should be carried out prior to their use in critical experiments.
In summary, our LNAzyme-based protocol has proven useful for the depletion of unwanted targets in mixtures of 16S rRNA genes and native 16S rRNA types. The method lends itself to analyses of highly uneven communities, whose dominant members can easily be identified prior to the selection or design of suitable LNAzymes. Depending on the choice of primers for the PCR steps (Fig. 2), nearly full-length sequences can be obtained. Furthermore, the possibility of starting the approach with either DNA or RNA as the initial template offers much flexibility for applications in microbial ecology. For example, stable isotope probing (SIP) of RNA is a sensitive tool to identify uncultured microbes that take up and assimilate a specific substrate (58). The resulting stable isotope-labeled RNA fraction, however, may be dominated by rRNA from the largest or most active population that used the offered tracer. In such a situation, the specific cleavage and depletion, by LNAzymes, of this dominant rRNA would facilitate the analysis of less abundant rRNA types and thus provide a more complete picture of the functionally relevant microbial community members. Recently, we used such a combination of RNA-SIP and LNAzymes to study interactions of heterotrophic bacteria with autotrophic nitrifiers (data not shown). If DNA-SIP is more appropriate, the same LNAzymes may be used with few modifications of the protocol, as shown in Fig. 2. Another application for LNAzymes could be purity checks of microbial isolates. Microscopy-based tools such as FISH are insufficient for this purpose, because rare contaminants would be overlooked due to the high detection limit of 1,000 to 10,000 cells per ml (59). Even PCR and DGGE analysis of 16S rRNA genes may not detect populations whose abundance is below 1% of the total community (17). The use of a 16S rRNA-directed LNAzyme, which is specific for the organism to be purified, would deplete this most abundant target and facilitate the detection of other organisms. As the application of LNAzymes does not require expensive or specialty equipment, the method should be usable in most laboratories where basic infrastructure for molecular work is available.
Recently developed high-throughput sequencing techniques allow the detection of the “rare biosphere” by providing large numbers of sequence reads per sample (e.g., see references 4 and 5). These methods undoubtedly mark a new era of microbial diversity analyses. However, their use for routine applications and smaller projects is still limited by the computational efforts needed to analyze the massive amounts of data and to compensate for methodical biases (60, 61) and by the relatively high costs per sequencing run. Moreover, current deep-sequencing techniques cannot be applied if (nearly) full-length rRNA gene sequences are needed for phylogenetic analyses or probe and primer design and if new probes or primers must be evaluated in experiments using the cloned target genes. In such cases, the LNAzyme-based approach complements the deep-sequencing methods, because it facilitates the cloning of full-length rRNA genes of rare organisms. Moreover, diversity analyses by deep sequencing of uneven microbial communities could benefit from LNAzyme-mediated depletion of the most dominant sequence types prior to the sequencing step. This would increase the number of sequence reads from rarer organisms and thus improve the coverage of the rare portion of the community.
The characterization of rare biosphere organisms is a difficult but important challenge for microbial ecology. Despite their low abundance, these microbes can be key players in ecosystems, like a rare Desulfosporosinus species that represented only 0.006% of the total microbial community but turned out to be an important sulfate reducer in a peatland (62). LNAzymes are a promising tool that offers more direct access to these often overlooked community members and thus will help to achieve a more complete understanding of their functions.
Supplementary Material
ACKNOWLEDGMENTS
We thank Eva Spieck for providing “Ca. Nitrospira defluvii” enrichment biomass, Christian Baranyi for technical assistance, and David Berry for critically reading the manuscript.
J.D. and I.L. were funded by the Graduate School Symbiotic Interactions of the University of Vienna. This work was also partly funded by the Austrian Science Fund (FWF) (grants S10002-B17 and I44-B06) and by the Vienna Science and Technology Fund (WWTF) (grant LS09-040).
Footnotes
Published ahead of print 21 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03392-12.
REFERENCES
- 1. Amann RI. 1995. In situ identification of micro-organisms by whole cell hybridization with rRNA-targeted nucleic acid probes, p 1–15 In Akkeman ADC, van Elsas JD, de Bruigin FJ. (ed), Molecular microbial ecology manual, vol 3.3.6 Kluwer Academic Publishers, Dordrecht, Netherlands. [Google Scholar]
- 2. Giovannoni SJ, Britschgi TB, Moyer CL, Field KG. 1990. Genetic diversity in Sargasso Sea bacterioplankton. Nature 345:60–63 [DOI] [PubMed] [Google Scholar]
- 3. Hugenholtz P, Pace NR. 1996. Identifying microbial diversity in the natural environment: a molecular phylogenetic approach. Trends Biotechnol. 14:190–197 [DOI] [PubMed] [Google Scholar]
- 4. Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc. Natl. Acad. Sci. U. S. A. 103:12115–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Webster NS, Taylor MW, Behnam F, Lücker S, Rattei T, Whalan S, Horn M, Wagner M. 2010. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 12:2070–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Achtman M, Wagner M. 2008. Microbial diversity and the genetic nature of microbial species. Nat. Rev. Microbiol. 6:431–440 [DOI] [PubMed] [Google Scholar]
- 7. Hugenholtz P, Goebel BM, Pace NR. 1998. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180:4765–4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rappé MS, Giovannoni SJ. 2003. The uncultured microbial majority. Annu. Rev. Microbiol. 57:369–394 [DOI] [PubMed] [Google Scholar]
- 9. Schloss PD, Handelsman J. 2004. Status of the microbial census. Microbiol. Mol. Biol. Rev. 68:686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Braker G, Fesefeldt A, Witzel K-P. 1998. Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl. Environ. Microbiol. 64:3769–3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Minz D, Flax JL, Green SJ, Muyzer G, Cohen Y, Wagner M, Rittmann BE, Stahl DA. 1999. Diversity of sulfate-reducing bacteria in oxic and anoxic regions of a microbial mat characterized by comparative analysis of dissimilatory sulfite reductase genes. Appl. Environ. Microbiol. 65:4666–4671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Purkhold U, Pommering-Röser A, Juretschko S, Schmid MC, Koops H-P, Wagner M. 2000. Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys. Appl. Environ. Microbiol. 66:5368–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Semrau JD, Chistoserdov A, Lebron J, Costello A, Davagnino J, Kenna E, Holmes AJ, Finch R, Murrell JC, Lidstrom ME. 1995. Particulate methane monooxygenase genes in methanotrophs. J. Bacteriol. 177:3071–3079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zehr JP, Jenkins BD, Short SM, Steward GF. 2003. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ. Microbiol. 5:539–554 [DOI] [PubMed] [Google Scholar]
- 15. Farrelly V, Rainey FA, Stackebrandt E. 1995. Effect of genome size and rrn gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial species. Appl. Environ. Microbiol. 61:2798–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hugenholtz P, Pitulle C, Hershberger KL, Pace NR. 1998. Novel division level bacterial diversity in a Yellowstone hot spring. J. Bacteriol. 180:366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muyzer G, de Waal EC, Uitterlinden AG. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Green SJ, Minz D. 2005. Suicide polymerase endonuclease restriction, a novel technique for enhancing PCR amplification of minor DNA templates. Appl. Environ. Microbiol. 71:4721–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. von Wintzingerode F, Landt O, Ehrlich A, Gobel UB. 2000. Peptide nucleic acid-mediated PCR clamping as a useful supplement in the determination of microbial diversity. Appl. Environ. Microbiol. 66:549–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Breaker RR, Joyce GF. 1994. A DNA enzyme that cleaves RNA. Chem. Biol. 1:223–229 [DOI] [PubMed] [Google Scholar]
- 21. Santoro SW, Joyce GF. 1997. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. U. S. A. 94:4262–4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlosser K, Li Y. 2009. Biologically inspired synthetic enzymes made from DNA. Chem. Biol. 16:311–322 [DOI] [PubMed] [Google Scholar]
- 23. Silverman SK. 2005. In vitro selection, characterization, and application of deoxyribozymes that cleave RNA. Nucleic Acids Res. 33:6151–6163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cairns MJ, King A, Sun LQ. 2003. Optimisation of the 10-23 DNAzyme-substrate pairing interactions enhanced RNA cleavage activity at purine-cytosine target sites. Nucleic Acids Res. 31:2883–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vester B, Wengel J. 2004. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry 43:13233–13241 [DOI] [PubMed] [Google Scholar]
- 26. Schubert S, Gul DC, Grunert HP, Zeichhardt H, Erdmann VA, Kurreck J. 2003. RNA cleaving ‘10-23′ DNAzymes with enhanced stability and activity. Nucleic Acids Res. 31:5982–5992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vester B, Hansen LH, Lundberg LB, Babu BR, Sorensen MD, Wengel J, Douthwaite S. 2006. Locked nucleoside analogues expand the potential of DNAzymes to cleave structured RNA targets. BMC Mol. Biol. 7:19 doi:10.1186/1471-2199-7-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vester B, Lundberg LB, Sorensen MD, Babu BR, Douthwaite S, Wengel J. 2004. Improved RNA cleavage by LNAzyme derivatives of DNAzymes. Biochem. Soc. Trans. 32:37–40 [DOI] [PubMed] [Google Scholar]
- 29. Santoro SW, Joyce GF. 1998. Mechanism and utility of an RNA-cleaving DNA enzyme. Biochemistry 37:13330–13342 [DOI] [PubMed] [Google Scholar]
- 30. Suenaga H, Liu R, Shiramasa Y, Kanagawa T. 2005. Novel approach to quantitative detection of specific rRNA in a microbial community, using catalytic DNA. Appl. Environ. Microbiol. 71:4879–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spieck E, Hartwig C, McCormack I, Maixner F, Wagner M, Lipski A, Daims H. 2006. Selective enrichment and molecular characterization of a previously uncultured Nitrospira-like bacterium from activated sludge. Environ. Microbiol. 8:405–415 [DOI] [PubMed] [Google Scholar]
- 32. Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhu K, Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Lüßmann TLR, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH. 2004. ARB: a software environment for sequence data. Nucleic Acids Res. 32:1363–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gieseke A, Purkhold U, Wagner M, Amann R, Schramm A. 2001. Community structure and activity dynamics of nitrifying bacteria in a phosphate-removing biofilm. Appl. Environ. Microbiol. 67:1351–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. 2007. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Daims H, Nielsen JL, Nielsen PH, Schleifer KH, Wagner M. 2001. In situ characterization of Nitrospira-like nitrite-oxidizing bacteria active in wastewater treatment plants. Appl. Environ. Microbiol. 67:5273–5284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lebedeva EV, Alawi M, Maixner F, Jozsa PG, Daims H, Spieck E. 2008. Physiological and phylogenetical characterization of a new lithoautotrophic nitrite-oxidizing bacterium ‘Candidatus Nitrospira bockiana’ sp. nov. Int. J. Syst. Evol. Microbiol. 58:242–250 [DOI] [PubMed] [Google Scholar]
- 37. Lebedeva EV, Off S, Zumbragel S, Kruse M, Shagzhina A, Lucker S, Maixner F, Lipski A, Daims H, Spieck E. 2011. Isolation and characterization of a moderately thermophilic nitrite-oxidizing bacterium from a geothermal spring. FEMS Microbiol. Ecol. 75:195–204 [DOI] [PubMed] [Google Scholar]
- 38. Hengesbach M, Meusburger M, Lyko F, Helm M. 2008. Use of DNAzymes for site-specific analysis of ribonucleotide modifications. RNA 14:180–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abdo Z, Schuette UM, Bent SJ, Williams CJ, Forney LJ, Joyce P. 2006. Statistical methods for characterizing diversity of microbial communities by analysis of terminal restriction fragment length polymorphisms of 16S rRNA genes. Environ. Microbiol. 8:929–938 [DOI] [PubMed] [Google Scholar]
- 40. Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. 2005. At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl. Environ. Microbiol. 71:7724–7736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Daims H, Stoecker K, Wagner M. 2005. Fluorescence in situ hybridisation for the detection of prokaryotes, p 213–239 In Osborn AM, Smith CJ. (ed), Molecular microbial ecology. Bios-Garland, Abingdon, United Kingdom [Google Scholar]
- 42. Daims H, Wagner M. 2007. Quantification of uncultured microorganisms by fluorescence microscopy and digital image analysis. Appl. Microbiol. Biotechnol. 75:237–248 [DOI] [PubMed] [Google Scholar]
- 43. Daims H, Lücker S, Wagner M. 2006. daime, a novel image analysis program for microbial ecology and biofilm research. Environ. Microbiol. 8:200–213 [DOI] [PubMed] [Google Scholar]
- 44. Purkhold U, Wagner M, Timmermann G, Pommerening-Röser A, Koops HP. 2003. 16S rRNA- and amoA-based phylogeny of 12 novel betaproteobacterial ammonia oxidizing isolates: extension of the data set and proposal of a new lineage within the nitrosomonads. Int. J. Syst. Evol. Microbiol. 53:1485–1494 [DOI] [PubMed] [Google Scholar]
- 45. Lücker S, Wagner M, Maixner F, Pelletier E, Koch H, Vacherie B, Rattei T, DamstÉ JS, Spieck E, Le Paslier D, Daims H. 2010. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Natl. Acad. Sci. U. S. A. 107:13479–13484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fuchs BM, Glöckner FO, Wulf J, Amann R. 2000. Unlabeled helper oligonucleotides increase the in situ accessibility to 16S rRNA of fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 66:3603–3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pedros-Alio C. 2006. Marine microbial diversity: can it be determined? Trends Microbiol. 14:257–263 [DOI] [PubMed] [Google Scholar]
- 48. Nikolausz M, Marialigeti K, Kovacs G. 2004. Comparison of RNA- and DNA-based species diversity investigations in rhizoplane bacteriology with respect to chloroplast sequence exclusion. J. Microbiol. Methods 56:365–373 [DOI] [PubMed] [Google Scholar]
- 49. Liles MR, Manske BF, Bintrim SB, Handelsman J, Goodman RM. 2003. A census of rRNA genes and linked genomic sequences within a soil metagenomic library. Appl. Environ. Microbiol. 69:2684–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fluiter K, Frieden M, Vreijling J, Koch T, Baas F. 2005. Evaluation of LNA-modified DNAzymes targeting a single nucleotide polymorphism in the large subunit of RNA polymerase II. Oligonucleotides 15:246–254 [DOI] [PubMed] [Google Scholar]
- 51. Hartmann M, Widmer F. 2008. Reliability for detecting composition and changes of microbial communities by T-RFLP genetic profiling. FEMS Microbiol. Ecol. 63:249–260 [DOI] [PubMed] [Google Scholar]
- 52. Suzuki MT, Giovannoni SJ. 1996. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62:625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Donini S, Clerici M, Wengel J, Vester B, Peracchi A. 2007. The advantages of being locked. Assessing the cleavage of short and long RNAs by locked nucleic acid-containing 8-17 deoxyribozymes. J. Biol. Chem. 282:35510–35518 [DOI] [PubMed] [Google Scholar]
- 54. Fuchs BM, Wallner G, Beisker W, Schwippl I, Ludwig W, Amann R. 1998. Flow cytometric analysis of the in situ accessibility of Escherichia coli 16S rRNA for fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 64:4973–4982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fuchs BM, Syutsubo K, Ludwig W, Amann R. 2001. In situ accessibility of Escherichia coli 23S rRNA to fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 67:961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Loy A, Maixner F, Wagner M, Horn M. 2007. probeBase—an online resource for rRNA-targeted oligonucleotide probes: new features 2007. Nucleic Acids Res. 35:D800–D804 doi:10.1093/nar/gkl856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Loy A, Arnold R, Tischler P, Rattei T, Wagner M, Horn M. 2008. probeCheck—a central resource for evaluating oligonucleotide probe coverage and specificity. Environ. Microbiol. 10:2894–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Manefield M, Whiteley AS, Griffiths RI, Bailey MJ. 2002. RNA stable isotope probing, a novel means of linking microbial community function to phylogeny. Appl. Environ. Microbiol. 68:5367–5373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Amann RI, Ludwig W, Schleifer K-H. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. 2010. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12:118–123 [DOI] [PubMed] [Google Scholar]
- 61. Quince C, Lanzen A, Curtis TP, Davenport RJ, Hall N, Head IM, Read LF, Sloan WT. 2009. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods 6:639–641 [DOI] [PubMed] [Google Scholar]
- 62. Pester M, Bittner N, Deevong P, Wagner M, Loy A. 2010. A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 4:1591–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.