

Abstract
Saccharomyces cerevisiae cannot utilize cellobiose, but this yeast can be engineered to ferment cellobiose by introducing both cellodextrin transporter (cdt-1) and intracellular β-glucosidase (gh1-1) genes from Neurospora crassa. Here, we report that an engineered S. cerevisiae strain expressing the putative hexose transporter gene HXT2.4 from Scheffersomyces stipitis and gh1-1 can also ferment cellobiose. This result suggests that HXT2.4p may function as a cellobiose transporter when HXT2.4 is overexpressed in S. cerevisiae. However, cellobiose fermentation by the engineered strain expressing HXT2.4 and gh1-1 was much slower and less efficient than that by an engineered strain that initially expressed cdt-1 and gh1-1. The rate of cellobiose fermentation by the HXT2.4-expressing strain increased drastically after serial subcultures on cellobiose. Sequencing and retransformation of the isolated plasmids from a single colony of the fast cellobiose-fermenting culture led to the identification of a mutation (A291D) in HXT2.4 that is responsible for improved cellobiose fermentation by the evolved S. cerevisiae strain. Substitutions for alanine (A291) of negatively charged amino acids (A291E and A291D) or positively charged amino acids (A291K and A291R) significantly improved cellobiose fermentation. The mutant HXT2.4(A291D) exhibited 1.5-fold higher Km and 4-fold higher Vmax values than those from wild-type HXT2.4, whereas the expression levels were the same. These results suggest that the kinetic properties of wild-type HXT2.4 expressed in S. cerevisiae are suboptimal, and mutations of A291 into bulky charged amino acids might transform HXT2.4p into an efficient transporter, enabling rapid cellobiose fermentation by engineered S. cerevisiae strains.
INTRODUCTION
Ethanol production from cellulosic biomass not only is sustainable but also does not cause ethical issues like corn- or sugar-based ethanols do (1). The economic production of ethanol from cellulosic biomass requires substantial improvements in all unit operations, such as physical pretreatment, chemical or enzymatic depolymerization, and fermentation. The efficient hydrolysis of cellulose and the utilization of mixed sugars (glucose and xylose) present in cellulosic hydrolysates are especially necessary for producing cellulosic ethanol on a commercial scale (2–4).
Fungal cellulases, which are widely used for degrading cellulose, exhibit weak β-glucosidase activity. The low β-glucosidase activity can lead to an accumulation of cellobiose because of its slow degradation of cellobiose into glucose during simultaneous saccharification and fermentation (SSF). The accumulation of cellobiose during SSF reduces the overall rate and efficiency of cellulose hydrolysis because cellobiose is an inhibitor of cellulases. Therefore, supplementation with bacterial or fungal β-glucosidase is necessary to enhance cellulose hydrolysis in spite of the disadvantages in the cost of doing so. In order to reduce the enzyme costs from supplementation with β-glucosidase, engineered Saccharomyces cerevisiae strains capable of directly fermenting cellobiose have been developed (5–7). Because native S. cerevisiae cannot utilize cellobiose, the introduction of cellodextrin transporter (cdt-1) and intracellular β-glucosidase (gh1-1) from Neurospora crassa, the display of β-glucosidase on the surface of yeast cells, or the secretion of β-glucosidase is necessary to facilitate cellobiose assimilation by yeast (5, 8, 9). These cellobiose-fermenting S. cerevisiae strains are expected to enable SSF processes without the need to supplement with β-glucosidase.
In addition to the cost benefit of eliminating β-glucosidase during SSF, the direct fermentation of cellobiose rather than the fermentation of glucose after cellobiose hydrolysis might facilitate efficient and rapid fermentation of the mixed sugars present in cellulosic hydrolysates (10). Most cellulosic hydrolysates contain both glucose and xylose as major sugars. While S. cerevisiae can be engineered to ferment xylose, the ethanol yields and productivity from xylose by engineered S. cerevisiae strains are substantially lower than those from glucose fermentation (11–13). Moreover, the sequential utilization of glucose and xylose by engineered S. cerevisiae strains has been reported when mixtures of glucose and xylose were used, because glucose inhibits xylose transport in engineered S. cerevisiae (14). This sequential utilization of glucose and xylose may result in even lower xylose fermentation rates because of the high ethanol concentrations produced by glucose fermentation when xylose is converted into ethanol. In order to overcome this drawback from the sequential utilization of cellulosic sugars, we proposed simultaneous cofermentation of cellobiose and xylose by engineered S. cerevisiae strains as a solution (6). Additionally, simultaneous cofermentation by engineered yeast strains of cellobiose and galactose, which are prevalent in the hydrolysates of marine plant biomass, has been demonstrated (7).
Three cellodextrin transporters (cdt-1, cdt-2, and NCU00809) from N. crassa were identified and functionally expressed in S. cerevisiae (5). However, cellobiose fermentation rates by engineered strains expressing each cellodextrin transporter and gh1-1 were much different (i.e., the rate for the cdt-1-expressing strain was higher than that for the one expressing cdt-2, which was higher than that for the strain expressing NCU00809) (6). This indicates that cellodextrin transport might be a limiting step for efficient cellobiose fermentation by engineered S. cerevisiae strains. In addition to cellodextrin transporters from N. crassa, two cellobiose transporters from Scheffersomyces stipitis and Kluyveromyces lactis were identified and were functionally expressed in S. cerevisiae. Slow cellobiose fermentation by an engineered S. cerevisiae strain expressing a putative hexose transporter (HXT2.4) and β-glucosidase (BGL5) from S. stipitis has been reported (15). Also, the coexpression of LAC12, coding for lactose permease from K. lactis, and cepA, coding for cellobiose phosphorylase from Clostridium stercorarium, resulted in slow cellobiose utilization in S. cerevisiae (16).
In this study, we also observed poor cellobiose fermentation by an engineered S. cerevisiae strain expressing HXT2.4 and the gene coding for intracellular β-glucosidase (gh1-1). However, we were able to isolate a mutant HXT2.4 that improved cellobiose fermentation drastically compared to the wild-type HXT2.4, by performing serial subcultures on cellobiose medium. Expression of the mutant HXT2.4(A291D) not only led to a higher cellobiose consumption rate than expression of the wild-type HXT2.4 did, but also resulted in less accumulation of cellodextrin than that resulting from expression of cdt-1. We further characterized mutant variants obtained through saturation mutagenesis of the A291 residue and deduced the relationships between different amino acid substitutions at position A291 of HXT2.4 and rates of cellobiose fermentation.
MATERIALS AND METHODS
Strains and plasmid constructions.
S. cerevisiae CEN.PK2-1D (MATα leu2 trp1 ura3 his3 MAL2-8C SUC2) (17) and S. cerevisiae D452-2 (MATα leu2 his3 ura3 can1) (18) were used for engineering cellobiose metabolism in yeast. Escherichia coli DH5 (F− recA1 endA1 hsdR17 [rK− mK+] supE44 thi-1 gyrA relA1) (Invitrogen, Gaithersburg, MD) was used for gene cloning and manipulation. In order to overexpress HXT2.4 in S. cerevisiae, HXT2.4 was cloned from S. stipitis (GenBank accession no. XM_001387720) and ligated with the pRS426 expression vector. HXT2.4 was amplified by PCR with the forward primer HXT2.4-F (5′-GGCGGATCCAAAAATGTCTGACAAACTTCACAACATCAAG-3′) and the reverse primer HXT2.4-R (5′-GGCGTCGACATAATCAGGTATAATTTATTGACTAAAGCTTAG-3′) (the introduced BamHI and SalI restriction sites of each primer are underlined). The phosphoglycerokinase (PGK1) promoter and the CYC1 terminator were used to overexpress HXT2.4, as we previously constructed pRS426 cdt-1 for overexpressing cdt-1(6). pRS425-gh1-1 containing gh1-1 under the control of the PGK1 promoter and the CYC1 terminator was coexpressed to enable the intracellular utilization of cellobiose (6).
Medium and culture conditions.
E. coli was grown in Luria-Bertani medium; 50 μg/ml of ampicillin was added to the medium when required. Yeast strains were routinely cultivated at 30°C in YP medium (10 g/liter Bacto yeast extract and 20 g/liter Bacto peptone) supplemented with 20 g/liter of glucose. To select transformants using amino acid auxotrophic markers, we used yeast synthetic complete (YSC) medium, which contained 6.7 g/liter of a yeast nitrogen base plus 20 g/liter of glucose; 20 g/liter of agar; and CSM-Trp (Bio 101, Vista, CA), which supplied the appropriate nucleotides and amino acids.
Fermentation experiments.
Yeast cells were grown in YP medium containing 20 g/liter of cellobiose to prepare the inocula for cellobiose fermentation experiments. Cells at mid-exponential phase from YP medium containing cellobiose were harvested and inoculated after being washed twice with sterilized water. All of the flask fermentation experiments were performed using 50 ml of YP medium containing 80 g/liter of cellobiose in a 250-ml flask at 30°C, with an initial optical density at 600 nm (OD600) of ∼1.0 under oxygen-limited conditions. All flask fermentation experiments were independently repeated twice. The variation between independent fermentations was less than 5%. The fermentation profiles shown in Fig. 1 to 5 are from one representative fermentation. An evolutionary engineering approach to improve the cellobiose fermentation rate of an HXT2.4-expressing strain was performed using serial subcultures on cellobiose medium. When initial cellobiose (80 g/liter) was almost consumed, small amounts of cells were transferred to a new medium to make an initial OD600 of ∼0.01. These serial subcultures were repeated nine times for 50 days. After the ninth serial subculture, a single colony was isolated from YSC medium agar plates containing 20 g/liter of cellobiose. The improved phenotypes were confirmed by the isolated single colony, and then plasmids were isolated and sequenced. For cellodextrin fermentations, cellobiose, cellotriose, or cellotetraose was used as a sole carbon source to monitor cell growth in 200 μl of YP medium using a 96-well plate. A 50-μl aliquot of mineral oil was added to the top of the medium to prevent evaporation during fermentations. The initial OD600 was ∼0.2. A Synergy H4 hybrid microplate reader (BioTek Instruments Inc., Winooski, VT) was used with continuous mixing. Cellotriose and cellotetraose were obtained from the Seikagaku Biobusiness Corporation (Tokyo, Japan).
Fig 1.

Cellobiose fermentation profiles by the CEN-CDT1-BGL, CEN-HXT2.4-BGL, and evolved CEN-HXT2.4-BGL strains. Fermentation experiments were performed on YP medium containing 80 g/liter of cellobiose under oxygen-limited conditions. Symbols: CEN-CDT1-BGL (●), CEN-HXT2.4-BGL (■), and evolved CEN-HXT2.4-BGL (▲).
Fig 5.
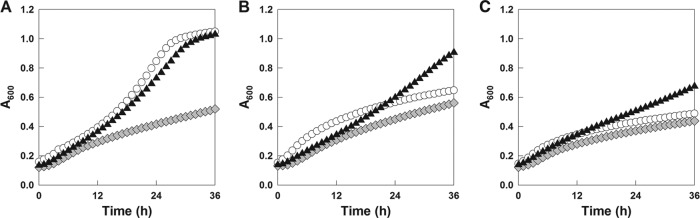
Growth assay of the cdt-1-expressing control strain, the WT HXT2.4 transformant, and the mutant HXT2.4(A291D) transformant on cellobiose (A), cellotriose (B), or cellotetraose (C) medium. Symbols: D452-CDT1-BGL (○), D452-HXT2.4-BGL ( ), and D452-HXT2.4(A291D)-BGL (▲).
), and D452-HXT2.4(A291D)-BGL (▲).
Yeast transformation and plasmid isolation.
Transformations of expression cassettes for constructing cellobiose metabolic pathways were performed using the EZ-Yeast transformation kit (Bio 101, Vista, CA). Transformants were selected on YSC medium containing 20 g/liter of glucose or cellobiose. Amino acids and nucleotides were added as necessary. Plasmid isolation from an evolved engineered S. cerevisiae strain was performed using Zymoprep yeast plasmid miniprep I kit (Zymo Research, Inc., Orange, CA) and the isolated plasmids were amplified through E. coli transformation. The isolated plasmid from the evolved strain was sequenced using primers (see Table S1 in the supplemental material).
Saturation mutagenesis.
Saturation mutagenesis was carried out using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) to generate all possible substitutions at the A291 residue of HXT2.4. A library of random mutants [HXT2.4(A291X)] was synthesized with a set of degenerate mutagenic primers, HXT_A291X-1 (5′-GAAAAGTTATATNNNAGCTCTTCTTAC-3′) and HXT_A291X-2 (5′-GTAAGAAGAGCTNNNATATAACTTTTC-3′), using pRS426-HXT2.4 as a template. PCR was performed on a C1000 thermal cycler (Bio-Rad) under the following conditions: an initial denaturation step for 30 s at 98°C followed by 16 repeating cycles of 20 s at 98°C, 30 s at 50°C, and 5 min at 72°C, and a final step of 10 min at 72°C.
Measurement of HXT2.4 expression levels via fluorescence.
For the expression of green fluorescent protein (GFP)-tagged wild-type (WT) HXT2.4 or mutant HXT2.4(A291D), the GFP-containing pRS426 plasmid with the PGK1 promoter and CYC1 terminator was used (5). When the OD600 reached ∼10 during fermentations, yeast cells were harvested and washed twice with sterilized water. A 200-μl aliquot of the cell suspension was transferred to a Corning 96-well clear-bottomed plate (Corning, NY). Fluorescence intensities were measured with a BioTek Synergy HT spectrophotometer (Winooski, VT) using excitation and emission wavelengths of 485 and 528 nm, respectively.
Determination of [3H]cellobiose transport assays and kinetic parameters.
Cellobiose transport assays were performed using a modification of the oil-stop method (19). Engineered yeast strains expressing transporter genes fused to GFP were grown to the mid-exponential phase in selective medium, washed 3× with assay buffer (30 mM morpholineethanesulfonic acid [MES]-NaOH [pH 5.6] and 50 mM ethanol), and resuspended to an OD600 of 40. To start transport reactions, 50 μl of cells was added to 50 μl of [3H]cellobiose layered over 100 μl of silicone oil (Sigma 85419). Reactions were stopped by spinning cells through oil for 1 min at 17,000 × g, tubes were frozen in ethanol and dry ice, and tube bottoms containing the cell pellets were clipped off into 1 ml of 0.5 M NaOH. The pellets were solubilized overnight, 5 ml of Ultima Gold scintillation fluid was added, and the counts per minute (cpm) were determined in a Tri-Carb 2900TR scintillation counter. [3H]cellobiose was purchased from Moravek Biochemicals, Inc., and had a specific activity of 4 Ci/mmol and a purity of >99%. Kinetic parameters were determined by measuring the linear rate of [3H]cellobiose uptake over 3 min for cellobiose concentrations between 0.5 and 400 μM. Vmax and Km values were determined by fitting a single rectangular 2-parameter hyperbolic function to a plot of rates versus cellobiose concentration by using a nonlinear regression in SigmaPlot. Vmax values were normalized for differences in transporter abundance by measuring the GFP fluorescence from 200 μl of cells at an OD600 of 40 immediately before beginning transport assays. Kinetic parameters in the text are reported as the means ± the standard errors of the mean (SEM) from three separate experiments.
Analytical methods.
Cell growth was monitored by OD600 using a UV-visible spectrophotometer (BioMate 5; Thermo Spectronic, NY). Cellobiose, cellodextrin, glucose, glycerol, acetate, and ethanol concentrations were determined using a high-performance liquid chromatograph (HPLC) (1200 series; Agilent Technologies) equipped with a refractive index detector using a Rezex ROA-Organic Acid H+ (8%) column (Phenomenex Inc., Torrance, CA). The column was eluted with 0.005 N H2SO4 at a flow rate of 0.6 ml/min at 50°C.
RESULTS
Introduction of HXT2.4 from S. stipitis into S. cerevisiae with intracellular β-glucosidase (gh1-1) resulted in poor cellobiose fermentation.
Eight putative cellodextrin transporters, which have been annotated as lactose transporters (LAC1, LAC2, and LAC3) and putative hexose transporters (HXT2.1, HXT2.3, HXT2.4, HXT2.5, and HXT2.6), were identified from a BLAST search using CDT-1 and CDT-2 from N. crassa as query proteins. Those eight transporters showed high amino acid sequence identities with CDT-1 (29% to 32%) and CDT-2 (29% to 36%). Among the identified putative transporters, we selected HXT2.4 as a strong candidate because HXT2.4 has high sequence identities with both CDT-1 (31%) and CDT-2 (36%). HXT2.4 was cloned and ligated with pRS426 vector under the control of the PGK1 promoter and the CYC1 terminator, as we previously constructed pRS426-cdt-1 for overexpressing cdt-1 (6). After overexpression of either HXT2.4 or cdt-1 along with gh1-1 in S. cerevisiae (CEN.PK2-1D), cellobiose fermentation rates by the resulting strains (CEN-HXT2.4-BGL and CEN-CDT1-BGL) were investigated (see Fig. S1 in the supplemental material). The CEN-HXT2.4-BGL strain consumed 72 g/liter of cellobiose and produced 29 g/liter of ethanol for 54 h, whereas the CEN-CDT1-BGL strain consumed 79 g/liter of cellobiose and produced 31 g/liter of ethanol within 36 h. Although the ethanol yields (0.39 to 0.40 g/g) by the CEN-HXT2.4-BGL and CEN-CDT1-BGL strains were similar, the cellobiose consumption rate (1.33 g cellobiose/liter · h) and ethanol productivity (0.54 g ethanol/liter · h) of the CEN-HXT2.4-BGL strain were 40% lower than those (2.19 g cellobiose/liter · h and 0.86 g ethanol/liter · h) of the CEN-CDT1-BGL strain.
Isolation of a mutant HXT2.4 strain facilitates higher cellobiose fermentation rates through an evolutionary engineering approach.
Serial subcultures of the CEN-HXT2.4-BGL strain on cellobiose medium were performed to increase the rate of cellobiose fermentation. The specific growth rates of the CEN-HXT2.4-BGL strain growing on a cellobiose medium as a sole carbon source increased drastically (from 0.029 h−1 to 0.080 h−1) during serial transfers (see Fig. S2 in the supplemental material). After the ninth serial subculture where the specific growth rate of the culture did not improve further, the evolved cells were plated onto the synthetic complete (SC) medium containing 20 g/liter of cellobiose as a sole carbon source in order to isolate a single colony. The cellobiose fermentation rate of the single colony (evolved CEN-HXT2.4-BGL) was determined using YP medium containing 80 g/liter of cellobiose. The evolved CEN-HXT2.4-BGL strain consumed 75 g/liter of cellobiose and produced 32 g/liter of ethanol within 36 h, resulting in an ethanol yield of 0.43 g/g (Fig. 1). Compared to its parental strain (CEN-HXT2.4-BGL), the evolved CEN-HXT2.4-BGL strain showed a higher cellobiose consumption rate (1.33 g/liter · h versus 2.08 g/liter · h) and greater ethanol productivity (0.54 g/liter · h versus 0.88 g/liter · h). Cell growth, cellobiose consumption rate, and ethanol production rate of the evolved CEN-HXT2.4-BGL strain were almost comparable to those of the CEN-CDT1-BGL strain. Interestingly, the amounts of accumulated cellodextrin (cellotriose and cellotetraose) of the evolved CEN-HXT2.4-BGL strain were much lower than those of the CEN-CDT1-BGL strain, resulting in a higher ethanol yield (0.43 g/g versus 0.39 g/g) (Fig. 1D).
Single amino acid substitution (A291D) in HXT2.4 in the isolated plasmid from the evolved CEN-HXT2.4-BGL strain.
Both plasmids (pRS426-HXT2.4 and pRS425-gh1-1) isolated from the evolved CEN-HXT2.4-BGL strain were sequenced to identify any putative mutations that were responsible for improved cellobiose fermentation by the evolved CEN-HXT2.4-BGL strain. There was no mutation in pRS425-gh1-1, but a single nucleotide mutation (C872A) was found in the coding region of HXT2.4 from pRS426-HXT2.4. This mutation was predicted to make an amino acid change from alanine to aspartate at position 291 (A291D) of the translated polypeptide. A protein structure modeling tool (Phyre2) (20) predicted that the A291D mutation might be located in the cytoplasmic area between the sixth and seventh domains among 12 membrane-spanning domains (see Fig. S3 in the supplemental material). No other mutations were found in other regions, such as the promoter, the origin of plasmid replication, or auxotrophic markers of the isolated plasmids from the evolved CEN-HXT2.4-BGL strain. We also confirmed the effect of the A291D mutation in the open reading frame of HXT2.4 by retransforming not only the isolated plasmids into the parental strain but also a created plasmid containing the A291D mutation via site-directed mutagenesis. The resulting strains showed cellobiose fermentation rates almost identical to those of the evolved CEN-HXT2.4-BGL strain (see Fig. S4 in the supplemental material), suggesting that there was no chromosomal mutation contributing to the improved cellobiose fermentation of the evolved CEN-HXT2.4-BGL strain.
Substitutions of alanine with negatively charged amino acids (aspartate and glutamate) at position 291 of HXT2.4 led to improved cellobiose fermentation.
The effects of amino acid substitutions at position 291 of HXT2.4(A291X) on cellobiose fermentation were investigated through saturation mutagenesis. A library of random mutants (pRS426-HXT2.4_A291X) was constructed using a QuikChange site-directed mutagenesis kit with degenerated primers (21). The diversity of the mutant library was confirmed by sequencing the isolated plasmids from 10 randomly picked clones. Ten sequenced plasmids all contained different nucleotides (GAA, GGG, CAT, AAA, TTC, TTA, CTA, TCT, TCA, and TAT) at positions 871 to 873, with glutamate, glycine, histidine, lysine, leucine, serine, or tyrosine substituted for alanine. After introduction of the plasmids containing mutated amino acids at position A291 of HXT2.4, along with pRS425-gh1-1, into S. cerevisiae D452-2, cellobiose consumption rates and ethanol production rates of the transformants were compared with those of the control strain with the WT HXT2.4 (Table 1; see also Fig. S5 in the supplemental material). As expected, all resulting strains showed varied cellobiose fermentation rates depending on the mutation at position 291 of HXT2.4(A291X). The substitution of alanine with glutamate (A291E) in HXT2.4 enabled the fastest cell growth, highest cellobiose consumption rate, and greatest ethanol production among the 10 transformants. The mutant HXT2.4(A291E) transformant consumed 78 g/liter of cellobiose within 24 h, whereas the WT HXT2.4 transformant consumed only 56 g/liter of cellobiose over 60 h. Interestingly, the substitution of alanine with serine or glycine (A291S or A291G) resulted in slower cellobiose consumption than that of the WT HXT2.4 transformant.
Table 1.
Sequencing results of 10 randomly picked mutant HXT2.4(A291X) strains selected from the library of mutant HXT2.4(A291X) strains based on growth on cellobiose
| Nucleotides at positions 871–873 | Amino acid substitution at position 291 | Cellobiose consumption rate (g/liter · h)a | Ethanol production rate (g/liter · h)a |
|---|---|---|---|
| GCC | Alanineb | 0.94 | 0.15 |
| GAA | Glutamate | 3.18 | 1.16 |
| AAA | Lysine | 2.14 | 0.88 |
| CTA | Leucine | 1.55 | 0.41 |
| TTA | Leucine | 1.09 | 0.20 |
| TTG | Leucine | 0.77 | 0.14 |
| TAT | Tyrosine | 0.91 | 0.15 |
| TCT | Serine | 0.53 | 0.06 |
| TCA | Serine | 0.51 | 0.05 |
| GGG | Glycine | 0.42 | 0.01 |
| CAT | Histidine | No growth |
After expression of the mutant HXT2.4(A291X) and gh1-1 in Saccharomyces cerevisiae, cellobiose fermentation rates were compared with those of the WT HXT2.4 transformant.
Alanine is the amino acid in the wild-type HXT2.4 transformant.
The random library of mutant HXT2.4(A291X) strains was transformed into S. cerevisiae D452-2 along with pRS425-gh1-1 to isolate a mutant HXT2.4 strain, enabling even faster cellobiose fermentation than one with the A291D mutation. Nineteen fast-growing colonies on minimum medium plates containing 20 g/liter of cellobiose as a sole carbon source were isolated. As expected, all isolated transformants showed higher cellobiose fermentation rates than that of the control strain (see Fig. S6 in the supplemental material). Plasmids containing mutant HXT2.4 from the selected 19 transformants were isolated and sequenced. Sequence analysis of the isolated plasmids revealed that three transformants with the substitution of glutamate or aspartate for alanine (A291E or A291D) exhibited the fastest cell growth, highest cellobiose consumption rate, and greatest ethanol production among the 19 transformants.
In order to characterize further the effects on cellobiose fermentation of amino acid substitutions at position 291 of HXT2.4(A291X), we compiled all fermentation results obtained in this study (Fig. 2). Amino acid substitutions of negatively charged amino acids (glutamate or aspartate) for alanine enabled the highest cellobiose fermentation capabilities. Additionally, substitutions of positively charged amino acids (lysine or arginine) for alanine exhibited improved cellobiose fermentation, although they were slightly less effective than the negatively charged amino acids. Amino acid substitutions of valine or leucine for alanine resulted in slight improvements in cellobiose fermentation. Interestingly, those improved cellobiose fermentation rates through amino acid substitutions at position 291 of HXT2.4 were inversely proportional to the maximum amounts of accumulated cellodextrin (Fig. 2; see also Fig. S7 in the supplemental material). Cellobiose fermentation profiles of the representative transformants containing various HXT2.4 mutants clearly showed that position 291 of HXT2.4 is a key amino acid residue affecting the cellobiose fermentation rates of engineered S. cerevisiae strains (Fig. 3).
Fig 2.
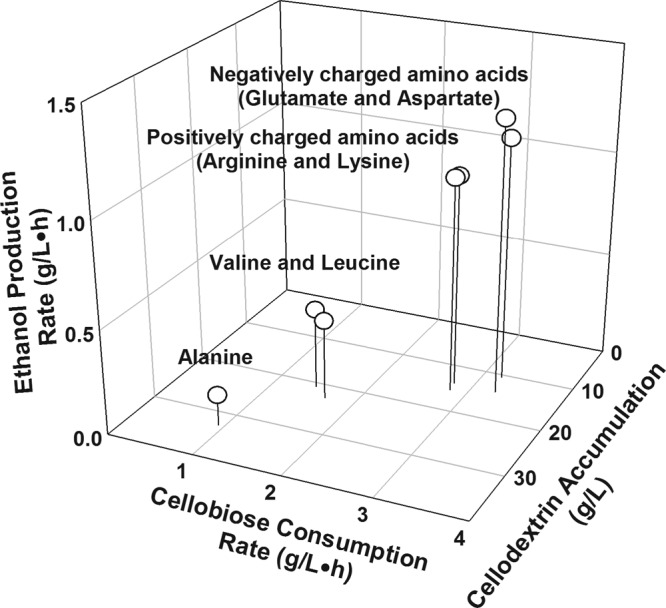
Comparison of cellobiose consumption rates, ethanol production rates, and maximum amounts of accumulated cellodextrin by the D452-HXT2.4(A291X)-BGL mutant transformants and the WT HXT2.4 transformant (D452-HXT2.4-BGL). Fermentation experiments were performed on YP medium containing 80 g/liter of cellobiose under oxygen-limited conditions.
Fig 3.
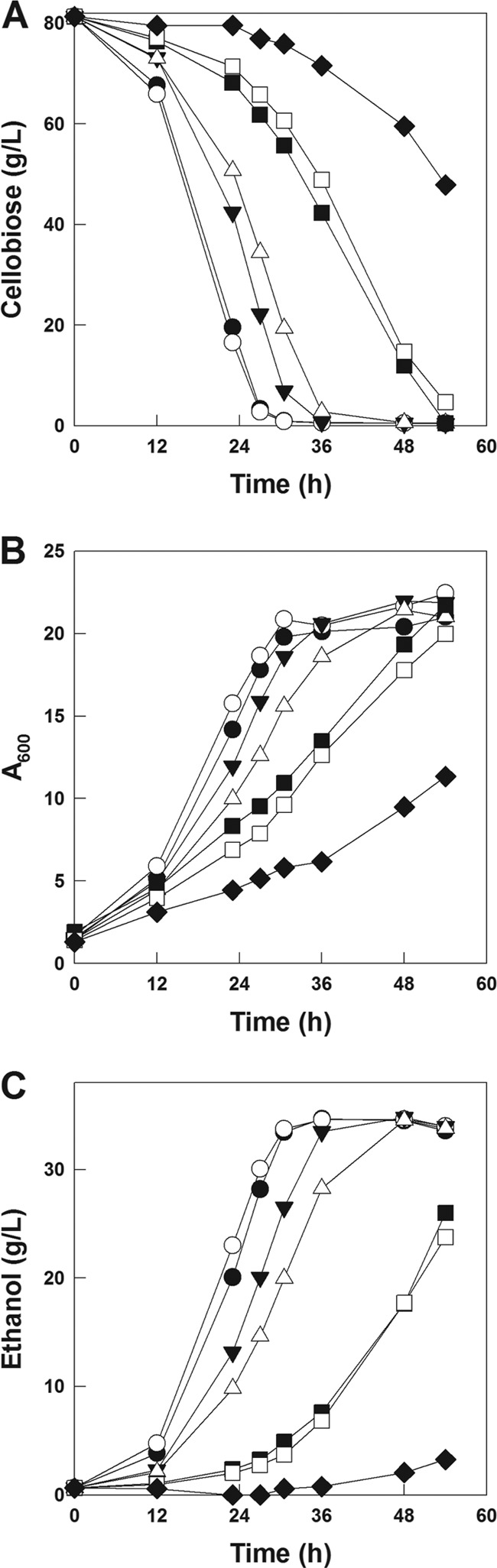
Comparison of cellobiose consumption (A), cell growth (B), and ethanol production (C) rates by the transformants, each containing different amino acid substitutions at A291. Fermentation experiments were performed on YP medium containing 80 g/liter of cellobiose under oxygen-limited conditions. Symbols: D452-HXT2.4(A291D)-BGL strain (●), D452-HXT2.4(A291E)-BGL strain (○), D452-HXT2.4(A291K)-BGL strain (▼), D452-HXT2.4(A291R)-BGL strain (▽), D452-HXT2.4(A291V)-BGL strain (■), D452-HXT2.4(A291L)-BGL strain (□), and D452-HXT2.4(wild-type)-BGL strain (◆).
Comparisons of expression levels or kinetic properties between WT HXT2.4 and mutant HXT2.4(A291D) strains.
The amino acid substitution at position 291 of HXT2.4 may cause better protein folding or localization in the plasma membrane, leading to fast cellobiose fermentation. In order to determine whether the mutant HXT2.4(A291D) was folded better in the plasma membrane, we overexpressed GFP-tagged HXT2.4_GFP or HXT2.4(A291D)_GFP in S. cerevisiae along with gh1-1. The localization of heterologous sugar transporters on membranes can be determined using a GFP-tagged sugar transporter and fluorescence microscopy (22). We also confirmed membrane localizations of the GFP-tagged HXT2.4 (HXT2. 4_GFP) and HXT2.4(A291D) using fluorescence microscopy (see Fig. S8 in the supplemental material). When OD600 reached 10.0 during cellobiose fermentation, cells were harvested, and GFP fluorescence levels were measured. GFP fluorescence levels of HXT2.4_GFP and HXT2.4(A291D)_GFP strains were similar within less than 10% variation (15,668 ± 155 and 16,559 ± 147 absorbance units [AU]), suggesting that expression levels of the wild-type HXT2.4 and mutant HXT2.4 were similar. We also determined the kinetic properties of WT HXT2.4 and mutant HXT2.4(A291D) (Fig. 4). The mutant HXT2.4(A291D) exhibited four-fold higher maximal cellobiose transport rates (Vmax) and 1.5-fold higher Km than those of WT HXT2.4.
Fig 4.
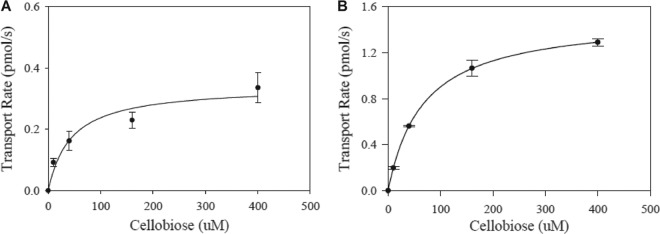
Transport kinetics of WT HXT2.4 (A) and mutant HXT2.4(A291D) (B). The linear rate of [3H]cellobiose uptake into yeast strains with WT HXT2.4 or mutant HXT2.4(A291D) was determined using various concentrations of cellobiose. All values were normalized to 10 million GFP fluorescence.
We further characterized the wild-type and mutant HXT2.4(A291D) transformants as to whether they can transport cellodextrins by growth assays using cellobiose, cellotriose, or cellotetraose in a 96-well plate. S. cerevisiae expressing cdt-1 was used as a positive control because cdt-1 is reported to have cellodextrin transport capabilities (5). The control strain expressing cdt-1 and the mutant HXT2.4(A291D) transformant grew well in cellobiose medium, while the WT HXT2.4 transformant showed poor growth, as we expected (Fig. 5A). Interestingly, the mutant HXT2.4(A291D) transformant showed even better cell growth than cdt-1- or WT HXT2.4-expressing S. cerevisiae strains in medium containing cellodextrins (cellotriose or cellotetraose) (Fig. 5B and C). These results suggest that an amino acid substitution at position 291 of HXT2.4 not only alters the cellobiose transport capability of HXT2.4 but also facilitates longer cellodextrin (cellotriose and cellotetraose) transport by engineered S. cerevisiae strains.
DISCUSSION
S. stipitis has been known to ferment cellobiose and produce ethanol with a yield of 0.30 to ∼0.41 g/g and a productivity of 0.15 to ∼0.21 g/liter · h (23, 24). Therefore, it was hypothesized that S. stipitis may have cellobiose transporters and intracellular β-glucosidase. Among the putative cellobiose transporters from S. stipitis that show high sequence homologies with CDT-1 and CDT-2, we focused on evaluating the capability of HXT2.4 for cellobiose fermentation for two reasons. First, HXT2.4 showed high protein sequence homologies with both CDT-1 and CDT-2 and HXT2.4 is located next to the genes coding for cellulose hydrolysis enzymes, such as endo-1,4-β-glucanase (EGC2) and β-glucosidase (BGL5), in the genome (25). Second, a previous report showed slow fermentation of cellobiose by an engineered S. cerevisiae strain expressing HXT2.4 and BGL5 (15). In this study, we confirmed that HXT2.4 transports cellobiose when expressed in S. cerevisiae. However, the HXT2.4-expressing S. cerevisiae strain not only consumed cellobiose much more slowly but also produced ethanol more slowly than the cdt-1-expressing S. cerevisiae strain, even though the same β-glucosidase (gh1-1) was expressed. Therefore, we undertook an evolutionary engineering approach to isolate the fast-growing strain expressing gh1-1 and HXT2.4 from the cellobiose medium.
Numerous attempts to improve the phenotypes through evolutionary engineering approaches have been reported (26). The simplest method is to use growth rate as a selection pressure by performing serial subcultures of a target strain under specific culture conditions (27, 28). This approach was applied for isolating S. cerevisiae mutants that show improved resistance against stress conditions, such as oxidative, freeze-thawing, high-temperature, and ethanol stresses (29). To improve pentose sugar (xylose or arabinose) utilization by S. cerevisiae, evolutionary engineering was used under various conditions (30–32). When we performed serial subcultures of the CEN-HXT2.4-BGL strain on a cellobiose medium, we observed that the specific growth rate increased 2.5-fold just after the first cycle of subculture. This drastic improvement after the first transfer suggests that a simple mutation might have caused improved cellobiose fermentation. Indeed, we found only one mutation (A291D) in HXT2.4 and we were able to reproduce the improved cellobiose fermentation phenotypes after reintroducing the mutant HXT2.4(A291D) into S. cerevisiae.
Interestingly, the evolved CEN-HXT2.4-BGL strain exhibited a higher ethanol yield (0.43 versus 0.39 g ethanol/g cellobiose) than the CEN-CDT1-BGL strain due to reduced cellodextrin accumulations (Fig. 1). We also previously observed cellodextrin accumulations during cellobiose fermentation (6). While we do not understand how small amounts of cellodextrin accumulate in the medium, we speculate that the transglycosylation activity of intracellular β-glucosidase (GH1-1) is responsible for the accumulated cellodextrin (33, 34). In this study, we showed that cellodextrin accumulation can be altered when cellobiose transport capabilities are changed through different cellobiose transporters, or through mutant transporters in S. cerevisiae. In addition, mutant HXT2.4(A291D) facilitated cellodextrin utilization, in comparison to strains expressing cdt-1 or WT HXT2.4. The greater cellodextrin transport capability of mutant HXT2.4(A291D) would be helpful in an SSF-type ethanol production process because cellodextrins are intermediates of the hydrolysis reaction from cellulose to cellobiose or glucose. The efficient cellodextrin utilization could lead to faster cellulose utilization, as well as to lower enzyme dosages, because glucose and cellodextrin are inhibitors of cellulase enzymes.
According to comparisons of expression levels and kinetic properties of WT HXT2.4 and mutant HXT2.4(A291D), we were able to conclude that the improved cellobiose fermentation of mutant HXT2.4(A291D) was not because of better protein folding or localization in the plasma membrane but because the kinetic properties of the mutant HXT2.4 were better than those of the WT HXT2.4. After compiling the relationships between cellobiose fermentation rates and amino acid substitutions at position 291, we can speculate that substitutions for alanine at position 291 with positively charged amino acids, such as glutamate or aspartate, can enhance cellobiose transport. A similar case was reported previously; E. coli melibiose (d-Gal-α[1→6]-d-Glc) permease (MelB) was inactivated by replacing a positively charged amino acid (arginine) with neutral amino acids (cysteine or glutamine) (35). Point mutations of transporters (HXT7, GXS1, or XUT3) can alter sugar (glucose or xylose) uptake rates drastically in engineered S. cerevisiae strains (36, 37). In order to verify more detailed mechanisms, the crystallization of HXT2.4 or predictions of structural changes depending on particular amino acid substitutions should be studied further, because no protein structure of HXT2.4 is available.
In this study, we were able to transform a poor cellobiose transporter into an efficient cellobiose transporter using single amino acid substitutions of alanine with charged amino acids at position 291 of HXT2.4. The amino acid substitution of alanine with aspartate caused improved transport capabilities of cellodextrins and cellobiose. In addition, this substitution (A291D) in HXT2.4 caused no different protein folding or localization but resulted in better kinetic properties when expressed in engineered S. cerevisiae strains. We envision that this finding might contribute to further engineering of sugar transporters for efficient biofuel production from cellulosic biomass.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by funding from the Energy Biosciences Institute to Yong-Su Jin.
Footnotes
Published ahead of print 21 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03253-12.
REFERENCES
- 1. Wyman CE. 2007. What is (and is not) vital to advancing cellulosic ethanol. Trends Biotechnol. 25:153–157 [DOI] [PubMed] [Google Scholar]
- 2. Carroll A, Somerville C. 2009. Cellulosic biofuels. Annu. Rev. Plant Biol. 60:165–182 [DOI] [PubMed] [Google Scholar]
- 3. Stephanopoulos G. 2007. Challenges in engineering microbes for biofuels production. Science 315:801–804 [DOI] [PubMed] [Google Scholar]
- 4. Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ, Jr, Hallett JP, Leak DJ, Liotta CL, Mielenz JR, Murphy R, Templer R, Tschaplinski T. 2006. The path forward for biofuels and biomaterials. Science 311:484–489 [DOI] [PubMed] [Google Scholar]
- 5. Galazka JM, Tian C, Beeson WT, Martinez B, Glass NL, Cate JH. 2010. Cellodextrin transport in yeast for improved biofuel production. Science 330:84–86 [DOI] [PubMed] [Google Scholar]
- 6. Ha SJ, Galazka JM, Kim SR, Choi JH, Yang X, Seo JH, Glass NL, Cate JH, Jin YS. 2011. Engineered Saccharomyces cerevisiae capable of simultaneous cellobiose and xylose fermentation. Proc. Natl. Acad. Sci. U. S. A. 108:504–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ha SJ, Wei Q, Kim SR, Galazka JM, Cate JH, Jin YS. 2011. Cofermentation of cellobiose and galactose by an engineered Saccharomyces cerevisiae strain. Appl. Environ. Microbiol. 77:5822–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saitoh S, Hasunuma T, Tanaka T, Kondo A. 2010. Co-fermentation of cellobiose and xylose using beta-glucosidase displaying diploid industrial yeast strain OC-2. Appl. Microbiol. Biotechnol. 87:1975–1982 [DOI] [PubMed] [Google Scholar]
- 9. van Rooyen R, Hahn-Hägerdal B, La Grange DC, van Zyl WH. 2005. Construction of cellobiose-growing and fermenting Saccharomyces cerevisiae strains. J. Biotechnol. 120:284–295 [DOI] [PubMed] [Google Scholar]
- 10. Kim SR, Ha SJ, Wei N, Oh EJ, Jin YS. 2012. Simultaneous co-fermentation of mixed sugars: a promising strategy for producing cellulosic ethanol. Trends Biotechnol. 30:274–282 [DOI] [PubMed] [Google Scholar]
- 11. Jeffries TW, Jin YS. 2000. Ethanol and thermotolerance in the bioconversion of xylose by yeasts. Adv. Appl. Microbiol. 47:221–268 [DOI] [PubMed] [Google Scholar]
- 12. Jeffries TW, Jin YS. 2004. Metabolic engineering for improved fermentation of pentoses by yeasts. Appl. Microbiol. Biotechnol. 63:495–509 [DOI] [PubMed] [Google Scholar]
- 13. Hahn-Hägerdal B, Karhumaa K, Jeppsson M, Gorwa-Grauslund MF. 2007. Metabolic engineering for pentose utilization in Saccharomyces cerevisiae. Adv. Biochem. Eng. Biotechnol. 108:147–177 [DOI] [PubMed] [Google Scholar]
- 14. Lee WJ, Ryu YW, Seo JH. 2000. Characterization of two-substrate fermentation processes for xylitol production using recombinant Saccharomyces cerevisiae containing xylose reductase gene. Process Biochem. 35:1199–1203 [Google Scholar]
- 15. Nelson SS, Headman Van Vleet JR, Jeffries TW. 2010. Engineered cellobiose utilization in Saccharomyces cerevisiae by expression of Pichia stipitis genes. 32nd Symposium on Biotechnology for Fuels and Chemicals, Clearwater Beach, FL [Google Scholar]
- 16. Sadie C, Rose SH, den Haan R, van Zyl WH. 2011. Co-expression of a cellobiose phosphorylase and lactose permease enables intracellular cellobiose utilisation by Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 90:1373–1380 [DOI] [PubMed] [Google Scholar]
- 17. Entian KD, Kotter P. 2007. Yeast genetic strain and plasmid collections. Methods Microbiol. 36:629–666 [Google Scholar]
- 18. Hosaka K, Nikawa J, Kodaki T, Yamashita S. 1992. A dominant mutation that alters the regulation of INO1 expression in Saccharomyces cerevisiae. J. Biochem. 111:352–358 [DOI] [PubMed] [Google Scholar]
- 19. Arendt CS, Ri K, Yates PA, Ullman B. 2007. Genetic selection for a highly functional cysteine-less membrane protein using site saturation mutagenesis. Anal. Biochem. 365:185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 21. Myers RM, Lerman LS, Maniatis T. 1985. A general method for saturation mutagenesis of cloned DNA fragments. Science 229:242–247 [DOI] [PubMed] [Google Scholar]
- 22. Du J, Li S, Zhao H. 2010. Discovery and characterization of novel d-xylose-specific transporters from Neurospora crassa and Pichia stipitis. Mol. Biosyst. 6:2150–2156 [DOI] [PubMed] [Google Scholar]
- 23. Parekh SR, Parekh RS, Wayman M. 1988. Fermentation of xylose and cellobiose by Pichia stipitis and Brettanomyces clausenii. Appl. Biochem. Biotechnol. 18:325–338 [Google Scholar]
- 24. Parekh S, Wayman M. 1986. Fermentation of cellobiose and wood sugars to ethanol by Candida shehatae and Pichia stipitis. Biotechnol. Lett. 8:597–600 [Google Scholar]
- 25. Jeffries TW, Van Vleet JR. 2009. Pichia stipitis genomics, transcriptomics, and gene clusters. FEMS Yeast Res. 9:793–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steipe B. 1999. Evolutionary approaches to protein engineering. Curr. Top. Microbiol. Immunol. 243:55–86 [DOI] [PubMed] [Google Scholar]
- 27. Sauer U. 2001. Evolutionary engineering of industrially important microbial phenotypes. Adv. Biochem. Eng. Biotechnol. 73:129–169 [DOI] [PubMed] [Google Scholar]
- 28. Cadière A, Ortiz-Julien A, Camarasa C, Dequin S. 2011. Evolutionary engineered Saccharomyces cerevisiae wine yeast strains with increased in vivo flux through the pentose phosphate pathway. Metab. Eng. 13:263–271 [DOI] [PubMed] [Google Scholar]
- 29. Cakar ZP, Seker UOS, Tamerler C, Sonderegger M, Sauer U. 2005. Evolutionary engineering of multiple-stress resistant Saccharomyces cerevisiae. FEMS Yeast Res. 5:569–578 [DOI] [PubMed] [Google Scholar]
- 30. Kuyper M, Toirkens MJ, Diderich JA, Winkler AA, van Dijken JP, Pronk JT. 2005. Evolutionary engineering of mixed-sugar utilization by a xylose-fermenting Saccharomyces cerevisiae strain. FEMS Yeast Res. 5:925–934 [DOI] [PubMed] [Google Scholar]
- 31. Wisselink HW, Toirkens MJ, Wu Q, Pronk JT, van Maris AJA. 2009. Novel evolutionary engineering approach for accelerated utilization of glucose, xylose, and arabinose mixtures by engineered Saccharomyces cerevisiae strains. Appl. Environ. Microbiol. 75:907–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sonderegger M, Sauer U. 2003. Evolutionary engineering of Saccharomyces cerevisiae for anaerobic growth on xylose. Appl. Environ. Microbiol. 69:1990–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prade H, Mackenzie LF, Withers SG. 1997. Enzymatic synthesis of disaccharides using Agrobacterium sp. β-glucosidase. Carbohydr. Res. 305:371–381 [Google Scholar]
- 34. Blanchard JE, Withers SG. 2001. Rapid screening of the aglycone specificity of glycosidases: applications to enzymatic synthesis of oligosaccharides. Chem. Biol. 8:627–633 [DOI] [PubMed] [Google Scholar]
- 35. Abdel-Dayem M, Basquin C, Pourcher T, Cordat E, Leblanc G. 2003. Cytoplasmic loop connecting helices IV and V of the melibiose permease from Escherichia coli is involved in the process of Na+-coupled sugar translocation. J. Biol. Chem. 278:1518–1524 [DOI] [PubMed] [Google Scholar]
- 36. Kasahara T, Kasahara M. 2010. Identification of a key residue determining substrate affinity in the yeast glucose transporter Hxt7: a two-dimensional comprehensive study. J. Biol. Chem. 285:26263–26268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Young EM, Comer AD, Huang H, Alper HS. 2012. A molecular transporter engineering approach to improving xylose catabolism in Saccharomyces cerevisiae. Metab. Eng. 14:401–411 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.