

Abstract
Acinetobacter baumannii is an emerging nosocomial, opportunistic pathogen that survives desiccation and quickly acquires resistance to multiple antibiotics. Escherichia coli gains antibiotic resistances by expressing genes involved in a global response to DNA damage. Therefore, we asked whether A. baumannii does the same through a yet undetermined DNA damage response akin to the E. coli paradigm. We found that recA and all of the multiple error-prone DNA polymerase V (Pol V) genes, those organized as umuDC operons and unlinked, are induced upon DNA damage in a RecA-mediated fashion. Consequently, we found that the frequency of rifampin-resistant (Rifr) mutants is dramatically increased upon UV treatment, alkylation damage, and desiccation, also in a RecA-mediated manner. However, in the recA insertion knockout strain, in which we could measure the recA transcript, we found that recA was induced by DNA damage, while uvrA and one of the unlinked umuC genes were somewhat derepressed in the absence of DNA damage. Thus, the mechanism regulating the A. baumannii DNA damage response is likely different from that in E. coli. Notably, it appears that the number of DNA Pol V genes may directly contribute to desiccation-induced mutagenesis. Sequences of the rpoB gene from desiccation-induced Rifr mutants showed a signature that was consistent with E. coli DNA polymerase V-generated base-pair substitutions and that matched that of sequenced A. baumannii clinical Rifr isolates. These data strongly support an A. baumannii DNA damage-inducible response that directly contributes to antibiotic resistance acquisition, particularly in hospitals where A. baumannii desiccates and tenaciously survives on equipment and surfaces.
INTRODUCTION
Acinetobacter baumannii is a Gram-negative coccobacillus that has quickly become a major nosocomial pathogen in hospitals worldwide, particularly infecting critically ill and immunocompromised patients in intensive care units (1, 2). With its tenacious resistance to desiccation and disinfectants (1), it is able to live on hospital equipment, including plastics, fabrics, and dry surfaces, for long periods of time (3–7). Due to A. baumannii's ability to readily gain multiple antibiotic resistances (2, 8), there is now a high incidence of multidrug-resistant strains in many hospitals, which are sometimes resistant to every antibiotic available to clinicians (9–13). Therefore, there is an increasing need to understand the underlying mechanisms that permit A. baumannii to readily evolve in the hospital environment. Though horizontal gene transfer and homologous recombination are important for A. baumannii to gain antibiotic resistance (2, 14), it is unclear how A. baumannii regulates, if at all, systems that govern recombination and mutagenesis.
A well-understood mechanism by which Escherichia coli and possibly other bacteria can become resistant to antibiotics is through the elevated expression of gene products that increase mutagenesis (15, 16). The E. coli SOS response, a well-characterized global transcriptional response triggered by DNA damage, replication stress, or antibiotics (17, 18), ultimately helps cells survive poor environmental conditions. The SOS response induces over 40 genes (19) involved in DNA repair (17), mutagenesis (16, 17, 20, 21), homologous recombination (22), virulence (23), and tolerance and persistence to fluoroquinolones (24).
In E. coli, DNA damage (17) or other effectors, such as nucleotide starvation (25), trigger DNA replication fork arrest, which in turn signals induction of the SOS gene response. RecA initiates the response by coating single-stranded DNA that accumulates at stalled replication forks, forming a nucleoprotein filament. Also known as RecA*, this filament promotes autocleavage of LexA, the global transcriptional repressor of the SOS gene network, through an endowed coprotease activity. It is LexA proteolysis which ultimately permits the expression of SOS-regulated genes (17). RecA is also necessary for homologous recombination (26) and participates in the DNA damage tolerance pathway by forming complexes with translesion synthesis (TLS) DNA polymerases DinB (or DNA polymerase IV [Pol IV] [27]) and DNA Pol V (27–29). A. baumannii encodes a predicted recA gene that when knocked out sensitizes it to DNA damage and a number of different stressors (30). Moreover, recA and ddrR (encoding a protein of unknown function) are induced upon UV irradiation in Acinetobacter baylyi ADP1 (31, 32), a nonpathogenic strain of Acinetobacter, suggesting a key role for RecA in mechanisms involved in stress survival. Nevertheless, efforts to identify a global DNA damage response in Acinetobacter have not been pursued. The lack of a LexA homologue in this genus has undoubtedly hindered efforts to identify such a response (33).
Damaged DNA must be either repaired or tolerated for a cell to survive. UvrA is one of the first gene products in which elevated expression can be detected upon DNA damage in the E. coli DNA damage response (19). This enzyme is part of the nucleotide excision repair (NER) pathway that detects DNA-distorting lesions, e.g., those produced by UV irradiation (34), and recruits the NER components to repair them. The E. coli DNA damage response also induces error-prone Y-family TLS DNA polymerases, Pol V and DinB, as well as B-family DNA Pol II, to perform DNA synthesis past replication-stalling lesions that have been left behind on the template DNA. These lesions stall DNA replication because they cannot be used as the template by replicative DNA polymerases. Y-family DNA polymerases have a relatively open active site compared to replicative DNA polymerases, permitting the accommodation of damaged bases. In addition, they lack an exonuclease activity, which enables other DNA polymerases to proofread DNA synthesis. Because of these features, Y-family DNA polymerases are generally more error prone on undamaged DNA than replicative, high-fidelity DNA polymerases (21, 35–38). This low-fidelity DNA synthesis increases mutagenesis and can lead to acquisition of antibiotic resistance through the modification of certain gene products (15, 16). The mutation signatures of DNA Pol V and DinB are base-pair substitutions and −1 frameshifts, respectively (27, 39, 40). Notably, sequenced clinical A. baumannii strains from different locations worldwide have multiple mutations that result in quinolone resistance (41–43), possibly the result of base-pair substitutions made by mutagenic Y-family DNA polymerases.
Y-family DNA polymerases are evolutionarily conserved from bacteria to humans (44). DNA Pol V (UmuD′2C) is composed of the catalytic enzyme UmuC and a homodimer of the accessory protein UmuD′. UmuD′ is the product of the coprotease activity of RecA* on UmuD; it is a 24-residue amino-terminal truncation of full-length UmuD. The error-prone DNA Pol V is known to bypass UV-induced DNA lesions, and it is responsible for most UV-induced mutagenesis; because of this, umuD and umuC are highly regulated in E. coli to minimize the intracellular concentration of active DNA Pol V (17). A. baumannii is capable of UV-induced mutagenesis, and it has also been observed that it carries multiple umuD and umuC genes (45, 46). It has been assumed that these genes are responsible for the mutagenesis. However, since there are multiple umuD and umuC genes, it is not yet known whether one or all of them are expressed upon DNA damage.
Therefore, we sought to assess whether a common response to DNA damage exists in A. baumannii by determining whether E. coli canonical DNA damage genes (e.g., recA, uvrA), as well as the multiple error-prone DNA polymerase genes, are induced upon DNA damage. We also investigated induced mutagenesis, an output of the DNA damage response, and assessed the impact of having multiple umuD and umuC genes. In this report, we present evidence that supports the existence of an A. baumannii inducible DNA damage response in which RecA plays a major regulatory role. We demonstrate that this response increases mutagenesis and is one of the mechanisms used by A. baumannii to acquire antibiotic resistances under clinically relevant DNA-damaging conditions.
MATERIALS AND METHODS
Strains and growth conditions.
A. baumannii ATCC 17978 (47) and ATCC 19606 (48) were purchased from the American Type Culture Collection (ATCC). The isogenic A. baumannii ATCC 17978 recA-deficient mutant (recA::Km) was the generous gift of the G. Bou lab (Universitario A Coruña, Spain). All GenBank accession numbers, including those of strains used for in silico analyses, are shown in Table 2. A. baumannii and E. coli cultures were routinely grown at 37°C in Luria broth (LB) or on LB agar. MICs were determined using a standard liquid broth dilution method (49). For all strains, 100 μg ml−1 of rifampin (Rif; Calbiochem) and 30 μg ml−1 of kanamycin (Km; Sigma) were used.
Table 2.
Comparison of number of putative TLS DNA polymerase genes from select isolates of A. baumannii
| A. baumannii strain | GenBank accession no. | No. of putative genes |
|||
|---|---|---|---|---|---|
| umuC | umuD | dinB | polB | ||
| ATCC 17978 | CP000521 | 4 | 3 | 1 | 0 |
| TCDC-AB0715 | CP002522 | 3 | 2 | 1 | 0 |
| AB059 | ADHB00000000 | 3 | 2 | 1 | 0 |
| ATCC 19606 | ACQB00000000 | 2 | 2 | 1 | 0 |
| AB0057 | CP001182 | 2 | 2 | 1 | 0 |
| AB058 | ADHA00000000 | 2 | 1 | 1 | 0 |
| ABNIH3 | AFTB00000000 | 2 | 1 | 1 | 0 |
| ACICU | CP000863 | 2 | 1 | 1 | 0 |
| AYE | CU459141 | 1 | 1 | 1 | 0 |
| MDR-ZJ06 | CP001937 | 1 | 1 | 1 | 0 |
Homology searches and sequence alignments.
A. baumannii protein sequences were obtained from the NCBI protein-protein BLAST search engine (50) using E. coli protein sequences as query. Genomic sequences that were not annotated were hand curated accordingly. The genomic organizations of ATCC 17978 umuDC operons were determined by finding the predicted open reading frames (ORFs) of the genes of interest in the available genome sequence. Protein sequences were aligned using the multiple-sequence-alignment tool of CLC Main Workbench (CLC Bio). Gene locus tags for these A. baumannii ATCC 17978 genes are as follows: umuD(A1S_0636) and umuC(A1S_0637), umuD(A1S_1174) and umuC(A1S_1173), umuD(A1S_1389), umuC(A1S_2008), umuC(A1S_2015), and dinB(A1S_0186).
Construction of A. baumannii ATCC 17978 dinB::Km.
The dinB::Km insertion knockout was created using a method developed by Aranda et al. (30) with some modifications. An amplicon of approximately 3,000 bp was constructed by splicing by overlap extension PCR (51). This fragment contains a kanamycin resistance gene insertion at bp 414 to 612 (resulting in a 198-bp deletion) of the A. baumannii ATCC 17978 dinB gene (see Table 1 for oligonucleotide sequences). The Km resistance gene was amplified by PCR from pUA66 (52) using the kan-F and kan-R oligonucleotides (Table 1). dinB-int-R and dinB-nest-F (Table 1) were used to amplify the 5′ end of the dinB gene and approximately 550 bp upstream of dinB. dinB-int-F and dinB-nest-R (Table 1) were used to amplify the 3′ end of the dinB gene and approximately 500 bp downstream of dinB. Finally, using dinB-nest-F and dinB-nest-R (Table 1), the three pieces were joined together by PCR. All PCRs were carried out using GoTaq 2× master mix (Promega). This 3,000-bp product was ligated into the pGEM-T Easy vector (Promega) using T4 DNA ligase (Promega), and the resulting dinB::Km plasmid was introduced into A. baumannii ATCC 17978 cells by electroporation at 1.8 mV for 5 ms following standard E. coli protocols (53). A. baumannii dinB::Km colonies were confirmed by sequencing (Tufts Core Facility) using chromosomal flanking oligonucleotides dinB-up-F with dinB-down-R, dinB-up-F with kan-R, and dinB-down-R with kan-F (Table 1). Kanamycin was used at 35 μg ml−1 for selection in A. baumannii and plasmid maintenance in E. coli.
Table 1.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′ to 3′) |
|---|---|
| umuDC(0636–0637)-F | GGCTGAAAATCCAGATTAC |
| umuDC(0636–0637)-R | CATTGCCATCATTCGAGG |
| umuDC(1173–1174)-F | CGTTATGTTGATGAACAATG |
| umuDC(1173–1174)-R | GTCAATGGCTTAAAGCAG |
| umuD(1389)-F | GTGAAATGGAGGCGATATGCCAAAG |
| umuD(1389)-R | CGTTGTTCGGATGAACCTGCTGTATC |
| umuC(2008)-F | GCAGATTTCAGTTAATGAGTAAGGG |
| umuC(2008)-R | CGTGAGACCACACATCCATC |
| umuC(2015)-F | CGAATTTTTGCACTCGTTGAC |
| umuC(2015)-R | GGTTCACCCATCTTAATTCC |
| dinB-F | ATGCGCAAAATCATTCATATCG |
| dinB-R | CTCATGGACATGGCAGAGCG |
| uvrA-F | TGAGCCAAAGTCATATCCGTATTCG |
| uvrA-R | GCCGAAAGTGATTCGACATAACG |
| recA-F | GCATTACAAGCCGCTTTGAGCC |
| recA-R | CTCAGCATCAATGAAGGCACATGTAC |
| 16S-337F | GACTCCTACGGGAGGCAGCAG |
| 16S-518R | GTATTACCGCGGCTGCTGG |
| rpoB-1441F | GAGCGTGCTGTTAAAGAGCG |
| rpoB-2095R | CTGCCTGACGTTGCATGT |
| dinB-up-F | GCGACTGAAGGCGGTGATTATA |
| dinB-down-R | CAGTTCCGGCTTCAGCAAGTAAGC |
| dinB-int-F | CTCGCTTGGACTCCTGTTGATGAAGAAGCTGTTTTAGTTCAC |
| dinB-int-R | AGCTGGCAATTCCGACGTCTCGAGGCTGTCAGTCCGGTTTG |
| dinB-nest-F | GGTTAAAAGCACGCGAACATGG |
| dinB-nest-R | CTACACTGGTGTCATCAGCGAG |
| kan-F | AGACGTCGGAATTGCCAGCT |
| kan-R | ATCAACAGGAGTCCAAGCGAG |
UV, MMS, and ciprofloxacin treatment.
Saturated cultures of A. baumannii ATCC 17978 (∼109 cells; parental) and A. baumannii ATCC 17978 dinB::Km were diluted 1:1,000 in LB and grown for 2.5 h. They were then subcultured for 2 h three consecutive times by diluting cultures 1:50 each time to ensure that cells were in exponential phase. A. baumannii ATCC 17978 recA::Km cultures were grown similarly, with the exception that the final growth cycle was 4 h. For UV treatment, 10-ml saturated cultures were spun down and resuspended in an equal volume of SMO (100 mM NaCl, 20 mM Tris-HCl, pH 7.5), and 2-ml samples were evenly placed in a sterile glass petri dish. Samples were irradiated in the dark under a UV germicidal lamp with 270 J m−2 for the parental and dinB::Km strains or 5 J m−2 for the recA::Km strain, resulting in approximately 2 to 20% survival. Parallel samples of the parental strain were also irradiated with 100 J m−2.
For methyl methanesulfonate (MMS) and ciprofloxacin treatments, cultures were grown to exponential phase as described for UV treatment. MMS (25 mM, 1× MIC; Sigma) or 6 μg ciprofloxacin ml−1 (10× MIC; Sigma) were used to treat the parental and dinB::Km cultures for 1 h. In addition, parental strain cultures were treated for 2 and 3 h with ciprofloxacin. For A. baumannii ATCC 17978 recA::Km cultures, 0.8 mM MMS (1× MIC) or 1 μg ciprofloxacin ml−1 (10× MIC) was used. After treatment, which resulted in 10-fold killing for all strains used after 1 h, cells were spun down and washed in SMO two times.
Semiquantitative RT-PCR.
UV-treated samples were incubated for 1 h prior to RNA extraction to allow gene expression. Total RNA was obtained by following the RNA Protect and RNeasy protocols (Qiagen). Absence of DNA was verified by carrying out a PCR with GoTaq 2× master mix (Promega) and the same oligonucleotide sets described below for reverse transcription-PCR (RT-PCR) (Table 1) at the highest concentration of total RNA used for RT-PCR (100 ng). The total RNA concentration was measured by a spectrophotometer at A260 (NanoDrop 2000; Thermo Scientific). Equal amounts of total RNA (100 ng) from treated and untreated samples were 10-fold serially diluted and used as the template for the SuperScript III one-step RT-PCR system with Platinum Taq (Life Technologies) kit. The concentrations of the serially diluted total RNA were measured within the NanoDrop spectrophotometer's limit of detection of 1 ng μl−1 and were determined to be within approximately 10% of the predicted concentration. PCR conditions were followed per the manufacturer's recommendations. Oligonucleotides (Table 1) were designed to be specific for amplifying either the unique junctions between umuD and umuC in the umuDC operons or to the unlinked umuC, umuD, dinB, uvrA(A1S_3295), recA(A1S_1962), and 16S rRNA (A1S_r01) open reading frames. cDNA was separated by electrophoresis in 1% agarose (SeaKem) gels. Gel images were analyzed using ImageJ (version 1.46r) software (Wayne Rasband, NIH). The software provides a measurement of the thickness and intensity of the separated electrophoresis bands. The area of each band was determined to learn the specific mRNA concentration present at each dilution from treated and untreated samples; the concentration was in turn divided by the total RNA dilution factor. Changes in relative expression were thus calculated.
Spontaneous and induced mutagenesis.
For all mutagenesis assays, bacterial cultures were started with ≤100 cells to reduce the probability of preexisting mutants in the starting inoculum. For UV-induced mutagenesis, samples were treated as described for UV treatment (270 J m−2 for the parental strain), with the exception that cultures were grown one time at a 1:50 dilution from the starting saturated culture. After treatment, samples were immediately diluted 1:10 in LB medium-containing flasks wrapped in tin foil and grown to saturation. Then, the appropriate cell dilutions were deposited on LB plates with and without rifampin to assess, respectively, the number of rifampin-resistant (Rifr) mutants and the total number of CFU. Colonies were counted after 24 h of incubation. Mutation frequency was calculated by dividing the number of Rifr mutants by the total number of CFU. Spontaneous Rifr mutants from untreated saturated cultures were determined as described above. Statistical significance was calculated using a Student t test.
For MMS-induced mutagenesis, cultures were grown and treated as described for MMS treatment, with the exception that cultures were grown one time at a 1:50 dilution directly from the saturated cultures. After the 1-h treatment, washed cultures were diluted 1:3 in LB medium and grown to saturation. Rifr mutation frequency was determined as described above.
The protocol used for desiccation-induced mutagenesis is a modification of the one used by Aranda et al. (30). Samples of saturated cultures (0.5 ml) were deposited onto sterile 0.45-μm-pore-size, black-gridded 47-mm filters (Millipore) by filtration. Filters were dried inside a closed, sterile petri dish at 37°C for 24 h (recA+ strains) or 6 h (recA::Km strains). Three- to 5-fold killing was observed for the A. baumannii ATCC 17978, A. baumannii ATCC 17978 dinB::Km, and A. baumannii ATCC 19606 strains, and 15-fold killing was observed for the recA::Km strain. In addition, an exponential-phase culture of A. baumannii ATCC 17978 was desiccated as described above for 24 h, which resulted in 15-fold killing.
Sequencing of Rifr mutants.
Colony PCR was performed according to the GoTaq 2× master mix (Promega) protocol on 32 individual desiccation-induced Rifr mutants from 6 independent A. baumannii ATCC 17978 recA+ experiments and also on 10 individual dinB::Km Rifr mutants from 5 independent A. baumannii ATCC 17978 dinB::Km experiments. Oligonucleotides rpoB-1441F and rpoB-2095R (Table 1) amplify a 654-bp region of rpoB (locus A1S_0287) where Rifr-inducing base-pair substitutions are frequently located (54). Sequencing (Tufts Core Facility) was carried out using the same oligonucleotide set. The data obtained were analyzed using CLC Main Workbench (CLC Bio).
Immunoblotting.
Cells were spun down and lysed with Bugbuster reagent (Novagen) after UV treatment. The total protein concentration was determined for each sample with the Bradford reagent (Bio-Rad) following the manufacturer's protocol. Equal amounts of total protein per sample mixed 1:1 with Laemmli sample buffer (2×; Sigma) were separated by SDS-PAGE on a 4 to 12% bis-Tris gel (Life Technologies) with 1× MOPS (morpholinepropanesulfonic acid) buffer (Life Technologies). After electrophoresis, proteins were transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore), and incubation with primary and secondary antibodies was carried out according to published procedures (53). Bound antibodies were detected with Luminata Crescendo Western horseradish peroxidase substrate (Millipore), followed by autoradiography or imaging on a Typhoon 8600 analyzer (GE Healthcare) using ImageQuant (version 5.2) software (Molecular Dynamics). Gel images were analyzed using ImageJ (version 1.46r) software (Wayne Rasband, NIH; see the previous section). The relative fold change in expression was determined by dividing the obtained intensities by the intensity of the untreated sample.
Polyclonal rabbit anti-DinB antibody, the generous gift of Takehiko Nohmi (55), was affinity purified (56) and diluted 1:100. Polyclonal rabbit anti-UvrA antibody (Covance) was generated using purified UvrA protein (the generous gift of Ben Van Houten) and used at a 1:10,000 dilution. Rabbit polyclonal anti-RecA antibody (Abcam, Cambridge, MA) was used at a 1:10,000 dilution, while the mouse monoclonal anti-RpoB antibody (Abcam, Cambridge, MA) was used at a 1:5,000 dilution.
RESULTS
Most A. baumannii genomes encode multiple error-prone DNA polymerase genes organized either as operons or as unlinked genes.
We wanted to know if A. baumannii regulates the error-prone translesion synthesis (TLS) DNA polymerases in response to DNA damage or environmental stress, because this would account for a yet undetermined mechanism of genomic evolution and antibiotic resistance acquisition in this organism.
To gain insights into the expression, genetic context, and relevance of these predicted TLS DNA polymerase genes in A. baumannii ATCC 17978, we searched the sequenced genomes of 10 independent A. baumannii isolates (Table 2) for genes whose products show similarity with the E. coli TLS DNA polymerases UmuC, DinB, and DNA Pol II and the accessory protein UmuD. This was done using the standard protein-protein BLAST search engine made available by NCBI (50); genomic sequences that were not annotated were hand curated accordingly. Interestingly, we found no polB genes (encoding TLS DNA polymerase II) in these genomes (Table 2). As in E. coli, A. baumannii isolates have only one putative dinB gene. DinB homologues from A. baumannii share sequence similarity with E. coli DinB, with E values being less than or equal to 2 × 10−69, and were found to have nearly 100% sequence conservation between A. baumannii isolates (see Fig. S1 in the supplemental material). Not surprisingly, we discovered that A. baumannii DinB is also recognized by E. coli polyclonal antibody (see below and Fig. 4).
Fig 4.
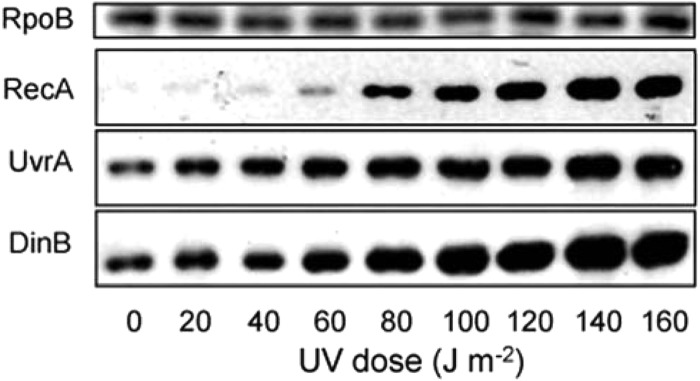
Intracellular concentrations of A. baumannii ATCC 17978 DNA damage-inducible proteins increase upon UV irradiation. At 160 J m−2, there was 40-fold more RecA protein, 2.5-fold more UvrA, and 3-fold more DinB than for the untreated samples, while RpoB remained constant. A. baumannii cultures were grown to exponential phase, as indicated in Materials and Methods, and irradiated with increasing amounts of UV (J m−2). Equal amounts of whole-cell lysates per treatment were probed with polyclonal anti-RecA, polyclonal anti-UvrA, polyclonal anti-DinB, and monoclonal anti-RpoB antibodies (refer to Materials and Methods). Antibodies used were raised against the E. coli proteins. A comparative experiment using the isogenic recA::Km strain could not be performed due to its extreme sensitivity to UV irradiation.
Because E. coli DNA Pol V (composed of UmuD′2C) is extensively regulated to minimize unnecessary mutagenesis (17), it is very surprising that the majority of A. baumannii genomes encode multiple, putative umuC and umuD homologues (Table 2). There is even one isolate, A. baumannii ATCC 17978, with four putative umuC homologues and three umuD homologues. We found that isolates have acquired different combinations of the number of umuC and umuD genes (Table 2) both on the chromosome and on plasmids (e.g., strain ACICU; Table 2). The total intracellular concentration of active DNA Pol V depends on the expression of these multiple umuC and umuD genes. However, even if an isolate has acquired numerous umuC genes, A. baumannii DNA Pol V activity likely depends on enough supporting umuD gene products (57). Because A. baumannii ATCC 17978 has more copies of both umuC and umuD genes, it may have the potential for more DNA damage-induced (or DNA Pol V-induced) mutagenesis than the other isolates listed (Table 2).
Conserved catalytic residues of the active site (58) were used to validate A. baumannii ATCC 17978 umuC gene products' homology to E. coli UmuC (see Fig. S2 in the supplemental material). Each of the putative UmuC homologues, annotated in GenBank as either RumB, DNA-directed DNA polymerase, or DNA repair protein, shares sequence similarity with E. coli UmuC throughout the protein sequences, with E values being less than or equal to 7 × 10−82 (see Fig. S2 in the supplemental material). Similar E values were found for all putative A. baumannii umuC genes listed in Table 2. UmuD protein sequences of all A. baumannii isolates share sequence similarity with E. coli UmuD, with E values being less than or equal to 7 × 10−18, in agreement with previous reports (46, 59).
In A. baumannii ATCC 17978, we found that the four umuC genes are uniquely organized and that the organization is different from that in E. coli. Figure 1 diagrams the arrangements of the two umuDC operons, the two unlinked umuC operons, and the one unlinked umuD gene of A. baumannii ATCC 17978. There are interesting differences between A. baumannii and E. coli even within the umuDC operons: for instance, in E. coli, umuD and umuC genes overlap by 1 nucleotide (Fig. 1A) (17). In contrast, we found that the ORF of umuC(A1S_0637) overlaps the ORF of umuD(A1S_0636) by 20 nucleotides and the ORF of umuC(A1S_1173) does not overlap the umuD(A1S_1174) ORF at all. Instead, the umuC(A1S_1173) ORF starts 3 nucleotides after the stop codon of umuD (Fig. 1). In E. coli, the −1 frameshift within the ORF of the umuDC operon is part of the regulation of expression of the umuD and umuC gene products, resulting in significantly less translation of umuC than umuD and, thus, a low intracellular concentration of DNA Pol V molecules (17). Therefore, it is likely that these gene arrangements in A. baumannii would influence the synthesis of their gene products as well.
Fig 1.

The A. baumannii ATCC 17978 predicted umuC and umuD genes are organized differently in A. baumannii ATCC 17978 than they are in E. coli. (A) There is one umuDC operon in the E. coli (Ec) chromosome in which the umuD ORF is expressed approximately 10-fold better than umuC due to a −1 frameshift between the two ORFs (17). This frameshift in the gene is depicted as overlapping arrows. (B) A. baumannii ATCC 17978 (Ab) has two putative umuDC operons in an organization similar to the one in E. coli, but within the umuDC(0636 to 0637) operon there is an overlap between the umuD and umuC genes of 20 nucleotides (depicted by overlapping arrows). In the umuDC(1174 to 1173) operon, we find no overlap between the two predicted genes. There are also two unlinked predicted umuC genes and one unlinked predicted umuD gene. For easier identification, locus tags (“A1S_” is not included before the numbers) are included as part of each A. baumannii gene name. Arrows represent predicted ORFs, and white boxes represent promoter (P) or putative promoter (P*) regions.
Predicted TLS DNA polymerase and other DNA damage response genes are expressed in A. baumannii ATCC 17978.
We wanted to ascertain whether the predicted multiple umuC and umuD genes and the single dinB gene are expressed in A. baumannii ATCC 17978, since this isolate has acquired the most TLS DNA polymerases of those sequenced (Table 2). To also examine the role of RecA, if any, in gene expression, we obtained an isogenic A. baumannii ATCC 17978 strain with a kanamycin resistance gene cassette inserted within recA (recA::Km [30]), rendering its gene product functionally inactive. We hypothesized that RecA would play a key role in the induction of the aforementioned genes as well as other DNA damage response genes in A. baumannii, despite lacking a discernible LexA. We measured mRNA transcript levels by semiquantitative RT-PCR to determine basal-level gene induction (Fig. 2). Total RNA was purified from untreated A. baumannii cells; then, the same amount of starting RNA template was used for subsequent RT-PCRs. The relative mRNA expression levels were thus obtained using gel electrophoresis image analysis (refer to Materials and Methods). Each gene's basal level of expression was calculated as a percentage of the level of expression of 16S rRNA, a standard housekeeping gene, in both the recA+ and recA::Km strains. This analysis permits the assessment of any differences in the relative basal level of expression between the examined genes. It should be noted here that we were able to measure recA expression in the recA::Km strain because of the kanamycin resistance cassette insertion (30). The recA oligonucleotides are specific to the 5′ end of the gene (first 260 bp), a region that remains intact on the chromosome of the recA::Km strain.
Fig 2.

Representative, evolutionarily conserved DNA damage response genes are expressed in A. baumannii ATCC 17978. The predicted genes encoding DNA damage response genes are all expressed in the recA+ strain, though at different levels. The relative expression of each gene is shown as a percentage of the level of expression of 16S rRNA, a standard housekeeping gene. In the recA::Km strain, most genes analyzed had no detectable change in relative basal-level gene expression. Some genes showed modest detectable decreases and modest to moderate increases in expression, which suggests a role for RecA in gene regulation. Semiquantitative RT-PCR was performed on total RNA purified from untreated cultures of A. baumannii ATCC 17978. See Materials and Methods for details of this experimental procedure. Gene-specific RT-PCR primers were used to amplify approximately 300 bp of either the unique junctions between the umuD and umuC genes organized as operons or unique sequences of the unlinked genes. Locus tags from the A. baumannii ATCC 17978 genome (“A1S_” is not included before the numbers) are included as part of the umuD and umuC names. Data from a representative experiment are shown.
We found that the A. baumannii umuDC operons, the unlinked umuD and umuC operons, dinB, uvrA, and recA are expressed because we detected their respective transcripts (Fig. 2). Notably, umuD(1389) and recA had the highest relative basal level of expression in the recA+ strain (Fig. 2). The umuDC(0636 to 0637) operon, unlinked umuC(2008), uvrA, and dinB had the second-highest level of relative expression in the recA+ strain. Lastly, the umuDC(1174 to 1173) operon and unlinked umuC(2015) had the lowest relative basal level of expression in the recA+ strain, suggesting that these genes may be the most tightly regulated of those analyzed in A. baumannii ATCC 17978. In the recA::Km strain, we found a similar gene expression profile; however, one surprising difference was evident: umuC(2015) and uvrA had markedly higher relative basal levels of expression in the recA::Km strain than in the recA+ strain (Fig. 2). This suggests a role for RecA in the regulation of these genes, possibly an involvement in repression.
A. baumannii TLS DNA polymerases are upregulated as part of a RecA-mediated DNA damage response.
Escherichia coli and other bacteria manage genomic instability in response to DNA damage or environmental stress by regulating a globally induced response, the SOS gene network (17, 21). The lack of an identifiable LexA homologue has made it difficult to characterize a similar damage response in Acinetobacter (33, 46). In the classic E. coli DNA damage response, the orchestrated upregulation of stress-response proteins is controlled at the level of transcription (19, 23, 60). We assessed whether we could detect changes in gene expression after treatment with three different DNA-damaging agents: MMS, ciprofloxacin, and UV. These agents are known to induce the DNA damage regulatory system in E. coli through various mechanisms. MMS is a cytotoxic DNA alkylating agent that produces replication fork-stalling 3-methyladenine (3-meA) lesions (61). Ciprofloxacin is an antibiotic that is a strong inducer of the SOS response in E. coli (24, 62); it causes replication stress because it traps the gyrase-DNA complex and blocks DNA replication, potentiating DNA double-strand breaks (63). UV irradiation is also classically used as a strong inducer of the SOS response (64) because it produces fork-stalling DNA lesions such as thymine-thymine dimers (17). Like E. coli, A. baumannii is sensitive to killing by UV, and the recA::Km strain is extremely sensitive, as predicted (30) (data not shown). A. baumannii ATCC 17978 recA+ or recA::Km strains were each treated with MMS at their respective MICs, and ciprofloxacin was used at a clinically relevant concentration of 10× the MIC. To compare strains with dramatically different sensitivities to DNA-damaging agents, we used doses of drugs or UV treatments in which they had the same viability. Otherwise, cells would either die (e.g., if a UV dose typically used for a recA+ strain was used for a recA::Km strain) or the treatment would not elicit a response (e.g., if a UV dose typically used for a recA::Km strain was used for a recA+ strain).
We determined first whether there was induction of A. baumannii DNA Pol V genes upon treatment with DNA-damaging agents. In the recA+ strain, the levels of expression for all umuDC operons and unlinked umuD and umuC loci increased upon all three treatments (Fig. 3A, black bars). The gene expression profiles differed for each treatment, but the umuDC(1174 to 1173) operon had the highest fold increase in expression in each case. We also saw drastic differences in induction between treatments. For instance, umuC(2015) was only modestly upregulated upon MMS treatment (∼1.5-fold), but upon ciprofloxacin and UV treatment, its expression increased ∼10- and ∼4-fold, respectively (Fig. 3A). umuD(1389) gene expression was induced ∼10-fold for MMS and ciprofloxacin treatment and only 2-fold upon UV treatment (Fig. 3A).
Fig 3.

The predicted A. baumannii TLS DNA polymerases and other DNA damage response genes are induced by DNA damage and regulated by RecA. (A) Expression of putative DNA polymerase V genes. All umuD and umuC loci are upregulated upon MMS, ciprofloxacin, or UV light treatment in the recA+ strain. In the recA::Km strain, most genes have no change in expression, which is denoted as a fold change of 1. We also observed increased expression for some of the genes, though it was lower than that in the recA+ strain. (B) Expression of other DNA damage response genes. The three DNA-damaging conditions examined resulted in upregulation of recA and uvrA in the recA+ strain. uvrA is regulated by RecA, as shown by its high expression in the recA+ strain. Notably, a large increase in recA expression is seen in the UV-treated recA::Km strain. There is no increase in expression of dinB or the 16S rRNA control in either strain. The recA+ and recA::Km strains were treated with 25 mM and 1.5 mM MMS, respectively, for 1 h; 6 μg ml−1 and 1 μg ml−1 of ciprofloxacin, respectively, for 1 h; and 270 J m−2 and 5 J m−2 of UV light, respectively. Semiquantitative RT-PCR was performed on total RNA purified from treated and untreated cultures as described in the Fig. 2 legend and Materials and Methods. Locus tags from the A. baumannii ATCC 17978 genome (“A1S_” is not included before the numbers) are included as part of the gene names. Data from a representative experiment are shown.
Conversely, it is apparent that in the recA::Km strain, induced levels were either greatly reduced compared to those in the recA+ strain or not induced at all (Fig. 3A, white bars). Remarkably, we found that some genes were induced even in the absence of recA, as exemplified by the umuDC(0636 to 0637) operon during ciprofloxacin and UV treatment and the umuDC(1174 to 1173) operon during UV treatment (Fig. 3A).
We next examined the induction of two DNA damage response genes, recA and uvrA, and the other Y-family DNA polymerase, DinB (or DNA Pol IV). Like the DNA Pol V genes (Fig. 3A), there was induction of expression of recA and uvrA in the recA+ strain (Fig. 3B, black bars). The induction of recA upon ciprofloxacin treatment was quite dramatic (34-fold; Fig. 3B), suggesting that RecA is likely an important part of this response and that ciprofloxacin is a strong inducer of the A. baumannii DNA damage response, as it is for E. coli (24, 62).
Because we were able to measure recA expression in the A. baumannii recA::Km strain, we were able to see that its expression in the recA::Km strain was almost equal to that in the recA+ strain during MMS treatment (Fig. 3B). Upon ciprofloxacin treatment, recA was expressed at levels comparatively higher than those of other genes in the recA::Km strain (Fig. 3B). It was also expressed approximately one-third as much as was seen in the recA+ strain. In contrast, upon UV treatment, recA was significantly induced (∼30-fold) in the recA::Km strain compared to the recA+ strain (∼5-fold; Fig. 3B), suggesting deregulation of recA in the absence of RecA.
No significant changes in expression were observed for dinB in either the recA+ or recA::Km strain, which was similar to the results for the 16S rRNA control (Fig. 3B). In a time course with ciprofloxacin or UV treatment, we found no detectable differences in the levels of induction of many genes, including dinB, in comparison to the results shown in Fig. 3 (recA+ strain; data not shown).
In response to persistent DNA damage or replication stress, transcript upregulation should coincide with an increase in protein levels (17, 19). We tested for a change in abundance of three DNA damage-inducible proteins in response to UV-induced damage. We selected RecA, UvrA, and DinB, because each of these is encoded by a single gene in A. baumannii ATCC 17978. DinB was of particular interest since we were unable to see a detectable increase in transcript level. We predicted that antibodies raised against the E. coli proteins would recognize the respective A. baumannii homologues, given the similarity in their predicted primary sequences.
Increasing levels of all three proteins in response to increasing doses of UV irradiation were observed in A. baumannii ATCC 17978 (Fig. 4). The relative increase in RecA protein expression at 160 J m−2 compared to that under the untreated condition was 40-fold; for UvrA it was 2.5-fold, and for DinB it was 3-fold (Fig. 4). No change was observed in the housekeeping protein RpoB, the RNA polymerase β subunit (Fig. 4). Although the use of different antibodies precludes comparison of the amplitude of induction of the three proteins, the simultaneous increase in abundance of all three in response to DNA damage is consistent with a DNA damage regulatory program in A. baumannii. While a change in the expression of dinB at the level of transcription was undetectable (Fig. 3B), the observable increase in protein over time strongly suggests that DinB is induced upon DNA damage.
Taken together, these data provide evidence for a bona fide DNA damage-inducible response in A. baumannii with TLS as a key component. The induction of a host of genes, including the multiple DNA polymerase V components, was shown using a DNA alkylating agent, UV irradiation, and treatment with an antibiotic frequently used by clinicians at clinically relevant concentrations. High-level induction of these genes is dependent on RecA, but the data also suggest that the role of RecA in A. baumannii gene regulation is different from the E. coli paradigm. These results are also consistent with the hypothesis that A. baumannii may induce this DNA damage response as a possible mechanism for genomic evolution upon multiple stressors. We therefore sought to gain evidence for the role of the DNA damage response in A. baumannii-induced mutagenesis.
A. baumannii recA-dependent DNA damage response contributes to induced mutagenesis.
We set forth to test whether the A. baumannii recA-dependent DNA damage response is responsible for DNA damage-induced mutagenesis by using an established Rifr assay (65). Rifampin is an antibiotic frequently coupled with colistin and used by clinicians to treat multidrug-resistant A. baumannii infections (54). Rifampin targets the β subunit of the bacterial RNA polymerase holoenzyme, RpoB. Only base-pair substitutions, i.e., not frameshifts, in the rpoB gene lead to select residue changes in the target site of RpoB, decreasing the effectiveness of rifampin binding (65). These base-pair substitutions can be the result of error-prone DNA polymerase, such as DNA Pol V (39). A. baumannii clinical Rifr isolates have been shown to have mutations in rpoB (54), validating this assay for use in A. baumannii. Both parental strain A. baumannii ATCC 17978 and the recA::Km isogenic strains were tested for induced mutagenesis by selecting for Rifr mutants after exposure to UV and to the alkylating agent MMS. We also constructed an A. baumannii ATCC 17978 dinB::Km insertion knockout strain to assess the impact of DinB on induced mutagenesis. TLS DNA polymerase gene products are necessary in E. coli for both survival and induced mutagenesis in cells that have accumulated UV- and MMS-induced DNA lesions (17, 61, 62), and we know (see previous sections) that these genes are induced by treatment with these reagents in A. baumannii.
As shown in Fig. 5A, in the parental recA+ strain there was a dramatic increase (∼30-fold for UV; ∼400-fold for MMS) in the frequency of DNA damage-induced Rifr mutation frequency (UV, gray bars; MMS, black bars) compared to the spontaneous Rifr mutation frequency (white bars). No significant increase in the MMS- or UV-induced Rifr mutation frequency was observed for the recA::Km strain (Fig. 5A). Interestingly, a significantly lower spontaneous mutation frequency (3.5-fold; P < 0.01) was found for the dinB::Km strain than the parental strain (Fig. 5A), and it was also not statistically significantly different from that for the recA::Km strain (P > 0.05). UV- and MMS-induced mutation frequencies for the dinB::Km strain were the same as those for the parental strain; however, the fold increase of induced mutation frequencies compared to spontaneous mutation frequencies was larger than that for the dinB+ strain (70-fold for UV, 1,400-fold for MMS).
Fig 5.

Mutation frequency is elevated upon treatment with DNA-damaging agents or upon desiccation in a recA-dependent manner. (A) The A. baumannii ATCC 17978 strain has a higher frequency of rifampin-resistant mutants upon both UV and MMS treatment than untreated cultures. There is no significant increase in induced mutation frequency for the isogenic recA::Km strain. The isogenic dinB::Km strain shows a modest but significant decrease (3.5-fold) in spontaneous mutants than the parental strain but has the same frequency of induced rifampin-resistant mutants as the parental strain upon both treatments. There is also no significant difference in the spontaneous mutation frequency between untreated recA::Km and dinB::Km strains. Error bars represent the standard errors of the means for at least 3 independently tested cultures, and statistical significance was determined using a Student t test. A statistically significant increase in mutation frequency between treated and untreated cultures (P ≤ 0.02) is indicated (*). (B) A. baumannii ATCC 17978 has a dramatically increased frequency of rifampin-resistant mutants after desiccation only in a recA+ background. The A. baumannii ATCC 17978 recA::Km strain showed no difference in predesiccation to postdesiccation rifampin-resistant mutants (P = 0.2). A. baumannii ATCC 19606, a strain containing fewer isogenic umuD and umuC genes than the A. baumannii ATCC 17978 strain, has an increased desiccation-induced Rifr frequency but fewer Rifr mutants than the A. baumannii ATCC 17978 recA+ strain. A statistically significant increase in mutation frequency between postdesiccation and predesiccation cultures (P < 0.01) is indicated (*). A. baumannii ATCC 17978 and ATCC 19606 cells were desiccated for 24 h, resulting in 3- to 5-fold killing compared to that for nondesiccated cells. Cells were then rehydrated in LB medium, outgrown, and deposited on plates with rifampin (100 μg ml−1). The recA::Km strain, treated for 6 h, was killed 15-fold compared to the level of killing for nondesiccated cells. A. baumannii ATCC 17978 recA+ cultures at 15-fold killing showed no difference in mutation frequency compared to that for the cultures that resulted in 3- to 5-fold killing (not shown). Error bars represent the standard errors of the means for at least 5 independently tested cultures. Statistical significance was determined using a Student t test.
Together, these data demonstrate that rifampin resistance can be acquired through the recA-dependent DNA damage response in A. baumannii, likely resulting from DNA base-pair substitutions in the rpoB gene (54). The data for the dinB::Km strain also suggest a role for A. baumannii DinB in generating spontaneous mutations and emphasize that the multiple DNA Pol V enzymes likely have a greater role in induced mutagenesis.
Desiccation-induced mutagenesis is recA dependent.
From these data, we became intrigued by the possibility that A. baumannii may be able to mutate in the hospital setting as the result of environmental processes likely to produce DNA damage. It is known that A. baumannii is able to survive on hospital equipment for long periods of time and has considerable desiccation tolerance (3, 4). Desiccation and desiccation-rehydration cause various DNA lesions, including alkylation, oxidation, cross-linking, base removal, and strand breaks (66); and it has been reported that A. baumannii ATCC 17978 recA::Km cells are sensitive to desiccation stress (30). It is likely that A. baumannii cells on the surfaces of hospital equipment incur these types of desiccation-induced DNA lesions. We hypothesized that these DNA lesions would result in elevated mutagenesis when cells are rehydrated. We simulated desiccation-induced DNA damage by drying A. baumannii cells on filters for a period of time, resulting in standardized killing (see Materials and Methods). As expected and in agreement with previous findings (30), recA::Km cultures were more sensitive to drying than the parental cultures (data not shown). Cells were rehydrated and grown in rich liquid medium to assess the frequency of Rifr mutants. As seen in Fig. 5B, the mutation frequency postdesiccation (gray bars) compared to that predesiccation (white bars; spontaneously arising mutations only) was significantly increased (∼50-fold) in the A. baumannii ATCC 17978 recA+ strain. No significant increase in mutation frequency was observed in the A. baumannii ATCC 17978 strain lacking recA postdesiccation (P = 0.2). The postdesiccation mutation frequency of A. baumannii ATCC 17978 dinB::Km matches the frequency of A. baumannii ATCC 17978 (data not shown), and we can again infer that there is a lessened role for A. baumannii DinB in induced mutagenesis and a greater role for DNA Pol V enzymes. Together, these results correlate with the results of DNA alkylation-induced mutagenesis (MMS; Fig. 5A), since it is probable that cells incur DNA alkylation lesions from desiccation (66).
Because A. baumannii ATCC 17978 has 4 predicted umuC genes and 3 predicted umuD genes (Fig. 1 and Table 2), we expected that a strain with fewer TLS genes would result in fewer Rifr mutants upon desiccation rehydration. As a proof of concept, we used the strain A. baumannii ATCC 19606, which possesses 2 predicted umuC loci (HMPREF0010_03135 and HMPREF0010_00311) that are the same as those present in A. baumannii ATCC 17978 (A1S_1173 and A1S_2008, respectively). The 2 predicted A. baumannii ATCC 19606 umuD loci, HMPREF0010_00986 and HMPREF0010_03136, are also present in the A. baumannii ATCC 17978 genome as A1S_1389 and A1S_1174, respectively. Moreover, we have shown that these common loci were induced upon DNA damage (Fig. 3). We compared the frequency of Rifr mutants after desiccation between these two strains. Like A. baumannii ATCC 17978, we found that there were significantly more Rifr mutants for A. baumannii ATCC 19606 postdesiccation than predesiccation (∼7-fold; P < 0.01; Fig. 5B). Remarkably, this increase is significantly less (∼7-fold) than the increase observed for A. baumannii ATCC 17978 (Fig. 5B), even though both strains are comparably sensitive to desiccation. Therefore, these data suggest a correlation between the number of genes encoding error-prone DNA Pol V and the number of desiccation-induced Rifr mutants.
We then tested the hypothesis that the A. baumannii ATCC 17978 recA+ desiccation-induced Rifr mutants were the result of rpoB base-pair substitutions. The rpoB gene from 32 individual colonies was sequenced, and it was found that all isolates had indeed acquired mutations in this gene (Table 3). Sequence analysis revealed single base-pair substitutions that resulted in amino acid substitutions. Our data coincide with published data for clinical Rifr isolates containing amino acid substitutions for aspartic acid at position 525, histidine at position 535, serine at position 540, leucine at position 542, and isoleucine at position 581 (54). At these positions, we found the recognized D525Y, H535L, and S540Y substitutions (54), as well as a number of novel substitutions that are indicated in Table 3. We also found new substitutions of the glutamic acid in position 522 for lysine, leucine, or arginine.
Table 3.
Mutation signatures of desiccation-induced A. baumannii ATCC 17978 Rifr mutants
| Mutation type and rpoB nucleotide changed | RpoB amino acid substitution | Mutation frequency (%) (n = 42) |
|---|---|---|
| Transversions | ||
| 1564 CAG → AAG | 522 Gln → Lysa | 7 |
| 1565 CAG → CTG | 522 Gln → Leua | 10 |
| 1573 GAC → TAC | 525 Asp → Tyrb | 5 |
| 1574 GAC → GTC | 525 Asp → Valc | 5 |
| 1603 CAT → GAT | 535 His → Aspa | 2 |
| 1604 CAT → CTT | 535 His → Leub | 12 |
| 1619 TCT → TAT | 540 Ser → Tyrb | 12 |
| 1741 ATC → TTC | 581 Ile → Phea,b | 14 |
| Total | 67 | |
| Transitions | ||
| 1565 CAG → CGG | 522 Gln → Arga,b | 12 |
| 1603 CAT → TAT | 535 His → Tyra | 2 |
| 1619 TCT → TTT | 540 Ser → Phea | 7 |
| 1625 CTT → CCT | 542 Leu → Proa,b | 10 |
| 1696 CGT → TGT | 566 Arg → Cysa,c | 2 |
| Total | 33 |
Novel Acinetobacter substitution.
The mutation was also found in an A. baumannii ATCC 17978 dinB::Km strain.
The mutation was unique to A. baumannii ATCC 17978 dinB::Km strain.
Boldface and underscoring indicate the nucleotide changes.
In addition, the rpoB sequence from 10 A. baumannii dinB::Km desiccation-induced Rifr mutants was sequenced. Many of the same mutations as those in the dinB+ strain were found, including amino acid substitutions at positions 522, 525, 535, 540, 542, 566, and 581 (Table 3). Two mutations, D525V and R566C, were also found to be unique to the dinB::Km strain. The majority of mutations in the dinB::Km strain were transversions (7 out of 10), as were the majority of mutations in the dinB+ strain (21 out of 32). Analysis of the total sequences of the dinB+ and dinB::Km strains combined revealed the majority (67%; 28 out of 42) of base-pair substitutions to be transversions (Table 3), a signature of DNA Pol V in E. coli, and all but one listed substitution (A-to-G transition) are also known to be DNA Pol V generated (45).
DISCUSSION
A. baumannii is desiccation resistant, which permits long-term survival and transmission in hospital environments. It also quickly becomes multidrug resistant and has thus become a major worldwide health concern (1–4, 8). It is clear that homologous recombination, horizontal gene transfer, and plasmids play a role in antibiotic resistance acquisition (1, 2, 14), though the underlying regulatory mechanisms, if any, have remained unknown. A global response to DNA damage or harsh environmental conditions has been shown to play a key function in antibiotic and virulence acquisition in other organisms (23, 67), but it has been unclear whether such a response exists in A. baumannii. In this study, we present evidence for a bona fide A. baumannii global DNA damage-inducible response and identify this response to be one important mechanism of antibiotic resistance acquisition.
It has been unclear why A. baumannii isolates have acquired, most likely through horizontal gene transfer (46), multiple umuDC operons and unlinked umuC genes or umuD genes (Fig. 1 and Table 2). This is in stark contrast to E. coli, which highly regulates a single umuDC operon to minimize the intracellular concentration of active DNA Pol V (17). We found that these multiple DNA Pol V gene components are all expressed at different levels in A. baumannii ATCC 17978 (Fig. 2) and induced upon DNA damage (Fig. 3A). Different DNA-damaging agents caused distinct expression of the multiple umuD and umuC genes (Fig. 3A), consistent with an idea in which the multiple DNA Pol V enzymes may have different lesion-bypass abilities (and mutation signatures; Table 3). Thus, these possibly provide A. baumannii ATCC 17978 with multiple alternatives to cope with DNA damage. The unlinked umuD(1389) is ubiquitously present in all the A. baumannii genomes analyzed (Table 2). Its role in A. baumannii DNA damage response is likely similar to its role in E. coli. Indeed, this umuD gene product is most closely similar to the A. baylyi umuD gene product shown to be cleaved in E. coli in response to DNA damage (59), suggesting that its role in the DNA Pol V complex might be similar to that of E. coli UmuD′.
We found that DinB is also induced by DNA damage, based on a detectable increase in protein levels upon UV treatment (Fig. 4), but we were unable to detect increased dinB transcript levels (Fig. 3B) even over a time course of treatment (data not shown). We do not yet understand the reason for this discrepancy. We tested whether A. baumannii DinB activity is conserved, and we found that E. coli dinB::Km is complemented by A. baumannii dinB on a low-copy-number plasmid (see Fig. S3 in the supplemental material). Like E. coli DinB, which accurately bypasses N2-furfuryl-dG lesions generated by nitrofurazone (68, 69), complementation with plasmid-borne dinB rescues cells from nitrofurazone-induced death (see Fig. S3 in the supplemental material). In contrast and to our surprise, A. baumannii dinB does not complement E. coli dinB::Km upon treatment with alkylating agents (see Fig. S3 in the supplemental material). In addition, A. baumannii dinB::Km cells are neither sensitive to alkylating agents (data not shown) nor more or less mutagenic upon treatment than dinB+ cells (Fig. 5A). These results suggest that A. baumannii DinB has lesion bypass activities different from those of E. coli DinB and also provide more support for our hypothesis that mutagenesis and TLS are dominated by the DNA Pol V enzymes in A. baumannii ATCC 17978, especially considering that there is no DNA Pol II in the A. baumannii sequences analyzed (Table 2).
Here we provide evidence for RecA regulating the induction of A. baumannii DNA damage response genes (Fig. 3). RecA is essential to mount a DNA damage response in E. coli (17) and is necessary for A. baumannii to survive DNA damage and general stress (30). Interestingly, we also observed RecA-independent induction of some genes, and RecA may also have an autoregulatory role (Fig. 3B). The precise mechanistic role of RecA in the regulation of the A. baumannii DNA damage response remains unknown, as does the yet unidentified LexA-like transcriptional repressor. DNA damage responses vary from bacterium to bacterium (23, 32, 70, 71), so it is possible that (i) a protein unidentifiable by primary and secondary structure has evolved a similar function as LexA or (ii) there is no LexA-like repressor and the regulation in A. baumannii is different from that in E. coli. While both of these options are currently being investigated, this study suggests that the latter is the most likely. In agreement with this idea, in A. baumannii we found no homologue of DinI, a protein that turns off the SOS response in E. coli by inhibiting the LexA cleavage promoted by RecA nucleoprotein filament (21). We also failed to complement a lexA-deficient strain of E. coli with plasmids containing A. baumannii genes encoding LexA-like candidates, including umuD(1389) (A. MacGuire and V. G. Godoy, unpublished data). umuD(1389) may still have a regulatory role in Acinetobacter spp., as it does in A. baylyi, a notion put forth by Hare et al. (59).
A. baumannii is notorious for readily incorporating foreign DNA, such as transposons, insertion sequence elements, and antibiotic resistance-encoding islands, into its genome (1, 2, 45). Therefore, it has the ability to acquire antibiotic resistances possibly from a wide range of bacteria. In combination with error-prone DNA polymerases, both inherent and acquired through these means, A. baumannii could evolve new resistances when faced with environmental stress by generating base-pair substitutions in a variety of cellular targets (13, 42, 43). Our finding that A. baumannii mutates upon desiccation-rehydration (Fig. 5B) not only is novel but also has obvious implications in the clinical setting: improper disinfection of A. baumannii from surfaces could lead to desiccation-induced mutagenesis. Importantly, current methods of disinfection are lacking in their ability to kill A. baumannii or hinder further antibiotic resistance acquisitions. Use of UV light as a sterilizing agent in hospitals (72–75) may even promote mutagenesis (Fig. 5A) if not done properly. Incorporation of a RecA inhibitor (76, 77), for example, into new disinfectants may be a viable option in the near future as novel inhibitors continue to be discovered and patented (78, 79). This would impede the DNA damage response, suppressing both induced mutagenesis and homologous recombination in the hospital and thus limiting evolution of antibiotic resistance (80–82). Another intriguing use for a bacterial RecA inhibitor includes combining it with antibiotic treatment as a combination therapy, thereby increasing bacterial susceptibility and the therapeutic effects of the antibiotic (82).
In summary, we have uncovered a mechanism that may aid A. baumannii in genomic evolution and acquisition of antibiotic resistance. This global DNA damage response has hallmark features of responses that are well understood; however, it is clear that the system in place is by no means conventional. Elucidation of the more intricate details of this system will further efforts to combat this deadly opportunistic pathogen.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the 1RO1GM088230-01A1 award from NIGMS to V. G. Godoy.
We thank Marin Vulic for critical reading of the manuscript, the G. Bou lab for generously sending us the A. baumannii recA::Km strain, Ivan Matic for the E. coli ΔalkA tag dinB strain, and Ben Van Houten for the UvrA protein. We also thank Ashley MacGuire for providing her unpublished data and other members of the V. G. Godoy lab for helpful discussions.
Footnotes
Published ahead of print 11 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02176-12.
REFERENCES
- 1. Bergogne-Berezin E, Friedman H, Bendinelli M. 2008. Acinetobacter biology and pathogenesis, vol xvi Springer-Verlag, New York, NY [Google Scholar]
- 2. Gerischer U. 2008. Acinetobacter molecular biology. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 3. Jawad A, Seifert H, Snelling AM, Heritage J, Hawkey PM. 1998. Survival of Acinetobacter baumannii on dry surfaces: comparison of outbreak and sporadic isolates. J. Clin. Microbiol. 36:1938–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kramer A, Schwebke I, Kampf G. 2006. How long do nosocomial pathogens persist on inanimate surfaces? A systematic review. BMC Infect. Dis. 6:130 doi:10.1186/1471-2334-6-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neely AN. 2000. A survey of gram-negative bacteria survival on hospital fabrics and plastics. J. Burn Care Rehab. 21:523–527 [DOI] [PubMed] [Google Scholar]
- 6. Neely AN, Maley MP, Warden GD. 1999. Computer keyboards as reservoirs for Acinetobacter baumannii in a burn hospital. Clin. Infect. Dis. 29:1358–1360 [DOI] [PubMed] [Google Scholar]
- 7. Wendt C, Dietze B, Dietz E, Ruden H. 1997. Survival of Acinetobacter baumannii on dry surfaces. J. Clin. Microbiol. 35:1394–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lockhart SR, Abramson MA, Beekmann SE, Gallagher G, Riedel S, Diekema DJ, Quinn JP, Doern GV. 2007. Antimicrobial resistance among Gram-negative bacilli causing infections in intensive care unit patients in the United States between 1993 and 2004. J. Clin. Microbiol. 45:3352–3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bergogne-Berezin E. 1995. The increasing significance of outbreaks of Acinetobacter spp.: the need for control and new agents. J. Hosp. Infect. 30(Suppl):441–452 [DOI] [PubMed] [Google Scholar]
- 10. Bergogne-Berezin E. 2001. The increasing role of Acinetobacter species as nosocomial pathogens. Curr. Infect. Dis. Rep. 3:440–444 [PubMed] [Google Scholar]
- 11. Bergogne-Berezin E, Joly-Guillou ML. 1991. Hospital infection with Acinetobacter spp.: an increasing problem. J. Hosp. Infect. 18(Suppl A):250–255 [DOI] [PubMed] [Google Scholar]
- 12. Bergogne-Berezin E, Towner KJ. 1996. Acinetobacter spp. as nosocomial pathogens: microbiological, clinical, and epidemiological features. Clin. Microbiol. Rev. 9:148–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vila J, Marti S, Sanchez-Cespedes J. 2007. Porins, efflux pumps and multidrug resistance in Acinetobacter baumannii. J. Antimicrob. Chemother. 59:1210–1215 [DOI] [PubMed] [Google Scholar]
- 14. Snitkin ES, Zelazny AM, Montero CI, Stock F, Mijares L, Murray PR, Segre JA. 2011. Genome-wide recombination drives diversification of epidemic strains of Acinetobacter baumannii. Proc. Natl. Acad. Sci. U. S. A. 108:13758–13763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cirz RT, Romesberg FE. 2007. Controlling mutation: intervening in evolution as a therapeutic strategy. Crit. Rev. Biochem. Mol. Biol. 42:341–354 [DOI] [PubMed] [Google Scholar]
- 16. Petrosino JF, Galhardo RS, Morales LD, Rosenberg SM. 2009. Stress-induced beta-lactam antibiotic resistance mutation and sequences of stationary-phase mutations in the Escherichia coli chromosome. J. Bacteriol. 191:5881–5889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2006. DNA repair and mutagenesis, 2nd ed ASM Press, Washington, DC [Google Scholar]
- 18. Miller C, Thomsen LE, Gaggero C, Mosseri R, Ingmer H, Cohen SN. 2004. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science 305:1629–1631 [DOI] [PubMed] [Google Scholar]
- 19. Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Galhardo RS, Hastings PJ, Rosenberg SM. 2007. Mutation as a stress response and the regulation of evolvability. Crit. Rev. Biochem. Mol. Biol. 42:399–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sutton MD, Smith BT, Godoy VG, Walker GC. 2000. The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 34:479–497 [DOI] [PubMed] [Google Scholar]
- 22. Kogoma T. 1997. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 61:212–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelley WL. 2006. Lex marks the spot: the virulent side of SOS and a closer look at the LexA regulon. Mol. Microbiol. 62:1228–1238 [DOI] [PubMed] [Google Scholar]
- 24. Dorr T, Lewis K, Vulic M. 2009. SOS response induces persistence to fluoroquinolones in Escherichia coli. PLoS Genet. 5:e1000760 doi:10.1371/journal.pgen.1000760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Godoy VG, Jarosz DF, Walker FL, Simmons LA, Walker GC. 2006. Y-family DNA polymerases respond to DNA damage-independent inhibition of replication fork progression. EMBO J. 25:868–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM. 1994. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 58:401–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Godoy VG, Jarosz DF, Simon SM, Abyzov A, Ilyin V, Walker GC. 2007. UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol. Cell 28:1058–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rehrauer WM, Bruck I, Woodgate R, Goodman MF, Kowalczykowski SC. 1998. Modulation of RecA nucleoprotein function by the mutagenic UmuD′C protein complex. J. Biol. Chem. 273:32384–32387 [DOI] [PubMed] [Google Scholar]
- 29. Tang M, Bruck I, Eritja R, Turner J, Frank EG, Woodgate R, O'Donnell M, Goodman MF. 1998. Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA protein. Proc. Natl. Acad. Sci. U. S. A. 95:9755–9760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aranda J, Bardina C, Beceiro A, Rumbo S, Cabral MP, Barbe J, Bou G. 2011. Acinetobacter baumannii RecA protein in repair of DNA damage, antimicrobial resistance, general stress response, and virulence. J. Bacteriol. 193:3740–3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hare JM, Perkins SN, Gregg-Jolly LA. 2006. A constitutively expressed, truncated umuDC operon regulates the recA-dependent DNA damage induction of a gene in Acinetobacter baylyi strain ADP1. Appl. Environ. Microbiol. 72:4036–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rauch PJ, Palmen R, Burds AA, Gregg-Jolly LA, van der Zee JR, Hellingwerf KJ. 1996. The expression of the Acinetobacter calcoaceticus recA gene increases in response to DNA damage independently of RecA and of development of competence for natural transformation. Microbiology 142(Pt 4):1025–1032 [DOI] [PubMed] [Google Scholar]
- 33. Robinson A, Brzoska AJ, Turner KM, Withers R, Harry EJ, Lewis PJ, Dixon NE. 2010. Essential biological processes of an emerging pathogen: DNA replication, transcription, and cell division in Acinetobacter spp. Microbiol. Mol. Biol. Rev. 74:273–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang W. 2011. Surviving the sun: repair and bypass of DNA UV lesions. Protein Sci. 20:1781–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beuning PJ, Simon SM, Godoy VG, Jarosz DF, Walker GC. 2006. Characterization of Escherichia coli translesion synthesis polymerases and their accessory factors. Methods Enzymol. 408:318–340 [DOI] [PubMed] [Google Scholar]
- 36. Fuchs RP, Fujii S, Wagner J. 2004. Properties and functions of Escherichia coli: Pol IV and Pol V. Adv. Protein Chem. 69:229–264 [DOI] [PubMed] [Google Scholar]
- 37. Ling H, Boudsocq F, Woodgate R, Yang W. 2001. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell 107:91–102 [DOI] [PubMed] [Google Scholar]
- 38. Yang W, Woodgate R. 2007. What a difference a decade makes: insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. U. S. A. 104:15591–15598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maor-Shoshani A, Reuven NB, Tomer G, Livneh Z. 2000. Highly mutagenic replication by DNA polymerase V (UmuC) provides a mechanistic basis for SOS untargeted mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 97:565–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reuven NB, Tomer G, Livneh Z. 1998. The mutagenesis proteins UmuD′ and UmuC prevent lethal frameshifts while increasing base substitution mutations. Mol. Cell 2:191–199 [DOI] [PubMed] [Google Scholar]
- 41. Seward RJ, Towner KJ. 1998. Molecular epidemiology of quinolone resistance in Acinetobacter spp. Clin. Microbiol. Infect. 4:248–254 [DOI] [PubMed] [Google Scholar]
- 42. Vila J, Ruiz J, Goni P, Jimenez de Anta T. 1997. Quinolone-resistance mutations in the topoisomerase IV parC gene of Acinetobacter baumannii. J. Antimicrob. Chemother. 39:757–762 [DOI] [PubMed] [Google Scholar]
- 43. Vila J, Ruiz J, Goni P, Marcos A, Jimenez de Anta T. 1995. Mutation in the gyrA gene of quinolone-resistant clinical isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 39:1201–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R. 2001. The Y-family of DNA polymerases. Mol. Cell 8:7–8 [DOI] [PubMed] [Google Scholar]
- 45. Di Nocera PP, Rocco F, Giannouli M, Triassi M, Zarrilli R. 2011. Genome organization of epidemic Acinetobacter baumannii strains. BMC Microbiol. 11:224 doi:10.1186/1471-2180-11-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hare J, Bradley J, Lin CL, Elam T. 2012. Diverse responses to UV light exposure in Acinetobacter include the capacity for DNA damage-induced mutagenesis in the opportunistic pathogens Acinetobacter baumannii and Acinetobacter ursingii. Microbiology 158(Pt 3):601–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith MG, Gianoulis TA, Pukatzki S, Mekalanos JJ, Ornston LN, Gerstein M, Snyder M. 2007. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 21:601–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schaub IG, Hauber FD. 1948. A biochemical and serological study of a group of identical unidentifiable gram-negative bacilli from human sources. J. Bacteriol. 56:379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Amsterdam D. 1996. Susceptibility testing of antimicrobials in liquid media, p 52–111 In Lorian V. (ed), Antibiotics in laboratory medicine, 4th ed Williams & Wilkins, Baltimore, MD [Google Scholar]
- 50. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68 [DOI] [PubMed] [Google Scholar]
- 52. Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, Shavit S, Liebermeister W, Surette MG, Alon U. 2006. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods 3:623–628 [DOI] [PubMed] [Google Scholar]
- 53. Ausubel FM. 1994. Current protocols in molecular biology. John Wiley & Sons, Inc., Brooklyn, NY [Google Scholar]
- 54. Giannouli M, Di Popolo A, Durante-Mangoni E, Bernardo M, Cuccurullo S, Amato G, Tripodi MF, Triassi M, Utili R, Zarrilli R. 2012. Molecular epidemiology and mechanisms of rifampicin resistance in Acinetobacter baumannii isolates from Italy. Int. J. Antimicrob. Agents 39:58–63 [DOI] [PubMed] [Google Scholar]
- 55. Nohmi T. 2006. Environmental stress and lesion-bypass DNA polymerases. Annu. Rev. Microbiol. 60:231–253 [DOI] [PubMed] [Google Scholar]
- 56. Tang WJ. 1993. Blot-affinity purification of antibodies. Methods Cell Biol. 37:95–104 [DOI] [PubMed] [Google Scholar]
- 57. Woodgate R, Rajagopalan M, Lu C, Echols H. 1989. UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD′. Proc. Natl. Acad. Sci. U. S. A. 86:7301–7305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boudsocq F, Ling H, Yang W, Woodgate R. 2002. Structure-based interpretation of missense mutations in Y-family DNA polymerases and their implications for polymerase function and lesion bypass. DNA Repair (Amst.) 1:343–358 [DOI] [PubMed] [Google Scholar]
- 59. Hare JM, Adhikari S, Lambert KV, Hare AE, Grice AN. 14 June 2012. The Acinetobacter regulatory UmuDAb protein cleaves in response to DNA damage with chimeric LexA/UmuD characteristics. FEMS Microbiol. Lett. [Epub ahead of print.] doi:10.1111/j.1574-6968.2012.02618.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Markham BE, Harper JE, Mount DW, Sancar GB, Sancar A, Rupp WD, Kenyon CJ, Walker GC. 1984. Analysis of mRNA synthesis following induction of the Escherichia coli SOS system. J. Mol. Biol. 178:237–248 [DOI] [PubMed] [Google Scholar]
- 61. Bjedov I, Dasgupta CN, Slade D, Le Blastier S, Selva M, Matic I. 2007. Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics 176:1431–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Benson RW, Norton MD, Lin I, Du Comb WS, Godoy VG. 2011. An active site aromatic triad in Escherichia coli DNA Pol IV coordinates cell survival and mutagenesis in different DNA damaging agents. PLoS One 6:e19944 doi:10.1371/journal.pone.0019944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Malik M, Zhao X, Drlica K. 2006. Lethal fragmentation of bacterial chromosomes mediated by DNA gyrase and quinolones. Mol. Microbiol. 61:810–825 [DOI] [PubMed] [Google Scholar]
- 64. Schlacher K, Goodman MF. 2007. Lessons from 50 years of SOS DNA-damage-induced mutagenesis. Nat. Rev. Mol. Cell Biol. 8:587–594 [DOI] [PubMed] [Google Scholar]
- 65. Garibyan L, Huang T, Kim M, Wolff E, Nguyen A, Nguyen T, Diep A, Hu K, Iverson A, Yang H, Miller JH. 2003. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair (Amst.) 2:593–608 [DOI] [PubMed] [Google Scholar]
- 66. Potts M. 1994. Desiccation tolerance of prokaryotes. Microbiol. Rev. 58:755–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li B, Smith P, Horvath DJ, Jr, Romesberg FE, Justice SS. 2010. SOS regulatory elements are essential for UPEC pathogenesis. Microbes Infect. 12:662–668 [DOI] [PubMed] [Google Scholar]
- 68. Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, Walker GC. 2006. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature 439:225–228 [DOI] [PubMed] [Google Scholar]
- 69. Jarosz DF, Godoy VG, Walker GC. 2007. Proficient and accurate bypass of persistent DNA lesions by DinB DNA polymerases. Cell Cycle 6:817–822 [DOI] [PubMed] [Google Scholar]
- 70. Davis EO, Springer B, Gopaul KK, Papavinasasundaram KG, Sander P, Bottger EC. 2002. DNA damage induction of recA in Mycobacterium tuberculosis independently of RecA and LexA. Mol. Microbiol. 46:791–800 [DOI] [PubMed] [Google Scholar]
- 71. Yasbin RE, Cheo DL, Bayles KW. 1992. Inducible DNA repair and differentiation in Bacillus subtilis: interactions between global regulons. Mol. Microbiol. 6:1263–1270 [DOI] [PubMed] [Google Scholar]
- 72. Botzenhart K, Ruden H, Tolon M, von Scharfenberg KM. 1976. Clinical uses of ultraviolet light radiation. Prakt. Anaesth. 11:320–327 (In German) [PubMed] [Google Scholar]
- 73. Hart D. 1960. Bactericidal ultraviolet radiation in the operating room. Twenty-nine-year study for control of infections. JAMA 172:1019–1028 [DOI] [PubMed] [Google Scholar]
- 74. Riley RL, Kaufman JE. 1971. Air disinfection in corridors by upper air irradiation with ultraviolet. Arch. Environ. Health 22:551–553 [DOI] [PubMed] [Google Scholar]
- 75. Riley RL, Nardell EA. 1989. Clearing the air. The theory and application of ultraviolet air disinfection. Am. Rev. Respir. Dis. 139:1286–1294 [DOI] [PubMed] [Google Scholar]
- 76. Sexton JZ, Wigle TJ, He Q, Hughes MA, Smith GR, Singleton SF, Williams AL, Yeh LA. 2010. Novel inhibitors of E. coli RecA ATPase activity. Curr. Chem. Genomics 4:34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wigle TJ, Sexton JZ, Gromova AV, Hadimani MB, Hughes MA, Smith GR, Yeh LA, Singleton SF. 2009. Inhibitors of RecA activity discovered by high-throughput screening: cell-permeable small molecules attenuate the SOS response in Escherichia coli. J. Biomol. Screen. 14:1092–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Romesberg F, David NE, Cirz R. May 2009. Compositions and methods to reduce mutagenesis. U.S. patent US 2009/0136518 A1
- 79. Singleton SF. May 2012. Inhibitors of RecA activities for control of antibiotic-resistant bacterial pathogens. U.S. patent US 2012/0135092 A1
- 80. Cirz RT, Chin JK, Andes DR, de Crecy-Lagard V, Craig WA, Romesberg FE. 2005. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 3:e176 doi:10.1371/journal.pbio.0030176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lee AM, Ross CT, Zeng BB, Singleton SF. 2005. A molecular target for suppression of the evolution of antibiotic resistance: inhibition of the Escherichia coli RecA protein by N(6)-(1-naphthyl)-ADP. J. Med. Chem. 48:5408–5411 [DOI] [PubMed] [Google Scholar]
- 82. Thi TD, Lopez E, Rodriguez-Rojas A, Rodriguez-Beltran J, Couce A, Guelfo JR, Castaneda-Garcia A, Blazquez J. 2011. Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials. J. Antimicrob. Chemother. 66:531–538 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.