

Abstract
Escherichia coli cells normally require RNase E activity to form colonies (colony-forming ability [CFA]). The CFA-defective phenotype of cells lacking RNase E is partly reversed by overexpression of the related endoribonuclease RNase G or by mutation of the gene encoding the RNA helicase DeaD. We found that the carbon source utilization by rne deaD doubly mutant bacteria differs from that of rne+ cells and from that of cells mutated in deaD alone and that the loss of rne function in these bacteria limits conversion of the glycolytic pathway product phosphoenolpyruvate to the tricarboxylic acid (TCA) cycle intermediate oxaloacetic acid. We show that the mechanism underlying this effect is reduced production of the enzyme phosphoenolpyruvate carboxylase (PPC) and that adventitious overexpression of PPC, which facilitates phosphoenolpyruvate utilization and connects the glycolytic pathway with the TCA cycle, restored CFA to rne deaD mutant bacteria cultured on carbon sources that otherwise were unable to sustain growth. We further show that bacteria producing full-length RNase E, which allows formation of degradosomes, have nutritional requirements different from those of cells supplied with only the N-terminal catalytic region of RNase E and that mitigation of RNase E deficiency by overexpression of a related RNase, RNase G, is also affected by carbon source. Our results reveal previously unsuspected effects of RNase E deficiency and degradosome formation on nutrient utilization by E. coli cells.
INTRODUCTION
RNase E was initially discovered in Escherichia coli as an endoribonuclease that processes 9S rRNA (1) and later was shown to have a multifunctional role in the processing of various other structural RNAs (1, 2, 3, 4), the global degradation of mRNA (5, 6, 7, 8), the control of plasmid DNA replication (9), and the maturation/processing of a variety of small catalytic RNAs (9, 10). Commonly, the cleavage of the targeted RNA by RNase E is followed by digestion of RNase E-generated fragments by other ribonucleases as tightly coupled events (11). DNA sequence analysis has predicted that orthologs of E. coli RNase E exist among dozens of evolutionarily disparate bacterial species (12, 13). The ribonucleolytic activity of RNase E resides in its N-terminal half, N-Rne (14), whereas the C-terminal half (CTH) includes binding sites for several proteins that form a multicomponent complex termed the “degradosome” (15, 16). Although degradosome formation is not essential for E. coli growth (14, 17, 18), deletion of the CTH has been found to reduce the rate of decay of some RNase E substrates (19, 20).
In E. coli, the actions of RNase E are essential for colony-forming ability (CFA) (21), and transfer of liquid cultures of rne(Ts) mutant bacteria to a nonpermissive temperature prevents normal cell division and results in the formation of elongated filamentous structures containing connected cell bodies (22). Multiple studies aimed at elucidating the basis of RNase E essentiality have been carried out (6, 23, 24, 25, 26). While CFA can be restored to rne deletion mutants by overproduction or mutation of RNase G (6, 24, 25, 27), an RNase E paralog (2, 4) that structurally resembles the amino-terminal catalytic region of RNase E (14), such cells grow more slowly than rne+ bacteria and retain the filamentation phenotype characteristic of RNase E-deficient mutants in liquid culture (25). The filamentation of RNase G-overproducing Δrne mutants is reversed by overexpression of FtsZ; however, FtsZ-overproducing cells remain defective in their rate of division (25).
During recently reported investigations of insertion mutations that restore CFA to E. coli cells lacking RNase E (26), we observed that the survival of such Δrne cells was dependent on culture conditions: colony formation was observed when the cells were grown on Luria-Bertani (LB) medium but not when they were grown on M9 medium containing glucose as the sole carbon source. This finding prompted us to investigate nutritional factors affecting the growth of Δrne bacteria, and the ability of cells containing mutations in the deaD gene (26), which encodes an RNA helicase (28), to propagate enabled us to do this in the absence of any complementing RNase. The results we report here show that a combination of a glycolytic pathway sugar plus a tricarboxylic acid (TCA) cycle sugar is needed for propagation of Δrne deaD doubly mutant bacteria, whereas no such requirement was observed for cells mutated in deaD alone. They further indicate, as shown by Western blotting and confirmed genetically, that RNase E-deficient cells are defective in production of the enzyme phosphoenolpyruvate carboxylase (PPC), which converts phosphoenolpyruvate to oxaloacetic acid and connects the glycolytic pathway with the TCA cycle, establishing a previously unsuspected role for PPC in RNase E essentiality.
MATERIALS AND METHODS
Strains and plasmids.
The strains used in this study are listed in Table 1. MT4000 was constructed by the method described by Lee et al. (6). MT4001, MT4002, MT4003, MT2105, and MT2005 were constructed by transformation of MT4000 by pNRNE4, MT4000 by pRNG2S, MT4000 by pRNG2SΔH, CM2100 by pRNG2SΔH, and MT2000 by pRNG2SΔH, respectively. MT2113 was obtained by P1 transduction of deaD::Tn10 into CM2100, and colonies were selected on tetracycline (Tc) plates containing 0.1% arabinose (0.1% Ara) to induce adventitious production of RNase E in the Δrne strain (6, 26). P1(deaD::Tn10) was prepared from MT2013 as described previously (26). MT2103 was constructed by replacing pBAD-RNE of CM2100 with pLAC-RNE2 by the method described by Lee et al. (6). To test the effects of PPC overexpression, we introduced either a modified ASKA library plasmid (29) expressing PPC (i.e., ASKA-ppc) or an empty vector [pCA24N(Ap)] into the MT2113 strain, generating the MT2114 and MT2117 strains, respectively.
Table 1.
Bacterial strains and plasmids
| Strain or plasmid | Description | Reference(s) or source |
|---|---|---|
| Strains | ||
| MG1655 | ilvG rfb-50 rph-1 fnr-267 eut | E. coli Genetic Stock Center (CGSC 6300) |
| MG165D | Same as MG1655 but carrying deaD::Tn10 (clone 2) | This study |
| NCM3416 | Same as MG1655 but carrying rph+ and zib-207::Tn10 | 40 |
| MT4000 | Same as NCM3416 but carrying rne::cat and pBAD-RNE | This study |
| MT4001 | Same as MT4000 but carrying pNRNE4 | This study |
| MT4002 | Same as MT4000 but carrying pRNG2S | This study |
| MT4003 | Same as MT4000 but carrying pRNG2SΔH | This study |
| MT2000 | lacZ43(Fs) LAM− relA1 spoT1 thi-1 rne::cat; carries pBAD-RNE | 25 |
| MT2005 | Same as MT2000 but carrying pRNG2SΔH | This study |
| MT2013 | Same as MT2000 but carrying deaD::Tn10 (clone 2) | 26 |
| CM2100 | Same as MG1655 but carrying rne::cat and pBAD-RNE | This study |
| MT2103 | Same as MG1655 but carrying rne::cat and pLAC-RNE2 | This study |
| MT2105 | Same as CM2100 but carrying pRNG2SΔH | This study |
| MT2113 | Same as CM2100 but carrying deaD::Tn10 (clone 2) | This study |
| MT2114 | Same as MT2113 but carrying ASKA-ppc | This study |
| MT2115 | Same as MT2113 but carrying pACYC177 | This study |
| MT2116 | Same as MT2113 but carrying pMT177Z | This study |
| MT2117 | Same as MT2113 but carrying pCA24N(Ap) | This study |
| Plasmids | ||
| pBAD-RNE | pSC101 ori Kmr, rne under control of PBAD | 6, 25 |
| pLAC-RNE2 | pSC101 ori Apr, rne under control of lacUV5 promoter | 41 |
| pNRNE4 | P15A ori Apr, His-tagged N-rne under control of lacUV5 promoter | 25 |
| pRNG2 | P15A ori Apr, His-tagged rng (also N-terminally extended) under control of lacUV5 promoter | Gift from Kangseok Lee |
| pRNG2S | P15A ori Apr, His-tagged rng under control of lacUV5 promoter | Gift from Kangseok Lee |
| pRNG2SΔH | P15A ori Apr, natural short-form rng under control of lacUV5 promoter | Gift from Kangseok Lee |
| pCA24N(Ap) | ColE1 ori Apr | 26 |
| ASKA-ppc | ColE1 ori Apr, ppc under control of PT5-lac | This study |
| pACYC177 | P15A ori Kmr Apr | 42 |
| pMT177Z | P15A ori Apr, ftsZ under control of natural promoter | 25 |
pRNG2SΔH, which expresses the natural form of RNase G (i.e., no N-terminal or C-terminal extension), was constructed by ligating ClaI- and EcoRI-digested PCR products encoding the C-terminal region of RNase G lacking a hexahistidine tag into pRNG2S using the same restriction enzyme sites. The PCR primers used were rng-948F (5′-TGTCGGTCATCGCAATCT-3′) and 3′-rng-no-His (5′-AGACTAGTGAATTCACATCATTACGACGTCAAACTG-3′). pRNG2S, which expresses C-terminal His-tagged RNase G lacking any N-terminal extension, was constructed by ligating an NdeI- and NsiI-digested PCR product encoding the N-terminal region of RNase G containing its natural N-terminal sequence (ATGACGGCTGAA…) into pRNG2 using the same restriction enzyme sites. The PCR primers used were 5′-rng-nat (5′-ATACATATGACGGCTGAATTGTTAGTA-3′) and rng-595R (5′-AGACGCGTTTCAGATAAG-3′). pRNG2, which expresses C-terminal His-tagged RNase G lacking any N-terminal extension, was constructed by ligating NotI- and SpeI-digested PCR DNA encoding the region of rng containing an extended N-terminal sequence (ATGAGAAAAGGGATAAAC) into pNRNE4 (25) using the same restriction enzyme sites. The PCR primers used were 5′-rng-2 (5′-GGATCCGCGGCCGCTTTAAGAAGGAGATATACATATGAGAAAAGGGATAAAC-3′) and 3′-rng-His (5′-GTCTAGACTAGTGAATTCAGTGGTGGTGGTGGTGGTGCATCATTACGACGTGCAAACT-3′). The pRNG2SΔH, pRNG2S, and pRNG2 plasmids were kindly provided by Kangseok Lee (C. J. Moore, H. Go, K. Lee, and S. N. Cohen, unpublished data). Construction of an ASKA plasmid with an Ap resistance gene was described previously (26).
Media and culture conditions.
LB medium or M9 minimal medium (30) containing 0.1 mM CaCl2, 1 mM MgSO4, and appropriate carbon sources at the following concentrations were used: d-(+)-glucose, 0.2%; d-mannitol, 0.2%; d-(−)-fructose, 0.2%; glycerol, 0.5%; sodium pyruvate, 0.2%; sodium acetate, 0.2%; succinic acid, 0.5%; l-(−)-malic acid, 0.5%; fumaric acid, 0.5%; and Casamino Acids (CAA) (see BD Bionutrients Technical Manual), 0.05%. Where indicated, LB components at the following concentrations were used: yeast extract (Bacto Yeast Extract; BD), 5 g/liter; tryptone (Bacto Tryptone; BD), 10 g/liter; and sodium chloride (Invitrogen), 10 g/liter. Where indicated, antibiotics were used at the following concentrations: ampicillin, 50 μg/ml; chloramphenicol, 10 μg/ml; kanamycin, 20 μg/ml; tetracycline, 5 μg/ml; and streptomycin 50 μg/ml. For all experiments, agar plates were freshly prepared on the day used (never stored at 4°C or overnight on the bench), bacteria were streaked onto plates containing 0.1% l-arabinose from glycerol stock (containing 40% glycerol) and incubated at 37°C overnight, and colonies were picked for inoculations.
Plating experiments.
For plating experiments, cells were freshly grown from a single colony (never grown overnight) to mid-log phase (optical densities at 600 nm [OD600] ranged from 0.3 to 0.6) at 37°C in LB medium containing the indicated supplements and antibiotics. Cells were diluted 10−4 to 10−5, and 100 μl of diluted culture was spread on LB or M9 plates containing carbon sources, supplements, and antibiotics. The plates were incubated at 37°C for 2 to 7 days depending on the experiment and scanned, and colony number was determined. Different nutrient combinations resulted in different rates of colony growth, and where indicated, the incubation period was extended by up to 2 days to enable colonies to be better visualized. Incubation conditions for each experiment are described in the figure legends. The absence of detectable colony formation after 7 days of incubation was defined as a CFA-negative (CFA−) phenotype. Colony-forming efficiency (CFE) was determined by dividing the colony number observed under the indicated growth condition by the colony number observed under the same condition when an arabinose-inducible RNase E gene was expressed (26).
Western blotting.
Cultures of CM2100, MT2113, MT2114, or MT2117 cells were grown overnight at 37°C on LB plates containing 0.1% arabinose and the appropriate antibiotics. Randomly selected colonies from each plate were suspended in LB and diluted to an OD600 of 0.001 in fresh LB containing the appropriate selective antibiotics, with or without 0.1% arabinose (as indicated) and with 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG) (for strains MT2114 and MT2117). The cultures were grown to an approximate OD600 of 0.8, or for a minimum of 6 h, and then harvested and prepared for loading onto a polyacrylamide gel. The cells were diluted to an OD600 of 0.01 per μl in water and SDS loading buffer and then boiled. Lysate from CM2100, MT2113, or MT2117 cells at an OD600 of 0.24 cultured with inducing agents as indicated was loaded alongside lysate from MT2114 cells at an OD600 of 0.1, and the proteins were separated on a Criterion XL 3 to 8% Tris-acetate polyacrylamide gel (Bio-Rad). The samples were transferred to a 0.1-μm nitrocellulose membrane and probed with anti-PPC (Agrisera rabbit polyclonal), anti-RNase E (mouse monoclonal), and anti-S1 (rabbit polyclonal) antibodies, in that order, with intervening membrane stripping. The numbers provided represent densitometric measurements of each band as determined by the VersaDoc model 1000 imaging system (Bio-Rad), corrected for loading based on the S1 protein control and the difference in amount of total protein loaded between MT2114 and the other strains. Values were then normalized relative to the RNase E or PPC band from the CM2100 cells.
RESULTS AND DISCUSSION
RNase E is required for glycolysis in E. coli.
We recently reported that a Tn10 insertion mutation in the deaD gene, which encodes an RNA helicase, enables colony formation by E. coli N3433 cells lacking RNase E function (26). During those experiments, we observed that this ability was affected by the carbon source included in the medium. While N3433 is the E. coli strain in which RNase E was first discovered and has since been widely employed for study of RNase E essentiality, this strain contains multiple mutations that potentially affect nutritional requirements [i.e., lacZ43(Fs), relA1, spoT1, and thi-1] and consequently affect the ability of even rne+ cells to form colonies efficiently on some carbon sources (see Fig. S1 in the supplemental material); we thus chose to further investigate the nutritional effects of loss of RNase E function in E. coli strain MG1655, which is prototrophic at these loci, after confirming that a Δrne deaD derivative of MG1655 (i.e., strain MT2113) forms colonies on LB with 100% colony-forming efficiency relative to that of cells producing RNase E (Fig. 1A).
Fig 1.

Effects of individual LB components on CFA of a Δrne deaD::Tn10 strain. (A) A culture of MT2113 in LB containing 0.1% arabinose, which induces adventitious expression of plasmid-borne rne, was spread on LB containing or lacking 0.1% arabinose, LB containing the additional components of M9-glucose but lacking the inducer, or LB containing 0.2% glucose and no arabinose. (B) A culture of MT2113 was spread on M9-glucose lacking any additive or supplemented with yeast extract, a tryptic digest of casein (labeled as tryptone), or NaCl, all in the absence of arabinose. (C) Identification of amino acid requirements for CFA by a Δrne deaD::Tn10 strain cultured on M9-glucose. A culture of MT2113 was spread on M9-glucose containing a variety of amino acid mixtures, in the absence of arabinose. A mixture of glycolytic pathway-derived amino acids, i.e., Ser, Cys, Gly, Trp, Tyr, Phr, Ala, Val, Leu, Ile, and His, is defined as “glycolytic a.a.,” and a mixture of TCA cycle-derived amino acids, i.e., Lys, Met, Thr, Asn, Asp, Glu, Gln, Pro, Arg, and His, is defined as “TCA a.a.” The concentration of each amino acid was 160 μM. Plates were scanned after incubation for 2 days (A and B) or 4 days (C) at 37°C.
As seen in Fig. 1, in the absence of any arabinose to induce plasmid-borne RNase E, the Δrne deaD MT2113 strain formed colonies on LB medium or on M9 medium containing both glucose and a mixture of amino acids as carbon sources; however, no MT2113 colonies were observed when yeast extract or the mixture of amino acids was omitted (Fig. 1B). In contrast, in the absence of any amino acid supplement, strain MG165D, which is an MG1655 derivative containing an intact rne gene but mutated by Tn10 insertion into deaD, formed colonies on M9 medium containing any of a series of tested sugars as a carbon source (see Fig. S2 in the supplemental material), arguing that the failure of MT2113 to form colonies in media lacking any supplemental amino acids and supplied with glucose as the sole carbon source results from defective RNase E function. Testing of additives of individual amino acids (Fig. 1C) indicated that amino acids specifically synthesized through the glycolytic pathway did not enable CFA by MT2113 supplied with glucose, whereas addition of amino acids generated through the TCA cycle did. Moreover, the addition of malate, fumarate, or succinate, which are all TCA cycle components and can generate TCA cycle-dependent amino acids, also enabled CFA on M9-glucose (Fig. 2). Together, our results indicate that the pathway to TCA cycle-dependent amino acid biosynthesis is intact in the Δrne deaD::Tn10 mutant strain and suggest that a defect in the enzymatic connection between the glycolytic pathway and TCA cycle in the mutant bacteria prevents products of glycolysis from entering the TCA cycle.
Fig 2.

Effect of carbon source combination on CFA of a Δrne deaD::Tn10 strain. A culture of MT2113 in LB was spread on carbon sources as indicated. The presence or absence of 0.1% arabinose is indicated. Plates were scanned after incubation at 37°C for 4 days.
Functional interactions between RNase E and PPC.
The glycolytic pathway and TCA cycle are connected by the enzyme phosphoenolpyruvate carboxylase (PPC), which converts phosphoenolpyruvate to oxaloacetic acid (31). To test the hypothesis that deficiency of PPC expression occurs in Δrne deaD bacteria, we used Western blotting to examine the effects of RNase E expression on the expression of PPC encoded by either the endogenous gene or a plasmid-borne PPC gene (Fig. 3). As seen, the expression of PPC protein is highly dependent on the expression of RNase E. When full-length RNase E was depleted to undetectable levels by growth in the absence of arabinose, the PPC protein was reduced by 79 to 91%. To determine whether overexpression of PPC rescues CFA in bacteria that fail to produce RNase E, we tested the ability of the MT2114 strain to form colonies on M9-glucose. As seen in Fig. 4, overexpression of PPC restored CFA to bacteria cultured on M9-glucose medium. No restoration of CFA occurred in the absence of the deaD mutation (data not shown), which is consistent with the previously reported ability of mutation of deaD to suppress the need for RNase E in E. coli cells (26). Together, these findings suggest that while PPC production is necessary for CFA in cells lacking RNase E function, it is not sufficient, and thus that the CFA-promoting function mediated by mutation of deaD is distinct from the PPC-related function. Additionally, adventitious expression of PPC (Fig. 4) did not result in CFA on M9-malate medium, supporting the view that upregulation of PPC alone does not restore normality to Δrne deaD mutant cells. During these experiments, we noted that although faint “shadows” of colonies often appeared following plating of such bacteria on M9-malate plates (see also Fig. 5), these presumed minute colonies did not increase in size during further incubation and upon replating did not result in colony formation (data not shown). We also observed that overproduction of PPC protein from the ASKA-ppc plasmid was increased by RNase E deficiency, in contrast to what was observed in cells expressing PPC from the endogenous gene. As the ASKA-ppc plasmid expresses the open reading frame of the PPC gene but lacks the 5′ and 3′ untranslated regions, this result suggests that RNase E production may have a dual role in PPC synthesis by affecting both mRNA synthesis and translation.
Fig 3.
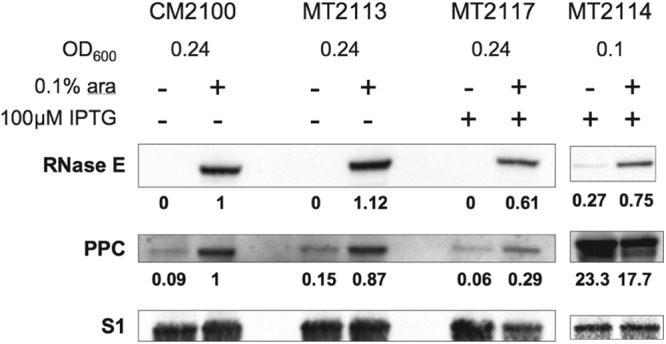
Effect of RNase E expression on PPC expression in a Δrne strain, a Δrne deaD::Tn10 strain, and a Δrne deaD::Tn10 strain complemented with ASKA-ppc or an empty vector. Cultures of CM2100, MT2113, MT2117, and MT2114 were grown with and without 0.1% arabinose to induce or deplete full-length RNase E from the pBAD-RNE plasmid (MT2114 and MT2117 were additionally grown with 100 μM IPTG), separated on a 3 to 8% Tris-acetate gel, transferred to a nitrocellulose membrane, and probed with the appropriate antibodies for the presence of RNase E, PPC, and S1 proteins. Due to the high level of PPC overexpression from ASKA-ppc in MT2114 when induced by 100 μM IPTG, a smaller amount of culture was loaded per lane than for the other strains (as described in Materials and Methods). Protein expression was determined as described in Materials and Methods.
Fig 4.
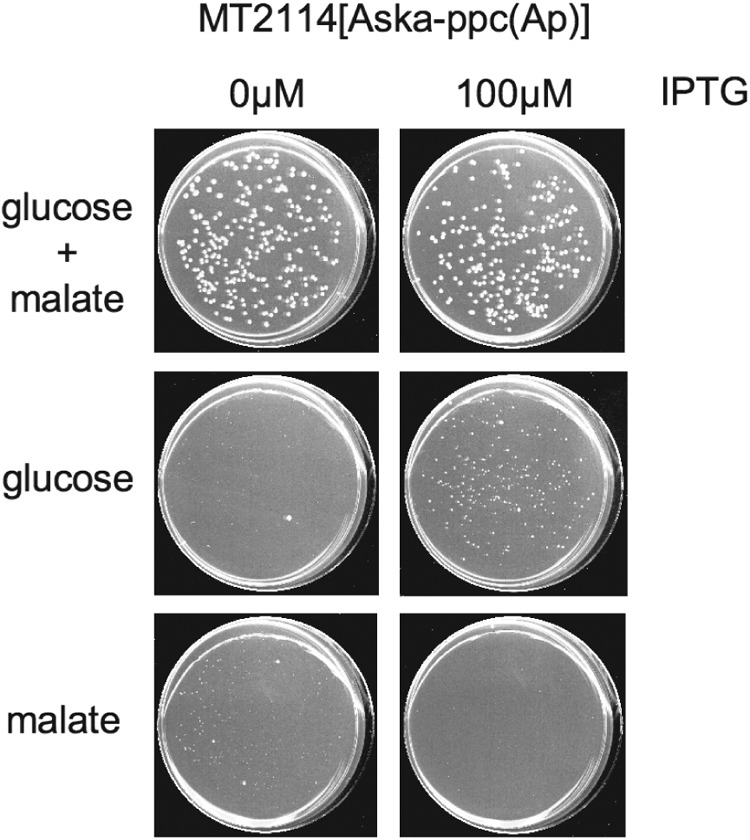
Effect of PPC complementation on CFA of a Δrne deaD::Tn10 strain cultured on M9-glucose or M9-malate. Cultures of MT2114 were spread on M9-glucose with 0.5% malate, M9-glucose, and M9-malate with or without 100 μM IPTG in the absence of arabinose. Plates were scanned after incubation at 37°C for 7 days.
Fig 5.

Effect of FtsZ complementation on CFA of a Δrne deaD::Tn10 strain on M9-glucose or M9-malate. Cultures of MT2115 and MT2116 were spread on M9 plates containing glucose and/or malate, as indicated, and containing or lacking 0.1% arabinose. Plates were scanned after incubation at 37°C for 4 days or, where indicated, after 2 additional days of incubation.
Interestingly, introduction of an FtsZ-producing plasmid (pMT177Z), which previously has been shown to reverse filamentation of RNase E-deficient cells (25) (see Fig. S3 in the supplemental material), into Δrne deaD::Tn10 bacteria enabled CFA on M9-malate but not on M9-glucose in the absence of amino acids whose synthesis is dependent on the TCA cycle (Fig. 5). As the genes expressed from the ftsQAZ operon are not known to contribute to glycolysis or gluconeogenesis, the effect of FtsZ on the nutritional requirements of RNase E-deficient cells suggests that an additional step in the glycolytic or gluconeogenetic pathway (32, 33) is impaired in these RNase E-deficient bacteria.
Role of the degradosome in carbon source utilization by E. coli.
The C-terminal half of RNase E functions as a binding site for multiple other proteins (15); one of these is the glycolytic pathway enzyme enolase, which converts 2-phosphoglycerate to phosphoenolpyruvate (34), which is the cellular substrate for PPC. Interaction between RNase E and enolase normally is not required for propagation of E. coli, as the N-terminal catalytic region of RNase E (N-Rne), which lacks the enolase binding site (35), is sufficient for colony formation by rne mutant bacteria (14). As both mannitol and glucose are upstream of the glycolytic pathway step that requires enolase (i.e., conversion of mannitol to pyruvate) and combinations of glucose plus pyruvate or mannitol plus pyruvate did not restore CFA to RNase-E deficient cells (Fig. 2), the sugar-related defect in CFA during RNase E deficiency is necessarily independent of enolase.
As PPC production is impaired in RNase E-deficient bacteria and a component of the RNase E-based degradosome generates the cellular substrate for PPC (i.e., phosphoenolpyruvate), we hypothesized that the presence of a scaffold region on RNase E may affect the carbon source utilization by E. coli cells. To test this notion, wild-type bacteria and a deaD+ rne deletion mutant strain complemented by either N-Rne or full-length Rne were tested for their ability to grow on M9 plates supplemented with specific sugars. As shown in Fig. 6, both wild-type bacteria (Fig. 6A) and a Δrne strain carrying a plasmid expressing full-length Rne (Fig. 6B) formed normal-size colonies on all tested carbon sources except acetate after 2 days of culture on M9 plates. However, Δrne bacteria carrying a plasmid expressing N-Rne [i.e., MT4001(pNRNE4)] did not form colonies on some of the carbon sources that are utilized effectively by full-length RNase E (i.e., fumarate and succinate), and colony growth on certain other carbon sources occurred much more slowly than was observed for Δrne cells complemented in trans by a plasmid expressing full-length RNase E (Fig. 6C and Table 2). Thus, while degradosome formation was not necessary for utilization of glucose by E. coli (Fig. 6C), the absence of the scaffold region of the enzyme affected utilization of other sugars. It should also be noted that overproduction of full-length RNase E in a Δrne strain (MT2103) did not enable formation of colonies on M9 medium containing 0.2% lactose, although wild-type NCM3416 bacteria were able to form colonies (data not shown). The basis for this effect currently is not known.
Fig 6.

(A and B) Growth of wild-type E. coli (A) or a full-length RNase E-complemented Δrne strain (B) on the indicated carbon sources. Cultures of NCM3416 or MT2103 were spread on LB or M9 plates as indicated (10 μM IPTG for MT2103). (C) Effect of RNase E degradosome formation on growth of E. coli on different carbon sources. Cultures of MT4001 were spread on LB or M9 plates containing or lacking 0.1% arabinose as indicated, and 100 μM IPTG was added as indicated. Plates were scanned after incubation at 37°C for 2 days and, where indicated, after 2 additional days of incubation.
Table 2.
Summary of plating experiments
| Strain | Resulta of plating on: |
|||||||
|---|---|---|---|---|---|---|---|---|
| LB medium | M9 with: |
|||||||
| Glucose | Glycerol | Pyruvate | Acetate | Malate | Fumarate | Succinate | ||
| NCM3416 | ND | ○ | ○ | ○ | △ | ○ | ○ | ○ |
| MT4001(pNRNE4) | ○ | ○ | △ | △ | × | △ | × | × |
| MT4002(pRNG2S) | ○ | ○ | 46% CFE, slow | △ | 37% CFE, slow | ○ | ○ | ○ |
| MT2105(pRNG2SΔH) | ○ | × | ND | ND | ND | × | ND | ND |
| MT2113 (deaD::Tn10) | △ | × | ND | △ | ND | × | ND | ND |
○, 100% CFE; △, 100% CFE but slow growth; ×, no growth.
Complementation of Δrne bacteria by different forms of RNase G results in differential sugar utilization.
Lee et al. reported that overproduction of His-tagged RNase G restores CFA to Δrne bacteria both on LB plates and in LB liquid medium (6). Subsequent investigations have shown that complementation of rne-defective mutants by RNase G is facilitated by unnatural extensions at the termini of the RNase G protein, and it was consequently concluded that RNase G function is constrained by the termini of the enzyme (24). In other instances, mutations in the catalytic region of RNase G have been reported to facilitate complementation of rne mutants (27). Given such information and the results we have described above, we were interested in learning whether Δrne cells complemented to viability by overproduction of the natural versus elongated forms of RNase G have different nutritional requirements.
As shown in Fig. 7, overproduction of the natural form of RNase G restored CFA when introduced into an MG1655-derived Δrne strain plated on LB medium but failed to enable CFA when cells were plated on M9 containing any tested sugar except for the combination of glucose and malate (Fig. 7A). In contrast, His-tagged RNase G enabled CFA with 100% efficiency, not only on LB but also on M9 medium containing glucose, pyruvate, malate, fumarate, or succinate, indicating that the nutritional requirements for such complementation resemble those of bacteria complemented by full-length RNase E (Fig. 7C). However, relative to cells complemented by plasmid-encoded RNase E, CFA occurred in Δrne cells at a reduced efficiency for His-tagged RNase G complementation during growth on glycerol (46% CFE) or acetate (37% CFE) (Fig. 7C). Complementation by the natural form of RNase G was not observed, even on LB, for the N3433-derived Δrne strain (Fig. 7B), which as noted above differs from MG1655 derivatives in carbon sources needed for CFA, suggesting that strain differences may account for the disparate results reported in investigations of complementation of RNase E deficiency by RNase G overexpression (6, 24, 27, 36, 37).
Fig 7.

Growth of a natural-form RNase G-complemented Δrne strain on LB and a variety of carbon sources. (A and B) Cultures of MT2105 (A) or MT2005 (B) were spread on LB or M9 plates containing or lacking 0.1% arabinose (as indicated), and 1 mM IPTG was added as indicated. Plates were scanned after incubation at 37°C for 2 days (A) and 4 days (B). (C) Growth of a Δrne strain expressing His-tagged RNase G. Cultures of MT4002 were spread on LB or M9 plates containing or lacking 0.1% arabinose and the indicated carbon sources, and 1 mM IPTG was added as indicated. Plates were scanned after incubation at 37°C for 2 days and, where indicated, after 2 additional days of incubation.
Proposed mechanism for the role of the glycolytic pathway and TCA cycle in RNase E essentiality.
The results reported here indicate that the absence of RNase E in E. coli made viable by a second-site Tn10 insertion mutation in deaD (26) results in defective production of PPC, which links glycolysis with the TCA cycle (Fig. 8) (31). We conclude that a deficiency of this enzyme results in the inability of cells to use glycolytic pathway carbon sources for the synthesis of amino acids generated by the TCA cycle. Consequently, whereas rne deaD mutant cells failed to grow solely on glucose or other glycolytic pathway intermediates, they formed colonies on minimal medium containing these sugars when the cellular connection between the glycolytic pathway and TCA cycle was enhanced by adventitious overexpression of a cloned ppc gene or when TCA cycle amino acids were added adventitiously. However, defective PPC synthesis is not the only nutritional consequence of RNase E deficiency, as cells overexpressing PPC remained unable to form colonies when supplied with certain sugars. Moreover, even the combination of glucose and malate did not support CFA in Δrne cells that lack an additional mutation of deaD. Thus, RNase E essentiality is not attributable to a single gene or pathway.
Fig 8.
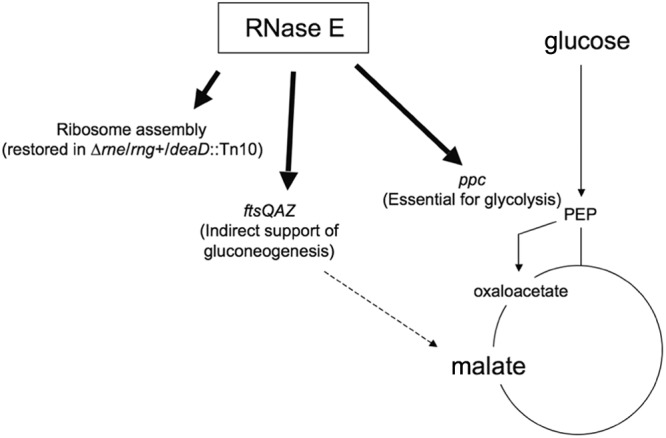
Proposed model for RNase E effects on the glycolytic pathway and TCA cycle. PEP, phosphoenolpyruvate.
Whereas the model we have proposed explains the bulk of the findings we have observed, some findings remain unexplained. (i) A Keio collection E. coli K-12 BW25113 strain deleted in ppc (JW3928-1) (38), as expected from the proposed model (see also reference 39), did not form colonies on M9 medium containing either glucose or pyruvate as a sole carbon source. However, rne deaD doubly mutant bacteria can form colonies on M9-pyruvate, as we reported previously (26). This finding is consistent with the conclusion that inactivation of PPC alone does not fully explain the modified metabolism observed in E. coli in the absence of RNase E. (ii) M9 minimal medium containing a CAA mixture as the sole carbon source did not confer CFA to rne deaD mutant cells, contrary to what was expected. As the combination of pyruvate (a glycolytic pathway intermediate) and malate (a TCA cycle intermediate) supported CFA (as shown in Fig. 2), all glycolytic pathway (and gluconeogenetic pathway) steps upstream from pyruvate should in principle be active in the cultured rne deaD mutant cells. Thus, we anticipated that providing a mixture of glycolytic pathway-derived and TCA cycle-derived amino acids (i.e., CAA) would fulfill the nutritional requirements for cell growth. The basis for the absence of CFA by rne deaD mutant bacteria cultured on medium containing such a mixture has not been determined.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kangseok Lee and Christine A. Miller for kindly providing strains and plasmids. We also thank Roberta Peterson for assistance with the manuscript.
This study was supported by NIH grants AI08619 and GM 54158 to S.N.C.
Footnotes
Published ahead of print 28 December 2012
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01558-12.
REFERENCES
- 1. Ghora BK, Apirion D. 1978. Structural analysis and in vitro processing to p5 rRNA of a 9S RNA molecule isolated from an rne mutant of E. coli. Cell 15:1055–1066 [DOI] [PubMed] [Google Scholar]
- 2. Li Z, Pandit S, Deutscher MP. 1999. RNase G (CafA protein) and RNase E are both required for the 5′ maturation of 16S ribosomal RNA. EMBO J. 18:2878–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ray A, Apirion D. 1980. Cloning the gene for ribonuclease E, an RNA processing enzyme. Gene 12:87–94 [DOI] [PubMed] [Google Scholar]
- 4. Wachi M, Umitsuki G, Shimizu M, Takada A, Nagai K. 1999. Escherichia coli cafA gene encodes a novel RNase, designated as RNase G, involved in processing of the 5′ end of 16S rRNA. Biochem. Biophys. Res. Commun. 259:483–488 [DOI] [PubMed] [Google Scholar]
- 5. Bernstein JA, Khodursky AB, Lin PH, Lin-Chao S, Cohen SN. 2002. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc. Natl. Acad. Sci. U. S. A. 99:9697–9702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee K, Bernstein JA, Cohen SN. 2002. RNase G complementation of rne null mutation identifies functional interrelationships with RNase E in Escherichia coli. Mol. Microbiol. 43:1445–1456 [DOI] [PubMed] [Google Scholar]
- 7. Ono M, Kuwano M. 1979. A conditional lethal mutation in an Escherichia coli strain with a longer chemical lifetime of messenger RNA. J. Mol. Biol. 129:343–357 [DOI] [PubMed] [Google Scholar]
- 8. Stead MB, Marshburn S, Mohanty BK, Mitra J, Castillo LP, Ray D, van Bakel H, Hughes TR, Kushner SR. 2011. Analysis of Escherichia coli RNase E and RNase III activity in vivo using tiling microarrays. Nucleic Acids Res. 39:3188–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin-Chao S, Cohen SN. 1991. The rate of processing and degradation of antisense RNAI regulates the replication of ColE1-type plasmids in vivo. Cell 65:1233–1242 [DOI] [PubMed] [Google Scholar]
- 10. Lundberg U, Altman S. 1995. Processing of the precursor to the catalytic RNA subunit of RNase P from Escherichia coli. RNA 1:327–334 [PMC free article] [PubMed] [Google Scholar]
- 11. Alifano P, Rivellini F, Piscitelli C, Arraiano CM, Bruni CB, Carlomagno MS. 1994. Ribonuclease E provides substrates for ribonuclease P-dependent processing of a polycistronic mRNA. Genes Dev. 8:3021–3031 [DOI] [PubMed] [Google Scholar]
- 12. Lee K, Cohen SN. 2003. A Streptomyces coelicolor functional orthologue of Escherichia coli RNase E shows shuffling of catalytic and PNPase-binding domains. Mol. Microbiol. 48:349–360 [DOI] [PubMed] [Google Scholar]
- 13. Schein A, Sheffy-Levin S, Glaser F, Schuster G. 2008. The RNase E/G-type endoribonuclease of higher plants is located in the chloroplast and cleaves RNA similarly to the E. coli enzyme. RNA 14:1057–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McDowall KJ, Cohen SN. 1996. The N-terminal domain of the rne gene product has RNase E activity and is non-overlapping with the arginine-rich RNA-binding site. J. Mol. Biol. 255:349–355 [DOI] [PubMed] [Google Scholar]
- 15. Miczak A, Kaberdin VR, Wei CL, Lin-Chao S. 1996. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl. Acad. Sci. U. S. A. 93:3865–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Py B, Higgins CF, Krisch HM, Carpousis AJ. 1996. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381:169–172 [DOI] [PubMed] [Google Scholar]
- 17. Lopez PJ, Marchand I, Joyce SA, Dreyfus M. 1999. The C-terminal half of RNase E, which organizes the Escherichia coli degradosome, participates in mRNA degradation but not rRNA processing in vivo. Mol. Microbiol. 33:188–199 [DOI] [PubMed] [Google Scholar]
- 18. Ow MC, Liu Q, Kushner SR. 2000. Analysis of mRNA decay and rRNA processing in Escherichia coli in the absence of RNase E-based degradosome assembly. Mol. Microbiol. 38:854–866 [DOI] [PubMed] [Google Scholar]
- 19. Khemici V, Poljak L, Toesca I, Carpousis AJ. 2005. Evidence in vivo that the DEAD-box RNA helicase RhlB facilitates the degradation of ribosome-free mRNA by RNase E. Proc. Natl. Acad. Sci. U. S. A. 102:6913–6918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morita T, Kawamoto H, Mizota T, Inada T, Aiba H. 2004. Enolase in the RNA degradosome plays a crucial role in the rapid decay of glucose transporter mRNA in the response to phosphosugar stress in Escherichia coli. Mol. Microbiol. 54:1063–1075 [DOI] [PubMed] [Google Scholar]
- 21. Apirion D. 1978. Isolation, genetic mapping and some characterization of a mutation in Escherichia coli that affects the processing of ribonucleic acid. Genetics 90:659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldblum K, Apririon D. 1981. Inactivation of the ribonucleic acid-processing enzyme ribonuclease E blocks cell division. J. Bacteriol. 146:128–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ow MC, Kushner SR. 2002. Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev. 16:1102–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deana A, Belasco JG. 2004. The function of RNase G in Escherichia coli is constrained by its amino and carboxyl termini. Mol. Microbiol. 51:1205–1217 [DOI] [PubMed] [Google Scholar]
- 25. Tamura M, Lee K, Miller CA, Moore CJ, Shirako Y, Kobayashi M, Cohen SN. 2006. RNase E maintenance of proper FtsZ/FtsA ratio required for nonfilamentous growth of Escherichia coli cells but not for colony-forming ability. J. Bacteriol. 188:5145–5152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tamura M, Kers JA, Cohen SN. 2012. Second-site suppression of RNase E essentiality by mutation of the deaD RNA helicase in Escherichia coli. J. Bacteriol. 194:1919–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chung DH, Min Z, Wang BC, Kushner SR. 2010. Single amino acid changes in the predicted RNase H domain of Escherichia coli RNase G lead to complementation of RNase E deletion mutants. RNA 16:1371–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toone WM, Rudd KE, Friesen JD. 1991. deaD, a new Escherichia coli gene encoding a presumed ATP-dependent RNA helicase, can suppress a mutation in rpsB, the gene encoding ribosomal protein S2. J. Bacteriol. 173:3291–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12:291–299 [DOI] [PubMed] [Google Scholar]
- 30. Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J. Bacteriol. 119:736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sauer U, Eikmanns BJ. 2005. The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol. Rev. 29:765–794 [DOI] [PubMed] [Google Scholar]
- 32. Chao YP, Patnaik R, Roof WD, Young RF, Liao JC. 1993. Control of gluconeogenic growth by pps and pck in Escherichia coli. J. Bacteriol. 175:6939–6944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krebs H. 1964. Gluconeogenesis. Proc. R. Soc. Lond. B Biol. Sci. 159:545–564 [DOI] [PubMed] [Google Scholar]
- 34. Spring TG, Wold F. 1971. The purification and characterization of Escherichia coli enolase. J. Biol. Chem. 246:6797–6802 [PubMed] [Google Scholar]
- 35. Callaghan AJ, Aurikko JP, Ilag LL, Gunter Grossmann J, Chandran V, Kuhnel K, Poljak L, Carpousis AJ, Robinson CB, Symmons MF, Luisi BF. 2004. Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J. Mol. Biol. 340:965–979 [DOI] [PubMed] [Google Scholar]
- 36. Ow MC, Perwez T, Kushner SR. 2003. RNase G of Escherichia coli exhibits only limited functional overlap with its essential homologue, RNase E. Mol. Microbiol. 49:607–622 [DOI] [PubMed] [Google Scholar]
- 37. Wachi M, Umitsuki G, Nagai K. 1997. Functional relationship between Escherichia coli RNase E and the CafA protein. Mol. Gen. Genet. 253:515–519 [DOI] [PubMed] [Google Scholar]
- 38. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J, Copley SD. 2007. Why metabolic enzymes are essential or nonessential for growth of Escherichia coli K12 on glucose. Biochemistry 46:12501–12511 [DOI] [PubMed] [Google Scholar]
- 40. Soupene E, van Heeswijk WC, Plumbridge J, Stewart V, Bertenthal D, Lee H, Prasad G, Paliy O, Charernnoppakul P, Kustu S. 2003. Physiological studies of Escherichia coli strain MG1655: growth defects and apparent cross-regulation of gene expression. J. Bacteriol. 185:5611–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee K, Cohen SN. 2001. Effects of 3′ terminus modifications on mRNA functional decay during in vitro protein synthesis. J. Biol. Chem. 276:23268–23274 [DOI] [PubMed] [Google Scholar]
- 42. Chang AC, Cohen SN. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 134:1141–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.