

Abstract
The orthologous penR and pntR genes from the pentalenolactone biosynthetic gene clusters of Streptomyces exfoliatus UC5319 and S. arenae TÜ469, respectively, were predicted to encode MarR/SlyA family transcriptional regulators, responsible for regulation of the biosynthesis of the sesquiterpenoid antibiotic pentalenolactone. The intrinsic target DNA sequences and small molecule ligands of purified recombinant PenR and PntR were identified by electrophoretic mobility shift assays. PenR bound to DNA from both the penR-gapN and penM-penH intergenic regions, while PntR bound only the corresponding pntR-gapR intergenic region. The targets of PenR and PntR were shown to be limited to conserved 37-bp DNA segments. Pentalenolactone and two late-stage biosynthetic intermediates, pentalenolactones D and F, act as ligands of both PenR and PntR, resulting in release of these proteins from their target DNA. The production of pentalenolactones was significantly decreased in the penR deletion mutant S. exfoliatus ΔpenR ZD27 but could be restored by complementation with either penR or pntR. Reverse transcription-PCR established that transcription of pentalenolactone biosynthetic and resistance genes decreased, while that of the penR gene itself increased in the penR deletion mutant S. exfoliatus ZD27 compared to the wild-type strain. The PenR protein thus serves as a positive regulator of pentalenolactone biosynthesis and self-resistance while acting as an autorepressor of penR.
INTRODUCTION
The MarR/SlyA family of transcriptional regulators is typified by Escherichia coli MarR, a repressor of genes that regulate multiple antibiotic resistance (Mar) and oxidative stress regulons (1, 2), and by Salmonella enterica serovar Typhimurium and E. coli SlyA, transcription regulators that are required for virulence and survival in the macrophage environment (3). Members of this family, which are widely distributed in bacteria and archaea, have been shown to regulate a wide variety of additional cellular processes, including catabolism of environmental aromatic compounds. MarR homologues are dimeric proteins harboring DNA-binding domains with a conserved winged helix-turn-helix fold that bind conserved 20- to 45-bp DNA sequences so as to regulate gene transcription either positively or negatively (4). Another common feature of MarR members is their interaction with specific anionic lipophilic ligands, such as salicylate (5), benzoate (6), uric acid (7), and nalidixic acid (8). These ligands attenuate the strength of binding of the MarR proteins with their cognate DNA targets and thereby modulate transcription. Crystal structures have been determined for several MarR regulators, including MarR itself (5), MexR (9), SlyA (10), and AdcR (11), either as apoproteins, in complex with their cognate DNA, or with their ligands, thus contributing to the elucidation of the molecular basis for transcriptional regulation by members of the MarR family.
Here, we describe the identification and characterization of the orthologous proteins PenR and PntR, new members of the MarR family that are shown to regulate biosynthesis of the antibiotic pentalenolactone in Streptomyces exfoliatus UC5319 and S. arenae TÜ469, respectively. Pentalenolactone (Fig. 1) is a widely occurring sesquiterpene metabolite that has been isolated from more than 30 species of Streptomyces (12–15). Its antibiotic action against bacteria, fungi, and protozoa is due to the presence of an electrophilic epoxylactone moiety that alkylates the active-site cysteine of the target glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (16–18). We have previously identified two homologous pentalenolactone biosynthetic gene clusters, the pen cluster from S. exfoliatus UC5319 (19) and the pnt cluster from S. arenae TÜ469 (19), and established the detailed biochemical role of each of the biosynthetic enzymes encoded by both clusters (20) (Fig. 1 and 2). The first committed step in the biosynthesis of pentalenolactone is the cyclization of the universal acyclic precursor farnesyl diphosphate (FPP) to pentalenene (21, 22). Six redox enzymes—the P450 PenI or PntI, the non-heme iron-, α-ketoglutarate-dependent dioxygenase PenH or PntH, the dehydrogenase PenF or PntF, the flavin-dependent Baeyer-Villiger monooxygenase PenE or PntE, the α-ketoglutarate-dependent dioxygenase PenD or PntD, and the P450 PenM or PntM—are then responsible for the multistep oxidative conversion of pentalenene to pentalenolactone in S. exfoliatus and S. arenae, respectively (19, 23–28). The biosynthesis of the isomeric metabolite neopentalenolactone in S. avermitilis is controlled by the closely related ptl gene cluster (20, 23, 29).
Fig 1.

Biosynthesis of pentalenolactone. The cyclization of farnesyl diphosphate to the tricyclic sesquiterpene hydrocarbon pentalenene and the oxidative conversion of pentalenene to pentalenolactone catalyzed by enzymes from the pentalenolactone producers S. exfoliatus UC5319 (designated as Pen proteins) or S. arenae TÜ469 (Pnt proteins) are depicted. See Fig. 2 for a description of the corresponding pen and pnt biosynthetic gene clusters.
Fig 2.

Pentalenolactone (PL) biosynthetic gene clusters and a model of regulation of PL biosynthesis. pen, PL biosynthetic gene cluster from S. exfoliatus UC5319; pnt, PL biosynthetic gene cluster from S. arenae TÜ469. Numbers indicate the sizes (bp) of intergenic regions. Other diagram items are defined as follows: arrow shapes, genes; wavy lines, mRNA; ovals, proteins; black arrows and shaded ovals, genes with TTA codons and their encoded proteins, respectively; X in an ellipse, other proteins involved in PL biosynthesis such as flavodoxin and flavodoxin reductase; lines with a single arrow, biochemical reactions or life process; lines with double arrowheads, positive regulation; lines with a short bar, negative regulation; lines with a triangle, ligands of regulators. All dotted lines and shapes are predicted relationships without direct experimental data in S. exfoliatus. PenR encoded by gene penR activates the transcription of the gapN-penMHGFEDBAI and penHGFEDBAI operons and represses the transcription of penR itself. PntR binds only in the pntR-gapR intergenic region and therefore is presumed to act as an autorepressor of pntR and activator of the single gapR-pntMHGFEDBAI biosynthetic operon, by analogy to penR. Pentalenolactone and late-stage biosynthetic intermediates at sufficiently high intracellular concentrations (>10 μM) can act as ligands of PenR or PntR to relieve DNA binding and thereby attenuate the activation or repression. PL biosynthesis may also be controlled by BldA at the translational level.
Very little is known about the regulation of terpenoid metabolism in bacteria. Both the S. exfoliatus and S. arenae biosynthetic clusters harbor orthologous resistance genes, gapN and gapR, respectively, that are located at the 5′ ends of each unidirectionally transcribed cluster and encode inducible, pentalenolactone-insensitive forms of GAPDH (19, 29–31). PenR from the pen gene cluster and PntR from the pnt gene cluster, as well as the homologous SAV_2989 from the closely related ptl gene cluster, represent three apparent MarR family transcriptional regulators whose parent genes are adjacent to and divergently transcribed from the GAPDH resistance genes in each biosynthetic gene cluster (Fig. 2). We have now expressed and purified recombinant PenR and PntR and established that each regulator binds to specific intergenic promoter regions within their respective biosynthetic operons. Pentalenolactone and other late stage biosynthetic intermediates can serve as ligands of both PenR and PntR that attenuate this binding by releasing the protein from its target DNA. Pentalenolactone biosynthesis is abolished in a penR deletion mutant but can be restored by complementation with penR or the homologous pntR. The PenR and PntR proteins also act as autorepressors of transcription of their parent genes.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Streptomyces and E. coli strains, plasmids, and cosmids are listed in Table S1 in the supplemental material. Primer sequences are listed in Table S2 in the supplemental material.
Materials.
Reagents and solvents purchased from Sigma-Aldrich or Fisher Scientific were of the highest quality available and were used without further purification. Restriction enzymes and T4 DNA ligase were purchased from New England BioLabs and used according to the manufacturer's specifications. IPTG (isopropyl-β-d-thiogalactopyranoside) was purchased from Invitrogen. Ni-NTA affinity resin was purchased from Qiagen. Amicon ultracentrifugal filter units (Amicon Ultra-15; 10,000 molecular weight cutoff) were purchased from Millipore. DNA primers were synthesized by Integrated DNA Technologies.
Methods.
The growth media and conditions used for E. coli and Streptomyces strains and standard methods for handling E. coli and Streptomyces in vivo and in vitro were as described previously (32, 33), unless otherwise noted. All DNA manipulations were performed following standard procedures (33). DNA sequencing was carried out at the U. C. Davis Sequencing Facility, Davis, CA. All proteins were handled at 4°C unless otherwise stated. Protein concentrations were determined according to the method of Bradford, using a Hewlett-Packard 8452A diode array UV/vis spectrophotometer with bovine serum albumin as the standard (34). Protein purity was estimated using SDS-PAGE and visualized using Coomassie brilliant blue stain. Gas chromatography-mass spectrometry (GC-MS) analyses were carried out on a Hewlett-Packard Series 2 GC-MSD at 70 eV electron impact (EI) operating in positive-ion mode, using a HP5MS capillary column (30 m by 0.25 mm) with a solvent delay of 3 min and a temperature program of 60°C for 2 min, followed by a temperature gradient of 60 to 280°C for 11 min at 20°C/min and a hold at 280°C for 2 min. Liquid chromatography-electrospray ionization-MS (LC-ESI-MS) analysis of recombinant proteins was carried out on a Thermo LXQ LC-ESI-MS equipped with Surveyor high-pressure liquid chromatography system and a Waters Symmetry C18 column (2.1 mm by 50 mm, 3.5 μm).
Expression and purification of recombinant PenR and PntR proteins.
Primers DQ108F/R and DQ109F/R were used to amplify pntR and penR, respectively, from cosmids G21 and 1E2 of the previously described S. exfoliatus and S. arenae genomic libraries (see Table S1 in the supplemental material) (19). The resultant amplicons were used for expression of PenR and PntR in E. coli. The individual 483-bp PCR products were each digested with NdeI and HindIII and then ligated into doubly digested pET-26b to give the expression plasmids pDQ71 and pDQ72, respectively, that were then individually introduced into E. coli BL21(DE3) by transformation. Each of the recombinant E. coli transformants was cultured in Luria-Bertani media containing 50 μg of kanamycin/ml at 37°C to an optical density at 600 nm of 0.6 to 0.8. After the addition of 0.4 mM IPTG, the culture was further incubated at 18°C overnight. The harvested cells were resuspended in lysis buffer (50 mM Tris-HCl, 10% glycerol, 300 mM NaCl, 0.1 mM dithiothreitol [DTT], 2.7 mM β-mercaptoethanol, 10 mM imidazole [pH 8.0]) containing 10 mg pepstatin/liter, 10 mg of phenylmethylsulfonyl fluoride/liter, and 0.2 mg of benzamidine/ml. After sonication, the cell debris was removed by centrifugation at 20,000 × g for 30 min, and the supernatant was loaded into a Ni-NTA column preequilibrated with lysis buffer. After the flowthrough was collected and washed with 20 mM imidazole in lysis buffer at a flow rate of 2 ml/min, PenR and PntR proteins were eluted with elution buffer (50 mM Tris-HCl, 10% glycerol, 300 mM NaCl, 0.1 mM DTT, 2.7 mM β-mercaptoethanol, 200 mM imidazole [pH 8.0]) at a flow rate of 1 ml/min. The fractions were analyzed by SDS-PAGE. The PenR and PntR proteins were concentrated and buffer-exchanged into storage buffer (50 mM NaH2PO4, 0.1 mM DTT [pH 7.0]) using an Amicon Ultra Centrifugal filter (Ultracel-10K) and a PD-10 gel filtration column. The yield of purified recombinant PenR was 46 mg/liter of culture, and the yield of purified PntR was 13 mg/liter of culture. ESI-MS data were as follows: PenR-His6 tag, m/z 18,469 (predicted for the full-length protein minus the N-terminal Met, m/z 18,464), and PntR-His6 tag, m/z 18,489 (predicted, m/z 18,484).
Isolation and purification of pentalenolactones.
Pentalenolactone D methyl ester (PL-D-Me) was isolated from S. exfoliatus penD mutant ZD20 as described previously (19). Pentalenolactone F methyl ester (PL-F-Me) was isolated from S. exfoliatus penM mutant ZD22 as described previously (24). Pentalenolactone methyl ester (PL-Me) was isolated from S. arenae TÜ469. The liquid medium used in fermentation of S. exfoliatus ZD20 and ZD22 contained 0.2% NaCl, 0.5% CaCO3, 1% corn gluten meal, 0.1125% Bacto dextrose, 0.2% blackstrap molasses, and 2% corn starch (pH 7.2) (35). The liquid medium used in fermentation of S. arenae TÜ469 contained 4% mannitol, 0.25% l-asparagine, 0.2% (NH4)2SO4, 0.1% NaCl, 0.3% K2HPO4·3H2O, 0.1% MgSO4·7H2O, 0.04% CaCl2·2H2O, 0.002% FeSO4·7H2O, and 0.001% ZnSO4·7H2O (pH 6.2) (13). After incubation at 30°C for 6 days, the cultures were acidified to pH 2.4 with H2SO4 or HCl and extracted with chloroform. The organic layer was dried over anhydrous Na2SO4, concentrated, and methylated with TMS-CHN2. The resulting mixtures of methyl esters were separated by flash column chromatography (silica gel, pentane-ethyl acetate, 10:1) (35, 36) to give purified pentalenolactone D methyl ester (12 mg/liter of culture), pentalenolactone F methyl ester (16 mg/liter of culture), and pentalenolactone methyl ester (2.1 mg/liter of culture), respectively. To obtain the corresponding free acids, each methyl ester was individually dissolved in 2.0 ml of tetrahydrofuran and 0.5 ml of water and then treated at 0°C with 52 μl of 2 N aqueous potassium hydroxide solution (0.10 mmol), followed by hydrolysis for 30 min to 4 h at room temperature until the corresponding methyl ester could no longer be detected by GC-MS. The solutions were acidified to pH 2.4 and extracted with chloroform. The combined organic layers were dried over anhydrous Na2SO4, concentrated in vacuo, and chromatographed on silica gel (chloroform-ethanol, 20:1) (37). The corresponding purified free acids were obtained in 25 to 50% yield.
Electrophoretic mobility shift assay (EMSA) of PenR and PntR binding to DNA and the effect of small molecule ligands on PenR-DNA and PntR-DNA complexes.
The binding of PenR or PntR to their putative target DNA regions was monitored by EMSA using the methods previously described (38, 39). Purified PntR or PenR was incubated with DNA fragments in a total volume of 20 μl at 30°C for 30 min. The binding buffer contained 20 mM Tris-HCl (pH 8.0), 50 mM KCl, 10 mM MgCl2, 5% glycerol, and 0.5 mM EDTA. DTT (1 mM) was added to the reaction mixture just before incubation. A 299-bp DNA fragment of hrdB, amplified with the consensus primer pair DQ110F and DQ110R by reverse-transcription PCR (RT-PCR), was used as a negative control. Primers used to amplify putative target region DNA fragments are listed in Table S2 in the supplemental material. The concentration of the double-stranded DNA fragment was fixed, while the concentration of protein was varied. The samples were applied to 2% agarose gel or 5% Tris-borate-EDTA nondenaturing polyacrylamide gel (Bio-Rad) and electrophoresed on ice. The gels were stained with ethidium bromide and imaged.
For the determination of apparent Kd (dissociation constant) values for PntR and PenR, increasing concentrations of PenR or PntR dimer from 0.5 to 250 nM were incubated for 30 min at 30°C with ca. 80 ng of DNA harboring the PenR or PntR target DNA regions. The bands of the EMSA gels were quantified using a Bio-Rad FX Plus molecular imager, and the percentage of shifted DNA was calculated. These values were plotted against the initial PenR or PntR dimer concentrations and the sigmoidal curves fit to the Hill equation for cooperative binding, F = Fmin + (Fmax − Fmin)[PenR]n/(Kdn + [PenR]n) and F = Fmin + (Fmax − Fmin)[PntR]n/(Kdn + [PntR]n), where F is the observed percent shifted DNA, calculated using Origin 8 software (Origin Lab Corp., Northampton, MA). Reported standard deviations in calculated Kd values represent the calculated statistical errors in the nonlinear, least-squares regression analysis generated by the Origin 8 package. No corrections were made for deviations between the initial and free protein concentrations at low overall concentrations of protein, which may result is a small overestimation of the actual value of the dissociation constant, Kd. All determinations were performed in triplicate.
The effect of pentalenolactone, pentalenolactone D, and pentalenolactone F on protein-DNA binding was also analyzed by EMSA. Purified PntR and PenR proteins (0.36 μM) were preincubated in binding buffer with their target DNA fragments for 15 min at 30°C, followed by the addition of individual pentalenolactone metabolites (18 μM) and further incubation for 15 min. Salicylate was used as a negative control. The samples were applied to 2% agarose gels and electrophoresed on ice. The gels were stained with ethidium bromide and imaged (see Fig. S5 in the supplemental material). For the determination of the apparent S0.5 values, the concentration of pentalenolactone at which 50% of the bound DNA ligand is released from its complex with PenR or PntR, increasing concentrations of pentalenolactone were incubated for 15 min at 30°C with preformed DNA-protein complex (ca. 80 ng of target DNA and 0.36 μM PenR or PntR homodimer). The bands of the EMSA gels were quantified as described above, and the percentage of shifted DNA was calculated. These values were plotted against pentalenolactone concentration and fit by nonlinear least-squares regression to the sigmoidal Hill equation, F = Fmin + (Fmax − Fmin)[PL]n/(S0.5n + [PL]n), as described above. The reported standard deviations in calculated S0.5 values represent the calculated statistical errors in the nonlinear, least-squares regression analysis generated by the Origin 8 package. All determinations were performed three times.
Construction of penR mutant S. exfoliatus ZD27 and penM-penR double mutant S. exfoliatus ZD28.
The method of PCR targeting in Streptomyces was used to generate penR mutants (40) (see Fig. S6 in the supplemental material). The ca. 6.7-kb BamHI DNA fragment, including the region immediately upstream of the pen gene cluster, through penR and gapN to partial penM, was recycled from cosmid G21 of the S. exfoliatus UC5319 genomic library (19) and inserted into the BamHI site of vector pDQ44 (19) to generate plasmid pDQ93. pDQ93 was transformed into E. coli BW25117/pIJ790. The 1,382-bp EcoRI/HindIII DNA fragment from pIJ773 carrying oriT and aac(3)IV was used as a template for PCR amplification with the primer pair DQ92F2 and DQ92R to give a 1,447-bp DNA fragment that was transformed into E. coli BW25113/pIJ790 containing pDQ93 to generate plasmid pDQ94 in which an internal 198 bp of penR from nucleotide (nt) 223 to 420 had been replaced by the 1,369-bp oriT and aac(3)IV. Plasmid pDQ94 was conjugated into both S. exfoliatus UC5319 and the S. exfoliatus penM mutant ZD22 (24). Apramycin (50 μg/ml) was used to select exconjugants. The double-crossover strains that had lost thiostrepton resistance were confirmed by PCR using primer pair DQ93F and DQ93R. The PCR products of the wild-type S. exfoliatus strain UC5319 and penM mutant ZD22 were 657 bp, while the PCR products of penR mutant ZD27 and penM-penR double mutant ZD28 were both 1,828 bp in which an internal 198 bp of penR in S. exfoliatus UC5319 and ZD22 from nt 223 to 420 had been replaced by the 1,369-bp fragment harboring oriT and aac(3)IV (see Fig. S6 in the supplemental material).
Complementation of penR mutant S. exfoliatus ZD27 and penM-penR double mutant S. exfoliatus ZD28 with penR or pntR.
Plasmid pHZ1358 (41) carrying the thiostrepton resistance gene tsr was used as a template for PCR amplification with the primer pair DQ113F and DQ113R to give a 1,072-bp DNA fragment harboring the tsr gene that was digested with EcoRI and inserted into the EcoRI site of pIB139 (42) to generate plasmid pDQ77. Primers DQ109R and DQ112R were used to amplify a DNA fragment of penR and its native promoter ca. 823 bp from the stop codon of penR to the start codon of gapN. The PCR product was inserted into the SmaI site of pIJ2925 (43) to generate plasmid pDQ88, which was sequenced to verify the integrity of the insert. pDQ88 was digested with BglII, and a 823-bp DNA fragment carrying penR was recycled and inserted into the BamHI site of pDQ77 to generate plasmid pDQ90. A 1,728-bp SmaI DNA fragment harboring partial orf-1, complete pntR, and partial gapR was recycled from pDQ20 (19) and inserted into pIJ2925 to generate plasmid pDQ89. pDQ89 was digested with BglII and the DNA fragment carrying pntR was recycled and inserted into the BamHI site of pDQ77 to generate plasmid pDQ91. pDQ90 and pDQ91 were each then conjugated into both the penR mutant S. exfoliatus ZD27 and the penM-penR double-mutant S. exfoliatus ZD28. The complementation strains S. exfoliatus ZD27::pDQ90 and ZD28::pDQ90 were confirmed by PCR using primer pair DQ109F and DQ109R that gave amplicons of 483 bp (not shown). The complementation strains S. exfoliatus ZD27::pDQ91 and ZD28::pDQ91were confirmed by PCR using primer pair DQ108F and DQ108R to give the expected 483-bp amplicons (data not shown).
GC-MS analysis of pentalenolactone production.
The liquid medium used in fermentation of S. exfoliatus UC5319, penR mutant ZD27, complementation strains S. exfoliatus ZD27::pDQ90 and ZD27::pDQ91, penM mutant S. exfoliatus ZD22, penM-penR double mutant ZD28, and complementation strains S. exfoliatus ZD28::pDQ90 and ZD28::pDQ91 was as previously described (35). After incubation at 30°C for 6 days, the cultures were acidified to pH 2.4 with H2SO4 and extracted with chloroform. The organic layer was dried over anhydrous Na2SO4, concentrated, methylated with TMS-CHN2, and analyzed by GC-MS. The identity of each of the resultant pentalenolactone metabolites was confirmed by direct comparison of retention time and mass spectra with pure authentic standards.
RT-PCR comparison of transcription levels of selected pen genes.
To compare time-dependent levels of transcription of selected genes from the pen cluster, total RNA was isolated using the RNeasy minikit (Qiagen, catalog no. 74104) from S. exfoliatus UC5319 and penR mutant S. exfoliatus ZD27 cultured in fermentation medium at 30°C, with samples drawn at 1-day intervals from days 2 through 5. The RNase-free DNase set (Qiagen, catalog no. 79254) was used to digest and remove DNA from RNA samples. The concentration of RNA was determined by measuring the UV absorbance at 260 nm, and sample pairs from the wild-type and mutant strains obtained on the same day were adjusted to identical concentrations of total RNA. A Qiagen OneStep RT-PCR kit (catalog no. 210210) was used with an initial incubation of 30 min at 50°C and 15 min at 95°C for the reverse transcriptase step, followed by 30 cycles of PCR amplification (30 s at 94°C, 30 s at 55°C, and 30 s at 72°C). The oligonucleotides used in each RT-PCR experiment are listed in Table S2 in the supplemental material. Control incubations (not shown), in which the reverse transcriptase step was omitted did not give any PCR-amplified product, thereby confirming that the observed RT-PCR products resulted exclusively from mRNA and not from residual gDNA contamination. All RT-PCR experiments were conducted in triplicate. As an internal reference for RT-PCR, a 299-bp fragment of S. exfoliatus hrdB was amplified using the primer pair, DQ110F and DQ110R, which was designed using the consensus DNA sequence of the known hrdB genes from three Streptomyces strains, scab24441 of S. scabiei, sgr1701 of S. griseus, and sco5820 of S. coelicolor. Since these consensus primers were not a perfect match for the actual target S. exfoliatus hrdB sequence, the resultant RT-PCR amplification levels are somewhat lower than that expected for an exact sequence match, but nonetheless showed no variation between wild-type and mutant S. exfoliatus samples at each time point sampled.
XylE (catechol 2, 3-dioxygenase) promoter probes.
The tsr gene of plasmid pHZ1358 was amplified by PCR using the primer pair DQ113F and DQ113R. The resultant 1,072-bp DNA fragment was digested with EcoRI and inserted into the EcoRI site of pHL133 carrying the xylE gene (44) to generate plasmid pDQ78. Cosmid G21 carrying the pen gene cluster was used as a template for PCR amplification with the primer pair DQ103F2 and DQ103R2 to give a 473-bp DNA product harboring a part of penM and the penM-penH intergenic region that was then inserted into the SmaI site of pIJ2925 to generate plasmid pDQ75. pDQ75 was digested with BglII and the penM-penH intergenic region was recycled and inserted into the BglII site of vector pDQ78 in the direction from penM to penH to generate plasmid pDQ80, which was then conjugated into S. exfoliatus UC5319.
A 548-bp DNA fragment carrying a part of penR and the penR-gapN intergenic region was amplified from cosmid G21 using the primer pair DQ84F and DQ112R and inserted into the SmaI site of vector pIJ2925 to generate plasmid pDQ108. pDQ108 was digested with BamHI, and the ∼540-bp BamHI DNA fragment was recycled and inserted into the BglII site of pDQ78 to generate plasmid pDQ109 with the penR-gapN intergenic region in the penR direction and plasmid pDQ110 with the penR-gapN intergenic region in the gapN direction, each of which was conjugated into S. exfoliatus UC5319.
S. exfoliatus UC5319::pDQ110, UC5319::pDQ109, and UC5319::pDQ80 and the control strain S. exfoliatus UC5319::pDQ78 lacking a promoter insert were each grown in fermentation medium at 30°C for 8 days. Samples were withdrawn each day, and the derived cell extracts were analyzed for catechol 2,3-dioxygenase activity, as previously described (32). Mycelium was harvested by centrifugation and rinsed with wash buffer (20 mM potassium phosphate [pH 7.5]). The washed mycelium was then suspended in cold sample buffer (100 mM phosphate buffer [pH 7.5], 20 mM sodium-EDTA [pH 8.0], 10% [vol/vol] acetone) and sonicated on ice. After the addition of 10 μl of 10% aqueous Triton X-100 solution per 1-ml sample to give 0.1% final concentration, the solution was kept on ice for 15 min and then centrifuged, and the supernatant was transferred to a fresh tube. A 50-μl portion was added to 0.5 ml of assay buffer (10 mM phosphate buffer [pH 7.5], 0.2 mM catechol) and 20 mM catechol stock solution prepared in ethanol and stored at −20°C, and the change in A375 was monitored. The slope of the linear part of the spectrophotometric output was used calculate specific activity based mU catechol dioxygenase (nmol/min = 30.03 × ΔA375/min). The protein content of each sample was determined by the Bradford assay. All activity determinations for each strain were based on analysis of three independent exconjugants.
RESULTS
Pentalenolactone biosynthetic gene clusters in Streptomyces harbor MarR/SlyA family transcriptional regulator genes.
Analysis of the two characterized Streptomyces pentalenolactone biosynthetic gene clusters has revealed that the farthest upstream, divergently transcribed genes in each cluster, designated penR and pntR, respectively, encode MarR-family transcriptional regulators (Fig. 2) with strong pairwise amino acid sequence identity (73 to 81%) and similarity (86 to 87%) over the full 151- to 153-amino-acid length (Fig. 3). The orthologous SAV_2989 protein from the closely related ptl neopentalenolactone biosynthetic gene cluster also shows comparable levels of identity and similarity. To investigate the function of the PenR and PntR proteins, the penR and pntR genes were each amplified by PCR, inserted into pET26b, and expressed in E. coli BL21(DE3) to give recombinant PenR and PntR, each carrying a C-terminal His6 tag. Each protein, which was purified to >95% purity on Ni2+-NTA resins (see Fig. S1 in the supplemental material), exhibited the predicted molecular mass MD upon ESI-MS analysis.
Fig 3.

Multiple sequence alignment of three homologous MarR family members, PenR from the pen gene cluster of S. exfoliatus, PntR from the pnt gene cluster of S. arenae, and SAV_2989 from the ptl (neopentalenolactone) gene cluster of S. avermitilis. The multiple protein sequence alignment was generated using BioEdit and secondary structures predicted using Phyre2 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index). α-Helices are represented as green boxes, and β-strands are represented as blue arrows. The region from residues 65 to 100 encompassing the α3 and α4 helices and the β1 and β2 strands corresponds to the established DNA-binding winged helix-turn-helix fold of MarR and related proteins.
Identification of target DNA regions bound by the PenR or PntR proteins.
Many transcriptional regulators are encoded adjacent to divergently transcribed genes that they regulate, with their binding sites residing in the intergenic region between the two genes (45, 46). Indeed, EMSAs using purified PenR protein and a 380-bp DNA fragment carrying the 358-bp penR-gapN intergenic region, obtained by PCR amplification using the primer pair DQ112F/DQ112R (see Table S2 in the supplemental material), showed concentration-dependent protein-DNA binding (Fig. 4A). Similarly, purified PntR also showed binding in EMSA experiments to a 162-bp DNA fragment harboring the 140-bp pntR-gapR intergenic region (Fig. 4B) that was obtained by PCR amplification using the primer pair DQ111F/DQ111R (see Table S2 in the supplemental material). In negative control experiments, neither PenR nor PntR bound to a nonspecific 299-bp DNA fragment obtained by PCR-amplification of an internal region of S. arenae TÜ469 hrdB (Fig. 4A-2 and B-2). In order to identify any additional targets for either PenR or PntR, each of the remaining intergenic regions from within both the pen and pnt biosynthetic gene clusters was individually amplified by PCR and analyzed by EMSA in combination with PenR or PntR. Unexpectedly, PenR also bound a second DNA fragment carrying the 260-bp penM-penH intergenic region. PenR did not bind to any other intergenic DNA fragments from the pen cluster, thus revealing that PenR has two target regions within the pen biosynthetic gene cluster (Fig. 4C). Interestingly, PntR did not bind to any intergenic region from the pnt cluster other than the above-mentioned pntR-gapR intergenic DNA, including the pntM-pntH intergenic segment.
Fig 4.

EMSA of DNA binding by PenR (A) and PntR (B). (A-1) PenR and 380-bp DNA fragment from the penR-gapN intergenic region; (A-2) negative control (PenR and 299-bp DNA fragment from the hrdB gene). (B-1) PntR and 162-bp DNA fragment from the pntR-gapR intergenic region; (B-2) PntR and 299-bp DNA fragment from hrdB. (C) PntR and PenR with additional intergenic regions of the pnt and pen gene clusters.
To refine further the identity of the actual DNA targets for PenR and for PntR, synthetic PCR primers were used to amplify different portions of the penR-gapN, penM-penH, and pntR-gapR intergenic regions (see Table S2 in the supplemental material). In this manner, EMSA experiments identified ca. 90-bp DNA tracts (see Fig. S2A, B, and C in the supplemental material) within each of the three intergenic regions that bound specifically to PenR or PntR, with no detectable binding to the remaining portions of the parent intergenic regions. Alignment of these three 90-bp DNA sequences revealed a highly conserved 34-bp DNA segment that was predicted to be the actual target of PenR and PntR (Fig. 5). These predictions were tested by Sau3AI digestion of the 380-bp PCR product carrying the 358-bp penR-gapN intergenic region to obtain two fragments, a 42-bp DNA segment harboring the predicted target of PenR, corresponding to a 34-bp segment of the penR-gapN intergenic region and the 3-bp start codon of penR, plus a second 324-bp DNA fragment. EMSA experiments confirmed that the 42-bp DNA band was retarded by increasing concentrations of PenR, while the mobility of the 324-bp DNA band was unchanged, thereby confirming specific binding of PenR only to the 42-bp fragment. Similarly, Sau3AI digestion of the PCR product carrying the 140-bp pntR-gapR intergenic region gave a an ∼42-bp DNA fragment harboring the predicted target of PntR, plus a second 110-bp fragment. In EMSA experiments, addition of PntR shifted the 42-bp DNA fragment, while the mobility of the 110-bp DNA band was unaffected. The apparent DNA-binding motifs for both PenR and PntR were thus narrowed to similar 37-bp regions within their respective intergenic binding partners. Interestingly, a similar 37-bp motif is also evident in the PenR-binding penM-penH intergenic region, as well as in the closely related sav2989-gap1 intergenic region, the predicted binding site for SAV_2989 in the ptl biosynthetic cluster. The four consensus 37-bp DNA sequences harbor two strongly conserved motifs represented by GAAAT(A/G)(T/C)ATCG and CTTAT(A/G)TA(A/G)GCT, the second of which displays a short and imperfect inverse repeat, CTTATATAAG (Fig. 5). Notably, the consensus binding motif is absent from all of the remaining intergenic regions within both the pen and the pnt clusters, as well as from all of the protein coding regions of both clusters.
Fig 5.

Multiple DNA sequence alignment of consensus binding sites of PenR, PntR, and SAV_2989 proteins. (A) Target DNA sequences of PenR and PntR and that predicted for the orthologous SAV_2989 protein from S. avermitilis, generated using Multalin (http://multalin.toulouse.inra.fr/multalin/multalin.html), showing GATCs in a dotted square (i.e., the Sau3AI sites): the CAT in a dotted square indicates the start codon of reverse gene penR, and the CAC in a dotted square indicates the start codon of the reverse gene pntR. The CTTATATAAG sequence with arrows indicates an inverted repeat. (B) Consensus DNA sequence logo (WebLogo [http://weblogo.berkeley.edu/logo.cgi]).
We next tested the relative binding affinity of PenR for each of its two distinct intergenic targets. Exploratory EMSA experiments using variable concentrations of protein revealed that the free DNA band from the penM-penH intergenic region was shifted by lower concentrations of PenR homodimer compared to the free DNA band from the penR-gapN intergenic region (see Fig. S3A and B in the supplemental material). Similarly, in direct competition experiments using a mixture of both DNA segments, the free DNA band for the penR-gapN intergenic region began to shift only at concentrations of PenR above those at which the penM-penH intergenic DNA was almost completely shifted (see Fig. S3C in the supplemental material). The apparent Kd for binding of the PenR homodimer to the penR-gapN target was determined by measuring the percentage of shifted DNA in the presence of varied concentrations (0.5 to 250 nM) of PenR protein. Fitting of the resulting sigmoidal binding plots to the Hill equation for cooperative binding gave a Kd of 31 ± 1 nM for binding of PenR to the penR-gapN target, nearly 2-fold higher than the value of 16 ± 1 nM determined for the somewhat stronger binding of PenR to the alternative penM-penH target. The corresponding apparent Kd value for binding of the PntR dimer to its cognate pntR-gapR DNA target was 9 ± 1 nM (Fig. 6). Each of the three measured Kd values for PenR- or PntR-DNA binding is within the reported low nanomolar range typical of MarR-DNA interactions (4), while the absence of detectable binding to regions other than the identified 42-bp tracts underscores the specificity of the observed protein-DNA interactions.
Fig 6.

Determination of Kd values for binding of PntR and PenR proteins to cognate DNA. Increasing concentrations of PntR and PenR were individually incubated with DNA fragments representing the one pnt intergenic region and the two pen intergenic binding regions, respectively. (A) Binding of PntR to the pntR-gapR intergenic region; (B) binding of PenR to the penR-gapN intergenic region; (C) binding of PenR to the penM-penH intergenic region. The error bars represent the means of three replicates. See Materials and Methods for details regarding data analysis and see Fig. S3 in the supplemental material for EMSA images.
Promoter probe assays of the PenR-binding intergenic DNA regions.
The specific binding of PenR to a 37-bp segment of the penR-gapN intergenic region immediately adjacent to the penR start codon raised the possibility that this intergenic region might harbor a bidirectional promoter allowing PenR to regulate transcription of both divergently transcribed flanking genes, penR and gapN (4) (Fig. 2). To investigate the predicted presence of promoters within each of the two penR-binding intergenic regions, we constructed promoter probes using catechol 2,3-dioxygenase activity as the reporter. Thus, the penR-gapN intergenic region was amplified by PCR and inserted in opposite directions into separate vector constructs upstream of xylE to generate plasmids pDQ109 (corresponding to the putative promoter of penR) and pDQ110 (corresponding to the promoter of gapN). Similarly, the penM-penH intergenic region was amplified and inserted upstream of xylE to give plasmid pDQ80. Each of the three plasmids was then conjugated into wild-type S. exfoliatus UC5319. The activity of catechol 2,3-dioxygenase in S. exfoliatus UC5319::pDQ80, harboring an integrated copy of the penM-penH intergenic region upstream of xylE, reached 15 to 30 mU/mg of protein on days 2 to 4 (Fig. 7). The catechol dioxygenase of S. exfoliatus UC5319::pDQ110, harboring the predicted promoter for gapN, reached a maximum of ca. 4 to 15 mU/mg of protein on day 4. In contrast, the catechol 2,3-dioxygenase activity of S. exfoliatus UC5319::pDQ109, harboring the promoter for penR, remained at a low but detectable levels (≤2 mU/mg of protein) throughout the fermentation period. The control strain, S. exfoliatus UC5319::pDQ78, which carried the same xylE construct without any promoter insert, showed no catechol dioxygenase activity throughout the 8-day test period. Expression of the S. exfoliatus pentalenolactone biosynthetic pen operon therefore appears to be under the control of two alternative promoters, both upregulated by PenR, one of which is located upstream of gapN and controls expression of both the resistance gene gapN and the entire set of structural genes of the biosynthetic pathway, and a second, downstream site under the control of a somewhat stronger promoter that is located adjacent to penH and that controls all of the biosynthetic genes save that for PenM, the P450 that catalyzes the final oxidative rearrangement step in the pathway. At the same time, PenR acts as an autorepressor of transcription in the opposite direction within the penR-gapN intergenic region, thereby reducing the transcription of its parent gene, penR.
Fig 7.

Catechol 2,3-dioxygenase activity in cell extracts of promoter probe strains. Symbols: black squares and line, S. exfoliatus UC5319::pDQ80, promoter of penH; red circles and line, S. exfoliatus UC5319::pDQ110, promoter of gapN; blue triangles and line, S. exfoliatus UC5319::pDQ109, promoter of penR; inverted blue triangles, control strain S. exfoliatus UC5319::pDQ78, lacking promoter insert upstream of xylE.
Small-molecule ligands of PenR and PntR.
The combined results strongly suggest that PenR, and presumably its S arenae orthologue PntR, is a positive activator of both pentalenolactone biosynthesis and self-resistance, while also acting as an autorepressor of its own expression, analogous to the observed mode of action of other MarR transcriptional regulators. Metabolites of biosynthetic pathways encoded by genes that are subject to transcriptional activation can often serve as native ligands for the transcriptional activator proteins themselves (45). In fact, EMSA experiments confirmed that addition of purified pentalenolactone released both PenR and PntR from their respective bound DNA targets (Fig. 8). The concentration of pentalenolactone required for release of 50% of target DNA from its complex with either PenR or PntR, defined as the apparent S0.5 constant, was determined by incubating increasing concentrations of pentalenolactone (0 to 95 μM) with preformed protein-DNA complexes. The resulting plots of the percent bound DNA versus the pentalenolactone concentration (Fig. 8) were fit to the standard binding equation to give an S0.5 of 8 ± 7 μM pentalenolactone for the release of PenR bound to its penR-gapN intergenic target DNA, an S0.5 of 8 ± 7 μM for the release of PenR from its penM-penH intergenic target, and an S0.5 of 6 ± 2 μM for the release of PntR from binding its pntR-gapR intergenic target. Interestingly, these values are all within an order of magnitude of the typical 5 to 50 μM extracellular concentrations of pentalenolactone that accumulate in the fermentation broth of cultures of S. exfoliatus and S. arenae over the usual 5- to 7-day fermentation period.
Fig 8.

Effect of pentalenolactone on binding of PenR and PntR to their cognate DNA binding segments. (A) Release of binding of PenR to the penR-gapN intergenic region with increasing concentration of pentalenolactone; (B) release of binding of PntR to the pntR-gapR intergenic region with increasing concentration of pentalenolactone; (C) release of binding of PenR to the penM-penH intergenic region with increasing concentration of pentalenolactone. The error bars represent the means of three replicates. See Materials and Methods for experimental details and the methods of data analysis for calculation of S0.5, the concentration of pentalenolactone at which 50% of bound DNA is released.
Additional EMSA experiments also established that pentalenolactone F, the penultimate intermediate in the pentalenolactone biosynthetic pathway, as well as its precursor, pentalenolactone D, were each able to reverse completely binding of either PenR and PntR to each of their cognate DNA binding targets (see Fig. S4 in the supplemental material). Although pentalenolactone F is expected to have GAPDH inhibitory activity, based on its characteristic epoxylactone pharmacophore, the biosynthetic intermediate pentalenolactone D carrying an α-methyl lactone is antibiotically inactive. Control experiments also showed that salicylate, a typical anionic lipophilic ligand of many MarR family proteins, had no effect on DNA binding by either PenR or PntR, thereby confirming that the inhibitory effect of the pentalenolactones on binding of both PenR and PntR to their cognate DNA partners is metabolite specific.
penR deletion mutant S. exfoliatus ZD27 and penM-penR double deletion mutant S. exfoliatus ZD28.
To corroborate the above-described regulatory model, we examined the effect on pentalenolactone production of deleting the penR gene from both the wild-type strain S. exfoliatus UC5319 and the previously described penM mutant S. exfoliatus ZD22 (24), which is blocked in the final P450-catalyzed oxidative rearrangement of pentalenolactone F to pentalenolactone. To construct the desired penR mutants, an internal 198 bp of penR from nt 223 to 420 was replaced by the 1,369-bp oriT and aac(3)IV in both the wild-type and mutant hosts so as to generate penR mutant S. exfoliatus ZD27 and the penM-penR double-mutant S. exfoliatus ZD28 (see Fig. S5 in the supplemental material). Plasmids pDQ90, carrying penR, and pDQ91, carrying pntR, both under the control of their native promoters, were then each conjugated back into each of the two penR mutants, S. exfoliatus ZD27 and ZD28, in order to obtain the corresponding complementation strains. GC-MS analysis of methylated extracts of 6-day fermentation broths of the penR deletion mutant S. exfoliatus ZD27 revealed that the levels of pentalenolactone methyl ester (PL-Me, retention time [r.t.] 13.3 min, m/z 290) and its biosynthetic intermediates, such as pentalenolactone D methyl ester (PL-D-Me, r.t. 12.9 min, m/z 278) and pentalenolactone F methyl ester (PL-F-Me, r.t. 13.2 min, m/z 292) were all at the lower limit of detection by GC-MS, while in the two derived penR and pntR complementation strains, S. exfoliatus ZD27::pDQ90 (ΔpenR::penR) and ZD27::pDQ91 (ΔpenR::pntR), the levels of production of pentalenolactones were restored to essentially those of the wild-type S. exfoliatus UC5319 (Fig. 9). Similarly, while the penM-penR double mutant S. exfoliatus ZD28 produced only minor amounts of pentalenolactone F, significantly less than the parent penM mutant, S. exfoliatus ZD22, the level of pentalenolactone F biosynthesis was restored to that of ZD22 in both complementation strains, S. exfoliatus ZD28::pDQ90 (ΔpenM-penR::penR) and ZD28::pDQ91 (ΔpenM-penR::pntR) (see Fig. S7 in the supplemental material). These results firmly establish that PenR, and by analogy its close orthologue PntR, is a required, positive regulator of pentalenolactone biosynthesis.
Fig 9.

GC-MS analysis of pentalenolactone production by wild-type and mutant strains of S. exfoliatus. The four panels show (from top to bottom) results for (i) wild-type S. exfoliatus UC5319, (ii) penR mutant S. exfoliatus ZD27, (iii) S. exfoliatus ZD27::pDQ90, with complementation by penR, and (iv) S. exfoliatus ZD27::pDQ91, with complementation by pntR. See Fig. S7 and S8 in the supplemental material for mass spectra of pentalenolactone and its intermediates and GC-MS analysis of the penM-penR double mutant of S. exfoliatus.
Role of penR in transcription of the pentalenolactone biosynthetic gene cluster.
We have also used RT-PCR to compare the relative, time-dependent levels of transcription in both wild-type and mutant backgrounds for three representative pen genes: the resistance gene gapN, the hydroxylase gene penH, and the penR transcription factor gene itself. Total RNA was isolated at 1-day intervals from parallel cultures of wild-type S. exfoliatus UC5319 and the penR mutant S. exfoliatus ZD27. After adjustment of total RNA titers to allow direct comparison of each sample pair, selected segments of the gapN, penH, and penR genes from both wild-type and mutant cultures were amplified by RT-PCR (Table S2 and Fig. 10). For each day, the expression levels of both gapN and penH were significantly reduced in the penR mutant compared to those in the wild-type strain sampled at the same time point, whereas the levels of mRNA derived from the residual portion of the partially deleted penR gene were consistently enhanced in the penR mutant compared to wild-type S. exfoliatus for each sample pair. Control experiments in which the constitutively expressed hrdB gene was amplified by a pair of consensus primers (see Table S2 in the supplemental material) showed no significant differences in levels of expression between the wild-type and the penR mutant host throughout the fermentation period. It is thus clearly established that PenR acts as a transcriptional activator of both the resistance gene gapN and the hydroxylase penH (as well, presumably, of all the remaining unidirectionally transcribed biosynthetic and transport genes of the pen pentalenolactone biosynthetic pathway), while PenR serves as an autorepressor of its own transcription (Fig. 2 and 10).
Fig 10.
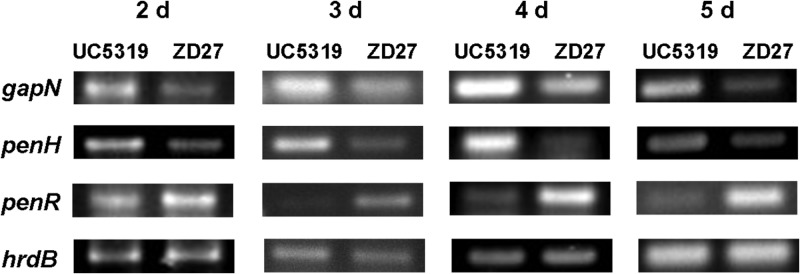
RT-PCR comparison of relative transcript levels in wild-type S. exfoliatus UC5319 and the penR mutant S. exfoliatus ZD27 of the pentalenolactone resistance gene gapN, the hydroxylase gene penH, and the transcription factor penR. Total RNA was isolated from parallel cultures of the wild-type and mutant S. exfoliatus at 1-day intervals starting on day 2 and ending on day 5. RNA concentrations were adjusted to allow direct comparison of each sample pair. The primer pair WYP9F/WYP9R was used to amplify a 152-bp DNA segment of the gapN gene, the primer pair WYP10F/WYP10R was used to amplify a 226-bp DNA segment from the penH gene, and the primer pair WYP7F/WYP7R was used to amplify a 184-bp DNA segment near the 5′ end of penR. The constitutive hrdB gene was also amplified by using a consensus primer pair. All incubations were sampled in triplicate. Controls lacking reverse transcriptase did not result in any detectable PCR amplification. Comparison of the relative intensities of pairs of RT-PCR bands is only valid for pairs that result from amplification of the same target gene in RNA samples isolated from the wild-type strain and the penR mutant at common time points. See Materials and Methods for details.
DISCUSSION
The combined experimental results establish for the first time that the MarR-like PenR protein of S. exfoliatus acts as a positive regulator that activates the transcription of the genes of both pentalenolactone biosynthesis and resistance, while serving as an autorepressor of transcription of its parent gene, penR. The pen cluster harbors two intergenic binding sites for the PenR protein, the penR-gapN region and the downstream penM-penH intergenic region, as established by in vitro EMSA analysis of protein-DNA binding. As expected, both of these intergenic regions also harbor functional promoters, as confirmed by the in vivo xylE promoter probe system. Using defined fragments from each intergenic region, the PenR-binding target has been refined to a pair of homologous 37-bp sequences, each harboring a two conserved 11- to 12-nt motifs, one of which exhibits a partial inverse repeat. Although further definition of the actual sites of PenR binding will require direct DNase I footprinting, it is significant that the orthologous PntR protein from the S. arenae pentalenolactone biosynthetic gene cluster has also been shown to bind to a similar conserved 37-bp sequence located in the pntR-gapR intergenic region, while a homologous motif is also evident in the corresponding sav2989-gap1 of the S. avermitilis ptl cluster, corresponding to a predicted binding site for the MarR-like SAV_2989 protein. This consensus binding motif is absent from all of the remaining intergenic regions of both the pen and pnt clusters, as well as within all of the protein coding sequences of each pentalenolactone biosynthetic cluster. Binding of both PenR and PntR to their cognate DNA sequences can be specifically reversed in vitro by low micromolar concentrations of the pathway end product pentalenolactone or either of its late-stage biosynthetic intermediates, pentalenolactones F and D.
Based on these results we suggest a model for the regulation of pentalenolactone biosynthesis, illustrated in Fig. 2. According to this picture, the pen and pnt biosynthetic gene clusters would be transcribed at most at very low levels at early stages of growth of the parent pentalenolactone-producing organism. With increasing cell growth, expression of PntR or PenR results in specific binding to specific intergenic regions of their biosynthetic gene clusters, thereby recruiting RNA polymerase to their respective promoters and an a consequent increase in the levels of transcription of the adjacent, unidirectionally transcribed, biosynthetic operon. Ribosomal translation of the resultant polycistronic mRNA would generate the full complement of functional biosynthetic pathway enzymes illustrated in Fig. 1 and 2, as well as the necessary pentalenolactone-resistant GAPDH. The presence of a second PenR binding site in the penM-penH intergenic region of S. exfoliatus indicates that the penHGFEDBAI mRNA also has its own PenR-activated promoter, a conclusion consistent with the above-described xylE promoter-probe experiments. The specific role of this second regulatory site is unclear, since a homologous PntR binding site is absent in the closely related pnt pentalenolactone biosynthetic operon. In both S. exfoliatus and S. arenae, the orthologous PntG and PenG proteins have been annotated as transmembrane efflux pumps that are thought to excrete pentalenolactone, thereby reducing the intracellular concentration of the antibiotic. At sufficiently high intracellular titers of 5 to 10 μM, however, pentalenolactone and its late-stage biosynthetic intermediates can release PenR from their target DNA, thereby suppressing transcription of the corresponding pnt and pen biosynthetic gene clusters, while derepressing the expression of PenR itself. Notably the endpoint extracellular concentrations of pentalenolactone typically reach the 10 μM level only at late stages of the fermentation process (5–7). Repression of antibiotic biosynthesis by binding of late-stage, antibiotically active metabolites to PenR may thus only occur near the end of the fermentation process or might well be a strategy for modulating the intracellular concentrations of antibiotic before it is actively excreted into the medium by the putative PenG efflux pump so as to control that these intracellular concentrations do not exceed the mM limits of the pentalenolactone-insensitive GAPDH resistance protein (29). The model of PenR-mediated regulation of pentalenolactone biosynthesis is strongly supported by the above-described promoter probe experiments, which confirm the presence of the predicted promoters in each of the intergenic regions shown by EMSA experiments to bind PenR, and by the RT-PCR results that establish that the expression of genes for both pentalenolactone biosynthesis and resistance is suppressed at each time point sampled for the penR deletion mutant compared to the wild-type production host. These results are further corroborated by the observation that production of pentalenolactone and its late-stage intermediates is reduced to near zero levels in penR deletion mutants of S. exfoliatus but can be restored to essentially wild-type titers by complementation with penR as well as its close orthologue pntR. Protein structural studies on PenR and PntR now under way should allow identification of the precise binding sites for both target DNA and the pentalenolactone ligands.
The MarR-like protein PenR is are thus shown to be responsible for local regulation of pentalenolactone biosynthesis in S. exfoliatus, while an analogous role is likely for the orthologous DNA-binding PntR protein which binds a similar pntR-gapR intergenic region in S. arenae and can also complement deletions of the penR gene in S. exfoliatus. On the other hand, the mechanism of global regulation of each biosynthetic cluster is still obscure. Interestingly, the pen and pnt gene clusters each harbor several structural genes that contain rare TTA codons, as do the predicted pentalenolactone biosynthetic cluster of S. bingchenggensis (47) and the closely related neopentalenolactone ptl cluster of S. avermitilis. Thus, the orthologous penM and pntM genes and the corresponding SBI_09672 gene of S. bingchenggensis have TTA codons at identical locations, whereas TTA codons are also found in penF in the pen gene cluster, in pntE and pntD in the pnt gene cluster, and in ptlH, ptlG, and ptlE in the ptl gene cluster (Fig. 2 and see Table S3 in the supplemental material). The only tRNA species in Streptomyces that is able to read the Leu TTA codon is encoded by the bldA gene (48). The corresponding bldA mutants are typically defective in both aerial mycelium formation and in antibiotic production. The presence of TTA codons in several pentalenolactone biosynthetic genes therefore suggests that these biosynthetic clusters may also be subject to global regulation mediated by a bldA-like gene (49). In S. griseus, Ohnishi and coworkers have recently reported that expression of the sesquiterpene cyclase caryolan-1-ol synthase (SGR2079, GcoA) is under the control of the well-studied A-factor (2-isocapyloyl-hydroxymethyl-γ-butyrolactone) regulatory cascade (50). In contrast, expression of a second sesquiterpene synthase, (+)-epicubenol synthase (SGR6065, GecA), is A-factor independent (51). The global regulatory systems for bacterial terpenoid biosynthesis therefore appear to utilize a variety of strategies, many of which remain to be elucidated.
Terpenes are a large and varied class of organic compounds, produced primarily by numerous microbes, plants, and some insects. Although much is now understood about the mechanisms of terpene biosynthesis, little has been known about the molecular and genetic details of the regulation of terpene bisosynthetic pathways, particularly in prokaryotes. We have described here for the first time examples of specific pathway regulation of sesquiterpene biosynthesis in two species of Streptomyces that produce the antibiotic pentalenolactone under the control of closely related MarR-like proteins. Further investigations will be aimed at establishing the molecular details of this transcriptional control, as well as the connection to cellular development and global regulatory networks.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tun-Li Shen of the Department of Chemistry, Brown University, for assistance with MS analysis and Ashish Garg for helpful advice on the preparation of the pentalenolactones.
This study was supported by National Institutes of Health grant GM30301 (D.E.C.) and by the National Natural Science Foundation of China, the Ministry of Science and Technology of China (973 Programs).
Footnotes
Published ahead of print 11 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02079-12.
REFERENCES
- 1. George AM, Levy SB. 1983. Amplifiable resistance to tetracycline, chloramphenicol, and other antibiotics in Escherichia coli: involvement of a non-plasmid-determined efflux of tetracycline. J. Bacteriol. 155:531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. George AM, Levy SB. 1983. Gene in the major cotransduction gap of the Escherichia coli K-12 linkage map required for the expression of chromosomal resistance to tetracycline and other antibiotics. J. Bacteriol. 155:541–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Libby SJ, Goebel W, Ludwig A, Buchmeier N, Bowe F, Fang FC, Guiney DG, Songer JG, Heffron F. 1994. A cytolysin encoded by Salmonella is required for survival within macrophages. Proc. Natl. Acad. Sci. U. S. A. 91:489–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilkinson SP, Grove A. 2006. Ligand-responsive transcriptional regulation by members of the MarR family of winged helix proteins. Curr. Issues Mol. Biol. 8:51–62 [PubMed] [Google Scholar]
- 5. Alekshun MN, Levy SB, Mealy TR, Seaton BA, Head JF. 2001. The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 Å resolution. Nat. Struct. Biol. 8:710–714 [DOI] [PubMed] [Google Scholar]
- 6. Egland PG, Harwood CS. 1999. BadR, a new MarR family member, regulates anaerobic benzoate degradation by Rhodopseudomonas palustris in concert with AadR, an Fnr family member. J. Bacteriol. 181:2102–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilkinson SP, Grove A. 2004. HucR, a novel uric acid-responsive member of the MarR family of transcriptional regulators from Deinococcus radiodurans. J. Biol. Chem. 279:51442–51450 [DOI] [PubMed] [Google Scholar]
- 8. Lomovskaya O, Lewis K, Matin A. 1995. EmrR is a negative regulator of the Escherichia coli multidrug resistance pump EmrAB. J. Bacteriol. 177:2328–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lim D, Poole K, Strynadka NC. 2002. Crystal structure of the MexR repressor of the mexRAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Biol. Chem. 277:29253–29259 [DOI] [PubMed] [Google Scholar]
- 10. Wu RY, Zhang RG, Zagnitko O, Dementieva I, Maltzev N, Watson JD, Laskowski R, Gornicki P, Joachimiak A. 2003. Crystal structure of Enterococcus faecalis SlyA-like transcriptional factor. J. Biol. Chem. 278:20240–20244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guerra AJ, Dann CE, 3rd, Giedroc DP. 2011. Crystal structure of the zinc-dependent MarR family transcriptional regulator AdcR in the Zn(II)-bound state. J. Am. Chem. Soc. 133:19614–19617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koe BK, Sobin BA, Celmer WD. 1956. PA 132, a new antibiotic. I. Isolation and chemical properties. Antibiot. Annu. 1956:672–675 [PubMed] [Google Scholar]
- 13. Keller-Schlerlein WLJ, Nyfeler R, Zähner H. 1970. Metabolic products of microorganisms. 105. Arenacemycin E, D, and C. Arch. Mikrobiol. 84:301–316 [PubMed] [Google Scholar]
- 14. Takeuchi S, Ogawa Y, Yonehara H. 1969. The Structure of pentalenolactone (PA-132). Tetrahedron Lett. 2737-2740. [DOI] [PubMed] [Google Scholar]
- 15. Martin DG, Slomp G, Mizsak S, Duchamp DJ, Chidester CG. 1970. The structure and absolute configuration of pentalenolactone (PA 132). Tetrahedron Lett. 4901-4904. [DOI] [PubMed] [Google Scholar]
- 16. Cane DE, Sohng JK. 1989. Inhibition of glyceraldehyde-3-phosphate dehydrogenase by pentalenolactone: kinetic and mechanistic studies. Arch. Biochem. Biophys. 270:50–61 [DOI] [PubMed] [Google Scholar]
- 17. Hartmann S, Neeff J, Heer U, Mecke D. 1978. Arenaemycin (pentalenolactone): a specific inhibitor of glycolysis. FEBS Lett. 93:339–342 [DOI] [PubMed] [Google Scholar]
- 18. Duszenko M, Balla H, Mecke D. 1982. Specific inactivation of glucose metabolism from eucaryotic cells by pentalenolactone. Biochim. Biophys. Acta 714:344–350 [DOI] [PubMed] [Google Scholar]
- 19. Seo MJ, Zhu D, Endo S, Ikeda H, Cane DE. 2011. Genome mining in Streptomyces: elucidation of the role of Baeyer-Villiger monooxygenases and non-heme iron-dependent dehydrogenase/oxygenases in the final steps of the biosynthesis of pentalenolactone and neopentalenolactone. Biochemistry 50:1739–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cane DE, Ikeda H. 2012. Exploration and mining of the bacterial terpenome. Acc. Chem. Res. 45:463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cane DE, Abell C, Harrison PH, Hubbard BR, Kane CT, Lattman R, Oliver JS, Weiner SW. 1991. Terpenoid biosynthesis and the stereochemistry of enzyme-catalyzed allylic addition-elimination reactions. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 332:123–129 [DOI] [PubMed] [Google Scholar]
- 22. Cane DE, Sohng JK, Lamberson CR, Rudnicki SM, Wu Z, Lloyd MD, Oliver JS, Hubbard BR. 1994. Pentalenene synthase. Purification, molecular cloning, sequencing, and high-level expression in Escherichia coli of a terpenoid cyclase from Streptomyces UC5319. Biochemistry 33:5846–5857 [DOI] [PubMed] [Google Scholar]
- 23. Jiang J, Tetzlaff CN, Takamatsu S, Iwatsuki M, Komatsu M, Ikeda H, Cane DE. 2009. Genome mining in Streptomyces avermitilis: a biochemical Baeyer-Villiger reaction and discovery of a new branch of the pentalenolactone family tree. Biochemistry 48:6431–6440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu D, Seo MJ, Ikeda H, Cane DE. 2011. Genome mining in Streptomyces: discovery of an unprecedented P450-catalyzed oxidative rearrangement that is the final step in the biosynthesis of pentalenolactone. J. Am. Chem. Soc. 133:2128–2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Quaderer R, Omura S, Ikeda H, Cane DE. 2006. Pentalenolactone biosynthesis: molecular cloning and assignment of biochemical function to PtlI, a cytochrome P450 of Streptomyces avermitilis. J. Am. Chem. Soc. 128:13036–13037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. You Z, Omura S, Ikeda H, Cane DE. 2006. Pentalenolactone biosynthesis: molecular cloning and assignment of biochemical function to PtlH, a non-heme iron dioxygenase of Streptomyces avermitilis. J. Am. Chem. Soc. 128:6566–6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. You Z, Omura S, Ikeda H, Cane DE. 2007. Pentalenolactone biosynthesis: molecular cloning and assignment of biochemical function to PtlF, a short-chain dehydrogenase from Streptomyces avermitilis, and identification of a new biosynthetic intermediate. Arch. Biochem. Biophys. 459:233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. You Z, Omura S, Ikeda H, Cane DE, Jogl G. 2007. Crystal structure of the non-heme iron dioxygenase PtlH in pentalenolactone biosynthesis. J. Biol. Chem. 282:36552–36560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tetzlaff CN, You Z, Cane DE, Takamatsu S, Omura S, Ikeda H. 2006. A gene cluster for biosynthesis of the sesquiterpenoid antibiotic pentalenolactone in Streptomyces avermitilis. Biochemistry 45:6179–6186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fröhlich K-U, Wiedmann M, Lottspeich F, Mecke D. 1989. Substitution of a pentalenolactone-sensitive glyceraldehyde-3-phosphate dehydrogenase by a genetically distinct resistant isoform accompanies pentalenolactone production in Streptomyces arenae. J. Bacteriol. 171:6696–6702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frohlich KU, Kannwischer R, Rudiger M, Mecke D. 1996. Pentalenolactone-insensitive glyceraldehyde-3-phosphate dehydrogenase from Streptomyces arenae is closely related to GAPDH from thermostable eubacteria and plant chloroplasts. Arch. Microbiol. 165:179–186 [DOI] [PubMed] [Google Scholar]
- 32. Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. The John Innes Foundation, Norwich, United Kingdom [Google Scholar]
- 33. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 34. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 35. Cane DE, Sohng JK, Williard PG. 1992. Isolation and structure determination of pentalenolactones A, B, D, and F. J. Org. Chem. 57:844–852 [Google Scholar]
- 36. Tillman AM, Cane DE. 1983. Pentalenolactone F, a new metabolite Isolated from Streptomyces: isolation and structure elucidation. J. Antibiot. 36:170–172 [DOI] [PubMed] [Google Scholar]
- 37. Danishefsky S, Hirama M, Gombatz K, Harayama T, Berman E, Schuda PF. 1979. Total Synthesis of dl-pentalenolactone. J. Am. Chem. Soc. 101:7020–7031 [Google Scholar]
- 38. Bussmann M, Baumgart M, Bott M. 2010. RosR (Cg1324), a hydrogen peroxide-sensitive MarR-type transcriptional regulator of Corynebacterium glutamicum. J. Biol. Chem. 285:29305–29318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wennerhold J, Krug A, Bott M. 2005. The AraC-type regulator RipA represses aconitase and other iron proteins from Corynebacterium under iron limitation and is itself repressed by DtxR. J. Biol. Chem. 280:40500–40508 [DOI] [PubMed] [Google Scholar]
- 40. Gust B, Challis GL, Fowler K, Kieser T, Chater KF. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U. S. A. 100:1541–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun Y, Zhou X, Liu J, Bao K, Zhang G, Tu G, Kieser T, Deng Z. 2002. “Streptomyces nanchangensis”, a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin, contains multiple polyketide gene clusters. Microbiology 148:361–371 [DOI] [PubMed] [Google Scholar]
- 42. Wilkinson CJ, Hughes-Thomas ZA, Martin CJ, Bohm I, Mironenko T, Deacon M, Wheatcroft M, Wirtz G, Staunton J, Leadlay PF. 2002. Increasing the efficiency of heterologous promoters in actinomycetes. J. Mol. Microbiol. Biotechnol. 4:417–426 [PubMed] [Google Scholar]
- 43. Janssen GR, Bibb MJ. 1993. Derivatives of pUC18 that have BglII sites flanking a modified multiple cloning site and that retain the ability to identify recombinant clones by visual screening of Escherichia coli colonies. Gene 124:133–134 [DOI] [PubMed] [Google Scholar]
- 44. Li W, Ying X, Guo Y, Yu Z, Zhou X, Deng Z, Kieser H, Chater KF, Tao M. 2006. Identification of a gene negatively affecting antibiotic production and morphological differentiation in Streptomyces coelicolor A3(2). J. Bacteriol. 188:8368–8375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Picossi S, Belitsky BR, Sonenshein AL. 2007. Molecular mechanism of the regulation of Bacillus subtilis gltAB expression by GltC. J. Mol. Biol. 365:1298–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schell MA. 1993. Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47:597–626 [DOI] [PubMed] [Google Scholar]
- 47. Wang XJ, Yan YJ, Zhang B, An J, Wang JJ, Tian J, Jiang L, Chen YH, Huang SX, Yin M, Zhang J, Gao AL, Liu CX, Zhu ZX, Xiang WS. 2010. Genome sequence of the milbemycin-producing bacterium Streptomyces bingchenggensis. J. Bacteriol. 192:4526–4527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lawlor EJ, Baylis HA, Chater KF. 1987. Pleiotropic morphological and antibiotic deficiencies result from mutations in a gene encoding a tRNA-like product in Streptomyces coelicolor A3(2). Genes Dev. 1:1305–1310 [DOI] [PubMed] [Google Scholar]
- 49. Higo A, Horinouchi S, Ohnishi Y. 2011. Strict regulation of morphological differentiation and secondary metabolism by a positive feedback loop between two global regulators AdpA and BldA in Streptomyces griseus. Mol. Microbiol. 81:1607–1622 [DOI] [PubMed] [Google Scholar]
- 50. Nakano C, Horinouchi S, Ohnishi Y. 2011. Characterization of a novel sesquiterpene cyclase involved in (+)-caryolan-1-ol biosynthesis in Streptomyces griseus. J. Biol. Chem. 286:27980–27987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nakano C, Tezuka T, Horinouchi S, Ohnishi Y. 2012. Identification of the SGR6065 gene product as a sesquiterpene cyclase involved in (+)-epicubenol biosynthesis in Streptomyces griseus. J. Antibiot. 65:551–558 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.