

Abstract
The quaking (qkI) gene encodes 3 major alternatively spliced isoforms that contain unique sequences at their C termini dictating their cellular localization. QKI-5 is predominantly nuclear, whereas QKI-6 is distributed throughout the cell and QKI-7 is cytoplasmic. The QKI isoforms are sequence-specific RNA binding proteins expressed mainly in glial cells modulating RNA splicing, export, and stability. Herein, we identify a new role for the QKI proteins in the regulation of microRNA (miRNA) processing. We observed that small interfering RNA (siRNA)-mediated QKI depletion of U343 glioblastoma cells leads to a robust increase in miR-7 expression. The processing from primary to mature miR-7 was inhibited in the presence QKI-5 and QKI-6 but not QKI-7, suggesting that the nuclear localization plays an important role in the regulation of miR-7 expression. The primary miR-7-1 was bound by the QKI isoforms in a QKI response element (QRE)-specific manner. We observed that the pri-miR-7-1 RNA was tightly bound to Drosha in the presence of the QKI isoforms, and this association was not observed in siRNA-mediated QKI or Drosha-depleted U343 glioblastoma cells. Moreover, the presence of the QKI isoforms led to an increase presence of pri-miR-7 in nuclear foci, suggesting that pri-miR-7-1 is retained in the nucleus by the QKI isoforms. miR-7 is known to target the epidermal growth factor (EGF) receptor (EGFR) 3′ untranslated region (3′-UTR), and indeed, QKI-deficient U343 cells had reduced EGFR expression and decreased ERK activation in response to EGF. Elevated levels of miR-7 are associated with cell cycle arrest, and it was observed that QKI-deficient U343 that harbor elevated levels of miR-7 exhibited defects in cell proliferation that were partially rescued by the addition of a miR-7 inhibitor. These findings suggest that the QKI isoforms regulate glial cell function and proliferation by regulating the processing of certain miRNAs.
INTRODUCTION
MicroRNAs (miRNAs) are noncoding short RNAs that regulate the expression of mRNAs by base-pairing to their 3′ untranslated regions (3′-UTR), leading to mRNA translational repression and/or mRNA degradation (1). At present, >1,500 miRNAs have been identified in the human genome (http://mirbase.org), where they are functionally involved in diverse cellular processes. Accumulating evidence suggests linking miRNA dysregulation with disease progression (2, 3). miRNAs are first transcribed by RNA polymerase II as long primary miRNAs (pri-miRNAs) in the nucleus, where they are subsequently processed into ∼70-nucleotide (nt) precursor miRNAs by the RNase III enzyme Drosha microprocessor complex (4–6). The precursor miRNAs are exported into the cytoplasm (7, 8), and they are processed into ∼22-nt RNA duplexes by the RNase III enzyme Dicer (9–12). One of the strands is assembled into RISC (RNA-induced silencing complex) to inhibit mRNA translation or to enhance the degradation of the mRNA (3). Interestingly, ∼50% of human miRNAs are located in the intronic regions of protein-encoding genes, with implications for the biogenesis of the miRNAs and their host mRNAs (13).
RNA binding proteins play important roles in the regulation of miRNA biogenesis. For example, the human immunodeficiency virus transactivating response RNA binding protein (TRBP) is an integral component of the Dicer-containing complex (14). Lin-28 was shown to regulate miRNA biogenesis by binding the miRNA terminal loop of Let-7 to block processing of primary to precursor miRNA (15). SF2/ASF enhances expression of a group of miRNAs by directly binding to a conserved double-stranded sequence in their primary miRNA stem region to assist Drosha cleavage (16). KSRP recognizes a conserved sequence in the terminal loop of Let-7 and several other miRNAs to promote their maturation by facilitating an association with Drosha in the nucleus or with Dicer in the cytoplasm (17).
The QKI proteins belong to the heteronuclear ribonucleoprotein particle K (hnRNPK) homology (KH) domain family of RNA binding proteins (18–20). The qkI gene expresses three major alternatively spliced mRNAs (5, 6, and 7 kb) encoding QKI-5, QKI-6, and QKI-7 that differ in their C-terminal 30 amino acids (21). The QKI-5 isoform contains a nuclear localization signal that renders this isoform predominantly nuclear (22). QKI-6 is cytoplasmic and nuclear, and QKI-7 is predominantly cytoplasmic (23). The QKI proteins selectively interact with a short sequence termed the QKI response element (QRE; ACUAAY[N1-20]UAAY) (24). A transcriptome-wide study using CLIP (cross-linking and immunoprecipitation) showed that QKI associates with intronic sequences and mature mRNAs (25). The QKI isoforms have been shown to function in various aspects of RNA processing during glial, oligodendrocyte, and Schwann cell differentiation (18, 19). Functional QREs have been identified in the myelin basic protein (MBP) (26–28), Krox-20 (Egr-2) (29), microtubule-associated protein 1B (30), the cyclin-dependent kinase inhibitor p27KIP1 mRNAs (31), actin-interacting protein 1 (AIP-1) (32), and hnRNPA1 mRNAs (33, 34).
We have recently shown that the QKI proteins interact with argonaute 2 (Ago2), a core component of RISC, during the stress response (35). To determine whether the QKI proteins regulate the expression of selected miRNAs, we monitored the expression of miRNAs by microarray analysis of small interfering RNA (siRNA)-mediated QKI depletion of U343 glioblastoma cells. The expression of miR-7 was identified as robustly upregulated with the loss of QKI. We noticed an increased association of pri-miR-7 with Drosha in the presence of the QKI isoforms, suggesting that the binding of pri-miR-7 to the QKI isoforms prevents its proper processing. Since miR-7 targets the epidermal growth factor (EGF) receptor (EGFR), these findings have implications for EGF signaling and cell proliferation in glial cells.
MATERIALS AND METHODS
Cells, siRNAs, transfections, and antibodies.
The HEK293, U343, and U87 cell lines were purchased from the American Type Culture Collection (Manassas, VA). Plasmids were transfected with Lipofectamine 2000 (Invitrogen), and siRNAs, miRNA mimics, and miRNA inhibitors were transfected with Lipofectamine RNAi MAX (Invitrogen) according to the manufacturer's instructions. The siRNAs and miRNA mimics were purchased from Dharmacon Inc. The siRNAs used were siLuciferase (siCTL, 5′-CGU ACG CGG AAU ACU UCG AdTdT-3′), siQKI (5′-GGA CUU ACA GCC AAA CAA CdTdT-3′) (35), siQKI-1 (5′-GAC GAA GAA AUU AGC AGA GUA dTdT-3′) (36), and siQKI-2 (5′-CCA GCU GGC CCU ACC AUA AdTdT-3′). miR-7 miRCURY LNA microRNA inhibitor and a negative control were purchased from Exiqon Inc. The anti-QKI panantibody has been described previously (37). Antitubulin (T6793) and anti-Myc (M4439) antibodies were purchased from Sigma Inc. Anti-EGFR antibodies (SC-03) were from Santa Cruz. Anti-extracellular signal-regulated kinase (anti-ERK; rabbit polyclonal no. 4695) and anti-phospho-ERK (anti-pERK; rabbit polyclonal no. 4377) antibodies were purchased from Cell Signaling Technology.
miRNA microarray and RT-qPCR.
U343 cells were transfected with 100 nM luciferase (siCTL) or siQKI with Lipofectamine RNAi MAX (Invitrogen). Forty-eight hours after transfection, the cells were transfected a second time, and they were harvested at 72 h. Total RNA was isolated using miRNAeasy minikits (Qiagen). miRNA microarray analysis was performed and the data were analyzed by LC Sciences using 3 biological replicates for siCTL- and siQKI-transfected U343 cells. Total RNA was treated with DNase (Promega) for 15 min at 37°C. One microgram of total RNA was used for the first-strand synthesis with reverse transcriptase II with oligo(dT) or random primers (Invitrogen). Real-time PCR was performed using a SYBR green PCR kit (Qiagen) or TaqMan 2× universal PCR master mix (Applied Biosystems, Foster City, CA) with gene-specific primers. The primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were from Qiagen (HS-GAPDH-1). For hnRNPK the primers were as follows: forward, 5′-ACT TGG GAC TCT GCA ATT GAC A-3′; reverse, 5′-CCC TGT GGT TCA TAA GCC ATT T-3′. TaqMan primers for human primary miR-7 came from Applied Biosystems: pri-miR-7-1, Hs03302860_pri; pri-miR-7-2, Hs03302865_pri; and pri-miR-7-3, Hs03302872_pri. Fifty nanograms of total RNA was used for miRNA first-strand synthesis using the TaqMan reverse transcription kit (Applied Biosystems), and real-time PCR was performed using TaqMan 2× Universal PCR master mix (Applied Biosystems). TaqMan miRNA-specific primers from Applied Biosystems were as follows: miR-7, 000268; miR-19b, 000396; miR-146a, 000468; miR-146b, 001097; and miR-338-5p, 002658. Real-time PCRs were performed on the 7300 real-time PCR system (Applied Biosystems). Data analysis was performed using real-time PCR software 7500, version 2.0.4 (Applied Biosystems). The relative concentrations of the genes of interest were determined using the comparative threshold cycle (ΔCT) method after normalization to the endogenous control (GAPDH).
Plasmids.
The hnRNPK minigene was generated as previously described (16). The mutated hnRNPK minigene, pEGFP/hnRNPK:mQRE, harbors UAAU/C to UAAG (mutated nucleotides are underlined) in the putative QREs and was generated by overlap extension PCRs using PCR primers that include two border primers for hnRNPK (5′-CGT CAT GAG TCG GGA GCT TC-3′ and 5′-GCA GGA CTC CTT CAG TTC TTC A-3′), primers for QRE-A (5′-CTT GCT TAA GGC TTC TTC CAG CAA GCT TAG TAT TC-3′ and 5′-CAA TAC TAA GCT TGC TGG AAG AAG CCT TAA GCA AG-3′), primers for QRE-B (5′-CGG AAT TGA AAG TTC TTA ATA TTT G-3′ and 5′-AAC TTT CAA TTC CGT TTT CTT AAG TCT G-3′), and primers for QRE-C (5′-GGA CCT TAG TAG AAC AGA CTT AAG-3′ and 5′-CTT AAG TCT GTT CTA CTA AGG TCC-3′).
An EGFR 3′-UTR reporter vector (pLuc:EGFR3′-UTR) was constructed by annealing and cloning a repeat flanking sequence from the EGFR 3′-UTR harboring a miR-7 targeting site (4309 to 4354, NM_005228), and the miR-7 seed sequence was optimized so as to be 100% complementary (underlined)—5′-CTG TGA GCA AGA CAA CAA AAT CAC TAG TCT TCC AGA GGA TGC TTG CTG TGA GCA AGA CAA CAA AAT CAC TAG TCT TCC AGA GGA TGC TTG-3′ and 5′-CAA GCA TCC TCT GGA AGA CTA GTG ATT TTG TTG TCT TGC TCA CAG CAA GCA TCC TCT GGA AGA CTT GGA AGA CTA GTG ATT TTG TTG TCT TGC TCA CAG-3′—and was inserted into the pMIR-REPORT Luciferase vector (Ambion). The constructs encoding myc-QKI-5, myc-QKI-6, myc-QKI-7, and myc-QKI-6:V-E were described previously (26, 37).
Luciferase assays.
HEK293 cells were cotransfected with either pMIR-REPORT Luciferase (pLuc) or the pEGFR 3′-UTR reporter (pLuc:EGFR3′-UTR) along with either the wild type (pEGFP/hnRNPK) or the mutated (pEGFP/hnRNPK:mQRE) minigene. pRLTK (Promega, Madison, WI) encoding Renilla luciferase was also included and used to control for transfection efficiency. The cell extracts were harvested after 48 h, and luciferase activity was assayed using a dual-luciferase reporter assay kit (Promega) and measured using a GloMax 20/20 luminometer (Promega). For the selection of stable clones, HEK293 cells were separately transfected with pEGFP, pEGFP/hnRNPK, or EGFP/hnRNPK:mQRE, and after 48 h, the cells were diluted 20 times, transferred to 15-cm dishes, and cultured in 500 μg/ml of G418 Dulbecco modified Eagle medium (DMEM) for 3 to 4 weeks. Clonal populations were selected for green fluorescent protein (GFP) and pri-miR-7 expression by quantitative reverse transcription-PCR (qRT-PCR) analysis.
In vivo RNA binding assay.
HEK293 cells stably expressing the hnRNPK minigene (pEGFP/hnRNPK) were transfected with myc-pcDNA or myc-QKI-5, -6, -7, or -6:V-E. Twenty-four hours after transfection, the cells were harvested in NP-40 lysis buffer (50 mM HEPES [pH 7.5], 150 mM KCl, 2 mM EDTA, 1 mM NaF, 0.5% [vol/vol] NP-40, 0.5 mM dithiothreitol [DTT], 0.01% SDS, complete EDTA-free protease inhibitor cocktail [Roche], and 0.5 U/μl of RNasin). The lysates were immunoprecipitated with anti-myc antibody, and the bound RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Reverse transcription assays were performed using SuperScript II reverse transcriptase (Invitrogen) with random primers. The sequences of the primers used for semiquantitative PCR were as follows: pri-miR-7-1, 5′-AAA ACT GCT GCC AAA ACC AC-3′ and 5′-GCT GCA TTT TAC AGC GAC CAA-3′, and unspliced hnrnpk, 5′-AGC TTT GTG TTA GCT TAT AC-3′ and 5′-GCT GCA TTT TAC AGC GAC CAA-3′. The hnRNPK splicing variants were detected as described previously (16), and 5′ GGG G AG GTT GTA CAG GTG AA 3′ and 5′ ATG ATG GTG AAA CGG TGA C 3′ were used for AIP-1. The primers used for pri-miR-7-1 quantitative real-time PCR were Hs03302860_pri from Applied Biosystems.
To evaluate in vivo protein-RNA interactions, U343 cells were incubated with 4-thiouridine (Sigma; T4509) at a final concentration of 100 μM 14 h prior to cross-linking. The cells were washed once with 1× phosphate-buffered saline (PBS), placed on ice, and irradiated uncovered with 0.15 J/cm2 of 355-nm UV light. Cells were harvested in NP-40 lysis buffer containing SDS (50 mM HEPES [pH 7.5], 150 mM KCl, 2 mM EDTA, 1 mM NaF, 0.5% [vol/vol] NP-40, 0.5 mM DTT, 0.125% SDS, complete EDTA-free protease inhibitor cocktail [Roche], and 0.5 U/μl of RNasin). Lysates were immunoprecipitated using 2 μg of either immunoglobulin (IgG) (Vector Laboratories) or anti-QKI-5 antibodies (Millipore) and complexes captured using protein A Sepharose beads (Sigma; P3391). The immunoprecipitates were treated with proteinase K buffer (100 mM Tris [pH 7.5], 50 mM NaCl, 10 mM EDTA, and 1 mg/ml of proteinase K) for 30 min at 55°C. The bound RNA was isolated using TRIzol reagent (Invitrogen) as per the manufacturer's protocol. Reverse transcription protocols and qRT-PCR were carried out as described above. Primers for pri-miR-7-1 and GAPDH are listed above. Primers for hypoxanthine phosphoribosyltransferase (HPRT) are as follows: 5′-TGA TAG ATC CAT TCC TAT GAC TGT AG-3′ and 5′-CAA GAC ATT CTT TCC AGT TAA AGT TG-3′.
Cell proliferation and cell cycle analysis.
U343 cells were transfected with 40 nM miRNA mimic, 40 nM siRNA, or 40 nM siRNA combined with 100 nM miRNA inhibitor according to the Invitrogen reversal transfection protocol using Lipofectamine RNAi MAX. The cells were counted every 24 h after transfection for 3 days using a Beckman Coulter Z2 cell counter. Similarly, the transfected cells were photographed 48 h after transfection. For bromodeoxyuridine (BrdU) incorporation analysis, 48 h after transfection, the cells were incubated with 10 μM BrdU for 1 h and then harvested and fixed with 75% ethanol for more than 2 h at −20°C. Cell cycle analysis was performed as described previously using a FACS Calibur flow cytometer (BD Biosciences) (38). The data were analyzed using BD CellQuest Pro software and FlowJo software. More than 20,000 cells per condition were analyzed.
Northern blotting.
For miR-7, 10 μg of total RNA was resolved on a 12% polyacrylamide–Tris base-boric acid-EDTA (TBE)–urea gel and blotted onto Hybond-N+ membranes (Amersham). RNA blots were hybridized with a miRCURY LNA miR-7 detection probe, 5′-ACA ACA AAA TCA CTA GTC TTC CA-3′ (Exiqion 38485-00), and antisense U6, 5′-ATA TGG AAC GCT TCA CGT AA-3′ labeled with 32P by using T4 polynucleotide kinase (Invitrogen). After hybridization, the membranes were developed by autoradiography.
Fluorescence in situ hybridization.
hnRNPK intron 15 was PCR amplified with 5′-AGC TTT GTG TTA GCT TAT ACA TAC TAA-3′ and 5′-AGA AAA GAA AAA AAG TGC G-3′ and cloned into pBluescript. Antisense pri-miR-7-1 probe was labeled with digoxigenin-UTP (Roche) by in vitro transcription with T7 RNA polymerase using a MEGA Script T7 kit (Ambion) according to the manufacturer's instructions. In situ hybridizations were performed on siRNA-treated U343 cells as described previously (35).
RESULTS
Identification of QKI-regulated miRNAs in glial cells.
Due to the QKI association with intronic regions (25), we sought to examine whether the QKI RNA binding proteins influence the expression of certain miRNAs in glial cells. The human U343 glioblastoma cell line, known to express the QKI-5, -6, and -7 isoforms, was transfected with siRNAs targeting luciferase (control) or the qkI mRNAs (35). The efficient knockdown of the QKI isoforms was confirmed by immunoblotting using pan-anti-QKI antibodies (37), while the anti-β-tubulin antibody was used as a loading control (Fig. 1A). Total RNA was isolated from control siRNA- and siQKI-treated cells and analyzed using miRNA microarrays. The expression of 10 miRNAs was significantly altered with decreased QKI expression (Fig. 1B). Differentially expressed miRNAs are detailed in Table 1 from three independent microarray experiments performed with three separate biological replicates. The expression of miR-204, -1979, -19b, -146a, -146b-5p, and -338-5p decreased, while the expression of miR-7, -611, and -1224-5p and let-7* increased (Fig. 1B). Those miRNAs with high signal intensity (>500) and the most significant change (>3-fold) were miR-19b, miR-146a, miR-146b-5p, miR-338-5p, and miR-7. Using quantitative real-time PCR, we confirmed the reduction in the expression of miR-146b-5p and miR-338-5p as well as the increase in miR-7 expression (Fig. 1C). However, no significant alteration in the expression of miR-19b and miR-146a was detected between the QKI-positive and -negative cell lines (Fig. 1C). The sequences of the primary RNAs encoding miR-146b-5p, miR-338-5p, and miR-7 were analyzed for the presence of QREs. miR-7 is encoded by three primary miRNA genes, termed miR-7-1, -2, and -3 (39). Interestingly, we identified three QKI binding sites in the primary sequence of miR-7-1 and miR-7-2 near the precursor stem-loop structure, but not miR-7-3 (see Fig. S1 in the supplemental material). The primary sequences of miR-146b-5p and miR-338-5p were devoid of any QREs, suggesting that they may be regulated indirectly by the QKI isoforms. Therefore, we focused on whether the QKI proteins could regulate miR-7 by associating with the QREs harbored in the primary RNAs. By monitoring the levels of miR-7-1, -2, and -3 in U343 cells by qRT-PCR, we ascertained that pri-miR-7-1 was abundantly expressed, while pri-miR–2 and -3 were essentially not expressed (see Fig. S2 in the supplemental material). The increased mature miR-7 expression observed in siQKI U343 cells was also confirmed by Northern blotting (see Fig. S3A in the supplemental material). We next confirmed the increase in miR-7 was not an off-target effect using two additional siRNAs directed against the QKI isoforms numbered siQKI-1 and siQKI-2. U343 cells transfected with siQKI, siQKI-1, and siQKI-2 all resulted in nearly a complete reduction in QKI isoforms, as assessed by immunoblotting and a significant increase in miR-7 levels, albeit to different levels (Fig. 1D).
Fig 1.

Identification of QKI regulated miRNAs in U343 glioblastoma cells. (A) Cell extracts prepared from U343 cells transfected with siCTL or siQKI were separated by SDS-PAGE and immunoblotted with anti-pan-QKI antibodies and β-tubulin as a loading control. (B) Total RNA purified from siCTL- and siQKI-transduced U343 cells was analyzed for miRNA expression using miRNA microarray analysis by LC Sciences. The heat map indicates the differential expression of miRNAs in the siCTL- versus siQKI-transduced cells. Each row represents a different mature miRNA sequence. Shown are median values from normalized, log ratio (base 2) data sets and plotted as a heat map from three biological and technical replicates. Green indicates decreasing miRNA expression, whereas red indicates increasing miRNA expression. (C) TaqMan qRT-PCR was performed to verify the changes observed in the microarray. miRNAs with signals of >500 and a log2 change (siQKI/siLuc) of <−1 or >1 were verified. The data shown represent the mean expression level of 3 different biological replicates calculated by ΔCT normalized to the endogenous GAPDH control of 3 independent experiments performed in triplicate. Error bars represent standard deviations of the means. (D) U343 cells were transfected with the indicated control or QKI siRNAs and the levels of miR-7 monitored by TaqMan qRT-PCR.
Table 1.
miRNA expression in siCTL- and siQKI-expressing U343 cellsa
| Reporter name | P value | Mean signal value |
Log2 (siQKI/siCTL) | Fold change | |
|---|---|---|---|---|---|
| siCTL | siQKI | ||||
| hsa-miR-204 | 2.26E−03 | 6,616 | 3,778 | −0.81 | 0.570382 |
| hsa-miR-146b-5p | 2.88E−03 | 1,395 | 266 | −2.39 | 0.190782 |
| hsa-miR-7 | 5.04E−03 | 296 | 4,016 | 3.76 | 13.54792 |
| hsa-miR-1979 | 6.29E−03 | 10,809 | 5,763 | −0.91 | 0.532185 |
| hsa-miR-338-5p | 7.19E−03 | 675 | 155 | −2.13 | 0.228458 |
| hsa-miR-146a | 7.34E−03 | 566 | 167 | −1.76 | 0.295248 |
| hsa-miR-1224-5p | 7.45E−03 | 149 | 479 | 1.69 | 3.226567 |
| hsa-miR-19b | 8.74E−03 | 515 | 134 | −1.94 | 0.260616 |
| hsa-miR-611 | 3.42E−03 | 42 | 79 | 0.90 | 1.880952 |
| hsa-let-7c* | 9.85E−03 | 30 | 56 | 0.89 | 1.866667 |
P < 0.01 by t test.
hsa-miR-7-1 is embedded in the last intron of the hnRNPK gene, which is alternatively spliced with duplicated 3′ splice sites. The three putative QREs, designated QRE-A, -B, and -C, are highlighted in Fig. 2A. The increased miR-7 observed in siQKI U343 cells could be the result of increased transcription of the hnRNPK host gene or an increase in pri-miR-7-1 transcription per se. Using qRT-PCR, we examined the expression of the hnRNPK and pri-miR-7-1 RNA levels. Interestingly, neither changed in the siQKI-transfected U343 cells compared to control cells (Fig. 2B and C). Moreover, QKI-deficient U343 cells did not exhibit defects in hnRNPK alternative splicing, as assessed by the usage of the proximal and distal 3′ splice sites (see Fig. S3B in the supplemental material). These findings suggest that QKI regulates the posttranscriptional processing of pri-miR-7.
Fig 2.
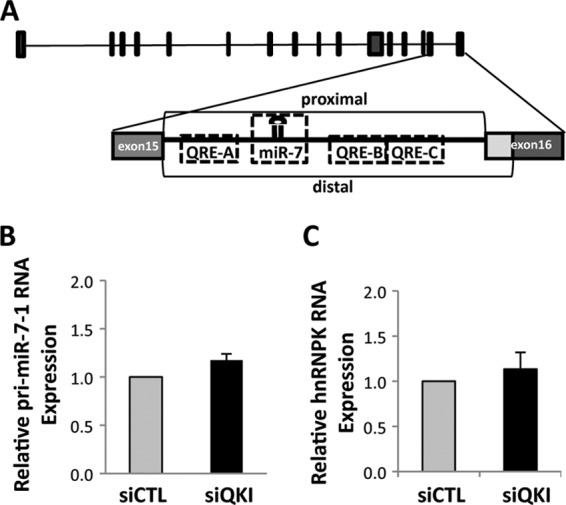
QKI does not affect the transcriptional regulation of pri-miR-7-1. (A) Schematic illustration of human hnRNPK gene with the pri-miR-7-1 located within intron 15 with its multiple QREs. The proximal and distal 3′ splice sites of the hnRNPK gene are shown. (B and C) RNA isolated from siCTL and siQKI U343 cells was analyzed by qRT-PCR for intronic pri-miR-7-1 and hnRNPK mRNA expression. The data for siCTL were normalized to 1.0; the data are shown as means and standard errors of the means.
QKI-5 and QKI-6 regulate the expression of miR-7.
We constructed a minigene as described previously (16) that encompasses the miR-7-1-containing intron of hnRNPK and its flanking exons downstream of an enhanced green fluorescent protein (EGFP) open reading frame (Fig. 3A). The use of the minigene avoids potential complications due to multiple endogenous miR-7 loci and unforeseen transcriptional or posttranscriptional regulations (16). A second hnRNPK minigene was constructed named pEGFP/hnRNPK:mQREs in which the 3 putative QREs located within the miR-7-1-containing intron were mutated as to prevent association with the QKI proteins. The epidermal growth factor receptor (EGFR) 3′-UTR is known to harbor two miR-7 seed sequences (40). We cloned the miR-7 binding sites and flanking sequences from the EGFR 3′-UTR into a firefly luciferase reporter that we named pLuc:EGFR3′-UTR (luciferase plus miR-7 target sequences). HEK293 cells were cotransfected with the hnRNPK minigene (pEGFP/hnRNPK) and the luciferase reporter (pLuc:EGFR3′-UTR). We examined whether the miR-7 RNAs were functional in these cells, and indeed, we observed a robust dose-dependent inhibition of the pLuc:EGFR3′-UTR but not the pLuc control reporter (Fig. 3B). The transfection of pEGFP/hnRNPK:mQRE led to similar findings (Fig. 3B). These findings confirm the production of mature functional miR-7 from both minigenes.
Fig 3.
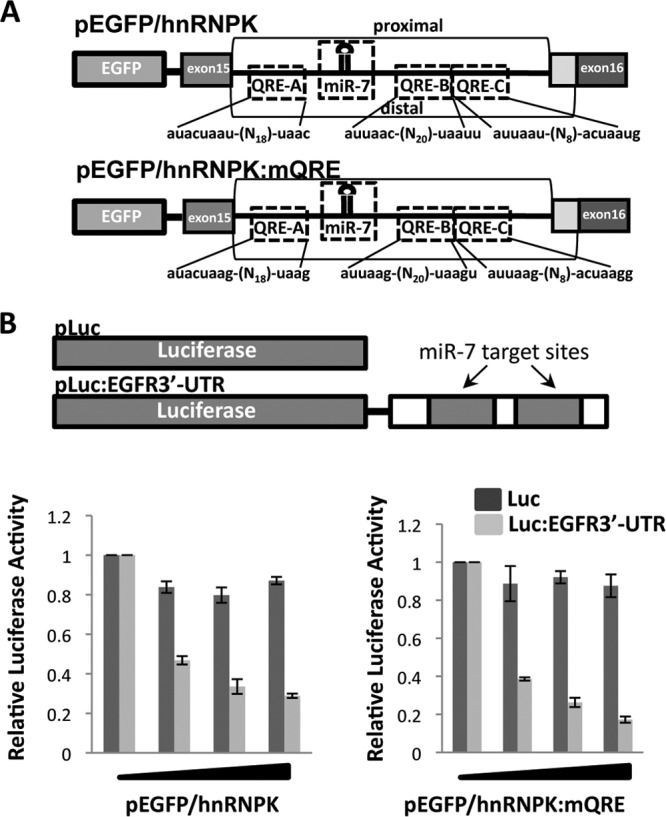
Generation of miR-7 expression vectors with or without QREs. (A) Schematic illustration of the hnRNPK minigene (pEGFP/hnRNPK). The putative QREs with their sequences are shown, as well as the location of miR-7. The mutated hnRNPK reporter gene is shown (pEGFP/hnRNPK:mQRE). (B) The pMIR-REPORT Luciferase (pLuc) and pMIR-REPORT Luciferase harboring sequences from the 3′-UTR of the EGF receptor with the miR-7 targeting sequences (pLuc:EGFR3′-UTR) are depicted. HEK293 cells were cotransfected with pLuc or pLuc:EGFR3′-UTR, along with increasing amounts of either the wild-type hnRNPK minigene (pEGFP/hnRNPK) or the mutated minigene (pEGFP/hnRNPK:mQRE), and assayed for luciferase activity normalized to that for Renilla.
We then generated cells stably expressing the pEGFP/hnRNPK and pEGFP/hnRNPK:mQRE minigenes. Cells expressing these minigenes were used to determine which of the QKI isoforms regulate the maturation of miR-7. The cells were lysed, and we observed a significant increase, ∼8-fold, in the production of miR-7 in cells transfected with pEGFP/hnRNPK, as measured by real-time RT-PCR (Fig. 4A). As the putative QREs of primary miR-7-1 do not reside within the precursor stem region or terminal loop or in the sequence of the mature miR-7, we hypothesized that the QKI isoforms may repress the maturation of miR-7-1 from the primary to precursor stage in the nucleus. To determine which of the QKI isoforms downregulate miR-7 expression, we transfected QKI-5, -6, or -7 in HEK293 cells stably expressing pEGFP/hnRNPK and pEGFP/hnRNPK:mQRE. Total RNA isolated from the transfected cells was resolved on denaturing gels, and the presence of the precursor and mature miR-7 RNAs was detected by Northern blotting. We observed that the ectopic expression of either QKI-5 or QKI-6 significantly reduced the expression of the mature miR-7 derived from pEGFP/hnRNPK-expressing cells (Fig. 4B, upper portion; compare lanes 2 and 4 with lane 1) but not cells expressing pEGFP/hnRNPK:mQRE (Fig. 4B, upper portion; compare lanes 7 and 9 with lane 6). QKI-7 and QKI-6:V-E, an RNA binding defect mutant of QKI-6 (26), did not reduce the expression of miR-7 (Fig. 4B, upper portion). Interestingly, the miRNA precursor levels did not parallel the decreased levels of mature miR-7, implying that QKI-5 and QKI-6 might also act at a post-Drosha-cleavage step such as during export or at the Dicer cleavage. The U6 snRNA was used as a loading control. The expression of the myc epitope-tagged QKI isoforms is shown in relation to the loading control β-tubulin (Fig. 4B, lower portion). These findings suggest that the QKI isoforms that localize to the nucleus (i.e., QKI-5 and QKI-6) are able to inhibit miR-7 maturation.
Fig 4.
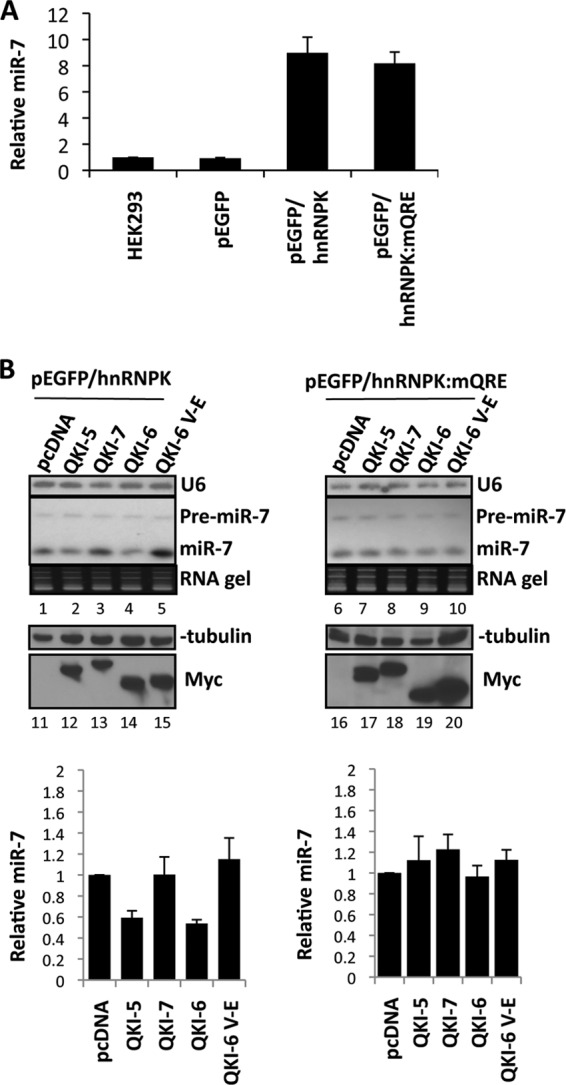
QKI-5 and QKI-6 abrogate mature miR-7 processing. (A) The levels of miR-7 were measure by real-time qRT-PCR in HEK293 or HEK293 cells stably expressing the indicated plasmids. (B) HEK293 cells stably expressing the indicated hnRNPK minigene were transfected with pcDNA empty vector or expression vectors encoding myc epitope-tagged QKI-5, QKI-6, QKI-7, or QKI-6:V-E (20 μg of plasmid in a 10-cm culture dish). After 48 h, the RNA was extracted and 10 μg of total RNA was loaded and resolved on 12% polyacrylamide TED-urea gels and transferred onto Hybond-N+ membranes. The membranes were hybridized with a U6 antisense and mature miR-7 antisense LNA (Exiqon) probe labeled at the 5′ end with 32P. The ethidium bromide-stained RNA gel is shown (lanes 1 to 10). The experiment was performed in triplicate, and results were quantified as the means and standard errors of the means. Protein extracts were also prepared and immunoblotted with anti-β-tubulin as a loading control and anti-Myc antibodies to visualize the QKI expression (lanes 11 to 20). The quantification is depicted at the bottom.
QKI-5 and QKI-6 bind the pri-miR-7-1.
To determine if the QKI isoforms associate directly with pri-miR-7-1, we examined the capacity of the QKI isoforms to coimmunoprecipitate with pri-miR-7. We also examined their ability to associate with the hnRNPK pre-mRNA and mRNA (Fig. 5). HEK293 cells stably expressing either the pEGFP/hnRNPK or pEGFP/hnRNPK:mQRE minigene were transiently transfected with QKI-5, -6, or 7 expression vectors. The QKI isoforms were immunoprecipitated and the bound RNAs monitored by semiquantitative RT-PCR and cross-linking qRT-PCR. We observed that QKI-5 and QKI-6 associated with the pri-miR-7-1 and with the unspliced host hnRNPK pre-mRNA. Some weak interaction was also observed with QKI-7 (Fig. 5B). As expected, no significant binding was observed in the absence of reverse transcriptase or in pcDNA and QKI-6:V-E controls (Fig. 5B). Moreover, we did not observe any association between the QKI isoforms and the hnRNPK mRNA (Fig. 5B). Binding of QKI-5, -6, and -7 to AIP-1 (32) served as a positive control (Fig. 5B). The binding between QKI and the pri-miR-7-1 RNA was direct and mediated by the QREs, as we did not observe an interaction between QKI-5/6 and pri-miR-7-1 when the three QREs were mutated in the pri-miR-7 (Fig. 5C). As association can occur after lysis (41), we labeled U343 cells with 4-thiouracil and performed UV cross-linking. The cells were lysed under harsh conditions, and immunoprecipitations were performed with either control immunoglobulin G (IgG) or anti-QKI-5 antibodies. The bound RNA was isolated, and the presence of the pri-miR-7 was verified by qRT-PCR. We observed ∼17-fold enrichment of pri-miR-7-1 bound to QKI-5 over the IgG control and ∼4-fold enrichment over GAPDH and HPRT negative controls (Fig. 5D). Taken together, these findings show that QKI isoforms associate with pri-miR-7.
Fig 5.
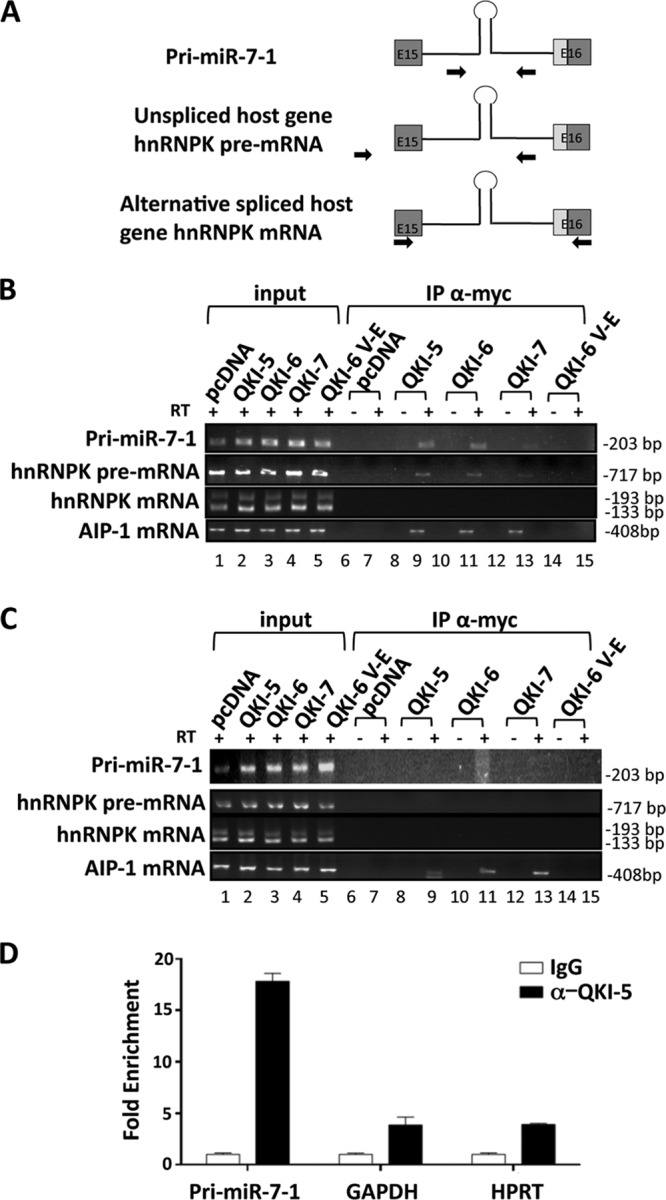
Association of the pri-miR-7 RNA with the QKI isoforms. (A) Schematic illustration of PCR primers used to detect the pri-miR-7-1 mRNA, the hnRNPK pre-mRNA, and mature mRNA. (B and C) HEK293 cells expressing pEGFP/hnRNPK (B) and pEGFP/hnRNPK:mQRE (C) were transfected with pcDNA or the indicated myc-QKI isoform. After 48 h, the cells were lysed and 10% of the fraction was used as input, while the remaining cellular extracts were immunoprecipitated (IP) with the anti-Myc antibody. The bound RNAs were purified and detected by semiquantitative RT-PCR to visualize the presence of the indicated RNAs. The samples were analyzed in the presence (+) or absence (−) of reverse transcriptase (RT) as indicated. The size of the DNA fragments is shown in base pairs on the right. (D) Cross-linked U343 cells were immunoprecipitated with either control IgG or anti-QKI-5 antibodies. The bound RNAs were isolated, and qRT-PCR was used to determine levels of bound pri-miR-7-1 or control mRNAs for GAPDH and HPRT.
The QKI isoforms cause accumulation of Drosha-associated pri-miR-7-1.
To examine if QKI affects the interaction of Drosha with pri-miR-7-1, we performed Drosha immunoprecipitations using U343 cells treated with control siRNA, siQKI, or siDrosha and monitored the levels of the associated pri-miR-7-1 by qRT-PCR. The respective siRNAs for QKI and Drosha led to decreased protein expression as observed by immunoblotting (Fig. 6A). We observed a strong interaction between Drosha and pri-miR-7-1 in control siRNA-treated cells, as expected, and this association was abolished in siQKI- and siDrosha-treated U343 cells (Fig. 6B), suggesting that the presence of QKI isoforms may alter the efficiency of pri-miR-7-1 processing by Drosha. To examine whether pri-miR-7-1 is retained in the nucleus, we performed in situ hybridization, and indeed, we detected the presence of pri-miR-7-1 within nuclear foci of control siRNA-treated cells but not siQKI-treated cells (Fig. 6C). These findings suggest that the QKI isoforms in U343 cells associate and sequester pri-miR-7-1 within the nucleus, preventing its proper maturation.
Fig 6.
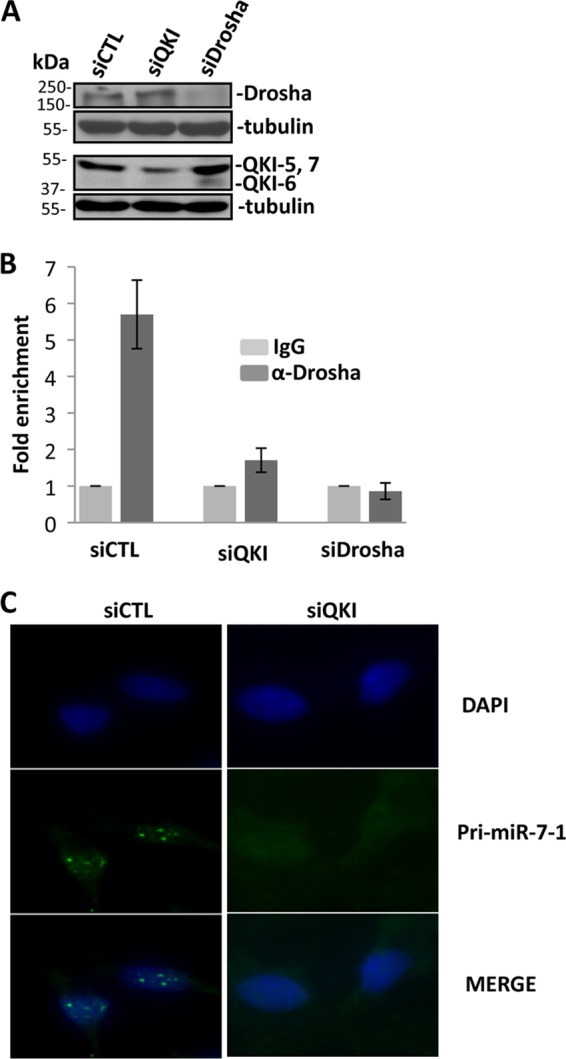
The QKI isoforms reduce pri-miR-7-1 processing by Drosha. (A) Cell lysates prepared from U343 cells transfected with siCTL, siQKI, or siDrosha and immunoblotted with anti-pan-QKI, anti-Drosha, or antitubulin antibodies. The migration of the molecular mass markers is shown on the left. (B) Cell lysates prepared from U343 cells transfected with siCTL, siQKI, or siDrosha were immunoprecipitated with anti-Drosha or IgG antibodies. The bound RNA was purified, and pri-miR-7-1 was detected by real-time qRT-PCR. (C) Fluorescence in situ hybridization performed using antisense pri-miR-7-1 as a probe on siCTL- or siQKI-transduced U343 cells. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), and the images were merged.
QKI deficiency reduces the expression of the EGFR and cell growth.
miR-7 is known to target the EGFR (40, 42). Thus, we examined the expression of the EGFR in U343 glioblastoma cells transfected with siCTL, siQKI-1, and siQKI-2. An miR-7 mimic was used as a positive control, while a negative-control mimic termed miR CTL was also used. The transfection of mimic miR-7 in U343 cells downregulated EGFR expression compared to miR CTL (Fig. 7A, lanes 1 and 2). siQKI-1 and siQKI-2 lowered the expression of the QKI isoforms, as expected, and also reduced the expression of the EGFR, similar to mimic miR-7 (Fig. 7A). Similar findings were obtained in U87 glioblastoma cells but to a lesser extent (see Fig. S4 in the supplemental material). We next examined whether QKI siRNA-transfected U343 cells had impaired extracellular signal-regulated kinase1/2 (ERK1/2) activation in response to EGF. Serum-starved U343 cells were stimulated with EGF for 15 min, and protein extracts were prepared and immunoblotted with anti-phospho-ERK antibodies. Robust ERK activation was observed in miR CTL and siCTL cells (Fig. 7B, lanes 2 and 6); however, this activation was impaired in miR-7-, siQKI-1-, and siQKI-2-transfected U343 cells (Fig. 7B, lanes 4, 8, and 10).
Fig 7.

QKI-deficient U343 cells have reduced EGFR expression, reduced EGF-dependent ERK activation, and reduced cell proliferation. U343 cells were transfected with 40 nM synthetic small RNAs: mimic miR negative control (miR CTL), mimic miR-7 (miR-7), siLuciferase (siCTL), and two siQKI siRNAs (siQKI-1 and siQKI-2), as described in Materials and Methods. In the rescue experiment, 100 nM miR-7 inhibitor or inhibitor control was cotransfected with the indicated siRNAs. (A) The cells were harvested at 48 h after transfection, and the total cell lysates were subjected to immunoblotting with anti-EGFR, anti-pan-QKI, and anti-β-tubulin antibodies as indicated. (B) The cells were starved overnight 48 h after transfection and then stimulated with 20 ng/ml of EGF for 15 min (+) or left untreated (−). Protein extracts were prepared, and pERK1/2, total ERK, and β-tubulin were detected by immunoblotting. (C) The cells were counted every 24 h after transfection using the Beckman Coulter Z2 cell counter. The cell numbers were normalized by the number of cells used for the transfection (day 0; between 190,000 and 260,000 cells). Two independent experiments were performed in duplicate. The data were expressed as averages ± standard deviations. **, P < 0.01; ***, P < 0.001 (Student t test). (D) The cells were photographed 48 h after transfection. The experiments were performed twice; one representative image is shown. (E) The cells were labeled with 10 μM BrdU for 1 h at 48 h after transfection and then harvested and stained with fluorescein isothiocyanate (FITC)-conjugated anti-BrdU antibody (green, FL1) and propidium iodide (red, FL2). The cell cycle was analyzed using FACS Calibur flow cytometry. Two independent experiments were performed; one representative FACS graph is shown. The averaged percentage of BrdU-positive cells was calculated from the two experiments. The FACS histograms and the percentage of cells at G0/G1, S, and G2/M phases are presented in Fig. S5 in the supplemental material.
Elevated miR-7 levels are known to suppress cell proliferation in glial cells (40, 42). To determine whether the increased miR-7 caused by the QKI siRNA altered the growth potential, we monitored cell proliferation and performed cell cycle analysis of U343 cells harboring miR CTL, miR-7, siCTL, and siRNAs targeting QKI. The transfection of miR-7, siQKI-1, and siQKI-2 prevented cell proliferation, while cells grew normally with miR CTL and siCTL, as assessed by cell counting using a Beckman Coulter Z2 cell counter (Fig. 7C). We reasoned that the growth inhibition observed in siQKI-1- and siQKI-2-transfected U343 cells was due to elevated miR-7 levels (Fig. 1D). Thus, we inhibited miR-7 using an miR-7 miRCURY LNA miRNA inhibitor. The transfection of the miR-7 inhibitor partially rescued the cell proliferation defect of siQKI-1- and siQKI-2-transfected U343 cells (Fig. 7C, right graph). Interestingly, cells depleted of the QKI isoforms using siQKI-1 and siQKI-2 had increased cell flattening (Fig. 7D), suggesting that these cells have an altered cytoskeleton. This phenotype was not observed in miR-7-treated cells (Fig. 7D), and it is reasonable to assume that the QKI-depleted U343 cells results in the alteration of other QKI targets in addition to miR-7. We also examined the cell proliferation defect of miR-7-, siQKI-1-, and siQK-2-transfected U343 cells by flow cytometry with propidium iodide (PI) (see Fig. S5 in the supplemental material), and with 2-color flow cytometry using PI and BrdU to label the cells in S phase (Fig. 7E). Indeed, the miR-7-, siQKI-1-, and siQKI-2-transfected cells were mainly in the G0/G1 phase of the cell cycle, with few cells in the S phase (Fig. 7E; see also Fig. S5). Taken together, our findings show that QKI-deficient U343 cells have altered EGFR expression and signaling. Moreover, these cells have defects in cell proliferation and are arrested in the G0/G1 phase, and this effect is partially attributed to elevated miR-7 levels.
DISCUSSION
In this work, we show that the depletion of the QKI isoforms in U343 glioblastoma cells leads to an miRNA imbalance. miR-7 was the miRNA that was the most elevated in QKI-depleted cells, and we pursued its characterization. We observed that pri-miR-7-1, and not pri-miR-7-2 or pri-miR-7-3, was the major primary miR-7 expressed in U343 cells, and interestingly, it harbors 3 putative QREs. The processing of pri-miR-7 to mature miR-7 was inhibited with the ectopic expression of the QKI-5 and QKI-6 isoforms, but not the cytoplasmic QKI-7 nor QKI-6 harboring an RNA binding-defective mutation (QKI-6:V-E). This regulation required the presence of the QREs within pri-miR-7-1. pri-miR-7-1 was tightly bound to the Drosha microprocesser complex in the presence of QKI, and this association was severely impaired in QKI-depleted U343 cells. These findings imply that the QKI isoforms associate with pri-miR-7-1 and may hinder Drosha processing. EGFR is a target of miR-7 (40), and indeed, QKI-depleted U343 cells, as well as U87 cells, had reduced EGFR expression and signaling to ERK. Elevated levels of miR-7 are known to inhibit cell proliferation, and indeed, we observed that siRNAs targeting QKI inhibited U343 cell proliferation and that this effect was partially reversed by inhibiting miR-7 activity. These findings suggest that the QKI isoforms regulate glial cell function by regulating the expression of specific miRNAs.
Glial cells such as oligodendrocytes are known to be regulated at multiple levels by epigenetics changes, including histone modifications and miRNAs (43, 44). The expression of >37 miRNAs is regulated during oligodendrocyte differentiation (45). Specifically, it was shown that miR-9 is able to target peripheral myelin protein gene pmp22 (45). In addition, miR-23 through targeting laminB1 is required for proper oligodendrocyte differentiation (46). These findings suggest a key role for miRNAs in oligodendrocyte differentiation. The role of miRNAs in oligodendrocyte function was further supported using a conditional allele of Dicer in mice (47, 48). The removal of Dicer in oligodendrocytes using CNP-Cre, Olig1-Cre, and Olig2-Cre resulted in mice with compromised myelin and increased immature progenitors (47, 48). miR-138, miR-219, and miR-338 have been shown to be upregulated during oligodendrocyte differentiation, and interfering with these miRNAs inhibits oligodendrocyte maturation. Overexpression of miR-219 and miR-338 was shown to be necessary to promote oligodendrocyte differentiation and to compensate for the loss of Dicer (47, 48). Since deficiency of QKI represses miR-338 in U343 cells (Fig. 1), it is likely that increased QKI expression during oligodendrocyte differentiation may contribute to the increased expression of miR-338 during oligodendrocyte maturation. Thus, QKIs regulate posttranscriptionally the expression of many genes directly by associating with QREs, and indeed, ∼2,500 transcripts were identified in vivo (25). Our findings suggest that the regulation of miRNAs by QKI isoforms observed herein provides an additional level of regulation in gene expression by altering miRNA levels.
Approximately 50% of human miRNAs are located within intronic regions of protein coding genes and the miRNA expression pattern often parallels that of the protein coding gene (49). pri-miR-7-1 is an miRNA located within intron 15 of the hnRNPK gene upstream of alternative splicing exons. We noted that QKI expression had no effect on the expression of pri-miR-7-1 or the expression of the hnRNPK gene or its 3′ splicing. Our results suggest that the QKI isoforms influence the efficiency of processing of pri-miR-7-1 and the export of the precursor miR-7. The presence of the QKI isoforms increases the association of pri-miR-7-1 with the Drosha microprocessor complex, as visualized by coassociation studies. It was shown that pri-miRNAs are processed cotranscriptionally (50, 51). Indeed, in QKI-depleted U343 cells, we observed a lack of nuclear retained pri-miR-7, consistent with it being processed more efficiently, resulting in elevated levels of miR-7.
The most frequent genetic alterations detected in glioblastomas include mutations within the TP53, CDK inhibitor p16INK4a, and phosphatase and tensin homologue deleted on chromosome 10 (PTEN) genes In addition, EGFR amplification, an increase in angiopoietin 1 (Ang 1), and vascular endothelial growth factor (VEGF-A) are frequently observed (52, 53). A study reported the loss of QKI mRNA expression in human glioblastomas and not other tumors (54). The qkI gene locus is located on 6q26-q27, a region commonly associated with abnormalities in human malignancies, including astroctytic tumors (55). The deletion of the qkI gene was observed in primary glioblastomas and anaplastic astrocytomas, defining qkI as a putative tumor suppressor gene (55). Another study identified glioblastoma cell line CRL2020 as a qkI knockout (56). In addition, the sequencing of 13,000 coding regions of colorectal and breast tumors identified 200 genes with mutations, including an R336Q mutation within qkI (57); however, it remains to be determined whether this mutation results in QKI loss of function. Recently, multidimensional cancer genome analysis has defined QKI as a putative tumor suppressor that is frequently (∼20%) deleted in glioblastomas (36). It was shown that QKI knockdown increases tumorigenicity, as assessed by increased growth in soft agar of glioma cells with stable clones containing shQKI and the increased incidence of tumors in flanks of nude mice harboring these QKI-deficient glioma cells (36). Although there is evidence suggesting that QKI may be a tumor suppressor, the loss of p53 in quaking viable mice did not accelerate tumor onset (58), and similarly, quaking viable mice harboring a patched1 null allele also did not have accelerated tumorigenesis (59). However, the quaking viable mice may not be an ideal model to study tumorigenesis, as these mice succumb to other defects when bred in the p53 null or patched1 background (58, 59).
A role for the QKI proteins was observed recently in miRNA function, as it was shown that QKI is downstream of p53 and regulates the stability of miR-20a, which, in turn, negatively regulates transforming growth factor β receptor 2 (TGF-βR2) (36). QKI was shown to associate with miR-20a via its KH domain, and this interaction is specific for miR-20a (36). The QKI/miR-20a interaction stabilizes miR-20a, which then suppresses TGF-βR2 (36). As miR-20a does not harbor a QRE, it remains unclear as to how QKI selectively recognizes miR-20a. We did not observe a variation in miR-20a in our microarray data (data not shown). Herein, we show a different mechanism by which QKI regulates miRNAs. We show that QKI associates with a QRE bearing pri-miR-7-1 and regulates its nuclear retention, such that the QKI-deficient cells have increased miR-7 production. We observe that robust QKI depletion of U343 cells using siRNAs alters cell morphology (Fig. 7D), leading to arrest in the G0/G1 phase of the cell cycle (Fig. 7C and E; see also Fig. S5 in the supplemental material). Moreover, this effect was partially rescued by the inhibition of miR-7 (Fig. 7C), suggesting that miR-7 contributes to the cell proliferation defect observed in QKI-depleted U343 cells.
miR-7 is a conserved miRNA that targets several genes. In Drosophila, miR-7 plays important roles against environmental fluctuations during development: it promotes photoreceptor differentiation of the eye and germ line stem cell lineage differentiation (60–62). In humans, ∼444 genes are predicted targets of miR-7 using Targetscan 6.1. Increasing evidence has indicated that miR-7 suppresses tumor generation in several human cancers by targeting multiple genes (63, 64). Moreover, miR-7 has been shown to inhibit the expression of EGFR and is downregulated in glioblastomas (40, 42). Since QKI regulates miRNA expression, it suggests that QKI also monitors gene expression during cell differentiation and tumor occurrence by altering miR-7 expression.
In conclusion, we identify a role for the QKI proteins in the regulation of miRNA processing in glial cells. We observed that the nuclear QKI isoforms associate with QREs within primary miR-7-1 and regulate the efficiency of its processing. These findings provide a new mechanism by which the QKI isoforms regulate glial cell proliferation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the Multiple Sclerosis Society of Canada (MSSOC). Y.W. was funded by a postdoctoral fellowship from the MSSOC.
Footnotes
Published ahead of print 14 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01604-12.
REFERENCES
- 1.Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136:215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mendell JT, Olson EN. 2012. MicroRNAs in stress signaling and human disease. Cell 148:1172–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Kouwenhove M, Kedde M, Agami R. 2011. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer 11:644–656 [DOI] [PubMed] [Google Scholar]
- 4.Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. 2004. Processing of primary microRNAs by the Microprocessor complex. Nature 432:231–235 [DOI] [PubMed] [Google Scholar]
- 5.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. 2004. The Microprocessor complex mediates the genesis of microRNAs. Nature 432:235–240 [DOI] [PubMed] [Google Scholar]
- 6.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. 2004. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 18:3016–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. 2002. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 21:4663–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U. 2004. Nuclear export of microRNA precursors. Science 303:95–98 [DOI] [PubMed] [Google Scholar]
- 9.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. 2001. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409:363–366 [DOI] [PubMed] [Google Scholar]
- 10.Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC. 2001. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106:23–34 [DOI] [PubMed] [Google Scholar]
- 11.Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD. 2001. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293:834–838 [DOI] [PubMed] [Google Scholar]
- 12.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. 2001. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 15:2654–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. 2004. Identification of mammalian microRNA host genes and transcription units. Genome Res. 14:1902–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. 2005. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436:740–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viswanathan SR, Daley GQ, Gregory RI. 2008. Selective blockade of microRNA processing by Lin28. Science 320:97–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, Sun S, Tu K, Gao Y, Xie B, Krainer AR, Zhu J. 2010. A splicing-independent function of SF2/ASF in microRNA processing. Mol. Cell 38:67–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG. 2009. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459:1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bockbrader K, Feng Y. 2008. Essential function, sophisticated regulation and pathological impact of the selective RNA-binding protein QKI in CNS myelin development. Future Neurol. 3:655–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chenard CA, Richard S. 2008. New implications for the QUAKING RNA binding protein in human disease. J. Neurosci. Res. 86:233–242 [DOI] [PubMed] [Google Scholar]
- 20.Ryder SP, Massi F. 2010. Insights into the structural basis of RNA recognition by STAR domain proteins. Adv. Exp. Med. Biol. 693:37–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebersole TA, Chen Q, Justice MJ, Artzt K. 1996. The quaking gene unites signal transduction and RNA binding in the developing nervous system. Nat. Genet. 12:260–265 [DOI] [PubMed] [Google Scholar]
- 22.Wu JI, Reed RB, Grabowski PJ, Artzt K. 2002. Function of quaking in myelination: regulation of alternative splicing. Proc. Natl. Acad. Sci. U. S. A. 99:4233–4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pilotte J, Larocque D, Richard S. 2001. Nuclear translocation controlled by alternatively spliced isoforms inactivates the QUAKING apoptotic inducer. Genes Dev. 15:845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galarneau A, Richard S. 2005. Target RNA motif and target mRNAs of the Quaking STAR protein. Nat. Struct. Mol. Biol. 12:691–698 [DOI] [PubMed] [Google Scholar]
- 25.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano MJ, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 14:129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larocque D, Pilotte J, Chen T, Cloutier F, Massie B, Pedraza L, Couture R, Lasko P, Almazan G, Richard S. 2002. Nuclear retention of MBP mRNAs in the Quaking viable mice. Neuron 36:815–829 [DOI] [PubMed] [Google Scholar]
- 27.Li Z, Zhang Y, Li D, Feng Y. 2000. Destabilization and mislocalization of the myelin basic protein mRNAs in quaking dysmyelination lacking the Qk1 RNA-binding proteins. J. Neurosci. 20:4944–4953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryder SP, Williamson JR. 2004. Specificity of the STAR/GSG domain protein Qk1: implications for the regulation of myelination. RNA 10:1449–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nabel-Rosen H, Volohonsky G, Reuveny A, Zaidel-Bar R, Volk T. 2002. Two isoforms of the Drosophila RNA binding protein, How, act in opposing directions to regulate tendon cell differentiation. Dev. Cell 2:183–193 [DOI] [PubMed] [Google Scholar]
- 30.Zhao L, Ku L, Chen Y, Xia M, LoPresti P, Feng Y. 2006. QKI binds MAP1B mRNA and enhances MAP1B expression during oligodendrocyte development. Mol. Biol. Cell 17:179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larocque D, Galarneau A, Liu HN, Scott M, Almazan G, Richard S. 2005. Protection of the p27KIP1 mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat. Neurosci. 8:27–33 [DOI] [PubMed] [Google Scholar]
- 32.Doukhanine E, Gavino C, Haines JD, Almazan G, Richard S. 2010. The QKI-6 RNA binding protein regulates actin-interacting protein-1 mRNA stability during oligodendrocyte differentiation. Mol. Biol. Cell 21:3029–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zearfoss NR, Clingman CC, Farley BM, McCoig LM, Ryder SP. 2011. Quaking regulates Hnrnpa1 expression through its 3′ UTR in oligodendrocyte precursor cells. PLoS Genet. 7:e1001269 doi:10.1371/journal.pgen.1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao L, Mandler MD, Yi H, Feng Y. 2010. Quaking I controls a unique cytoplasmic pathway that regulates alternative splicing of myelin-associated glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 107:19061–19066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Lacroix G, Haines J, Doukhanine E, Almazan G, Richard S. 2010. The QKI-6 RNA binding protein localizes with the MBP mRNAs in stress granules of glial cells. PLoS One 5:e12824 doi:10.1371/journal.pone.0012824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen AJ, Paik JH, Zhang H, Shukla SA, Mortensen R, Hu J, Ying H, Hu B, Hurt J, Farny N, Dong C, Xiao Y, Wan YA, Silver PA, Chin L, Vasudevan S, Depinho RA. 2012. STAR RNA-binding protein Quaking suppresses cancer via stabilization of specific miRNA. Genes Dev. 26:1459–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen T, Richard S. 1998. Structure-function analysis of Qk1: a lethal point mutation in mouse quaking prevents homodimerization. Mol. Cell. Biol. 18:4863–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Z, Chen T, Hébert J, Li E, Richard S. 2009. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol. Cell. Biol. 29:2982–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foà R, Schliwka J, Fuchs U, Novosel A, Müller RU, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter HI, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T. 2007. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129:1401–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kefas B, Godlewski J, Comeau L, Li Y, Abounader R, Hawkinson M, Lee J, Fine H, Chiocca EA, Lawler S, Purow B. 2008. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 68:3566–3572 [DOI] [PubMed] [Google Scholar]
- 41.Mili S, Steitz JA. 2004. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA 10:1692–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Webster RJ, Giles KM, Price KJ, Zhang PM, Mattick JS, Leedman PJ. 2009. Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7. J. Biol. Chem. 284:5731–5741 [DOI] [PubMed] [Google Scholar]
- 43.Huang J, Vogel G, Yu Z, Almazan G, Richard S. 2011. Type II arginine methyltransferase PRMT5 regulates gene expression of inhibitors of differentiation/DNA binding Id2 and Id4 during glial cell differentiation. J. Biol. Chem. 286:44424–44432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, Casaccia P. 2010. Epigenetic regulation of oligodendrocyte identity. Trends Neurosci. 33:193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lau P, Verrier JD, Nielsen JA, Johnson KR, Notterpek L, Hudson LD. 2008. Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J. Neurosci. 28:11720–11730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin ST, Fu YH. 2009. miR-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination. Dis. Model. Mech. 2:178–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dugas JC, Cuellar TL, Scholze A, Ason B, Ibrahim A, Emery B, Zamanian JL, Foo LC, McManus MT, Barres BA. 2010. Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron 65:597–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao X, He X, Han X, Yu Y, Ye F, Chen Y, Hoang T, Xu X, Mi QS, Xin M, Wang F, Appel B, Lu QR. 2010. MicroRNA-mediated control of oligodendrocyte differentiation. Neuron 65:612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griffiths-Jones S. 2007. Annotating noncoding RNA genes. Annu. Rev. Genomics Hum. Genet. 8:279–298 [DOI] [PubMed] [Google Scholar]
- 50.Kataoka N, Fujita M, Ohno M. 2009. Functional association of the microprocessor complex with the spliceosome. Mol. Cell. Biol. 29:3243–3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pawlicki JM, Steitz JA. 2008. Primary microRNA transcript retention at sites of transcription leads to enhanced microRNA production. J. Cell Biol. 182:61–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohgaki H, Kleihues P. 2009. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 100:2235–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wen PY, Kesari S. 2008. Malignant gliomas in adults. N. Engl. J. Med. 359:492–507 [DOI] [PubMed] [Google Scholar]
- 54.Li ZZ, Kondo T, Murata T, Ebersole TA, Nishi T, Tada K, Ushio Y, Yamamura K, Abe K. 2002. Expression of Hqk encoding a KH RNA binding protein is altered in human glioma. Jpn. J. Cancer Res. 93:167–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ichimura K, Mungall AJ, Fiegler H, Pearson DM, Dunham I, Carter NP, Collins VP. 2006. Small regions of overlapping deletions on 6q26 in human astrocytic tumours identified using chromosome 6 tile path array-CGH. Oncogene 25:1261–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mulholland PJ, Fiegler H, Mazzanti C, Gorman P, Sasieni P, Adams J, Jones TA, Babbage JW, Vatcheva R, Ichimura K, East P, Poullikas C, Collins VP, Carter NP, Tomlinson IPM, Sheer D. 2006. Genomic profiling identifies discrete deletions associated with translocations in glioblastoma multiforme. Cell Cycle 5:783–791 [DOI] [PubMed] [Google Scholar]
- 57.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. 2006. The consensus coding sequences of human breast and colorectal cancers. Science. doi:10.1126/science.1133427. [DOI] [PubMed]
- 58.Gavino C, Richard S. 2011. Loss of p53 in quaking viable mice leads to Purkinje cell defects and reduced survival. Sci. Rep. 1:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gavino C, Richard S. 2011. Patched1 haploinsufficiency impairs ependymal cilia function of the quaking viable mice, leading to fatal hydrocephalus. Mol. Cell. Neurosci. 47:100–107 [DOI] [PubMed] [Google Scholar]
- 60.Li X, Carthew RW. 2005. A microRNA mediates EGF receptor signaling and promotes photoreceptor differentiation in the Drosophila eye. Cell 123:1267–1277 [DOI] [PubMed] [Google Scholar]
- 61.Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW. 2009. A microRNA imparts robustness against environmental fluctuation during development. Cell 137:273–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pek JW, Lim AK, Kai T. 2009. Drosophila maelstrom ensures proper germline stem cell lineage differentiation by repressing microRNA-7. Dev. Cell 17:417–424 [DOI] [PubMed] [Google Scholar]
- 63.Saydam O, Senol O, Würdinger T, Mizrak A, Ozdener GB, Stemmer-Rachamimov AO, Yi M, Stephens RM, Krichevsky AM, Saydam N, Brenner GJ, Breakefield XO. 2011. miRNA-7 attenuation in Schwannoma tumors stimulates growth by upregulating three oncogenic signaling pathways. Cancer Res. 71:852–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skalsky RL, Cullen BR. 2011. Reduced expression of brain-enriched microRNAs in glioblastomas permits targeted regulation of a cell death gene. PLoS One 6:e24248 doi:10.1371/journal.pone.0024248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.