


Abstract
Type IIB restriction-modification systems, such as BcgI, feature a single protein with both endonuclease and methyltransferase activities. Type IIB nucleases require two recognition sites and cut both strands on both sides of their unmodified sites. BcgI cuts all eight target phosphodiester bonds before dissociation. The BcgI protein contains A and B polypeptides in a 2:1 ratio: A has one catalytic centre for each activity; B recognizes the DNA. We show here that BcgI is organized as A2B protomers, with B at its centre, but that these protomers self-associate to assemblies containing several A2B units. Moreover, like the well known FokI nuclease, BcgI bound to its site has to recruit additional protomers before it can cut DNA. DNA-bound BcgI can alternatively be activated by excess A subunits, much like the activation of FokI by its catalytic domain. Eight A subunits, each with one centre for nuclease activity, are presumably needed to cut the eight bonds cleaved by BcgI. Its nuclease reaction may thus involve two A2B units, each bound to a recognition site, with two more A2B units bridging the complexes by protein–protein interactions between the nuclease domains.
INTRODUCTION
Restriction-Modification (RM) systems use two enzyme activities to defend bacteria against foreign DNA, a restriction endonuclease (REase) and a modification methyltransferase (MTase) (1,2). The MTase transfers a methyl group from S-adenosylmethionine (SAM) to a cytosine or an adenine within a particular DNA sequence, the recognition site for that system, while the REase cleaves DNA with unmethylated sites (3). The majority of RM systems fall into either the Type I or Type II categories (4). Most Type I systems are oligomeric (R2M2S) proteins containing subunits for DNA cleavage (R), methylation (M) and sequence specificity (S) (2,5,6), though in some cases the R, M and S functions are all carried as domains within a single polypeptide (7). The cleavage activity is triggered by unmodified sites, though it often occurs distant from the site, due to the R subunits possessing an ATP-dependent DNA translocase. The main reaction of the MTase is to convert hemi-methylated sites, methylated in one strand only, to fully modified products methylated in both strands. Neither the fully nor the hemi-methylated DNA is cut by the REase. Consequently, DNA fully modified once by the MTase is never cleaved by the REase even after its semi-conservative replication.
Most Type II RM systems use separate proteins for restriction and modification that both bind to the same DNA sequence (1,8,9). The REase is commonly a homodimer that recognizes a palindromic sequence and cuts both strands at specified loci within the site: one subunit attacks the target phosphodiester bond in one strand, while the other cuts the scissile bond in the opposite strand (9,10). The MTase is usually a monomer whose main role is to transfer one methyl group onto DNA already methylated in one strand (8). But many Type II systems deviate considerably from these classical systems. They have been categorized into numerous subtypes—IIG, IIE, IIF, IIS and IIB, among others—on the basis of their genetic organization, their mode of action and their recognition sites and cleavage loci (4). Perhaps the only aspect common to all Type II REases, and the one that differentiates them from Type I systems, is that they cleave DNA at fixed positions relative to their recognition sites.
Variations in genetic organization include the Type IIG systems that, instead of separate REase and MTase proteins, possess a single polypeptide with both activities (11–14). The IIG systems require SAM not only as a co-factor for their methylation reactions but also to activate the nuclease.
Variations in mode of action are illustrated by the Type IIE and IIF systems (15,16). While the orthodox enzymes act at individual sites, the IIE and IIF REases need two copies of their target sequence for full activity. The IIE enzymes, often dimers, proceed to cut both DNA strands at one of the two sites; the other site acts as an allosteric activator (17,18). In contrast, the Type IIF enzymes tend to cut both strands at both sites before leaving the DNA (18,19): most operate as tetramers of identical subunits, with the four active sites, one in each subunit, each positioned to cleave one of the four target bonds (20–22). Enzymes that require two DNA sites can be identified by comparing their activities on DNA substrates with one or two cognate sites (19,22–24). Proteins that interact with two sites prefer sites in cis, where they loop out the intervening DNA, over sites in trans, where they must hold together two separate DNA molecules (16). Consequently, both Type IIE and IIF REases can cleave two-site substrates more rapidly than a one-site DNA. However, IIE enzymes cut two-site substrates one site at a time, whereas IIF enzymes cut both sites together (18).
Variations in recognition and cleavage loci are shown by the Type IIS REases, which bind asymmetric rather than palindromic sites and which cut the DNA at fixed loci away from the sequences, on one side of it (25,26). For example, FokI recognizes
and cuts both strands at the positions indicated (↓,↑), 9 and 13 nt away (3). An asymmetric sequence cannot be recognized in the same way as a dimeric enzyme recognizing a palindromic site, where each subunit contacts one half of the site (9,10). Instead, either a monomeric protein contacts the entire site, as is the case with FokI (27), or different subunits in an oligomeric protein each contact discrete parts of the sequence, as with BbvCI (28). If the nuclease is a monomer, it may have only one active site (26), yet it needs to cut both 5′–3′ and 3′–5′ strands. The use of the same active site to cut both strands is often—but not always—prohibited, as it requires the site to rotate not only around the DNA, from one strand to the other, but also perpendicular to the DNA, to match their antiparallel nature (29). To cut both strands, the monomer of FokI bound to the DNA associates with a second monomer (30–32) to give a dimeric unit with two active sites (33). The second active site can alternatively come from the isolated catalytic domain of the FokI protein, without its DNA recognition domain (30). The two catalytic centres each cut one specified strand: the DNA-bound monomer, the bottom strand 13 nt away and the recruited unit, the top strand 9 nt distant (34). However, the enzyme at the site associates more readily with another monomer bound elsewhere in the same DNA than with one in free solution, as proteins in cis cannot escape from each other (31,35). FokI thus attacks two-site substrates more rapidly than one-site DNA, although the two-site substrate is initially cut at just one site: the residual site is then cleaved at the same slow rate as the one-site DNA (23,31). The need for two recognition sites is common among the Type IIS REases: some, like FokI, act as IIE enzymes; others, like the tetramer BspMI, as IIF enzymes (36).
Another group that cleave outside their recognition sites are the Type IIB systems (37). Unlike the IIS enzymes that cut the DNA on one side of the site, the nucleases from the Type IIB systems make two double-strand breaks (DSBs) at each site, one either side, to excise a short fragment that carries the recognition sequence. Moreover, their recognition sequences usually consist of two specified but divergent blocks of sequence separated by a non-specific segment of fixed length, like many Type I sites (2,5). For example, BcgI, an archetype of the IIB enzymes (12,38–40), recognizes the sequence,
Its REase cuts both strands on both sides of this sequence, 10 and 12 nt away, to leave the site on a fragment 34 nt long in both strands (noted below as the 34-mer): its MTase targets the adenines marked above in italics. However, the BcgI nuclease has no activity when bound to a solitary site (41). Instead, like most Type IIB REases (24), it needs two sites (40), ideally in cis but alternatively in trans. After bridging the sites, it then proceeds to make two DSBs at both sites before releasing the final products, thus cutting a total of eight phosphodiester bonds within a single complex: hardly any of the partially cleaved intermediates accumulate during the course of the reaction (41).
BcgI, like many but not all Type IIB systems (37), is composed of two subunits: an A subunit of 71.6 kDa that carries both MTase and REase catalytic functions; a B subunit of 39.2 kDa that recognizes the target sequence (12,39). Like the Type IIG systems that have both activities in the same subunit, BcgI requires SAM not only for its MTase but also for its REase function: the nuclease also needs Mg2+ (38). Densitometry of coomassie-stained SDS gels yielded a 2:1 ratio of A:B subunits (12), albeit with the assumption—not always valid (42)—that the two polypeptides stain to the same extent. However, size exclusion chromatography of native BcgI gave apparent MW values of 250–300 kDa (39,43), larger than the 182 kDa expected for a protein with one B and two A subunits. Nevertheless, the size exclusion MWapp can be related to an A2B protomer if that unit has a large frictional ratio or if it self-associates (44).
The A subunit of BcgI resembles a fusion of R and M subunits from a Type I system without the ATP motor functions of R, while B is strikingly similar to an S subunit: it likewise possesses separate domains for recognizing each section of its bipartite target site (39). An A2B assembly for BcgI is thus a direct analogue of the R2M2S organization of Type I systems. Other Type IIB systems contain a single subunit with all three functions (13,45), akin to the single-polypeptide Type I enzymes (7). But while Type I REases generally make one DSB, usually about halfway between two recognition sites, BcgI makes four DSBs per reaction (41). How the BcgI RM protein is organized to hydrolyse eight phosphodiester bonds in a complex with two recognition sites is addressed here. In the following article (46), we show that the BcgI MTase transfers to an individual site in the DNA just one methyl group at a time. Hence, this protein may be organized differently for its REase and MTase activities.
MATERIALS AND METHODS
DNA
The plasmid pARG3-BcgI (a gift from New England Biolabs) over-expresses the native forms of bcgIA and bcgIB to yield the wild-type (WT) BcgI protein. The plasmid was subjected to site-directed mutagenesis (QuikChange II: Agilent) to replace Glu53 in the A subunit with Ala, to give pARG3-BcgIE53A [the E53A mutant has no nuclease activity (39)]. To construct the plasmid pAJJ1, the section of pARG3-BcgI encompassing bcgIA was amplified by PCR and the resultant fragment cloned between the NcoI and XhoI sites of pET28a (Merck).
The supercoiled (SC) forms of the plasmids pUC19 and pDG5 were 3H-labelled and purified as described previously (19,24). The following duplexes, all with HEX (hexachlorofluorescein) at the 5′-end of the top strand, were as before (41): HEX-42S, a 42 bp specific DNA with the recognition sequence for BcgI and both upstream and downstream cleavage loci; HEX-42NS, a 42 bp non-specific DNA that is identical to HEX-42S apart from single bp changes in each segment of the recognition sequence. HEX-24S and HEX-24NS are truncated versions containing the central 24 bp of HEX-42S and HEX-42NS respectively: HEX-24S thus has the recognition sequence but not the cleavage loci.
Proteins
Transformants of Escherichia coli XL1-Blue MFR with either pARG3-BcgI or pARG3-BcgIE53A, or E. coli Rosetta 2 with pAJJ1, were grown in 20 L L-broth with the appropriate antibiotics at 37°C in a Applikon Biotechnology bioreactor to an OD600 of 0.5–0.7. IPTG was added to 1 mM and the culture continued for 3–4 h before it was passed through a GEA Westphalia separator prior to cell harvesting by centrifugation. WT BcgI, the E53A mutant and the A protein were all purified by essentially the same procedure as described before for the WT protein (41), except that the initial extract—after cell disruption and the removal of the debris—was fractionated, first with polyethyleneimine and then with ammonium sulphate (47), prior to the column chromatography steps. As neither BcgIE53A nor the A subunit alone possess nuclease activity, the purification of these proteins was monitored by SDS-PAGE: the final preparations of all three proteins contained no detectable impurities (48). The identities of the purified proteins were confirmed by N-terminal amino-acid sequencing (W. Mawby, personal communication). Approximately 50% of the A subunit carried methionine at its N-terminus, as specified by its gene sequence, and 50% valine, the second residue in the sequence: the latter presumably arises from post-synthetic processing. Protein concentrations were assessed from A280 measurements using molar extinction coefficients calculated from amino acid compositions. Molarities of WT BcgI and E53A refer to the A2B assembly and that for the A protein to its monomeric state. Molecular masses encompass four possible states of the A protein (46): with and without the N-terminal methionine, and with or without bound SAM.
Native mass spectrometry
Aliquots of BcgIE53A were dialysed overnight against 200 mM AmAc (ammonium acetate), 10% glycerol, pH 8.4, and flash-frozen. When required, the samples were thawed on ice and buffer-exchanged twice with 200 mM AmAc using Micro Bio-Spin P6 columns (Bio-Rad) before being diluted in AmAc to the requisite concentration. (In this buffer, E53A still bound DNA specifically and the WT enzyme cleaved DNA when MgCl2 was added: data not shown.)
The protein sample was loaded into the mass spectrometer using nano-electrospray ionization (nano-ESI) in positive ion mode with an automated Advion Triversa inlet system (5 µm nozzle diameter, spray voltage 1.75 kV, gas pressure 0.25 psi). Mass to charge values were determined using a Waters Synapt 1 T-wave ion mobility quadrupole/time-of-flight tandem mass spectrometer with an 8 k quadrupole for medium-to-high m/z transmission. Experimental parameters were chosen to effect desolvation of high m/z sample ions while maintaining conditions that promote the transmission of intact protein complexes, with minimal disruption of native conformations (49): backing pressure, 5.4 mbar; source temperature, 25°C; sampling and extraction cone, 180 and 0 V, respectively; trap and transfer collision energies, 60 and 40 V; trap DC bias, 80 V. Data analysis was conducted using MassLynx 4.1 software (Waters) without background subtraction using smoothing by the Savitzky–Golay method (±50 channels).
Analytical ultracentrifugation
Sedimentation equilibrium experiments were carried out at 20°C in six-channel centrepieces for a Beckman XL-A ultracentrifuge. Sample chambers contained the protein—either WT BcgI, E53A or A protein—in 110 µl analytical ultracentrifugation (AUC) buffer (20 mM Tris–HCl, pH 8.4, 66 mM NaCl, 2 mM CaCl2, 20 µM SAM and 10% glycerol) and reference chambers 120 µl AUC buffer. During centrifugation, absorbancies were recorded along the centrifugal radius at 230, 280 or 292 nm, depending on the protein concentration. An initial scan was made directly on reaching 3000 rpm and the horizontal trace used to determine the protein concentration in the cell. Further scans were made after ≥16 h at speeds between 6000 and 15 000 rpm: at least two scans, ≥4 h apart, were taken at each speed. A final scan was recorded after 6 h at 40 000 rpm, to yield from the meniscus a baseline absorbance (B). Data sets were fitted in ORIGIN to Equation 1 for the distribution of a single ideal species after sedimentation to equilibrium:
| (1) |
where 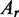 and
and 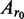 are the absorbance at radius
r and at the reference r0,
are the absorbance at radius
r and at the reference r0,
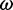 the centrifugal speed,
the centrifugal speed,
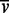 the partial specific volume and ρ the
solvent density: when applied to a single species, M denotes the
molecular mass of that species but when applied to a mixture of species of varied mass but
uniform
the partial specific volume and ρ the
solvent density: when applied to a single species, M denotes the
molecular mass of that species but when applied to a mixture of species of varied mass but
uniform 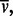 M denotes the mean weight-average MW (
M denotes the mean weight-average MW (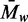 )
of all of the species in the cell (44,50). Multiple species were considered to be
present whenever the residuals between the best fit to Equation 1 varied systematically from the experimental data.
Further AUC experiments were carried out as above, but monitored at 539 nm, on samples
containing either the HEX-42S or the HEX-42NS duplex and excess BcgI.
)
of all of the species in the cell (44,50). Multiple species were considered to be
present whenever the residuals between the best fit to Equation 1 varied systematically from the experimental data.
Further AUC experiments were carried out as above, but monitored at 539 nm, on samples
containing either the HEX-42S or the HEX-42NS duplex and excess BcgI.
Reactions
DNA cleavage rates were generally measured by adding the requisite concentration of BcgI protein to 200 µl 3H-labelled DNA (5 nM) in buffer R (AUC buffer supplemented with 1 mM DTT and 200 µg/ml BSA, and with 10 mM MgCl2 in place of the CaCl2) at 37°C: the DNA was the SC form of either pUC19 or pDG5, which carry, respectively, one or two BcgI sites (24,41). Samples (10 µl) were taken before and at varied times after adding the enzyme, and mixed immediately with an EDTA quench prior to electrophoresis through agarose to separate the following DNA species: the intact SC substrate; the nicked open circle (OC) form; the linear (LIN) species with at least one DSB at one site and, for the two-site substrate, the final products with at least one DSB at both sites (L1 and L2). The concentration of each form was evaluated by scintillation counting. The reactions at each BcgI concentration were repeated at least three times, although occasionally with different sampling times for each test. For repeats with the same time points, the data at each time were averaged and the averages fitted to a single exponential decay with offset. For reactions sampled at different times, the decrease in SC DNA in each reaction was fitted to an exponential and the apparent rate constants then averaged.
Reactions that were too fast to measure by manual sampling were carried out by mixing, in a Hi-Tech RQF-63 quench-flow apparatus (TgK Scientific), equal volumes (80 µl) of 3H-labelled pDG5 (10 nM) and BcgI (at twice the required concentration), both in buffer R at 37°C. After the requisite delay time, the reaction was mixed with 80 µl EDTA (500 mM). Samples (100 µl) of the quenched reaction were then analysed by electrophoresis through agarose as above, to yield the residual concentration of the SC DNA.
For BcgI reactions in the presence of additional A protein, plasmid cleavage and the release of the 34-mer were measured in parallel (41). The reactions, in 200 µl buffer R* (as Buffer R but 5% [w/v] Ficoll 400 instead of the glycerol) at 37°C, contained 5 nM 3H-labelled pUC19 and specified concentrations of both native BcgI and BcgIA proteins. Samples (20 µl) were taken from the reactions at various times, mixed immediately with quench supplemented with 250 µg/ml proteinase K and incubated at 37°C for ≥30 min. Half of each sample was analysed by electrophoresis through agarose, as above, to evaluate the concentrations of the DNA species that could be resolved on agarose (the SC, OC and LIN forms of the plasmid). The other half was loaded onto a native 20% acrylamide gel, alongside an equivalent amount (5 nM) of HhaI-digested pUC19. The acrylamide gel was stained with SYBR Safe (Invitrogen) for 20 min, washed and its fluorescence recorded in a Typhoon PhosphorImager (Molecular Dynamics) using its 488 nm laser and 520 BP 40 emission filter. Images were analysed using ImageQuant software. The concentration of the 34-mer was determined relative to the known concentration of the 28 and 33 bp fragments from the HhaI-digested pUC19 control.
DNA binding
Tests to measure DNA binding by BcgI were carried out at 37°C in 10 µl buffer R*C (as Buffer R* but with 2 mM CaCl2 instead of the MgCl2) containing a HEX-labelled oligoduplex (20 nM) and varied concentrations of native BcgI. After adding the protein, samples were incubated for 5 min and then loaded directly onto 6% acrylamide gels in TBC (as TBE but with 2 mM CaCl2 in place of the EDTA) running at 4 V/cm. After loading, the voltage was increased to 12.5 V/cm. Gels were imaged in their plates in the PhosphorImager using its 532 nm laser and 555 BP 20 emission filter. Images were analysed in ImageQuant and the concentration of free DNA in each lane assessed relative to that in the lane without BcgI. The background was taken at an equivalent position in a lane lacking DNA.
RESULTS
Glu53 in the A subunit of the BcgI RM protein forms part of the PD… EXK motif common to many nuclease active sites (9) and its replacement with Ala abolishes the nuclease activity of BcgI (39). Hence, to enable studies of BcgI without its cleavage reaction, an over-producing strain for the E53A mutant of BcgIA was constructed (see ‘Materials and Methods’ section). Preparations of the E53A mutant from this strain yielded about twice the amount of protein as parallel preparations of the active WT enzyme (48). Over-expression of the WT nuclease may result in cell death due to cleavage of the chromosome at non-cognate sites, as seen with other restriction enzymes (51,52), although the BcgI RM protein carries the MTase that ought to protect the recognition sites on the cellular DNA (12,39). On the other hand, cells over-producing the inactive mutant might remain viable and so generate more of the protein. Experiments requiring high concentrations of the BcgI protein were thus facilitated by using the E53A protein rather than WT BcgI.
Mass spectrometry
Native mass spectrometry (MS) was carried out primarily on the E53A mutant, although equivalent spectra were obtained with WT BcgI albeit with lower signal-to-noise ratios (data not shown). Under conditions where non-covalent interactions are preserved, the spectrum in Figure 1 shows a series of peaks, which are assigned to the A and B subunits and their complexes with different stoichiometries. The predominant species (∼75% of the total intensity) is the A2B complex with an m/z of around 7000, with principal charge states of 28+ and 29+. The mass calculated for the A2B complex is ca. 182.4 kDa, as averaged over the four different species (±N-terminal methionine and ±bound SAM; see above), which are not resolved here. The major peak in Figure 1 yields an experimental mass of 182 434, which matches closely the A2B form of BcgI. In addition, further multiples of A2B assemblies were observed up to at least four such units, an (A2B)4 complex at around m/z 13 000 (Figure 1, insert). The relative ratios of these higher-order complexes in the sample cannot be directly measured from the spectrum, as the sensitivity of the ion detector decreases with increasing m/z in a non-linear way. The time-of-flight system has in theory no upper limit on the size of molecule it can detect but the transmission and the detection of ions depends on their mass. It is therefore difficult to determine the relative amounts of each species in the solution.
Figure 1.

Native MS. A nano-ESI mass spectrum of the E53A form of BcgI (27.5 µM) in 200 mM AmAc was recorded under non-denaturing conditions (see ‘Materials and Methods’ section). The insert shows the signals for the oligomeric states in the m/z range from 8500 to 14 500 magnified 5-fold. The charge state series assigned to each molecular species (in bold) are indicated with dashed brackets, and the charge and m/z value for the primary peak in each series is given.
In addition to the presence of these larger species, some separated components of the complexes were also observed. The masses of these smaller species, up to m/z 6000, correspond to the BcgIA and BcgIB subunits by themselves and to an AB assembly (Figure 1). These species are likely to be subunits that had dissociated from the BcgI endonuclease in solution, as suggested by the low charge states of these species (gas-phase dissociation products would have had disproportionally high charge states, similar to denatured proteins). In an A2B protomer, the BcgIB subunit would normally be expected to have the weakest non-covalent linkage to the assembly as its mass, and therefore surface area, is less than that of the A subunit: this ought to make it easier for BcgIB to leave the complex than BcgIA. Hence, the presence of an AB species, but not an A2 dimer, suggests that the two A subunits in the active BcgI protein both interact with B but not with each other: the A subunits presumably flank the B subunit, one on either side. If this suggestion is correct, then the first step in the disassembly of the protein should be the dissociation of one BcgIA subunit from the A2B unit to leave AB, as is seen in the spectra.
AUC
Native MS identified several assemblies of the BcgI protein containing multiple copies of the A2B protomer but it does not provide quantitative information about the amount of each species. Moreover, the native MS was done in a buffer designed to permit volatilization of the sample rather than that for optimal enzyme activity. To address these issues, AUC experiments were performed on WT BcgI under conditions similar to those used for its DNA cleavage assays, though in Ca2+ rather than Mg2+ so as to allow for DNA binding without cleavage (53). The inactive E53A mutant of BcgI behaved similarly to the WT in both the absence and presence of DNA and, in this case, with either Ca2+ or Mg2+ at hand (data not shown).
Sedimentation equilibrium experiments were carried out with varied concentrations of WT
BcgI. After centrifugation for ≥20 h to achieve equilibrium (as evidenced by
overlaying traces from successive scans ≥4 h apart), the radial distribution of the
protein was measured by absorbance at 280 nm for protein concentrations between 1 and 10
µM; or at 230 or 292 nm for lower and higher concentrations, respectively (Figures 2a and 2b). At each concentration, the
distribution was fitted to Equation
1, for the sedimentation of a single ideal species: the fits encompassed several
data sets from different times. At the lowest concentration of BcgI tested (0.4 µM),
the residuals between the experimental data and the best fit to Equation 1 were both small and random,
showing that the data can indeed be accounted for by a single species (Figure 2a). The AUC data at low BcgI
concentrations yielded a MW of 183 kDa, which corresponds closely to the 182.4 kDa
expected for the A2B protomer. In contrast, the fit of the data at high
concentrations to Equation 1 gave
large and non-random residuals (Figure 2b),
suggesting either the presence of multiple species and/or non-ideality (44). In this situation, the best fit to Equation 1 does not yield the MW of any
individual species but rather the mean weight–average MW
(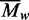 ) of all of the species in the sample (44,50).
) of all of the species in the sample (44,50).
Figure 2.

Sedimentation
equilibrium. Samples of BcgI in AUC buffer were centrifuged at 7500 rpm at 20°C
for the times noted below. The main panels show the absorbance (white circles) as a
function of the centrifugal radius: (a) 0.44 µM BcgI at 230 nm
after 30 h; (b) 18 µM BcgI at 292 nm after 44 h. The lines
through the data points in the main panels are the best fits of the following data
sets to Equation 1: (a) the
records after 26 and 30 h; (b) after 32, 38 and 44 h. The best fits were obtained
with values for M of 183 and 431 kDa in (a) and (b) respectively.
The plots above the main panels display the residuals between these fits and the
experimental data: the random variations in (a) show that the record can be
accounted for by a single ideal species with a unique value of M;
the systematic deviations in (b) demonstrate that more than one species is present,
which gives rise to a 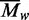 value.
value.
Values for 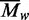 were obtained at each loading concentration
of BcgI tested (Figure 3). The
were obtained at each loading concentration
of BcgI tested (Figure 3). The
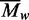 values increased with increasing levels of
BcgI: from that corresponding to a single A2B unit at low concentrations, then
to values consistent with dimers of A2B and finally to levels indicating
assemblies with more than two protomers. At the highest concentration tested (18 µM:
Figure 2b), the
values increased with increasing levels of
BcgI: from that corresponding to a single A2B unit at low concentrations, then
to values consistent with dimers of A2B and finally to levels indicating
assemblies with more than two protomers. At the highest concentration tested (18 µM:
Figure 2b), the
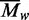 came to 431 kDa. As these mean values for
came to 431 kDa. As these mean values for
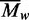 encompass the full range of protein
concentrations present in the centrifuge cell after sedimentation to equilibrium, from low
concentrations at the inside edge to high at the outside, a significant fraction of the
protein in this sample must be present in assemblies containing more than two protomers.
encompass the full range of protein
concentrations present in the centrifuge cell after sedimentation to equilibrium, from low
concentrations at the inside edge to high at the outside, a significant fraction of the
protein in this sample must be present in assemblies containing more than two protomers.
Figure 3.

Self-association of BcgI.
Varied concentrations of BcgI in AUC buffer were loaded into the AUC and sedimented
to equilibrium at 7500 rpm at 20°C. For each sample, the absorbance was measured
as a function of centrifugal radius and fitted to Equation 1. Apart from the data at the lowest BcgI
concentrations (viz. Figure
2a), the residuals between the fits and the experimental data displayed
non-random deviations (viz. Figure 2b), so the best fit at each initial concentration denotes an
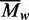 value. The
value. The
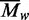 values are plotted against the
loading concentrations of BcgI. The values expected for the 182.4 kDa A2B
protomer and for the 365 kDa (A2B)2 dimer are marked with long
and short dashes, respectively.
values are plotted against the
loading concentrations of BcgI. The values expected for the 182.4 kDa A2B
protomer and for the 365 kDa (A2B)2 dimer are marked with long
and short dashes, respectively.
AUC was also used to analyse complexes of WT BcgI with either a specific duplex carrying
both recognition and cleavage sites (HEX-42S) or a non-specific DNA that lacked the
recognition sequence (HEX-42NS). Sedimentation was monitored at 539 nm, to detect the HEX
moiety on the DNA, either in its free state or when enzyme-bound. In the absence of BcgI,
MW values of 27.6 and 26.6 kDa were obtained for HEX-42S and HEX-42NS, respectively (data
not shown), close or equal to the expected MW, 26.6 kDa. When the non-specific duplex (2
µM) was centrifuged in the presence of excess BcgI (7.5 µM), the initial
uniform distribution of the HEX label throughout the cell (Figure 4a, black trace) was converted into the characteristic
profiles for an approach to sedimentation equilibrium (Figure 4a, coloured traces). The records at equilibrium (>22
h) yielded a 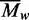 of 349 kDa, much larger than that for the
free DNA so all of the DNA must be bound to the protein. The
of 349 kDa, much larger than that for the
free DNA so all of the DNA must be bound to the protein. The
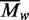 for the non-specific complex is similar to
that for the same concentration of free protein (Figure 3), which indicates that either two or more protomers can bind to one
duplex or that the DNA–protein complexes associate with themselves. Conversely,
after adding BcgI (7.5 µM) to the specific duplex (2 µM) and spinning the
sample at low velocity, the solution lacked virtually any absorbance at 539 nm (Figure 4b, black trace). Subsequent scans after
centrifugation at a higher speed revealed a progressive recovery in the
A539 signal (Figure
4b, coloured traces) but even after 54 h, the total absorbance in the cell
containing specific DNA was still lower than that containing non-specific DNA and had yet
to reach equilibrium. The complex of BcgI with specific DNA must precipitate out of
solution as soon as it is formed and then slowly redissolves back into solution. An 18 bp
duplex that contains the recognition site but not the cleavage loci also precipitated from
solution on the addition of WT BcgI, as did the specific 42 bp duplex with E53A in the
presence of either Ca2+ or Mg2+ (data not shown).
for the non-specific complex is similar to
that for the same concentration of free protein (Figure 3), which indicates that either two or more protomers can bind to one
duplex or that the DNA–protein complexes associate with themselves. Conversely,
after adding BcgI (7.5 µM) to the specific duplex (2 µM) and spinning the
sample at low velocity, the solution lacked virtually any absorbance at 539 nm (Figure 4b, black trace). Subsequent scans after
centrifugation at a higher speed revealed a progressive recovery in the
A539 signal (Figure
4b, coloured traces) but even after 54 h, the total absorbance in the cell
containing specific DNA was still lower than that containing non-specific DNA and had yet
to reach equilibrium. The complex of BcgI with specific DNA must precipitate out of
solution as soon as it is formed and then slowly redissolves back into solution. An 18 bp
duplex that contains the recognition site but not the cleavage loci also precipitated from
solution on the addition of WT BcgI, as did the specific 42 bp duplex with E53A in the
presence of either Ca2+ or Mg2+ (data not shown).
Figure 4.

AUC of BcgI–DNA complexes. The
samples, in AUC buffer at 20°C, contained WT BcgI protein (7.5 µM) and a
DNA duplex (2 µM) as follows: (a) HEX-42NS, a non-specific DNA
that lacks the recognition sequence; (b) HEX-42S, a specific DNA with
both recognition and cleavage loci. The samples were centrifuged in the AUC and the
absorbance across the cell monitored at 539 nm at the following stages after
initiating the run: the instrument was initially set to spin at 3000 rpm and an
absorbance scan recorded immediately on reaching that speed (black trace). The
velocity was then raised to 7500 rpm and scans taken after 14 h and after further 8
h delays. The records shown are after 14 h (green trace), 22 h (cyan trace), 38 h
(pink trace) and 54 h (blue trace). The data sets with the non-specific duplex (a)
recorded after ≥30 h were fitted to Equation 1: the best fit gave a
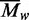 of 349 kDa.
of 349 kDa.
DNA cleavage
During its DNA cleavage reaction, the BcgI protein interacts with two copies of its recognition sequence and then proceeds to make four DSBs, one either side of each site, thus cutting a total of eight phosphodiester bonds in a single DNA–protein complex (41). It is highly unlikely that one A2B protomer can cut all eight bonds because it contains only two active sites for phosphodiester hydrolysis, one in each A subunit. Instead, it seems more likely that DNA cleavage is carried out by an assembly of A2B units, maybe like those seen by native MS and AUC. This possibility was addressed by analysing how the extent of cleavage of plasmids with one or two BcgI sites varied as the protein concentration was raised from below to above the level of BcgI sites in the DNA (Figure 5). BcgI can act with two recognition sites in cis or in trans, although, as is usual for interactions bridging two loci (16), it prefers sites in cis over those in trans. Two-site substrates are thus cleaved more readily than one-site DNA (24,40).
Figure 5.

DNA cleavage at low [BcgI]. Reactions in buffer R at 37°C contained one of the following plasmids (3H-labelled, initially ∼90% SC monomer) at 5 nM and BcgI protein at the concentrations noted below. The plasmids were (a) pUC19, which has one BcgI site; (b) pDG5, which has two sites. The enzyme concentrations for each reaction are marked on the right: 2 nM, white circles; 5 nM, black circles; 10 nM, white squares; 20 nM, black squares. Samples were taken at the indicated times and analysed by electrophoresis to separate the SC substrate from the cleaved products. The residual concentrations of the SC DNA were determined and are plotted as a function of time. The lines are the best fits to single exponential decays with floating offsets.
In previous studies (40), the extent of cleavage of plasmids with one or two BcgI sites was measured at varied BcgI concentrations after a fixed time (15 min): complete cleavage of the DNA within this time required a molar excess of protein over DNA, with more protein needed for the one-site than for the two-site DNA. As the extent of cleavage after a fixed time interval may reflect either the end point or the rate of the reaction, reactions at varied BcgI concentrations were monitored in this study over extended time bases (Figure 5). On the one-site plasmid, no—or little—cleavage was detected at BcgI levels below or equal to that of the DNA, even after prolonged incubation (Figure 5a). Instead, the reaction on the one-site substrate required enzyme concentrations in excess of the DNA. The extent of cleavage increased as the ratio of enzyme to recognition sites was raised above 1:1 but complete cleavage was only observed with a substantial excess of protein over DNA. The plasmid with two cognate sites might have been expected to require more enzyme per mole DNA to fill both sites but instead almost all of the two-site DNA was cleaved at equal levels of enzyme and DNA: i.e. with BcgI at half the concentration of recognition sites (Figure 5b).
Enzyme concentrations that gave complete cleavage of the DNA did not necessarily give maximal reaction rates: for instance, the two-site plasmid (present at 5 nM) was cleaved fully by both 10 and by 20 nM BcgI protein but the latter gave a faster rate (Figure 5b). Cleavage of both one- and two-site substrates was therefore studied across a range of BcgI concentrations far above that of the DNA (Figure 6). During these reactions, the concentrations of the SC DNA substrates fell exponentially to end points where essentially all of the DNA had been cleaved. With enzyme in excess over the DNA, the rate constants (kobs) from the exponential decline of the SC DNA denote the cleavage of at least one phosphodiester bond in the enzyme–DNA complex rather than the entire reaction ending with the release of the DNA cleaved at eight bonds. Phosphodiester hydrolysis by BcgI bound to the one-site substrate took ≥2 min to reach completion and could be monitored by manually mixing the reagents (Figure 6a). In contrast, BcgI bound to the two-site plasmid could take <0.2 min to cut all of the DNA, so in these cases the experiments were done in a rapid quench-flow device (Figure 6b). At BcgI concentrations that permitted both hand-mixing and quench-flow approaches, the two techniques yielded comparable results (data not shown).
Figure 6.

DNA cleavage at high [BcgI]. (a) The reactions, in buffer R at 37°C, contained 5 nM 3H-labelled SC pUC19 and BcgI protein at 75 nM, white circles; 250 nM, black circles; 750 nM, white squares. After manually mixing the reagents, samples were taken from the reactions by pipette and mixed immediately with an EDTA quench. The samples were then subjected to electrophoresis through agarose, to enable the concentration of SC DNA to be determined. The amount of SC DNA present in each sample is plotted against the time. (b) As (a) except that the DNA was SC pDG5 and that the reactions were carried out by quench-flow. (c) Apparent rate constants (kobs) from ≥3 independent experiments at each BcgI concentration were averaged and plotted against the concentration: for reactions on pUC19, white circles; for reactions on pDG5, black circles. Error bars denote standard deviations. The line drawn through each data set is the best fit to a hyperbolic function, kobs = (kmax × [BcgI])/(K½ + [BcgI]). The best fits were obtained with the following: for pUC19, kmax = 2.7 min−1 and K½ = 232 nM; for pDG5, kmax = 21 min−1 and K½ = 230 nM.
The values for kobs (Figures 6a and 6b: other data not shown) were plotted against the enzyme concentration (Figure 6c). For both one- and two-site plasmids, the rates increased with increasing BcgI concentrations in a hyperbolic manner (Figure 6c). The data with each substrate were fitted to an equation for a rectangular hyperbola to yield values for kmax (the rate constant for DNA cleavage at saturation of the DNA with protein) and for K½ (the protein concentration for half maximal rate). The two-site plasmid gave nearly a 10-fold higher value for kmax than the one-site DNA, 21 min−1 compared with 2.7 min−1. In contrast, the one- and two-site substrates gave the same value for K½, 230 nM in both cases. The faster rate of cutting the two-site over the one-site plasmid is thus due solely to the different kmax values. The DNA cleavage step in the BcgI reaction on sites in cis is thus faster than that on sites in trans.
The values for K½ merit two comments. Firstly, they indicate that remarkably high BcgI concentrations are required for its maximal reaction rate: cleavage of 5 nM DNA at 90% of its maximal rate requires an enzyme concentration of >2000 nM. Secondly, though proteins that bind two DNA sites nearly always have higher affinities for sites in cis than sites in trans (16), the equal K½ values show that reactions with two sites in trans feature the same affinity for BcgI protein as that during reactions with sites in cis. The K½ values may therefore reflect a weak protein–protein association in the assembly of the reaction complex rather than the DNA–protein association.
DNA binding
An alternative explanation for why DNA cleavage by BcgI requires such large amounts of protein is that the enzyme simply has a low affinity for its recognition sequence. To test this possibility, the binding of the BcgI protein to specific and non-specific DNA was examined directly by gel retardation (Figure 7). The binding studies were initially done with two 42 bp oligoduplexes, both labelled with HEX at one 5′ terminus. One, HEX-42S, is a potential substrate, as it carries both the recognition sequence and the cleavage loci both sides of the site. The other, HEX-42NS, differs only at 2 bp, one in each segment of the bipartite site so is effectively a non-specific sequence for BcgI (41). The buffer used here contained Ca2+ rather than Mg2+, to allow WT BcgI to bind to HEX-42S without cutting off its ends. Increasing concentrations of BcgI were added to a fixed amount of each DNA and the free DNA then separated by electrophoresis from the protein-bound species with retarded mobilities (Figures 7a and 7b). The extent of binding at each enzyme concentration was then determined by assessing the fluorescence from the band corresponding to the free DNA relative to that in samples without enzyme (Figure 7c). Similar results (not shown) were obtained with the inactive E53A mutant with either Ca2+ or Mg2+ and with no metal present.
Figure 7.

DNA binding. (a and b) The mixtures contained, in 10 µl buffer R*C, 20 nM DNA, either HEX-42S (a) or HEX-42NS (b), and BcgI at one of the following levels, from left to right: 0, 20, 40, 60, 80, 100, 150 or 200 nM. After 5 min at 37°C, the samples were subjected to electrophoresis through polyacrylamide and the fluorescence from each band recorded. On the left of gel images, F marks the position of the free DNA; C, the DNA–protein complexes that entered the gel; and W, the bottom of the wells. (c) From titrations of both HEX-42S and HEX-42NS with increasing amounts of BcgI protein, and from additional experiments with 24 bp duplexes (HEX-24S and HEX-24NS: gels not shown), the amount of free DNA in each sample was determined relative to the amount in a parallel lane without enzyme. The % DNA in the free form is shown as a function of the BcgI concentration: HEX-42S, white triangle; HEX-42NS, white squares; HEX-24S, black triangles; HEX-24NS, black squares. Mean values from three repeats are given: error bars show standard deviations. The red lines through the data with the specific duplexes denote stoichiometric binding where all of the added BcgI is bound to DNA until saturation of the DNA is achieved.
When increasing concentrations of BcgI were added to the specific duplex, HEX-42S, the concentration of the free DNA decreased linearly with the protein concentration until it had all been converted to DNA–protein complexes (Figure 7c). BcgI thus binds to DNA carrying its recognition sequence in a stoichiometric fashion, where all of the added BcgI is bound to the DNA until saturation of the DNA is achieved, as opposed to a dynamic equilibrium between free and bound proteins. However, saturation of the specific duplex seems to require 3–4 mole BcgI per mole DNA, as it took ∼70 nM BcgI to saturate 20 nM DNA (Figure 7c, intersection of red lines). BcgI also bound to the non-specific DNA, HEX-42NS, although with a lower affinity than the specific sequence (Figures 7b and 7c). Moreover, the specific and non-specific sequences gave rise to different complexes. The specific DNA was initially transformed to species that failed to enter the polyacrylamide gel and remained in the wells (Figure 7a): species that entered the gel were observed only after sufficient enzyme had been added to convert all of the DNA to complexes. The nature of the complexes that remain in the wells and those that enter the gel have yet to be established. They may be related to the complexes seen in the AUC, where the addition of BcgI to cognate DNA led initially to an insoluble complex which then slowly redissolved back into solution (Figure 4b). In contrast, all of the complexes formed with the non-specific DNA entered the gel (Figure 7b) but, as with the specific DNA, the species within the gel displayed progressively reduced mobilities with increasing amounts of BcgI, probably due to the binding of additional molecules of protein to each molecule of DNA.
Further experiments were carried out with 24 bp duplexes with or without the recognition sequence, HEX-24S and HEX-24NS: the former contains the recognition sequence but lacks the cleavage loci 10 and 12 nt away from the site. Nevertheless, BcgI bound to both of these 24 bp duplexes in the same manners (gels not shown) and with the same affinities (Figure 7c) as their 42 bp counterparts. The cleavage loci are therefore not needed for specific binding to the site.
A striking feature of the equilibrium binding of BcgI to its cognate sequence is that a much lower concentration of the protein is required to saturate binding to the site (Figure 7c) compared with the amount needed to saturate the DNA cleavage rate (Figure 6c): 70 cf. >2000 nM. A scheme that can account for this behaviour is a mechanism of the type seen with the FokI restriction enzyme (30–32),
In this scheme, one molecule of the enzyme (E) first binds to its DNA site (S) with an equilibrium dissociation constant KDNA but the resultant E.S complex has no activity until the DNA-bound protein associates with a second protein, with a constant Kprot for the protein–protein assembly: the complex containing two molecules of enzyme then proceeds to cleave the DNA at the kmax rate. In the case of FokI (31,32), the KD for the binding of the enzyme to its site (4 nM) is much smaller than that for the protein–protein association (in the range from 100 to 166 nM), so the level of FokI needed for its maximal DNA cleavage rate is much higher than that required to fill its binding sites on the DNA. The same seems to apply to BcgI, though the stoichiometry of the binding reaction (Figure 7c) implies that even its initial E.S complex must contain more than one A2B unit, and that it then proceeds to bind yet more A2B units. Indeed, if the K½ from its maximal cleavage rate (230 nM) corresponds to its value for Kprot, then the E.S complexes for BcgI and for FokI display similar affinities for their additional enzyme.
Activation by BcgIA
The monomer of FokI at its recognition site can be activated by associating not only with intact FokI protein but also with just its catalytic domain (30). The association of the intact protein and the catalytic domain generates a unit with two active sites that can then proceed to cut both DNA strands (33). The nuclease function of the BcgI protein resides in its A subunit while the sole function of B is DNA recognition (12,39). Hence, it might be possible to activate the BcgI REase at its recognition site by excess A protein, isolated from the B subunit. To test this idea, the bcgIA gene was cloned and over-expressed in E. coli cells that lacked bcgIB and the A protein purified to homogeneity (see ‘Materials and Methods’ section). In solution, BcgIA was found by AUC and native MS to exist primarily as a 71.5 kDa monomer (48). The fact that the isolated BcgIA protein exists as a monomer concurs with the native MS on the native BcgI protein, which had shown A monomers, AB and A2B assemblies, but no A2 dimers (Figure 1).
When the purified A protein was added by itself to the one-site plasmid pUC19, no DNA cleavage was observed even after long incubations with high BcgIA concentrations (Figure 8, red line). BcgIA was then added to pUC19 together with intact BcgI protein. However, the latter was present at the same concentration (5 nM) as the DNA, which is too low for it to cleave the DNA unaided (Figure 5a). Samples were taken from the mixture and a portion of each analysed by electrophoresis through agarose to separate the SC, OC and LIN forms of pUC19. Though neither protein alone was capable of cutting this DNA, together they rapidly converted most of the SC plasmid directly to LIN DNA, without accumulating nicked OC forms en route (Figure 8). The LIN DNA could have been cut in both strands on just one side of the BcgI site, to give a LIN species of 2686 bp, or on both sides to release the 34-mer product and a LIN DNA of 2652 bp. As these LIN forms are inseparable on agarose, a second portion from each sample was applied to a polyacrylamide gel to capture the 34-mer (41). The increase in the concentration of the 34-mer matched that for the LIN DNA (Figure 8). The A protein must therefore enable the intact protein to make DSBs on both sides of its site. Hence, both A subunits in the A2B protomer at the recognition site interact with free BcgIA monomers to give an assembly with two pairs of nuclease active sites, each of which makes a DSB. If the addition of A to A2B had resulted in only one unit for DSBs, it would have first cut one side of the site and only later the other side: the 34-mer would then have been liberated not at the same time as the LIN species but instead at some later time. Strikingly, extra A subunits also enhance the MTase activity of the BcgI protein (46) but, as the MTase methylates only one adenine at a time, the activation of the MTase almost certainly does not require the catalytic functions for methyl transfer in the free A subunit. In contrast, the liberation of the 34-mer shows that the extra A subunits contribute to the BcgI REase functional active sites for phosphodiester hydrolysis.
Figure 8.

Activation of BcgI by BcgIA. The reactions, in 200 µl buffer R* at 37°C, contained 100 nM BcgIA protein and 5 nM 3H-labelled pUC19 (initially 90% SC monomer), in either the absence or presence of 5 nM WT BcgI. Aliquots (20 µl) were removed at the times shown and immediately quenched before analysis by electrophoresis: one-half through agarose, to separate the SC, OC and LIN forms of the plasmid; the other half through polyacrylamide, to capture the 34-mer. The concentrations of the various forms of pUC19 were evaluated from the agarose gel and that for the 34-mer from the polyacrylamide gel (see ‘Materials and Methods’ section). For the reaction with BcgIA alone, only the level of SC DNA is shown: red unfilled triangles and line. For the reaction with both BcgIA and native BcgI, the following were assessed: intact SC DNA, white circles; nicked OC DNA, white squares; LIN DNA, with at least one DSB at the BcgI site, white inverted triangles; the 34-mer, black triangles. Mean values from three repeats are shown: error bars denote standard deviations.
The effect of the A protein was also examined at higher concentrations of native BcgI to see whether or not additional BcgIA could still enhance the cleavage rate. In reactions containing 25 nM WT BcgI and 5 nM pUC19, all of the DNA was cleaved readily (Figure 5a). When these reactions were supplemented with 100 nM BcgIA (as in Figure 8), no difference was observed in either the extent or the rate of the reaction relative to that in the absence of the A protein (data not shown). Hence, the additional catalytic domains that the REase at the recognition site needs for its nuclease reaction can come from either free A subunits or excess A2B protomers. Nevertheless, the activation requires a lower concentration of A2B units than free A subunits.
DISCUSSION
The substrate for the REase activity of the BcgI RM protein (Figure 9a) consists of two copies of its recognition sequence (24,40,41). In contrast to the many restriction enzymes that are most active when bound to two sites but which still have some activity when bound to a single site (22,54–56), the BcgI REase has an absolute requirement for two sites. It has no detectable activity when bound to a single site on a DNA held in physical isolation from any other DNA, but that isolated site is cleaved when a second specific DNA is added (41). At each site, it cuts both strands at two separate loci, one 10/12 nt upstream and the other 12/10 nt downstream (38). After binding to its substrate, the cleavage reaction yields none of the nicked species cut in one strand and only low levels of the intermediates with one DSB. Instead, most of the DNA is converted directly to the final products with two DSBs at both sites. Hence, after spanning two sites, the REase can cut all eight bonds within the lifetime of the synaptic complex (41). To achieve this feat, the REase activity of BcgI most likely requires an assembly containing eight catalytic centres for phosphodiester hydrolysis. Given the highly concerted nature of its reaction, it is unlikely that it sequentially moves its catalytic centres from bond to bond, especially not from a bond in the 5′–3′ strand to one in the 3′–5′ strand.
Figure 9.
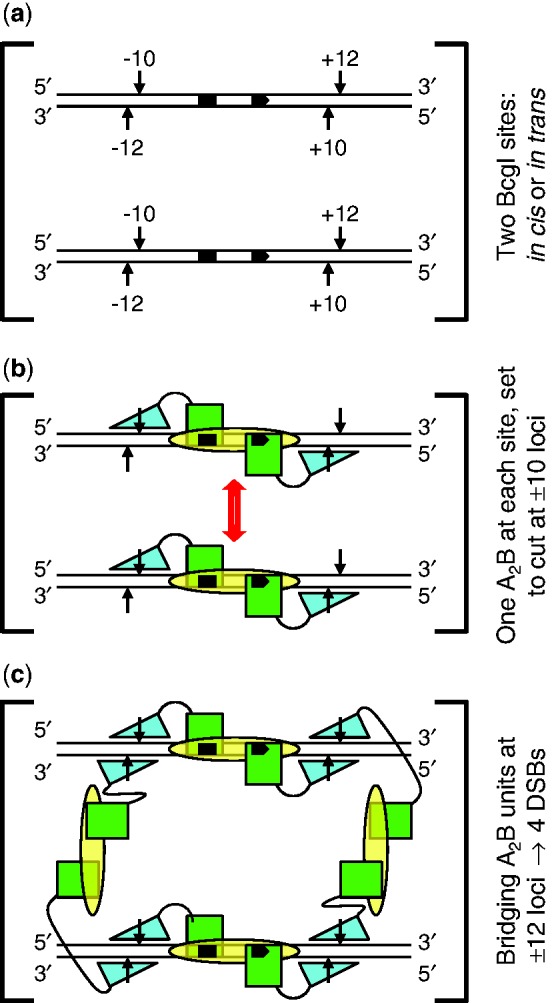
Scheme for action at eight phosphodiester bonds by BcgI. (a) The substrate for the REase activity of BcgI consists of two copies of its recognition sequence. The sites, shown here aligned in parallel, can be located either on the same DNA molecule, in cis, or on separate molecules, in trans. The bipartite recognition site is indicated by black segments, with an arrowhead to mark its 5′–3′ orientation: strand polarities are also indicated at DNA termini. Cleavage loci 10 and 12 nt distant from the site are marked with vertical arrows: upstream and downstream loci are noted with − or + signs, respectively. (b) The two sites in (a) are both shown bound to one A2B protomer of BcgI: an interaction between the protomers is suggested by the double-headed arrow (in red). The B subunit (yellow oval) is responsible for DNA specificity and is positioned spanning both segments of the recognition sequence. The A subunits carry both REase and MTase activities and are illustrated as separate domains connected by a flexible linker. The MTase domains (green squares) overlay the sites of methylation in the specified segments of the recognition sequence. The REase domains (cyan triangles) are placed against the target bonds at the −10 and +10 positions, upstream and downstream of the site in top and bottom strands, respectively: to mark their orientation on the DNA, the triangle points towards the 5′-end of each strand. At this stage, BcgI has yet to engage the scissile bonds at the −12 and +12 positions. (c) As (b) except that two additional A2B units from free solution now bridge the A2B units bound to each site via interactions between the nuclease domains of the free and the DNA-bound units. The two nuclease domains from one of the extra A2B units engage the scissile bonds at the −12 positions upstream of both recognition sites (on the left, as drawn in [c]) while those from the other occupy the downstream +12 positions in both sites (on the right). The scheme thus results in a dimer of nuclease domains, with two catalytic centres for phosphodiester hydrolysis at all four loci where BcgI makes a DSB.
The A subunit of the BcgI RM system carries both REase and MTase functions, while B resembles a DNA specificity subunit (HsdS) from a Type I RM system (12,39). HsdS subunits recognize bipartite sequences via separate domains for each segment, with each domain linked to a conserved region that associates with a MTase subunit in a pseudo-symmetric fashion (6,57). BcgIB appears to be organized similarly (39). Following SDS-PAGE, it appeared from protein staining that BcgI contained two mole A per mole B (K94), but gel stains are often far from quantitative (42). Nevertheless, native MS of the intact protein under conditions that minimize the disruption of non-covalent complexes (49) validated the earlier proposal and showed that BcgI is indeed composed of A2B units (Figure 1). Under these conditions, however, we also find some lone A and B subunits and an AB assembly but no A2 dimers. In all likelihood, the B subunit occupies the central position in the A2B assembly with A subunits symmetrically disposed on either side, both interacting with B but not with each other. This configuration is the same as that for the M2S structure of a Type I MTase (6,57) and a similar arrangement has been proposed for the AhdI MTase, a system that has a Type II REase but a Type I-like MTase (58). Native MS also revealed the presence of species containing multiple copies of the A2B protomer, from an (A2B)2 dimer to at least up to a (A2B)4 tetramer, albeit with progressively decreasing peak heights at higher masses (Figure 1). Yet, while native MS can identify these larger species, peak heights do not provide quantitative information about the amount of each species present in the sample as it was injected from the nano-ESI, since the sensitivity of the ion detector declines with increasing m/z values. Nonetheless the spectra indicated no preference for one oligomer over another, as the intensity of the peaks from the aggregates declined steadily with size.
AUC was therefore used to investigate the self-association of the BcgI protein (Figures 2 and 3). At sub-micromolar concentrations, BcgI existed in solution as a single ideal
species with a MW matching that for an individual protomer (Figure 2a). But at micomolar concentrations, of the order of those
required for maximal nuclease activity (Figure
6c), multiple aggregates were present (Figure
2b). The 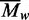 values increased with increasing
concentrations of BcgI, to levels above that for a dimeric (A2B)2 unit
(Figure 3). As the
values increased with increasing
concentrations of BcgI, to levels above that for a dimeric (A2B)2 unit
(Figure 3). As the
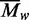 values reflect all of the species in the
cell, the mixtures must contain some of greater mass than the dimer. BcgI thus readily
associates with itself to give large assemblies containing several copies of the
A2B protomer. Its complex with non-specific DNA also contained multiple
protomers (Figure 4a). In contrast, the
addition of BcgI to a specific duplex gave an insoluble complex that precipitated
immediately from solution, as judged by its removal from solution on centrifugation at low
velocity (Figure 4b) and by its inability to
enter a polyacrylamide gel on electrophoresis (Figure
7a). The binding of BcgI to specific DNA thus generates highly aggregated states
that possibly link together several DNA molecules, but these then slowly dissociate to
release individual DNA–protein complexes back into solution.
values reflect all of the species in the
cell, the mixtures must contain some of greater mass than the dimer. BcgI thus readily
associates with itself to give large assemblies containing several copies of the
A2B protomer. Its complex with non-specific DNA also contained multiple
protomers (Figure 4a). In contrast, the
addition of BcgI to a specific duplex gave an insoluble complex that precipitated
immediately from solution, as judged by its removal from solution on centrifugation at low
velocity (Figure 4b) and by its inability to
enter a polyacrylamide gel on electrophoresis (Figure
7a). The binding of BcgI to specific DNA thus generates highly aggregated states
that possibly link together several DNA molecules, but these then slowly dissociate to
release individual DNA–protein complexes back into solution.
The cleavage of a plasmid with one BcgI site required excess protein over DNA (Figure 5a), which raises the possibility that this REase may carry out just one turnover. It had been suggested that further cleavage reactions were blocked by the excised 34-mer remaining bound to the enzyme while it was being methylated (12). Yet the fact that nuclease reactions on a plasmid with two sites proceed to completion when the enzyme is present at lower concentrations than sites on the DNA shows that the BcgI REase is capable of multiple turnovers, with each enzyme molecule participating in more than one cleavage event. BcgI is therefore not a suicide enzyme (59,60), one that is inactivated by its own reaction product. The need for excess enzyme for its reaction in trans on one-site DNA, despite its reactions in cis featuring multiple turnovers, can be accounted for if the BcgI proteins bound to both recognition sites have to recruit additional molecules of protein before they become active. In this situation, a reaction in trans will require two DNA molecules to be both loaded with enzyme at the recognition site and with the extra enzyme. But if the enzyme binds tightly to its cognate site as seems to be the case here (Figure 7c), fully loaded molecules of the one-site DNA carrying the extra enzyme will appear in solution only after adding a stoichiometric excess of enzyme over sites. Conversely, for a reaction in cis, the BcgI molecules bound to the two recognition sites in the same DNA molecule may interact with each other cooperatively (16), which could lead directly to the capture of the additional molecules of protein required for the cleavage reaction.
When bound to its recognition site, the A2B protomer is in a position to place its two nuclease domains against two of the four target phosphodiester bonds at that site (Figure 9b). This would give a structure similar to that for M.AhdI (58), an M2S MTase but which is thought from neutron scattering data to have arms extending from both M subunits. In the scheme shown in Figure 9b, the nuclease domains of the A subunits are placed against the −10 and +10 targets in the two strands, one upstream and one downstream of the site, but an equally plausible model would put these domains against the −12 and +12 bonds. On the other hand, it is improbable that an A2B unit places one nuclease centre against a bond 10 nt distant from the site and the other 12 nt away, as this would break the intrinsic symmetry of the system. Moreover, the positioning of one nuclease domain at a 10 position and the other at a 12 position calls for an implausible reconfiguration of one A subunit: an attack on both scissile bonds in the same strand (say −10 and +12) requires a 180° rotation of the nuclease domain in one A subunit relative to the other, to keep register with strand polarity; alternatively, to attack both strands at one cleavage locus (say −10 and −12), the distance between the nuclease and MTase domains in one A subunit would have to be massively different from that in the other. The synaptic complex of two A2B protomers and two recognition sites (Figure 9b) would thus seem to be incapable of making a DSB at any single cleavage locus. Indeed, it is probably incapable of cutting even one bond since no nicked species appear during DNA cleavage by BcgI (Figure 8: 41).
The monomer of the FokI REase binds to its asymmetric site via its DNA recognition domain (27) and then moves its catalytic domain to its target bond, 13 nt away in the bottom strand, but the REase remains inactive, not even nicking the DNA (32). It becomes active only after a second catalytic centre engages the target bond in the top strand. The second centre can come from another monomer of the FokI protein, either in free solution or bound elsewhere to the same DNA (31), or from just its catalytic domain (30). A similar scheme may also apply to the BcgI REase (Figure 9c). Much higher concentrations of BcgI protein are required to cleave DNA (Figure 6c) than to bind to specific DNA (Figure 7c) so the complex formed at the cognate site must remain inactive until it recruits additional molecules of the protein from free solution. BcgI bound to its site can also be activated by the A subunit alone (Figure 8). The function of either the additional A2B protein, or BcgIA alone, is presumably to associate transiently with the A subunits of the DNA-bound enzyme to position two catalytic centres for phosphodiester hydrolysis at each cleavage locus: the two centres can then cut both strands at that locus. Moreover, as the activation of the BcgI REase by the A protein alone leads directly to the excision of the 34-mer (Figure 8), both A subunits in the A2B protomer on the DNA must interact with free A protein before any reaction.
Figure 9 shows a proposal for the juxtaposition of eight catalytic centres against the eight target bonds in the BcgI substrate. In this scheme, one A2B protomer binds to each recognition site and positions its two centres for nuclease activity, one in each A subunit, against one particular set of phosphodiester bonds, illustrated here (Figure 9b) as the −10 and +10 targets. Two additional A2B protomers from free solution then become incorporated into the synaptic complex through interactions between their nuclease domains and the nuclease domains of the DNA-bound proteins (Figure 9c). The additional protomers bridge the two DNA sites, so that (in the configuration drawn) one of the extra protomers engages the −12 targets upstream of both sites and the other the +12 targets downstream of both sites. Alternative configurations for the bridging protomers are also possible: for example, they could span “diagonally” the upstream locus at one site and the downstream locus at the other site. Indeed, the insolubility of the specific DNA–protein complex for BcgI seen in the AUC (Figure 4b) could be due to the two bridging protomers spanning not the same two DNA-bound protomers to form the closed structure shown in Figure 9c but rather an open-ended structure with one DNA-bound protomer being linked via bridging units to two different DNA–protein complexes.
FUNDING
Biotechnology and Biological Sciences Research Council [BB/C513077/1]; Wellcome Trust [078794]. Funding for open access charge: Wellcome Trust.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank New England Biolabs for the over-producing plasmid for the native BcgI R-M system, and James McCullagh (Mass Spectrometry facility, Department of Chemistry, University of Oxford) for use of the Synapt MS.
REFERENCES
- 1.Wilson GG, Murray NE. Restriction and modification systems. Annu. Rev. Genet. 1991;25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
- 2.Murray NE. Immigration control of DNA in bacteria: self versus non-self. Microbiology. 2002;148:3–20. doi: 10.1099/00221287-148-1-3. [DOI] [PubMed] [Google Scholar]
- 3.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev SK, Dryden DTF, Dybvig K, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dryden DTF, Murray NE, Rao DN. Nucleoside triphosphate-dependent restriction enzymes. Nucleic Acids Res. 2001;29:3728–3741. doi: 10.1093/nar/29.18.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennaway CK, Taylor JE, Song CF, Potrzebowski W, Nicholson W, White JH, Swiderska A, Obarska-Kosinska A, Callow P, Cooper LP, et al. Structure and operation of the DNA-translocating Type I DNA restriction enzymes. Genes Dev. 2012;26:92–104. doi: 10.1101/gad.179085.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith RM, Diffin FM, Savery NJ, Josephsen J, Szczelkun MD. DNA cleavage and methylation specificity of the single polypeptide restriction-modification enzyme LlaGI. Nucleic Acids Res. 2009;37:7206–7218. doi: 10.1093/nar/gkp790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001;29:3784–3795. doi: 10.1093/nar/29.18.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perona JJ. Type II restriction endonucleases. Methods. 2002;28:353–364. doi: 10.1016/s1046-2023(02)00242-6. [DOI] [PubMed] [Google Scholar]
- 11.Janulaitis A, Petrusyte M, Maneliene Z, Klimasauskas S, Butkus V. Purification and properties of the Eco57I restriction endonuclease and methylase - prototypes of a new class. Nucleic Acids Res. 1992;20:6043–6049. doi: 10.1093/nar/20.22.6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kong H, Roemer SE, Waite-Rees PA, Benner JS, Wilson GG, Nwankwo DO. Characterization of BcgI, a new kind of restriction-modification system. J. Biol. Chem. 1994;269:683–690. [PubMed] [Google Scholar]
- 13.Piekarowicz A, Golaszewska M, Sunday AO, Siwinska M, Stein DC. The HaeIV restriction modification system of Haemophilus aegyptius is encoded by a single polypeptide. J. Mol. Biol. 1999;293:1055–1065. doi: 10.1006/jmbi.1999.3198. [DOI] [PubMed] [Google Scholar]
- 14.Morgan RD, Bhatia TK, Lovasco L, Davis TB. MmeI: a minimal Type II restriction-modification system that only modifies one DNA strand for host protection. Nucleic Acids Res. 2008;36:6558–6570. doi: 10.1093/nar/gkn711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mucke M, Kruger DH, Reuter M. Diversity of Type II restriction endonucleases that require two DNA recognition sites. Nucleic Acids Res. 2003;31:6079–6084. doi: 10.1093/nar/gkg836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halford SE, Welsh AJ, Szczelkun MD. Enzyme-mediated DNA-looping. Annu. Rev. Biophys. Biomol. Struct. 2004;33:1–24. doi: 10.1146/annurev.biophys.33.110502.132711. [DOI] [PubMed] [Google Scholar]
- 17.Yang CC, Topal MD. Nonidentical DNA-binding sites of endonuclease NaeI recognize different families of sequences flanking the recognition site. Biochemistry. 1992;31:9657–9664. doi: 10.1021/bi00155a019. [DOI] [PubMed] [Google Scholar]
- 18.Embleton ML, Siksnys V, Halford SE. DNA cleavage reactions by type II restriction enzymes that require two copies of their recognition sites. J. Mol. Biol. 2001;311:503–514. doi: 10.1006/jmbi.2001.4892. [DOI] [PubMed] [Google Scholar]
- 19.Wentzell LM, Nobbs TJ, Halford SE. The SfiI restriction endonuclease makes a four-strand DNA break at two copies of its recognition sequence. J. Mol. Biol. 1995;248:581–595. doi: 10.1006/jmbi.1995.0244. [DOI] [PubMed] [Google Scholar]
- 20.Deibert M, Grazulis S, Sasnauskas G, Siksnys V, Huber R. Structure of the tetrameric restriction endonuclease NgoMIV in complex with cleaved DNA. Nat. Struct. Biol. 2000;7:792–799. doi: 10.1038/79032. [DOI] [PubMed] [Google Scholar]
- 21.Vanamee ES, Viadiu H, Kucera R, Dorner L, Picone S, Schildkraut I, Aggarwal AK. A view of consecutive binding events from structures of tetrameric endonuclease SfiI bound to DNA. EMBO J. 2005;24:4198–4208. doi: 10.1038/sj.emboj.7600880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zaremba M, Owsicka A, Tamulaitis G, Sasnauskas G, Shlyakhtenko LS, Lushnikov AY, Lyubchenko YL, Laurens N, van den Broek B, Wuite GJL, et al. DNA synapsis through transient tetramerization triggers cleavage by Ecl18kI restriction enzyme. Nucleic Acids Res. 2010;38:7142–7154. doi: 10.1093/nar/gkq560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bath AJ, Milsom SE, Gormley NA, Halford SE. Many type IIs restriction endonucleases interact with two recognition sites before cleaving DNA. J. Biol. Chem. 2002;277:4024–4033. doi: 10.1074/jbc.M108441200. [DOI] [PubMed] [Google Scholar]
- 24.Marshall JJT, Gowers DM, Halford SE. Restriction endonucleases that bridge and excise two recognition sites from DNA. J. Mol. Biol. 2007;367:419–431. doi: 10.1016/j.jmb.2006.12.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szybalski W, Kim SC, Hasan N, Podhajska AJ. Class-IIS restriction enzymes - a review. Gene. 1991;100:13–26. doi: 10.1016/0378-1119(91)90345-c. [DOI] [PubMed] [Google Scholar]
- 26.Chan SH, Stoddard BL, Xu SY. Natural and engineered nicking endonucleases - from cleavage mechanism to engineering of strand-specificity. Nucleic Acids Res. 2011;39:1–18. doi: 10.1093/nar/gkq742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wah DA, Hirsch JA, Dorner LF, Schildkraut I, Aggarwal AK. Structure of the multimodular endonuclease FokI bound to DNA. Nature. 1997;388:97–100. doi: 10.1038/40446. [DOI] [PubMed] [Google Scholar]
- 28.Heiter DF, Lunnen KD, Wilson GG. Site-specific DNA-nicking mutants of the heterodimeric restriction endonuclease R.BbvCI. J. Mol. Biol. 2005;348:631–640. doi: 10.1016/j.jmb.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 29.Sasnauskas G, Zakrys L, Zaremba M, Cosstick R, Gaynor JW, Halford SE, Siksnys V. A novel mechanism for the scission of double-stranded DNA: BfiI cuts both 3′-5′ and 5′-3′ strands by rotating a single active site. Nucleic Acids Res. 2010;38:2399–2410. doi: 10.1093/nar/gkp1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc. Natl Acad. Sci. USA. 1998;95:10570–10575. doi: 10.1073/pnas.95.18.10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Catto LE, Ganguly S, Milsom SE, Welsh AJ, Halford SE. Protein assembly and DNA looping by the FokI restriction endonuclease. Nucleic Acids Res. 2006;34:1711–1720. doi: 10.1093/nar/gkl076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pernstich C, Halford SE. Illuminating the reaction pathway of the FokI restriction endonuclease by fluorescence resonance energy transfer. Nucleic Acids Res. 2012;40:1203–1213. doi: 10.1093/nar/gkr809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wah DA, Bitinaite J, Schildkraut I, Aggarwal AK. Structure of FokI has implications for DNA cleavage. Proc. Natl Acad. Sci. USA. 1998;95:10564–10569. doi: 10.1073/pnas.95.18.10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanders KL, Catto LE, Bellamy SR, Halford SE. Targeting individual subunits of the FokI restriction endonuclease to specific DNA strands. Nucleic Acids Res. 2009;37:2105–2115. doi: 10.1093/nar/gkp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rusling DA, Laurens N, Pernstich C, Wuite GJL, Halford SE. DNA looping by FokI: the impact of synapse geometry on loop topology at varied site orientations. Nucleic Acids Res. 2012;40:4977–4987. doi: 10.1093/nar/gks183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gormley NA, Hillberg AL, Halford SE. The type IIs restriction endonuclease BspMI is a tetramer that acts concertedly at two copies of an asymmetric DNA sequence. J. Biol. Chem. 2002;277:4034–4041. doi: 10.1074/jbc.M108442200. [DOI] [PubMed] [Google Scholar]
- 37.Marshall JJT, Halford SE. The Type IIB restriction endonucleases. Biochem. Soc. Trans. 2010;38:410–416. doi: 10.1042/BST0380410. [DOI] [PubMed] [Google Scholar]
- 38.Kong H, Morgan RD, Maunus RE, Schildkraut I. A unique restriction endonuclease, BcgI, from Bacillus coagulans. Nucleic Acids Res. 1993;21:987–991. doi: 10.1093/nar/21.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong H. Analyzing the functional organization of a novel restriction modification system, the BcgI system. J. Mol. Biol. 1998;279:823–832. doi: 10.1006/jmbi.1998.1821. [DOI] [PubMed] [Google Scholar]
- 40.Kong H, Smith CL. Does BcgI, a unique restriction endonuclease, require two recognition sites for cleavage? Biol. Chem. 1998;379:605–609. [PubMed] [Google Scholar]
- 41.Marshall JJT, Smith RM, Ganguly S, Halford SE. Concerted action at eight phosphodiester bonds by the BcgI restriction endonuclease. Nucleic Acids Res. 2011;39:7630–7640. doi: 10.1093/nar/gkr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gauci VJ, Wright EP, Coorssen JR. Quantitative proteomics: assessing the spectrum of in-gel protein detection methods. J. Chem. Biol. 2011;4:3–29. doi: 10.1007/s12154-010-0043-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marshall JJT. 2006. BcgI and other Type IIB restriction endonucleases. Ph.D. Thesis. University of Bristol. [Google Scholar]
- 44.Cantor CR, Schimmel PR. Biophysical Chemistry Part II: Techniques for the Study of Biological Structure and Function. New York, NY: W.H. Freeman; 1980. [Google Scholar]
- 45.Cesnaviciene EE, Petrusyte MM, Kazlauskiene RR, Maneliene Z, Timinskas A, Lubys A, Janulaitis A. Characterization of AloI, a restriction-modification system of a new type. J. Mol. Biol. 2001;314:205–216. doi: 10.1006/jmbi.2001.5049. [DOI] [PubMed] [Google Scholar]
- 46.Smith RM, Jacklin AJ, Marshall JJT, Sobott F, Halford SE. Organisation of the BcgI restriction-modification protein for the transfer of one methyl group to DNA. Nucleic Acids Res. 2012;41:405–417. doi: 10.1093/nar/gks1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgess RR. Protein precipitation techniques. Methods Enzymol. 2009;463:331–342. doi: 10.1016/S0076-6879(09)63020-2. [DOI] [PubMed] [Google Scholar]
- 48.Jacklin AJ. 2011. Structure and assembly of BcgI, a Type IIB restriction-modification system. Ph.D. Thesis. University of Bristol. [Google Scholar]
- 49.Sobott F, McCammon MG, Hernández H, Robinson CV. The flight of macromolecular complexes in a mass spectrometer. Philos. Transact. A: Math. Phys. Eng. Sci. 2005;363:379–389. doi: 10.1098/rsta.2004.1498. [DOI] [PubMed] [Google Scholar]
- 50.Tanford C. Physical Chemistry of Macromolecules. New York, NY: John Wiley; 1961. [Google Scholar]
- 51.Heitman J, Zinder ND, Model P. Repair of the Escherichia coli chromosome after in vivo scission by the EcoRI endonuclease. Proc. Natl Acad. Sci. USA. 1989;86:2281–2285. doi: 10.1073/pnas.86.7.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor JD, Goodall AJ, Vermote CL, Halford SE. Fidelity of DNA recognition by the EcoRV restriction/modification system in vivo. Biochemistry. 1990;29:10727–10733. doi: 10.1021/bi00500a003. [DOI] [PubMed] [Google Scholar]
- 53.Vipond IB, Halford SE. Specific DNA recognition by EcoRV restriction endonuclease induced by calcium ions. Biochemistry. 1995;34:1113–1119. doi: 10.1021/bi00004a002. [DOI] [PubMed] [Google Scholar]
- 54.Zaremba M, Sasnauskas G, Urbanke C, Siksnys V. Conversion of the tetrameric restriction endonuclease Bse634I into a dimer: Oligomeric structure-stability-function correlations. J. Mol. Biol. 2005;348:459–478. doi: 10.1016/j.jmb.2005.02.037. [DOI] [PubMed] [Google Scholar]
- 55.Bellamy SR, Milsom SE, Kovacheva YS, Sessions RB, Halford SE. A switch in the mechanism of communication between the two DNA-binding sites in the SfiI restriction endonuclease. J. Mol. Biol. 2007;373:1169–1183. doi: 10.1016/j.jmb.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laurens N, Rusling DA, Pernstich C, Brouwer I, Halford SE, Wuite GJL. DNA looping by FokI: the impact of twisting and bending rigidity on protein-induced looping dynamics. Nucleic Acids Res. 2012;40:4988–4997. doi: 10.1093/nar/gks184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kneale GG. A symmetrical model for the domain structure of Type I DNA methyltransferases. J. Mol. Biol. 1994;243:1–5. doi: 10.1006/jmbi.1994.1624. [DOI] [PubMed] [Google Scholar]
- 58.Callow P, Sukhodub A, Taylor JE, Kneale GG. Shape and subunit organisation of the DNA methyltransferase M.AhdI by small-angle neutron scattering. J. Mol. Biol. 2007;369:177–185. doi: 10.1016/j.jmb.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindahl T, Demple B, Robins P. Suicide inactivation of the E. coli O6-methylguanine-DNA methyltransferase. EMBO J. 1982;1:1359–1363. doi: 10.1002/j.1460-2075.1982.tb01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walsh CT. Suicide substrates, mechanism-based enzyme inactivators: recent developments. Annu. Rev. Biochem. 1984;53:493–535. doi: 10.1146/annurev.bi.53.070184.002425. [DOI] [PubMed] [Google Scholar]