

Abstract
Overwhelming experimental evidence accumulated over the past decade indicates that microRNAs (miRNAs) are key regulators of gene expression in animals and plants and play important roles in development, homeostasis and disease.
The miR-17~92 family of miRNA clusters is composed of three related, highly conserved, polycistronic miRNA genes that collectively encode for a total of fifteen miRNAs. Here we discuss recent studies demonstrating that these miRNAs are essential for vertebrate development and homeostasis. We also show how their mutation or deregulation contributes to the pathogenesis of a variety of human diseases, including cancer and congenital developmental defects. Finally, we discuss the current evidence suggesting how the different miRNAs encoded by these three clusters can functionally cooperate to fine-tune signaling and developmental pathways.
miRNAs and their biogenesis
A detailed description of the biogenesis of miRNAs has been is beyond the scope of this review (1), but a few aspects of it are important for our discussion. The life of the vast majority of miRNAs begins with the transcription, mediated by RNA polymerase II, of the pri-miRNA, a longer primary transcript that is capped and polyadenylated (2,3). The pri-miRNA then undergoes two sequential processing events that convert it into the mature miRNAs (4). First, while still in the nucleus, the pri-miRNA is cropped by the microprocessor complex (containing Drosha, DGCR8 and additional accessory factors) into a short hairpin, approximately 70 nt in length, known as the pre-miRNA (5-7). The pre-miRNA is then exported in the cytoplasm (8,9) where it is cleaved by the RNAse Dicer to generate a double-stranded short RNA 20-22 nucleotides in length (10-14). One of the two strands becomes the mature miRNA and is incorporated into the RNA-induced silencing complex (RISC) (15-17). The mature miRNAs allows the RISC complex to bind, via partial sequence complementarity, to target mRNAs, ultimately resulting in their degradation or translational repression (15,18-20).
Although the entire sequence of a miRNA can bind to the target, experimental and computational evidence strongly indicates that the nucleotides at position 2-7, the so-called “seed” sequence, are the key determinants of target specificity for a miRNA (21-23). Thus, miRNAs with the same seed sequence are predicted to target highly overlapping sets of genes and are therefore grouped in the same “miRNA family” (24,25).
miRNA clusters and polycistronic miRNAs
miRNA genes can be located in the context of non-coding transcription units or in the introns of protein-coding genes (26-28). Interestingly, many miRNAS are situated in polycistronic miRNA “clusters”, wherein multiple miRNA genes are generated from a single primary transcript (4,29). In fact, approximately 50% of D. melanogaster and at least one-third of human miRNA genes are clustered (26,27,30,31). The high conservation of miRNA clusters across species suggests evolutionary pressure to maintain such organization.
Although the multiple miRNAs belonging to a particular cluster are often highly related to one another, having emerged via duplication events, the occurrence of miRNAs belonging to distinct “seed” families within the same cluster is also commonly observed (32). The co-expression of miRNAs belonging to different “seed” families from the same cluster adds an additional layer of complexity and begs the question of whether these distinct miRNAs share common biological functions despite targeting different gene sets.
The miR-17~92 family of miRNA clusters
One of the best-characterized polycistronic miRNA clusters is miR-17~92. This cluster maps to human chromosome 13 and encodes for six individual miRNAs (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a). The organization and sequences of the miR-17~92 family is highly conserved among vertebrates, and gene duplication and deletion events during early vertebrate evolution have resulted in two mammalian paralogs: the miR-106b~25 cluster and the miR-106a~363 cluster (Figure 1a)(33). The miR-106b~25 cluster is located on human chromosome 7 and resides within the 13th intron of the MCM7 gene, while the miR-106a~363 is located on chromosome X. Both miR-17~92 and miR-106b~25 are highly expressed in a wide array of mouse tissues and are particularly abundant in embryonic stem cells and during embryogenesis, while miR-106a~363 is generally expressed at lower levels (34-37). The fifteen miRNAs encoded by miR-17~92 and its two paralogs can be grouped into four “seed” families (miR-17, miR-18, miR-19 and miR-92; Figure 1b). Although the miR-17~92 cluster shows excellent sequence conservation among vertebrates, obvious orthologs of the miR-17, miR18 and miR-19 seed families are not found outside of vertebrates (33). The exception is represented by the miR-92 seed family, for which homologs have been identified in D. melanogaster and C. elegans (33).
Figure 1.
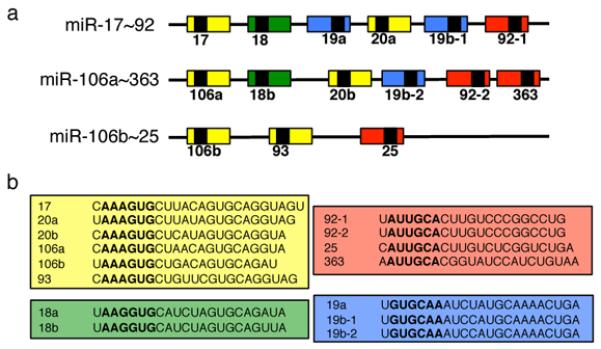
(a). Schematic representation of the three members of the miR-17~92 family of microRNA clusters. miRNAs sharing the same seed sequence are represented by boxes of the same color. (b) Mature miRNA sequences of the sixteen miRNAs encoded by the three clusters. The miRNAs are grouped into four seed families. Seed sequences are shown in bold.
Transcriptional regulation of miR-17~92
At the crux of miR-17~92 functions is its role as a direct transcriptional target of c-Myc (Figure 2). This oncogenic transcription factor binds to a conserved non-canonical E-box sequence located 1480 bp upstream of miR-17 (38). Although putative c-Myc binding sites are located within close proximity of miR-106a~363, direct binding has not been demonstrated and little is known about the transcriptional regulation of miR-106b~25, which is the third member of the family.
Figure 2.
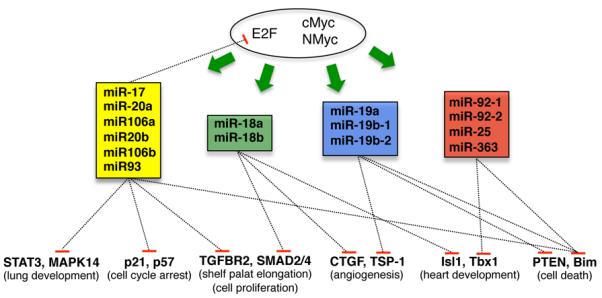
Schematic representation of targets regulated by specific seed family of the cluster miR-17~92 and paralogs. Dotted lines indicate which microRNA family has been shown to regulate a specific target.
Consistent with the proliferative functions associated with miR-17~92 as a downstream effector of c-Myc, the E2F family of transcription factors has also been found to directly bind to the promoter of miR-17~92 and to regulate its transcription (39,40). Interestingly, miR-17 and miR-20, which have identical seed sequences, directly inhibit translation of E2F1, E2F2, and E2F3, establishing an auto-regulatory loop within the E2F transcriptional network (38-40). Collectively, these studies suggest a model wherein c-Myc induces the cell’s proliferative machinery but establishes a threshold of E2F expression through repression by miR-17~92 (38). Furthermore, it has been suggested that because miR-17 and miR-20 target E2F1, which can act as a pro-apoptotic molecule, miR-17~92 shifts the balance from apoptosis to proliferation (40).
Post-transcriptional regulation of miR-17~92
Although initial studies focused on the transcriptional regulation of miR-17~92, more recent studies have begun to shed light on how members of the cluster are individually and uniquely regulated beyond transcription.
Recent studies have shown that miRNA processing can be modulated in different cellular settings. Because multiple steps are required to generate mature miRNAs, it is not surprising that the expression of a mature miRNA does not necessarily correlate with that of its pri-miRNA (41-43).
In the case of miR-17~92, the six miRNAs encoded by the cluster are often found to be expressed at different levels, which indicates that either they are processed with different efficiency or that their stability is different. It has been suggested that the primary transcript assumes a globular tertiary structure with the 3′ end of the transcript serving as the core domain (44). According to this model, the most exposed miRNAs (miR-17, -18a and -20a) are more accessible to the microprocessor complex and therefore are more easily processed. In vitro processing experiments are consistent with this model (44);however, in vivo miR-92a and miR-19b, two miRNAs that should be in the “core” of pri-miR-17~92 according to the model, are often among the most highly expressed members of the cluster ((45); CB, unpublished data).It is possible that additional factors dynamically affect the tertiary structure miR-17~92 assumes in vivo and thereby promote the processing of some components of the cluster over others. In addition, modulation at other stages of maturation, as well as different stability of the mature miRNAs, could play an important role. For example, the RNA-binding protein hnRNPA1 has been shown to specifically recognize the loop structure of miR-18a and facilitate its processing by Drosha (46).
miR-17~92 as an oncogene
An oncogenic role for miR-17~92 was first suggested by the finding, in 2004, of recurrent focal amplification of the cluster in diffuse large B cell lymphomas of recurrent focal amplifications (47). Shortly thereafter, He and colleagues demonstrated that ectopic expression of a truncated version of miR-17~92 (lackingmiR-92a) strongly cooperates with c-Myc in a mouse model of B-cell lymphoma (48), thus providing convincing experimental evidence that miR-17~92 is a bona fide oncogene.
These first results also suggested suppression of apoptosis as a possible mechanism through which miR-17~92 promotes transformation (48), but the key targets remained unknown. Equally unclear was how the different members of the cluster contributed to its overall oncogenic activity.
These questions were answered by subsequent gain-of-function and loss-of-function studies in the Eμ-Myc mouse model of B-cell lymphoma (49)(50). These studies demonstrated that the six miRNAs encoded by the cluster are not functionally equivalent when it comes to promoting cell survival and tumorigenesis. In fact, the two members of the miR-19 seed family were found to be necessary and sufficient to recapitulate the oncogenic activity of the full cluster (49)(50). These studies also demonstrated that the tumor suppressor Pten is a prominent miR-19 target and its suppression can at least partially explain the pro-survival effect of miR-19 (49,50). Interestingly, these findings were later found to also apply to a model of T-cell leukemias (51).
In a complementary line of investigation, Mu and colleagues used a conditional knockout allele of miR-17~92 to show that acute genetic deletion of miR-17~92 in Myc-driven lymphomas leads to increased cell death and reduced tumorigenicity (49). This finding is important because it suggests that sustained expression of this cluster may play an important role not only in tumor formation, but also in tumor maintenance; thus pharmacological inhibition of miR-17~92 may prove therapeutically useful.
A role for miR-17~92 in promoting tumorigenesis was also subsequently demonstrated outside the hematopoietic system. Overexpression of the cluster is observed in a variety of human cancers, including small-cell lung cancer, colon cancer, neuroblastomas, medulloblastoma and gastric cancer (52-58). Moreover, a causal role for miR-17~92 has been demonstrated in multiple mouse models of human cancer. The emerging picture suggests that miR-17~92 and its two paralogs play a widespread role in tumorigenesis, but the specific miRNAs involved and the key targets they regulate appear highly context-specific.
For example, MacPherson and colleagues (59) have reported overexpression and genomic amplification of miR-17~92 in mouse and human retinoblastomas. They also showed that its ectopic expression inRb−/− and p107−/− retinas results in increased proliferation and rapid onset of the disease. In this context, in contrast to leukemias and lymphomas, the oncogenic activity of the cluster appears largely due to the action of members of the miR-17 seed family (miR-17 and miR-20a), which directly target the cell cycle inhibitors p21CIP1 and p57KIP2(59).
Another example of miR-17~92’s tumor-promoting activities is its role in colon cancer, a context in which frequent overexpression of the cluster has been reported (48). In a tumor engraftment model, Dews and colleagues demonstrated that upregulation of miR-17~92 by Myc in colonocytes increases tumorigenicity by promoting angiogenesis. This effect was attributed to direct repression of anti-angiogenic factors thrombospondin-1 (TSP-1) and connective tissue growth factor (CTGF) by miR-18a and miR-19, respectively (60).
Another human cancer in which miR-17~92 is frequently overexpressed is medulloblastomas, specifically medulloblastomas with a constitutively active sonic hedgehog (SHH) signaling pathway. The cluster is also prominently expressed in proliferating mouse cerebellar granule neuron progenitors (GNPs), which are believed to be the cells of origin for these tumors (55,61). A role for miR-17~92 in medulloblastomas was confirmed by in vivo studies. Mice carrying homozygous deletion of Ink4c and heterozygous deletion of Patched (Ptch1), the receptor for Sonic Hedgehog (Shh), develop spontaneous medulloblastomas that closely resemble the human disease (55). Using GNP isolated from these mice at post-natal day 6 (P6), Uziel and colleagues showed that ectopic expression of miR-17~92 increases tumor formation upon orthotopic transplantation into immunocompromised mice (55). Importantly, the authors did not observe changes in expression levels of Ptenand E2f1in response to miR-17~92 overexpression, suggesting that a distinct set of targets is responsible for the oncogenic activity of miR-17~92 in this model.
Finally, overexpression of miR-17~92 has been reported in neuroblastomas where it is a poor prognostic indicator (62). Accordingly, inhibition of miR-17 strongly suppresses the growth of highly aggressive neuroblastoma cell lines (57).
Oncogenic roles of miR-106b~25 and miR-106a~363
Given the high degree of sequence similarity between miR-17~92 and its two paralogs, miR-106b~25 and miR-106a~363, it is not surprising that that these two highly related clusters share the ability to promote tumorigenesis with miR-17~92.
For example, high miR-106b~25 expression has been reported to confer poor prognosis in gastric cancer (56). Increased expression of E2F1 is also particularly common in gastric cancers (63), and, similar to miR-17~92, miR-106b~25 expression levels positively correlated with E2F1 levels. Furthermore, miR-106b and miR-93 appear to regulate E2F1 expression, establishing a negative feedback loop analogous to that observed in the E2F/miR-17/miR-20 axis (39,40). Another characteristic trait of gastric tumors is their resistance to TGF-β signaling (64,65), which may be partly due to high levels of miR-106b~25.
In another study, Poliseno and colleagues showed cooperation between miR-106b~25 and its host gene Mcm7 in promoting prostate cancer formation. Transgenic mice overexpressing Mcm7 and 106b~25 mice were found to develop varying degrees of prostatic hyperplasia and PINs (66). Presumably, as consequence of miR-106b~25 overexpression, Pten levels were reduced in the prostate of these mice. Interestingly, neither expression of Mcm7 nor miR-106b~25 alone was sufficient to induce this phenotype, an observation interpreted as evidence of functional cooperation between miR-106b~25 and its host gene (66).
Although miR-106a~363 is normally expressed at much lower levels compared to the other two paralogs, there is substantial evidence suggesting that this cluster can behave as an oncogene under specific circumstances. The most convincing evidence comes from retroviral insertional mutagenesis screens. Two groups have independently reported recurrent activating provirus integration sites immediately upstream of the miR-106a~363 locus in murine T cell lymphoma and T cell leukemia (67-70), and miR-106a~363 has been found overexpressed in nearly 50% of human T cell leukemia samples (67-70). Interestingly, miR-17~92 has also been reported to be similarly activated by retroviral insertions (67-70).
miR-17~92 in development and homeostasis
A close relationship often exists between the physiologic functions of a proto-oncogene and its roles in cancer. This has proven particularly true with respect to the miR-17~92 family of miRNA clusters. The generation of mice carrying loss-of-function (34) and gain-of-function alleles of miR-17~92 (71,72) has provided a unique opportunity to probe their role in development, homeostasis and disease. Here we will discuss the key results emerging from the characterization of these mice and how they translate to human development and disease.
The miR-17~92 cluster is broadly expressed from the earliest stages of development (ES cells) to adulthood, with the mature miRNAs detectable in virtually all tissues analyzed, albeit at variable levels (34-37). Interestingly, while miR-106b~25 and miR-17~92 are usually expressed at comparable levels in most tissues examined, miR-106a~363 is often expressed at very low levels as measured by a variety of techniques including high throughput sequencing (34).
The difference in expression levels can at least partially explain the different phenotypes observed in mice carrying targeted deletion of the various members of this family of miRNA clusters. Deletion of the miR-106a~363 or of miR-106b~25 clusters does not result in any obvious developmental abnormality in mice. Even the concomitant deletion of both clusters results in viable and fertile animals (34). In contrast, homozygous deletion of miR-17~92 alone has dramatic developmental consequences and leads to fully penetrant perinatal lethality (34). By mid-gestation, miR-17~92−/− mice exhibit smaller size and die at birth, likely due to severe lung hypoplasia and cardiac defects. Remarkably, simultaneous deletion of miR-106b~25m miR-106a~363 and miR-17~92 elicits a much more severe phenotype, with embryonic lethality before E15 caused by severe cardiovascular defects and extensive apoptosis across the body. This result indicates the existence of substantial functional redundancy among the paralogs during development (34).
The lung hypoplasia seen in miR-17~92 null mice, and the complementary phenotype – hyper-proliferation of the lung epithelium– reported in transgenic mice overexpressing miR-17~92 (72) provides an interesting link between the physiologic functions of this cluster and its proposed oncogenic effect in lung cancer (52,54,73). Beyond proliferation, miR-17~92 has been implicated in lung morphogenesis, and the miR-17 family has been proposed to modulate branching of the lung epithelium through the regulation of STAT3 and MAPK14 (74).
Analogously, the oncogenic role of miR-17~92 in B lymphomagenesis is mirrored in miR-17~92-null mice by a block of B cell differentiation at the pro-B to pre-B transition (34,75) and by the observation that mice overexpressing miR-17~92 in the lymphoid compartment develop a lymphoproliferative disease and autoimmunity (71). Although our understanding of how miR-17~92 modulates hematopoiesis is still incomplete, both gain-of-function and loss-of-function studies implicate modulation of apoptosis by miR-17~92.
It is important to notice that although miR-17~92-overexpressing mice display a lymphoproliferative disorder characterized by polyclonal expansion of the B and T cell compartments, they do not develop full blown neoplasia. This suggests that miR-17~92 overexpression alone is not sufficient for full transformation and that additional oncogenic lesions are required.
miR-17~92 haploinsufficiency, skeletal development, and Feingold syndrome
The role of miR-17~92 in human disease is not limited to cancer. A recent paper reported the identification of hemizygous germline deletions involving the miR-17~92 locus in a subset of families affected by a rare autosomal dominant condition known as Feingold syndrome (76). Patients with this condition present a wide array of skeletal defects, the most common being short stature, digital anomalies –brachymesophalangy of the 2ndand 5th fingers in particular – and microcephaly. Affected individuals may also have learning disabilities and defective development of the gastrointestinal system, albeit with lower penetrance (77).
The genetic lesions identified by de Pontual and colleagues in two families affected by the syndrome are microdeletions of the region of chromosome 13 that contains the miR-17~92 locus (MIR17HG). In both cases, the microdeletion leads to loss of the entire miR-17~92 cluster but also of the first exon of GPC5, a closely linked gene encoding for Glypican-5 (76). To determine the roles of these two genes in the phenotype, the authors analyzed miR-17~92+/− mice, in which the Gpc5 locus is intact. Strikingly, these animals almost exactly phenocopied the skeletal anomalies observed in individuals affected by Feingold syndrome, including brachymesophalangy of the 5th finger, short stature and microcephaly (76). Based on these findings, the authors concluded that haploinsufficiency of miR-17~92 is responsible for the skeletal defects observed in these families. Notably, in mice in which both copies of miR-17~92 were deleted, the skeletal defects were even more pronounced; this observation suggests that this cluster plays an essential role in skeletal development in mammals.
The fact that even a relatively modest reduction in the expression levels of miR-17~92 can have such profound phenotypic consequences further highlights the key importance of fine tuning gene expression, especially in the context of developmental processes that require the timely and coordinated activation and inactivation of complex transcriptional programs.
Although exceedingly rare, Feingold syndrome had been the subject of substantial interest because about 70% of affected individuals harbor monoallelic germline loss-of-function mutations of the proto-oncogene MYCN (77,78). The discovery that either MYCN or miR-17~92 haploinsufficiency can lead to the very same developmental anomalies in humans suggests that these two genes may be components of the same pathway. Indeed, de Pontual and colleagues show that, similar to MYC, MYCN can also directly bind to the miR-17~92 promoter and activate its transcription.
The identification of miR-17~92 mutations in Feingold syndrome patients is important not only because it is the first example of a miRNA mutation causing a developmental syndrome in humans, but also because it prompted the discovery of a previously unappreciated role of the MYCN-miR-17~92 axis in skeletal development.
Functional cooperation among miR-17~92 members
The analysis of genetically engineered mice carrying gain-of-function and loss-of-function mutations of miR-17~92 has been essential to furthering our understanding of the biological functions of this important miRNA cluster. Yet, the exact molecular mechanisms through which these miRNAs fine-tune developmental processes and how their deregulation leads to disease remain poorly understood. In particular, it is unclear whether the six miRNAs encoded by these clusters are functionally equivalent or control different molecular pathways, and the majority of their functionally relevant targets still remain to be identified.
As previously suggested, the miRNAs encoded by the miR-17~92 cluster can be group into four different “seed” families, whose members are predicted to target distinct sets of genes (Figure 1A). Although there is substantial evidence indicating that various members of the cluster differ in their oncogenic potential, there is also evidence that in some settings multiple members of the cluster can act synergistically, either by converging on the same targets or by targeting multiple nodes in the same pathway or biological output (Figure 2).
For example, miR-18 and miR-19 have been shown to directly repress the anti-angiogenic factors TSP-1 and CTGF (60), as previously discussed. Another occurrence of this type of cooperation is represented by the role of miR-17, miR-20a and miR-92 in the regulation of Isl1 and Tbx1 during cardiac development (79).
But the clearest example of different members of the miR-17~92 cooperatively modulating a signaling pathway is provided by the role of miR-17~92 in TGF-β signaling (56,62,80,81). TGF-β signaling regulates a multitude of cellular processes (82), and is particularly relevant not only during development, but also in cancer. In the canonical pathway of TGF-β signaling, TGF-β ligand binds to type I (TGFBRI) and type II (TGFBRII) receptors on the cell surface, resulting in TGFBRI phosphorylation (83). Propagation of the signal is exerted through subsequent phosphorylation of Smad proteins (Smad 1, 2, 3, 5, and 8), resulting in their homotrimerization and in heteromerization with Smad4 (83). Smad complexes then translocate into the nucleus, ultimately regulating the transcription of target genes.
Members of the miR-17~92 cluster have been shown to modulate TGF-β signaling at multiple levels. miR-17 and miR-20a directly target TGFBRII (62,81) while miR-18a targets Smad2 and Smad4. Activation of TGF-β signaling exerts cytostatic effects at least in part mediated by p21 and Bim (84,85), and these two genes are also bona fide targets of the miR-17~92 cluster. The miR-17 seed family modulates p21 levels, while the miR-19 and miR-92 seed families modulate Bim levels (56).
Accordingly, it has been reported that miR-17~92 expression globally affects genes responsive to TGF-β in a number of cell types, including neuroblastoma, glioblastoma, and hepatocellular carcinoma cell lines (62,81). More recently, the relationship between miR-17~92 and TGF-β has also been demonstrated in palatal development (80). These studies collectively present compelling evidence that miR-17~92 is a general attenuator of TGF-β signaling and demonstrate the ability of different members of the same miRNA cluster to cooperate in modulating a specific pathway in both cancer and normal development.
Perspectives and Conclusion
Although much has been learned about miR-17~92’s role in cancer and development in recent years, we are only beginning to uncover the vast regulatory functions this cluster and its two paralogs exert in mammals. A careful dissection of the individual contributions of members of the miR-17~92 family towards various biological outputs will be important to determine the degree of functional overlap and cooperation between the various components of this family of miRNA clusters, particularly in the context of development and tumorigenesis. Ultimately, the generation of more sophisticated mouse models, combined with high-throughput approaches to map miRNA-mRNA interaction in a systematic and unbiased way (86,87), will be needed to fully answer these important questions.
Acknowledgements
We apologize to all colleagues whose work could not be cited owing to space limits. We thank all the members of Ventura lab for critical discussion and J. Hollenstein for editing the manuscript. C.B. is supported by the American Italian Cancer Research Foundation fellowship. Work in our lab is funded by grants from NIH-NCI (grant R01CA149707), the Gabrielle’s Angel Foundation, the Starr Consortium and the Geoffrey Beene Cancer Research Foundation.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Kim VN. Nature reviews. Molecular cell biology. 2005;6(5):376–385. doi: 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- 2.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. EMBO J. 2004;23(20):4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai X, Hagedorn CH, Cullen BR. RNA. 2004;10(12):1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. EMBO J. 2002;21(17):4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basyuk E, Suavet F, Doglio A, Bordonne R, Bertrand E. Nucleic Acids Res. 2003;31(22):6593–6597. doi: 10.1093/nar/gkg855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. Nature. 2003;425(6956):415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 7.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. Nature. 2004;432(7014):235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 8.Yi R, Qin Y, Macara IG, Cullen BR. Genes Dev. 2003;17(24):3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Science. 2004;303(5654):95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Nature. 2001;409(6818):363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 11.Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC. Cell. 2001;106(1):23–34. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 12.Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. Science. 2001;293(5531):834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 13.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Genes Dev. 2001;15(20):2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knight SW, Bass BL. Science. 2001;293(5538):2269–2271. doi: 10.1126/science.1062039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hutvagner G, Zamore PD. Science. 2002;297(5589):2056–2060. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- 16.Khvorova A, Reynolds A, Jayasena SD. Cell. 2003;115(2):209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Cell. 2003;115(2):199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 18.Zeng Y, Cullen BR. RNA. 2003;9(1):112–123. doi: 10.1261/rna.2780503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng Y, Wagner EJ, Cullen BR. Mol Cell. 2002;9(6):1327–1333. doi: 10.1016/s1097-2765(02)00541-5. [DOI] [PubMed] [Google Scholar]
- 20.Doench JG, Petersen CP, Sharp PA. Genes Dev. 2003;17(4):438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wightman B, Ha I, Ruvkun G. Cell. 1993;75(5):855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 22.Lai EC. Nat Genet. 2002;30(4):363–364. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- 23.Stark A, Brennecke J, Russell RB, Cohen SM. PLoS Biol. 2003;1(3):E60. doi: 10.1371/journal.pbio.0000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Cell. 2003;115(7):787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 25.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. Mol Cell. 2007;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Science. 2001;294(5543):853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 27.Lau NC, Lim LP, Weinstein EG, Bartel DP. Science. 2001;294(5543):858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Genome Res. 2004;14(10A):1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cullen BR. Mol Cell. 2004;16(6):861–865. doi: 10.1016/j.molcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G. Genes Dev. 2002;16(6):720–728. doi: 10.1101/gad.974702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, Brownstein MJ, Tuschl T, Margalit H. Nucleic Acids Res. 2005;33(8):2697–2706. doi: 10.1093/nar/gki567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim VN, Nam JW. Trends Genet. 2006;22(3):165–173. doi: 10.1016/j.tig.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Tanzer A, Stadler PF. J Mol Biol. 2004;339(2):327–335. doi: 10.1016/j.jmb.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 34.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Cell. 2008;132(5):875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomson JM, Parker J, Perou CM, Hammond SM. Nat Methods. 2004;1(1):47–53. doi: 10.1038/nmeth704. [DOI] [PubMed] [Google Scholar]
- 36.Houbaviy HB, Murray MF, Sharp PA. Dev Cell. 2003;5(2):351–358. doi: 10.1016/s1534-5807(03)00227-2. [DOI] [PubMed] [Google Scholar]
- 37.Suh MR, Lee Y, Kim JY, Kim SK, Moon SH, Lee JY, Cha KY, Chung HM, Yoon HS, Moon SY, Kim VN, Kim KS. Dev Biol. 2004;270(2):488–498. doi: 10.1016/j.ydbio.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 38.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. Nature. 2005;435(7043):839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 39.Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, Ferbeyre G, Chartrand P. J Biol Chem. 2007;282(4):2135–2143. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 40.Woods K, Thomson JM, Hammond SM. J Biol Chem. 2007;282(4):2130–2134. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- 41.Obernosterer G, Leuschner PJ, Alenius M, Martinez J. RNA. 2006;12(7):1161–1167. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wulczyn FG, Smirnova L, Rybak A, Brandt C, Kwidzinski E, Ninnemann O, Strehle M, Seiler A, Schumacher S, Nitsch R. FASEB J. 2007;21(2):415–426. doi: 10.1096/fj.06-6130com. [DOI] [PubMed] [Google Scholar]
- 43.Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Genes Dev. 2006;20(16):2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chaulk SG, Thede GL, Kent OA, Xu Z, Gesner EM, Veldhoen RA, Khanna SK, Goping IS, MacMillan AM, Mendell JT, Young HS, Fahlman RP, Glover JN. RNA Biol. 2011;8(6):1105–1114. doi: 10.4161/rna.8.6.17410. [DOI] [PubMed] [Google Scholar]
- 45.Manni I, Artuso S, Careccia S, Rizzo MG, Baserga R, Piaggio G, Sacchi A. FASEB J. 2009;23(11):3957–3966. doi: 10.1096/fj.09-131847. [DOI] [PubMed] [Google Scholar]
- 46.Guil S, Caceres JF. Nat Struct Mol Biol. 2007;14(7):591–596. doi: 10.1038/nsmb1250. [DOI] [PubMed] [Google Scholar]
- 47.Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M. Cancer Res. 2004;64(9):3087–3095. doi: 10.1158/0008-5472.can-03-3773. [DOI] [PubMed] [Google Scholar]
- 48.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. Nature. 2005;435(7043):828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, de Stanchina E, D’Andrea A, Sander C, Ventura A. Genes Dev. 2009;23(24):2806–2811. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, Li QJ, Lowe SW, Hannon GJ, He L. Genes Dev. 2009;23(24):2839–2849. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, Zuber J, James T, Khan AA, Leslie CS, Parker JS, Paddison PJ, Tam W, Ferrando A, Wendel HG. Nat Cell Biol. 2010;12(4):372–379. doi: 10.1038/ncb2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y, Takahashi T. Cancer Res. 2005;65(21):9628–9632. doi: 10.1158/0008-5472.CAN-05-2352. [DOI] [PubMed] [Google Scholar]
- 53.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. Proc Natl Acad Sci U S A. 2006;103(7):2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsubara H, Takeuchi T, Nishikawa E, Yanagisawa K, Hayashita Y, Ebi H, Yamada H, Suzuki M, Nagino M, Nimura Y, Osada H, Takahashi T. Oncogene. 2007;26(41):6099–6105. doi: 10.1038/sj.onc.1210425. [DOI] [PubMed] [Google Scholar]
- 55.Uziel T, Karginov FV, Xie S, Parker JS, Wang YD, Gajjar A, He L, Ellison D, Gilbertson RJ, Hannon G, Roussel MF. Proc Natl Acad Sci U S A. 2009;106(8):2812–2817. doi: 10.1073/pnas.0809579106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I, Iliopoulos D, Pilozzi E, Liu CG, Negrini M, Cavazzini L, Volinia S, Alder H, Ruco LP, Baldassarre G, Croce CM, Vecchione A. Cancer Cell. 2008;13(3):272–286. doi: 10.1016/j.ccr.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 57.Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M, Boldrini R, Donfrancesco A, Federici V, Giacomini P, Peschle C, Fruci D. PloS one. 2008;3(5):e2236. doi: 10.1371/journal.pone.0002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schulte JH, Horn S, Otto T, Samans B, Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L, Buettner R, Schramm A, Christiansen H, Eilers M, Eggert A, Berwanger B. International journal of cancer. Journal international du cancer. 2008;122(3):699–704. doi: 10.1002/ijc.23153. [DOI] [PubMed] [Google Scholar]
- 59.Conkrite K, Sundby M, Mukai S, Thomson JM, Mu D, Hammond SM, MacPherson D. Genes Dev. 2011;25(16):1734–1745. doi: 10.1101/gad.17027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Nat Genet. 2006;38(9):1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gilbertson RJ, Ellison DW. Annu Rev Pathol. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- 62.Mestdagh P, Bostrom AK, Impens F, Fredlund E, Van Peer G, De Antonellis P, von Stedingk K, Ghesquiere B, Schulte S, Dews M, Thomas-Tikhonenko A, Schulte JH, Zollo M, Schramm A, Gevaert K, Axelson H, Speleman F, Vandesompele J. Mol Cell. 2010;40(5):762–773. doi: 10.1016/j.molcel.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suzuki T, Yasui W, Yokozaki H, Naka K, Ishikawa T, Tahara E. International journal of cancer. Journal international du cancer. 1999;81(4):535–538. doi: 10.1002/(sici)1097-0215(19990517)81:4<535::aid-ijc5>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 64.Park K, Kim SJ, Bang YJ, Park JG, Kim NK, Roberts AB, Sporn MB. Proc Natl Acad Sci U S A. 1994;91(19):8772–8776. doi: 10.1073/pnas.91.19.8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ju HR, Jung U, Sonn CH, Yoon SR, Jeon JH, Yang Y, Lee KN, Choi I. Cancer Lett. 2003;196(2):197–206. doi: 10.1016/s0304-3835(03)00237-4. [DOI] [PubMed] [Google Scholar]
- 66.Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, Sportoletti P, Varmeh S, Egia A, Fedele G, Rameh L, Loda M, Pandolfi PP. Sci Signal. 2010;3(117):ra29. doi: 10.1126/scisignal.2000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang CL, Wang BB, Bartha G, Li L, Channa N, Klinger M, Killeen N, Wabl M. Proc Natl Acad Sci U S A. 2006;103(49):18680–18684. doi: 10.1073/pnas.0609030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lum AM, Wang BB, Li L, Channa N, Bartha G, Wabl M. Retrovirology. 2007;4:5. doi: 10.1186/1742-4690-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Landais S, Landry S, Legault P, Rassart E. Cancer Res. 2007;67(12):5699–5707. doi: 10.1158/0008-5472.CAN-06-4478. [DOI] [PubMed] [Google Scholar]
- 70.Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M, Lagcher W, Sie D, Tanger E, Cox T, Reinders M, Hubbard TJ, Rogers J, Jonkers J, Wessels L, Adams DJ, van Lohuizen M, Berns A. Cell. 2008;133(4):727–741. doi: 10.1016/j.cell.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. Nat Immunol. 2008;9(4):405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. Dev Biol. 2007;310(2):442–453. doi: 10.1016/j.ydbio.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ebi H, Sato T, Sugito N, Hosono Y, Yatabe Y, Matsuyama Y, Yamaguchi T, Osada H, Suzuki M, Takahashi T. Oncogene. 2009;28(38):3371–3379. doi: 10.1038/onc.2009.201. [DOI] [PubMed] [Google Scholar]
- 74.Carraro G, El-Hashash A, Guidolin D, Tiozzo C, Turcatel G, Young BM, De Langhe SP, Bellusci S, Shi W, Parnigotto PP, Warburton D. Dev Biol. 2009;333(2):238–250. doi: 10.1016/j.ydbio.2009.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, Jensen K, Cobb BS, Merkenschlager M, Rajewsky N, Rajewsky K. Cell. 2008;132(5):860–874. doi: 10.1016/j.cell.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 76.de Pontual L, Yao E, Callier P, Faivre L, Drouin V, Cariou S, Van Haeringen A, Genevieve D, Goldenberg A, Oufadem M, Manouvrier S, Munnich A, Vidigal JA, Vekemans M, Lyonnet S, Henrion-Caude A, Ventura A, Amiel J. Nat Genet. 2011;43(10):1026–1030. doi: 10.1038/ng.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marcelis CL, Hol FA, Graham GE, Rieu PN, Kellermayer R, Meijer RP, Lugtenberg D, Scheffer H, van Bokhoven H, Brunner HG, de Brouwer AP. Hum Mutat. 2008;29(9):1125–1132. doi: 10.1002/humu.20750. [DOI] [PubMed] [Google Scholar]
- 78.van Bokhoven H, Celli J, van Reeuwijk J, Rinne T, Glaudemans B, van Beusekom E, Rieu P, Newbury-Ecob RA, Chiang C, Brunner HG. Nat Genet. 2005;37(5):465–467. doi: 10.1038/ng1546. [DOI] [PubMed] [Google Scholar]
- 79.Wang J, Greene SB, Bonilla-Claudio M, Tao Y, Zhang J, Bai Y, Huang Z, Black BL, Wang F, Martin JF. Dev Cell. 2010;19(6):903–912. doi: 10.1016/j.devcel.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li L, Shi JY, Zhu GQ, Shi B. J Cell Biochem. 2011 doi: 10.1002/jcb.23457. [DOI] [PubMed] [Google Scholar]
- 81.Dews M, Fox JL, Hultine S, Sundaram P, Wang W, Liu YY, Furth E, Enders GH, El-Deiry W, Schelter JM, Cleary MA, Thomas-Tikhonenko A. Cancer Res. 2010;70(20):8233–8246. doi: 10.1158/0008-5472.CAN-10-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Siegel PM, Massague J. Nat Rev Cancer. 2003;3(11):807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 83.Shi Y, Massague J. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 84.Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Proc Natl Acad Sci U S A. 1995;92(12):5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohgushi M, Kuroki S, Fukamachi H, O’Reilly LA, Kuida K, Strasser A, Yonehara S. Mol Cell Biol. 2005;25(22):10017–10028. doi: 10.1128/MCB.25.22.10017-10028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr., Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Cell. 2010;141(1):129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chi SW, Zang JB, Mele A, Darnell RB. Nature. 2009;460(7254):479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]