

Abstract
Historically known for its role in blood coagulation and bone formation, Vitamin K (VK) has begun to emerge as an important nutrient for brain function. While VK involvement in the brain has not been fully explored, it is well known that oxidative stress plays a critical role in neurodegenerative diseases. It was recently reported that VK protects neurons and oligodendrocytes from oxidative injury and rescues Drosophila from mitochondrial defects associated with Parkinson’s Disease. In this study, we take a chemical approach to define the optimal and minimum pharmacophore responsible for the neuroprotective effects of VK. In doing so, we have developed a series of potent VK analogs with favorable drug characteristics that provide full protection at nanomolar concentrations in a well-defined model of neuronal oxidative stress. Additionally, we have characterized key cellular responses and biomarkers consistent with the compounds’ ability to rescue cells from oxidative stress induced cell death.
INTRODUCTION
An increasing amount of experimental evidence implicates oxidative stress as one of the major causes of delayed cell death in a variety of neurodegenerative diseases, as well as in stroke, trauma, and seizures. The areas of the nervous system that undergo this delayed cell death represent a critical target for therapeutic interventions, and the recognition of this has stimulated extensive interest in understanding and targeting the responsible underlying processes. In many of these diseases and disorders, mitochondrial generated reactive oxygen species react with and damage cellular components, resulting in caspase independent cell death.1–3 In addition, one of the hallmarks of oxidative stress is a decrease in the reduced form of the major cellular antioxidant, glutathione (GSH), which has been suggested to play a key role in the degeneration of dopaminergic neurons.4, 5 Neuronal GSH synthesis is largely dependent on the exchange of intracellular glutamate for extracellular cystine via the cystine/glutamate antiporter. Concentrations of extracellular glutamate as low as 100 μM inhibit this antiporter,6 and it has been previously reported that extracellular levels of glutamate in the central nervous system (CNS) can reach concentrations as high as 10 mM following injury.7 This depletion of GSH leads to a unique form of mitochondrial driven programmed necrotic cell death (necroapoptosis or oxytosis), which does not depend on caspase activation.8 Recent studies have shown programmed necrotic cell death to be a tightly controlled process involving multiple inter-connected kinases, RIP1, RIP3, MLKL, and the mitochondrial phosphatase, phosphoglycerate mutase family member 5 (PGAM5), via its regulation of dynamin-related protein 1 (Drp-1) and subsequent mitochondrial fragmentation. In addition, PGAM5 has been shown to be at the convergent point of multiple cell death pathways. Knock-down of PGAM5 prevents both extrinsic (Tumor-necrosis factor-α and Fas ligand) and intrinsic (tert-butyl hydroperoxide and calcium ionophore) induced cell death.9
Vitamin K (VK) is a group of structurally similar, fat soluble vitamins that play well known roles in the post-translational modification of proteins required for blood coagulation and bone metabolism.10, 11 There are two forms of naturally occurring VK, phylloquinone (VK1) and the menaquinones (VK2). A synthetic form of VK, menadione (VK3), is also available and used in animal feeds and supplements. All forms of VK possess a common 2-methyl-1,4-naphthoquinone core structure, but individual forms differ in the length and degree of saturation of an aliphatic side chain attached to the 3′ position. VK1, found primarily in green leafy vegetables,12 is a single compound containing a saturated side chain consisting of four isoprenoid subunits. While it is the major dietary source of VK, post mortem and animal studies have indicated that concentrations are significantly lower in the brain and other tissues compared with the VK2.13 There are several forms of VK2 that are classified based on the length of the unsaturated 3′ side chain. The major form of VK2 (>90%) found in animal tissues has a four isoprenoid unit (geranylgeranyl) side chain. Although there is a small dietary presence of VK2, it is primarily obtained in situ by removal of the phytyl group of VK1 followed by a subsequent geranylgeranylation by the mitochondrial prenyltransferase UBIAD1.14–16 This has been shown to occur in cultured primary brain slices and neurons, indicating an as of yet unknown but important function for VK2 in brain function.17 In addition, evidence supporting the role of VK in brain function has been steadily accumulating, including roles in development and aging,18 brain sulfotransferase activity,19 mitochondrial electron transfer,20 and a potential role in the pathogenesis of Alzheimer’s21 and Parkinson’s Disease.20 Furthermore, investigations into the neuroprotective capacity of VK have indicated that VK has the capability to prevent oxidative stress induced cell death in cultured neurons and oligodendrocytes. This protection has been established in multiple models involving the depletion of glutathione, including exposure to methylmercury, L-buthionine sulfoximine (BSO), elevated levels of extracellular glutamate, and cystine depletion.22, 23
In this paper, we present the results of in vitro evaluations of various derivatives of VK aimed at exploring the structural requirements for efficient neuroprotective activity, without the manifestation of in vitro cytotoxicity. We report the development and facile synthesis of compounds with favorable drug-like properties for CNS applications (e.g. M.W.< 400, ClogP < 5.0, and tPSA < 60)24 that, based on their capacity to inhibit oxidative-stress mediated neuronal cell death, were found to be potent neuroprotective agents without obvious neurotoxicity in vitro, and with activity exceeding that of VK2 greater than 10 fold. Additionally, standard blood count and chemistry testing revealed no blood or major organ toxicity in mice over three weeks of treatments. We further investigated the mechanisms and biomarkers underlying the neuroprotective effects of VK2 and the most promising compounds.
RESULTS AND DISCUSSION
Vitamin K Analog Synthesis
2-amino-1,4-naphthoquinone (1d) was synthesized as previously described.25 We have scaled up the synthesis of this compound in our laboratory and have found the reported synthesis to be amenable to both milligram and multi-gram scale reactions. The other 2-amino substituted 1,4-napthoquinones were synthesized following a basic procedure as previously described with only minor modifications made as needed, which are described in detail in the experimental section (Table 2).26, 27 Briefly, to a solution of 2-bromo-1,4-naphthoquinone in ethanol was added an excess of the corresponding amine, and the reaction was stirred at room temperature and monitored by TLC. We found this reaction to provide decent yields, be applicable to a variety of amine substrates, and spectroscopically pure product was frequently obtained by simple vacuum filtration. The inexpensive cost of reagents, simple and environmentally friendly reaction and purification conditions, coupled with the extremely high potency, neuroprotective efficacy, and lack of in vitro neurotoxicity, makes these compounds an attractive and promising option for the development of neuroprotective agents.
TABLE 2.
In Vitro Neuroprotective Activity of 2-amino-1,4-naphthoquinones.
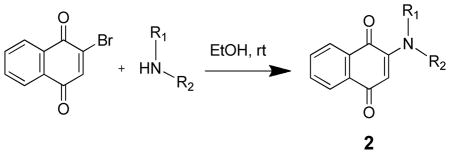
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | Protectiona | Toxicityb | Safety Index |
|
| |||||
| PC50 (nM) | TC50 (nM) | TC50/PC50 | |||
| VK2 | - | - | 432 | >100,000 | 231 |
|
| |||||
| 2a | -H | -Me | 1346 | >100,000 | 74 |
| 2b | -H | -Et | 761 | 81,000 | 106 |
| 2c | -Me | -Me | 246 | >100,000 | 407 |
| 2d | -H |
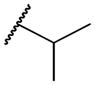
|
1703 | 46,000 | 27 |
| 2e | -H |
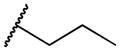
|
328 | 30,000 | 91 |
| 2f | -H |
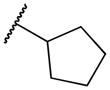
|
409 | 44,000 | 108 |
| 2g | -H |
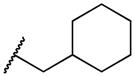
|
54 | >100,000 | 1,852 |
| 2h | -H |
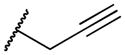
|
120 | 53,000 | 442 |
| 2i | -H |

|
64 | 7,000 | 109 |
| 2j | -H |
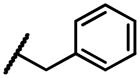
|
88 | >100,000 | 1,136 |
| 2k | -Me |
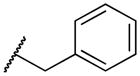
|
128 | >100,000 | 781 |
| 2l | -H |
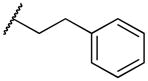
|
584 | >100,000 | 171 |
| 2m | -H |
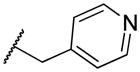
|
72 | 80,000 | 1,111 |
| 2n | -H |
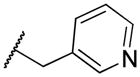
|
215 | 37,000 | 172 |
| 2o | -H |
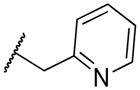
|
933 | 50,000 | 54 |
| 2p | -H |
|
59 | >100,000 | 1,695 |
| 2q | -H |
|
31 | >100,000 | 3,226 |
| 2r | -H |
|
66 | >100,000 | 1,515 |
| 2s | -H |
|
114 | >100,000 | 877 |
| 2t | -H |
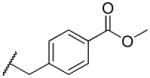
|
232 | >100,000 | 431 |
| 2u | -H |
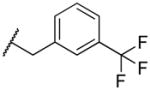
|
45 | >100,000 | 2,222 |
| 2v | -H |
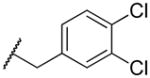
|
46 | >100,000 | 2,174 |
In vitro neuroprotective activity and
neurotoxicity assessed by treating HT22 cells with various concentrations of compounds with or without 10 mM glutamate for 24 hrs. Cell viability was estimated by treating cells with MTS and measuring absorbance at 490 nM. PC50, concentration producing 50% protection, values calculated using GraphPad Prism based on 12 point titrations, n ≥ 4; TC50, concentration producing 50% toxicity, values calculated using GraphPad Prism based on 7 point titrations, n ≥ 3.
In synthesizing the 2-amido substituted derivatives, when starting from compound 1d, we found the 2′ amine to be highly deactivated and not amenable to several standard peptide coupling procedures. Activations with triethylamine and tert-butyllithium and subsequent additions of acyl chlorides also proved to be unsuccessful. Desired products were obtained in reasonable yields by dissolving compound 1d and 4.2 equivalents of sodium hydride in dry THF, followed by dropwise addition of the corresponding acyl chloride (Supplementary Table S1). 2-ureyl substituted derivatives were synthesized as previously described (Supplementary Table S2).28 Briefly, to a stirred solution of Compound 1d dissolved in dimethylformamide was added the corresponding isocyanate followed by a catalytic amount of triethylamine. The reaction was then heated to 80°C and monitored using TLC. The non-redox chromone based analogs, 5c and 5d, were also synthesized by dissolving the appropriate carboxylic acid in dimethylformamide (Supplementary Table S3). The solution was then cooled to 0°C, and two equivalents of thionyl chloride were added and the mixture stirred for 30 minutes, followed by the addition of aniline. The following day, the reaction was quenched with an excess of saturated sodium bicarbonate, and the resulting precipitate was filtered and crystallized with hot ethyl acetate.
Neuroprotection Structure-Activity Relationships
The neuroprotective properties of VK and its derivatives were estimated through their protective effects against cell death in mouse neuronal HT22 cells induced by exposure to high levels of glutamate, which recapitulates a hallmark of the extracellular environment found in several neurodegenerative diseases and CNS injuries and is a well established model of neuronal oxidative stress.6–8, 29–31 We began this investigation by comparing the relative abilities of the two predominant naturally occurring forms of VK, VK1 and VK2, to prevent oxidative cell death. Consistent with the reported literature utilizing primary cortical neurons,23 we found that not only was VK2 many times more potent than VK1, but neither exhibited any signs of cellular toxicity (Supplementary Figure S1). Since both VK1 and VK2 rescued HT22 cells from oxidative cell death, we hypothesized that it was the 1,4-naphthoquinone core, and not the isoprenoid side chain, that was responsible for the neuroprotective effects. We then tested several 1,4-naphthoquinones with various substitutions at the 2′ and 3′ positions to determine the minimum structural requirement of the system and what the optimum scaffold for future developmental efforts and biological validation would be (Figure 1B and Table 1). Consistent with our hypothesis, un-substituted 1,4-naphthoquinone does exhibit neuroprotective activity at micromolar concentrations. Substitution by a single methyl group at the 2′ position (VK3) improves the compound’s ability to protect against cell death. Various other simple substituents at the 2′ and 3′ positions give the naphthoquinone structure the ability to prevent oxidative cell death at nanomolar concentrations (Table 1 and Supplementary Figure S2). Of the tested structures, it was found that the presence of a single amine group at the 2′ position provided the greatest potency and the lowest non-specific cellular toxicity, so this structure (1d) was chosen as the scaffold for further modification.
Figure 1.
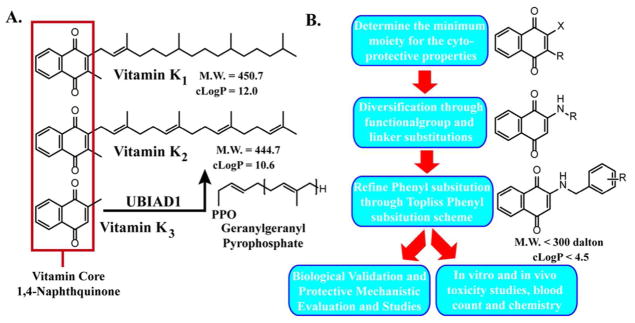
A. Structures of VK1, VK2, and VK3. VK3 is a pro-vitamin, and UBIAD1 converts VK3 or cleaved VK1 into VK2 in situ through geranylgeranylation. Defects in UBIAD1 have been shown to be a dominant enhancer of Parkinson’s related PINK1 mutations. B. Experimental flow. Synthetic approach and selective criteria used to generate more potent and non-toxic VK analogs.
TABLE 1.
In Vitro Neuroprotective Activity of 1,4-naphthoquinones substituted at the 2′ and 3′ positions.
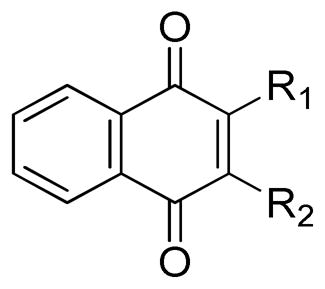
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | Protectiona | Toxicityb | Safety Index |
|
| |||||
| PC50 (nM) | TC50 (nM) | TC50/PC50 | |||
| VK2 | - | - | 432 | >100,000 | 231 |
|
| |||||
| 1a | -H | -H | 1541 | 19,000 | 12 |
| 1b | -Me | -H | 797 | 19,000 | 24 |
| 1c | -Me | -Me | 118 | 30,000 | 254 |
| 1d | -NH2 | -H | 61 | 49,000 | 803 |
| 1e | -NH2 | -Me | 1740 | 54,000 | 31 |
| 1f | -COOH | -H | 716 | >100,000 | 140 |
| 1g | -OH | -H | 1005 | >100,000 | 100 |
In vitro neuroprotective activity and
neurotoxicity assessed by treating HT22 cells with various concentrations of compounds with or without 10 mM glutamate for 24 hrs. Cell viability was estimated by treating cells with MTS and measuring absorbance at 490 nM. PC50, concentration producing 50% protection, values calculated using GraphPad Prism based on 12 point titrations, n ≥ 4; TC50, concentration producing 50% toxicity, values calculated using GraphPad Prism based on 7 point titrations, n ≥ 3.
We then began to explore the effects of modifying the 2′ amine group. Amino, amido, and ureyl derivatives were also synthesized and their effects tested. We found that for the most part, both the amido and ureyl derivatives demonstrated decreased potency relative to compound 1d, and many exhibited significant non-specific toxicity (Supplementary Table S1 and S2). The same can be said for most of the amino derivatives with short alkyl or cycloalkyl substituents, although in the case of the methyl and dimethyl groups (2a & 2c; Table 2), cellular toxicity was abolished. However, we found that the addition of a benzyl group to the 2′ amine completely abolished all toxicity associated with the molecule, while maintaining a low nanomolar PC50 (2j; Table 2).
Therefore, we selected compound 2j as our new lead compound. The effects of varying the number of carbons in the linker region between the quinone and the phenyl ring were explored by synthesizing and testing compounds 2i and 2l. While compound 2i, with the linker region removed, did show increased potency, it also manifested toxicity. Compound 2l, with a single extra methylene group added to the linker region, demonstrated no toxicity, but it suffered from a significant decrease in potency. Compound 2k, with the 2′ tertiary amine, was then found to exhibit decreased potency, indicating a potential hydrogen bond interaction occurring at this position. The nitrogen heterocycles 2m–2o also all exhibited toxicity. Next, we synthesized several analogs according to a Topliss scheme in order to maximize the potency of the molecule by determining the optimum substitution of the benzene ring.32 None of these compounds manifested any in vitro toxicity, and each of them exhibited increased potency. Compound 2q, with the chlorine atom located in the meta position, was the most potent. Compound 2q displayed a PC50 of 31 nM, which represents an almost 3 fold increase over compound 2j, and a more than 10 fold increase over VK2.
Cellular Biological Evaluation
Although we have previously shown that some naphthoquinones are capable of selectively inhibiting histone deacetylases (HDACs), the lead compounds used in this study do not directly possess that capacity.33 Since the first intracellular step in the initiation of the oxidative glutamate induced cell death is the depletion of GSH via the blockade of cystine import through the cystine/glutamate antiporter,8 we first tested whether the compounds had any effect on the depletion of GSH. We found that VK2, compounds 2q and 2j, Necrostatin-1 (Nec-1, inhibitor of necroapoptosis via inhibition of RIP1 kinase; only inhibits extrinsic death signaling), Idebenone (Ideb, synthetic coenzyme Q10 antioxidant), and Trolox (chemical antioxidant), did not prevent the depletion of GSH, even when treated at levels much higher than required for cellular protection (Figure 2A and Supplementary Figure S1). GSH depletion due to exposure of cells to high concentrations of glutamate results in the accumulation and production of free radicals. According to reports, and consistent with our own data (data not shown), this occurs in a time dependent manner. There is an initial linear increase in free radical accumulation that parallels GSH depletion over approximately the first four hours of glutamate treatment followed by a sharp, exponential increase in free radicals that occurs between six and eight hours.34, 35 Thus we tested whether the compounds were able to prevent free radical accumulation after eight hours of glutamate treatment using two distinct fluorogenic dyes that exhibit intense fluorescence when oxidized by a variety of reactive oxygen species (ROS) and reactive nitrogen species (RNS). We found that at concentrations consistent with their protective capacity, VK2 and compounds 2q and 2j completely prevented the increased accumulation of intracellular free radicals, with 2q and 2j being more effective at lower concentrations (Figure 2B and 2C).
Figure 2.

HT22 cells treated for 8 hrs with 10 mM glutamate. A. Depletion of total cellular GSH occurs in HT22 cells treated with glutamate. Co-treatment with VK2, 2j, or 2q (500 nM) does not prevent GSH depletion. Nec-1 (50 μM), Ideb (5 μM), and Trolox (25 μM) also did not prevent GSH depletion. B. Free radical accumulation measured using Rho123. Co-treatment with 250 nM 2j and 2q prevent the accumulation of free radicals in response to glutamate treatment, with VK2 being less effective. One-way ANOVA with Bonferroni’s posttest was used to compare mean intensities. Drug treatments were all significantly less than glutamate treatment, with 2j and 2q treatments being statistically similar to control, p<.01. C. Free radical accumulation is visualized with CM-H2DCFDA. Results are consistent with those found with Rho123.
We next examined the possibility of a direct antioxidant interaction between the compounds and free radicals. To evaluate the antioxidant potential of the compounds, we tested the ability of the compounds to quench the stable free radical 2,2-diphenyl-1-picrylhydrazyl (DPPH), a widely applied free radical scavenging assay.36, 37 In the presence of VK2 or compounds 2q or 2j, at concentrations far exceeding those needed for cellular protection, the optical absorbance of a solution of DPPH remained constant, while the known antioxidants Trolox and ascorbic acid rapidly scavenged DPPH, as evidenced by the loss of DPPH absorbance (Figure 3A).
Figure 3.
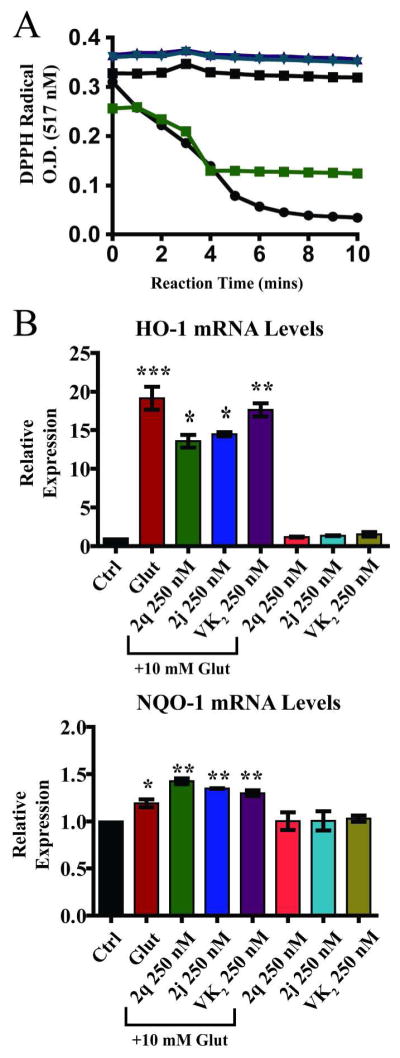
A. Free radical scavenging capacity determined by monitoring the disappearance of the optical absorbance of the stable free radical DPPH. Known free radical scavengers vitamin C (
 ) and Trolox (●) used as controls. VK2 (■), 2j (
) and Trolox (●) used as controls. VK2 (■), 2j (
 ), and 2q (
), and 2q (
 ) did not show direct antioxidant capacity. All compounds tested at 20 μM B. Expression of antioxidant response genes. Significant cellular antioxidant responses are elicited in HT22 cells after 8 hours of glutamate treatment with significant increase in HO-1 and NQO-1 gene expression. VK2, 2q, and 2j significantly decreased HO-1 expression but did not affect NQO-1 expression. One-way ANOVA with Bonferroni’s posttest was used to compare mean levels (n = 3), p<.01.
) did not show direct antioxidant capacity. All compounds tested at 20 μM B. Expression of antioxidant response genes. Significant cellular antioxidant responses are elicited in HT22 cells after 8 hours of glutamate treatment with significant increase in HO-1 and NQO-1 gene expression. VK2, 2q, and 2j significantly decreased HO-1 expression but did not affect NQO-1 expression. One-way ANOVA with Bonferroni’s posttest was used to compare mean levels (n = 3), p<.01.
The data presented thus far indicates that the protective activity of VK2 and the derivatives occurs intracellularly and downstream of the cystine/glutamate antiporter and the depletion of GSH, is mediated by the inhibition of the accumulation of intracellular free radicals, and does not occur via a direct free radical scavenging interaction. This suggests that the protection is likely a result of one of the following: the activation of an endogenous intracellular antioxidant response, the interference with a critical component of a cell death signaling pathway, or the inhibition of the production of free radicals.
A major mechanism by which cells defend themselves against oxidative stress is through the increased expression of genes whose protein products are involved in the removal of free radical species.38 Activation of the antioxidant response by VK2 and compounds 2q and 2j was evaluated using qRT-PCR to measure the mRNA levels of two of the antioxidant response genes, heme oxygenase 1 (HO-1) and NAD(P)H:quinone oxidoreductase (NQO-1).39 We found that treatment with glutamate significantly increased the mRNA expression of both enzymes. Co-treatment with VK2 and compounds 2q and 2j decreases the mRNA levels of HO-1, but not NQO-1, relative to the glutamate only treatment. However, even with this decrease, HO-1 mRNA levels in the drug and glutamate co-treatment conditions were still significantly higher than controls (Figure. 3B). Treatment with the compounds in the absence of glutamate did not affect the mRNA levels of either enzyme. Since VK2 and its analogs contain a naphthoquinone moiety, it is interesting to note that expression of NQO-1 is not up-regulated by treatment with VK2, 2q, or 2j, indicating these naphthoquinones are not likely to be substrates of NQO-1 and do not elicit phase 2 detoxification enzyme up-regulation. Enzyme levels of HO-1 and NQO-1 were also assessed, and minimum up-regulation was observed (Supplementary Figure S3). This data suggests that while the cellular antioxidant response is intact in HT22 cells; it is insufficient to prevent glutamate induced cell death, and the decreased activation in the co-treated cells likely reflects a lessening of overall cellular oxidative stress.
In order to investigate the hypothesis that protection is mediated by the inhibition of the production of free radicals, we sought to identify the source of the glutamate-induced free radicals. Mitochondria are a major source of cellular free radicals, primarily through the generation of superoxide, which is produced as a normal byproduct of oxidative phosphorylation. Mitochondrial dysfunction and subsequent increases in mitochondrial superoxide production have been implicated as important preceding events that ultimately result not only in the glutamate-induced cell death in HT22 cells,40 but also in several disease states, including neurodegenerative diseases such as Parkinson’s, Alzheimer’s, and amyotrophic lateral sclerosis (ALS),41 and brain injuries such as stroke and traumatic brain injury that are associated with cerebral hypoxia and ischemia.42–44 Mitochondrial dysfunction in cell death is characterized by a decline in mitochondrial membrane potential, respiratory defects, an increase in superoxide production, changes in ATP levels, and the release of apoptogenic factors, including cytochrome c and apoptosis-inducing factor.45 MitoSOX Red is a novel fluorogenic dye that can be used to selectively detect the production of superoxide by mitochondria. The reagent permeates live cells and selectively targets mitochondria. It is rapidly oxidized to a highly fluorescent product by superoxide but not by other ROS or reactive nitrogen species. Mitotracker Deep Red is a fluorogenic dye that is used to label mitochondria in living cells. Using these two dyes, we have shown that increased mitochondrial superoxide generation and mitochondrial fragmentation occurs in HT22 cells exposed to high levels of extracellular glutamate, and co-treatment with VK2, 2q, or 2j, prevented these events (Figure 4). This result is consistent with the recent discovery of the ability of eukaryotic cells to use VK2 as an alternative electron carrier that is capable of alleviating mitochondrial complex defects due to gene mutations, thereby reducing superoxide generation from the electron transport chain (ETC).20
Figure 4.

A. Glutamate treatment increases superoxide generation within mitochondria. MitoTracker DR (green) stains for active mitochondria and MitoSOX (red) localizes in the mitochondria and selectively reacts with superoxide. Co-localization of MitoTracker and MitoSox supports that the superoxide is likely generated by mitochondria. B. Mitochondrial morphology. Mitochondria under normal cellular conditional exhibits a complex network morphology (teal arrows). Under glutamate injury, mitochondrial fragmentation occurs (red arrows) and VK2 treatment maintains normal mitochondrial morphology.
In light of the findings that the protection against glutamate induced cell death provided by VK2, 2q, and 2j, are at least partially mediated by the attenuation of the increased production of mitochondrial superoxide, we also examined the effects of these compounds on tert-butylhydroperoxide (t-BuOOH) induced cell death. t-BuOOH is a short chain analog of the lipid hydroperoxides formed from peroxidation reactions during oxidative stress that is cytotoxic to cells.46 VK2, 2q, and 2j are capable of protecting HT22 cells from t-BuOOH, while the antioxidants Trolox and co-enzyme Q10 are less effective (Supplementary Figure S4). This result indicates that a specific molecular pathway is also involved in this protection. Emerging evidence indicates that mitochondrial fission and fragmentation, processes wherein the thread-like, tubular mitochondrial networks are split into small, isolated organelles, plays an active role in cell death. It is well documented that cells undergo rapid and extensive fragmentation in the early stages of cell death.47–51 Recent studies have shown that mitochondrial fragmentation is regulated by the translocation of dynamin-related protein 1 (Drp1) from the cytosol to the mitochondria, where it assembles to form spirals at division sites.52 Although the exact molecular mechanism is still not well studied, the activity of Drp1 is regulated by the phosphorylation and dephosphorylation of specific serine residues by protein kinases and phosphatases such as calcineurin and the PGAM5.9, 53 PGAM5 was recently identified as a key signaling protein that sits at the convergent point of multiple cell death pathways, and the silencing of PGAM5 prevents cell death caused by stimulation of both extrinsic (Tumor necrosis factor-α and Fas ligand) and intrinsic pathways (t-BuOOH and calcium ionophore).9 It has also been indicated that inhibition of this pathway is capable of preventing cell death in vitro and providing neuroprotection in vivo with a potentially extended therapeutic window.54, 55 Since VK2, 2q, and 2j were able to protect HT22 cells from both glutathione depletion and t-BuOOH induced cell deaths, we chose to investigate whether VK2 and our synthesized derivatives were able to affect this specific mitochondrial death pathway and maintain mitochondrial homeostasis. We found that when exposed to high levels of glutamate for 16 hours, phosphorylation of Drp1 at serine residue 637 decreased considerably (Figure 5). Co-treatment with VK2 at 500 nM completely prevented this de-phosphorylation. To our knowledge, this is the first report of this finding. Co-treatment with our derivatives, at much lower concentrations (125 nM), was also able to maintain Drp1 phosphorylation (Figure 5A). The effects of the compounds on cellular PGAM5 are interesting. We found that in response to glutamate treatment, a dramatic increase in the lower band of PGAM5 occurred. This response is similar to the tumor necrosis factor-alpha induced necroapoptotic response, and the lower band is thought to be a cleavage product of the PGAM5-L isoform.9 Furthermore, this increase is completely attenuated by VK2 at 500 nM and compounds 2q and 2j at 125 nM.
Figure 5.
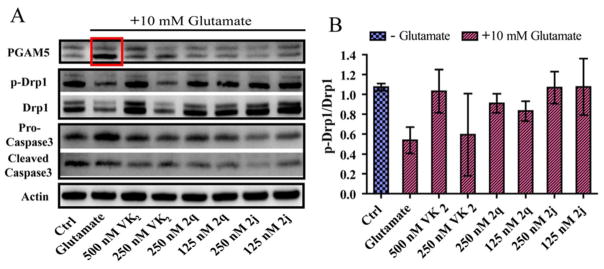
A. Western blot analysis of HT22 cells treated with 10 mM glutamate for 16 hrs. Glutamate treatment causes a dramatic increase in the lower band of PGAM5, as well as a decrease in phosphorylation of Drp1 at residue Ser 637. VK2, 2q, and 2j prevent PGAM5 cleavage and activation and subsequent dephosphorylation of Drp1. B. Quantification of p-Drp1 (Ser 637) relative to Drp1. Densitometric analysis confirms that there is a significant decrease in phosphorylation of Drp1 at residue Ser 637 with 10 mM glutamate treatment for 16 hrs, and the phosphorylation state is maintained by co-treatment with 500 nM VK2 and compounds 2q or 2j at 250 and 125 nM. Co-treatment with 250 nM VK2 was less effective. One-way ANOVA with Bonferroni’s posttest was used to compare mean levels (n = 3), p<.01.
Chronic Toxicity Evaluation
Preliminary in vivo toxicity was assessed for compounds 2q and 2j in mice. Although the compounds exhibit nanomolar efficacy in vitro, because of their complete lack of in vitro toxicity, we chose to use very high, chronic doses (50 mg/kg) for our preliminary in vivo toxicity evaluation. Adult mice were injected intraperitoneally (i.p.) daily. During the course of their treatments, animals exhibited no weight loss or outward signs of pain or distress. After three weeks of high dose injections, animals were sacrificed and standard blood count and chemistry analyses performed 2 hours after the final injection. All measured parameters of the treatment groups were consistent with those of the vehicle control group (Supplementary Tables S4 and S5), indicating that there is not likely to be any blood or major organ toxicity associated with chronic, high dose i.p. administration of these compounds.
CONCLUSIONS
In summary, we started with the natural product VK2, which has an emerging role in brain function and health, and systematically generated a number of synthetic derivatives with favorable CNS drug properties, which exhibited neuroprotective activity at low nanomolar concentrations and that demonstrated no in vitro neurotoxicity. We confirmed that the naphthoquinone core is the structural motif responsible for the neuroprotective activity and found that amine substitution at the 2′ carbon greatly enhanced the protective activity. The further addition of a benzyl group to the 2′ amine improved the safety index of the compound by completely abolishing any in vitro neurotoxicity, and chloro substitution at the meta position of the aromatic ring further improved the protective potency of the compound.
Our preliminary efforts to investigate the mechanism by which VK2 and its derivatives provide their protection against oxidative stress led us to the discovery that both VK2 and our derivatives prevent the dysregulation of mitochondrial function under injurious conditions, potentially via their ability to influence the critical PGAM5-Drp1 mitochondrial death signaling pathway. Based on the shared naphthoquinone motif, and the recent report that eukaryotic cells are able to use VK2 for this purpose,20 it is possible that our compounds are preventing stress induced mitochondrial dysfunction and subsequent superoxide generation by acting as enhanced or alternative electron carriers. Further investigation is needed to reveal why this parallel mechanism of electron transfer chain exists and why it appears to function more efficiently under injurious conditions.
During the preparation of this manuscript, a study by Sekine et al. identified the key protease, Presenilins-associated rhomboid-like protease (PARL), which is responsible for the activation and cleavage of PGAM5. This suggests that VK2, 2q, and 2j, are likely to act on a PARL regulatory mechanism. The study claims that PARL is activated by mitochondrial membrane depolarization, which would be consistent with our mitochondrial dysfunction hypothesis, although the exact mechanism of PARL activation in this situation is remains unclear. The identification of the exact mechanism by which this occurs and the involvement of VK2 and our derivatives in this pathway are currently under investigation in our lab.
EXPERIMENTAL
Cell Culture
The HT22 neuronal cell line is a subclone of HT4, derived from the mouse hippocampus56. They do not express active ionotropic glutamate receptors and are not subject to excitotoxicity.57 The HT22 cells used in this study were kindly provided by Dr. David Schubert (The Salk Institute for Biological Studies, La Jolla, CA, USA). The cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM/high glucose) supplemented with 10% fetal bovine serum (Hyclone) and 5 mL Antibiotic-Antimycotic (Amphotericin B, Penicillin, and Streptomycin; Invitrogen) at 37°C in 5% CO2.
Cell Viability Assay
Cell viability was assessed by the ability of the viable cells to metabolize 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)2-H-tetrazolium, inner salt (MTS), as described previously58. The metabolism of tetrazolium salts is often used to measure cellular proliferation, but in the system used here, it has been previously shown to correlate well with cell viability as determined by trypan blue exclusion and colony-forming assays.59 Briefly, HT-22 cells were seeded onto 96-well plates at 5.0 × 103 cells per well in 75 μL of medium and maintained at 37°C in 5% CO2 overnight prior to the initiation of experimental treatments. For glutamate toxicity testing, cells were subsequently treated with 25 μL of medium containing glutamate (monosodium glutamate, Sigma, 1 M stock concentration in media, to achieve a final concentration of 10 mM) plus inhibitors (stock in DMSO) and maintained at 37°C in 5% CO2. For t-BuOOH toxicity testing, cells were subsequently treated with 25 μL of medium containing t-BuOOH (Sigma, 10 mM stock concentration, to achieve a final concentration of 50 μM) plus inhibitors (stock in DMSO) and maintained at 37°C in 5% CO2 for 3 hours, after which the media was removed, cells washed gently with HBSS, the media replaced with standard culture media, and the cells returned to incubation at 37°C in 5% CO2. For both glutamate and t-BuOOH treatments, 10 μl of MTS solution was added to each well 24 hrs after initial treatment, and the cells were maintained in growth medium for 2 h at 37°C. Absorbance at 490 nm was subsequently measured by a SpectraMax 190 plate reader (Molecular Devices). The growth medium without cells in the presence of MTS solution was used as solution background and untreated cells were considered the controls. Cell viability was calculated as a percentage compared with untreated controls. EC50 determinations were based on 12-point titrations using GraphPad Prism, and each experiment was repeated at least four times. Morphology of HT22 cells following treatments was determined by phase-contrast microscopy. Digital images of cells grown on cell culture plates were captured at 10x magnification.
Glutathione Determination
The content of total glutathione (reduced and oxidized) in the supernatant of cell homogenate was measured using the glutathione assay kit (Cayman Chemical) in microtiter plate assay, which utilizes an enzymatic recycling method using glutathione reductase for the quantification of glutathione. The sulfhydryl group of glutathione reacts with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) and produces a yellow-colored 5-thio-2-nitrobenzoic acid (TNB), which was measured at 405 nm.
Antioxidant Activity Assay
Antioxidant activity of various reagents was assayed as previously described by monitoring the disappearance of the optical absorbance of the stable-free radical DPPH on reaction with test compounds.36 The rate of the reaction represents the antioxidant potency of test agents. The known free radical scavenger Trolox and ascorbic acid were used as positive controls. Briefly, 10 μL of test reagents at final concentrations of 20 μM were added to 200 μL of 100 μM DPPH in methanol. Optical absorbance of DPPH at 517 nm was immediately monitored for 10 min.
Intracellular Free Radical Measurement and Imaging
Intracellular accumulation of free radicals was evaluated by spectrofluorometry using the membrane-permeable compound dihydrorhodamine 123 (Rho123; Invitrogen), and images were collected using 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA; Invitrogen). Briefly, HT-22 cells were seeded onto black walled 96-well plates at 5.0 × 103 cells per well in 75 μL of medium and maintained at 37°C in 5% CO2 overnight prior to the initiation of experimental treatments. Cells were subsequently treated with 25 μL of medium containing glutamate (monosodium glutamate, Sigma, 1 M stock concentration in media, to achieve a final concentration of 10 mM) plus inhibitors (stock in DMSO) and maintained at 37°C in 5% CO2. After 8 hrs, Rho123 (5 μM) or CM-H2DCFDA (1 μM) was added to achieve the indicated final concentrations. Cells were allowed to incubate for 20 minutes and then washed twice with Hanks Balanced Salt Solution (HBSS; Hyclone; CaCl2 1.26 mM, MgCl2-6H2O 0.493 mM, MgSO4-7H2O 0.407 mM, KCl 5.33 mM, KH2PO4 0.441 mM, NaHCO3 4.17 mM, NaCl 137.93 mM, Na2HPO4 0.338 mM, D-Glucose 5.56 mM), with a final addition of 100 μL of HBSS in which the cells were visualized. Fluorescence of the oxidized Rho123 was measured using a Fluoroskan Ascent spectrofluorometer (Labsystems). Peak excitation and emission wavelengths were 500 and 536 nM, respectively. For fluorescent imaging of the oxidation product of CM-H2DCFDA, cells were immediately visualized using an InCell 2000 Analyzer (GE) with a 40× objective and a FITC excitation/emission filter set. Image processing was performed using ImageJ, and for all images, the microscope and image processing settings, such as levels, brightness, contrast, and exposure time were held constant across all treatment conditions.
Mitochondrial Morphology and Superoxide Generation
In order to evaluate the source of the free radical generation, fluorescence microscopy was used to image HT22 using the fluorogenic dyes Mitotracker Deep Red (DR) and MitoSOX Red (Invitrogen). Mitotracker DR is a cell permeant dye that accumulates in active, polarized mitochondria. MitoSOX Red is highly selective reagent for the detection of superoxide in the mitochondria of live cells. It is live-cell permeant and rapidly and selectively targeted to mitochondria, where it is readily oxidized by superoxide, but not other cellular free radicals, and exhibits red fluorescence. Briefly, HT22 cells were seeded onto black walled 96-well plates at 5.0 × 103 cells per well in 75 μL of medium and maintained at 37°C in 5% CO2 overnight prior to the initiation of experimental treatments. Cells were subsequently treated with 25 μL of medium containing glutamate (monosodium glutamate, Sigma-Aldrich, 1 M stock concentration in media, to achieve a final concentration of 10 mM) plus inhibitors (stock in DMSO) and maintained at 37°C in 5% CO2. After 8 hrs, MitoSOX Red (5 μM) and Mitotracker DR (10 nM) were added to achieve the indicated final concentrations. Cells were allowed to incubate for 20 minutes and then washed twice with Hanks Balanced Salt Solution (HBSS; Hyclone; CaCl2 1.26 mM, MgCl2-6H2O 0.493 mM, MgSO4-7H2O 0.407 mM, KCl 5.33 mM, KH2PO4 0.441 mM, NaHCO3 4.17 mM, NaCl 137.93 mM, Na2HPO4 0.338 mM, D-Glucose 5.56 mM), with a final addition of 100 μL of HBSS in which the cells were visualized. Cells were immediately visualized using an InCell 2000 Analyzer (GE) with a 40× objective and a Cy5/Cy5 excitation/emission filter set for Mitotracker DR and a YFP/Cy3 filter set for MitoSOX Red. Image processing was performed using ImageJ, and for all images, the microscope and image processing settings, such as levels, brightness, contrast, and exposure time were held constant across all treatment conditions.
Real-time Polymerase Chain Reaction
HT22 cells were seeded onto cell culture treated 6-well plates at 3.0 × 105 cells per well in 2 mL of medium and maintained at 37°C in 5% CO2 overnight prior to the initiation of experimental treatments. Cells were subsequently treated with 1 mL of medium containing glutamate (monosodium glutamate, Sigma-Aldritch, 1 M stock concentration in media, to achieve a final concentration of 10 mM) plus inhibitors (stock in DMSO) and maintained at 37°C in 5% CO2. After 16 hrs, the media was collected and the cells harvested by trypsanization. After centrifugation, pelleted cells were washed with cold Hanks’ Balanced Salt Solution (HBSS Ca2+/Mg2+ Free; Invitrogen; KCl 5.33 mM, KH2PO4 0.441 mM, NaHCO3 4.17 mM, NaCl 137.93 mM, Na2HPO4 0.338 mM, D-Glucose 5.56 mM), and qRT-PCR was used to detect HO-1, NQO-1, and PGAM5 mRNAs in the HT22 cells. Consistent with reports utilizing serum starvation models, the mRNA levels of both Actin and GAPDH were found to be strongly affected by glutamate treatment, but β2-microglobulin remained fairly stable. Therefore β2-microglobulin was used as a reference gene.60 HT22 cells were cultured as described for 16 hrs. Total RNAs were extracted using the RNEasy kit (Qiagen), and transcripts of interest were amplified by PCR using the 1-step Syber Green qRT-PCR kit (Quanta Biosciences) and the following specific primers (Harvard PrimerBank; Integrated DNA Technologies): HO-1, 5′-GCCACCAAGGAGGTACACAT-3′ and 5′-GCTTGTTGCGCTCTATCTCC-3′; PGAM5, 5′-TGACACCATTAGGTCGGGAACT-3′ and 5′-TACTGCACGGGTCATAGAGGA-3′; NQO-1, 5′-AGGATGGGAGGTACTCGAATC-3′ and 5′-AGGCGTCCTTCCTTATATGCTA-3′; and β2-microglobulin, 5′-ACCCGCCTCACATTGAAATCC-3′ and 5′-GGCGTATGTATCAGTCTCAGT-3′. Linearity of the primers was verified before use, and fold changes were calculated as previously described using the Livak method.60
Western Blot and Densitometric Analysis
HT-22 cells were cultured and treated as described above for the qRT-PCR experiments. After 16 hrs, the media was collected and the cells harvested by trypsanization. After centrifugation, pelleted cells were washed with cold HBSS Ca2+/Mg2+ Free, followed by lysis with a low salt lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 0.5 Triton X-100, 1X protease and phosphatase inhibitor cocktail) for 30 min, followed by a 15-s sonication pulse at 30 W. For Western blot analysis, the cell lysates were denatured with LDS loading buffer (Invitrogen) and run on Nupage 4–12% Tris-glysine gradient gels (Invitrogen) after centrifugation. Phosphorylated Drp1 (Ser637) (Cell Signaling) and total Drp1 (C5) (Santa Cruz), PGAM5 (K16) (Santa Cruz Biotechnology), Actin (Sigma-Aldritch) and alpha-Tubulin (Santa Cruz Biotechnology) were assessed with primary antibody, follow by rabbit, mouse, or goat IgG-HRP secondary antibody. The blot was developed using Pierce Thermo Dura-ECL reagent (Thermo-Fisher) and visualized using an ImageQuant LAS 4000 (GE). Digital images of membranes were quantified by computer-assisted densitometry using Image J 1.42q (National Institutes of Health, MD).
Animals and Chronic Toxicity Evaluation
Adult (12 weeks of age) mice (Balb/cByJ, 17–20g, Jackson Laboratory) were housed in temperature-controlled conditions under a light/dark photocycle with food and water supplied ad libitum. Mice were divided randomly into three groups. The first group (n=3) was injected i.p. with compound 2q at 50 mg/kg dissolved in sterile 5% DMSO, 95% Neobee61 (Spectrum Chemical). The second group (n=3) was injected i.p. with compound 2j at 50 mg/kg dissolved in sterile 5% DMSO, 95% Neobee. The third (vehicle control group) (n=2) was injected i.p. with an equivalent volume of sterile 5% DMSO, 95% Neobee. All animals were injected between 11:00 a.m. and 12:00 p.m. Animals were sacrificed by CO2 after three weeks of injections. Blood was collected by cardiac puncture and standard blood count and chemistry was evaluated by the MUSC/VA Veterinary Diagnostic Laboratory (Charleston, SC). All animal and treatment protocols were in compliance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health (Institute of Laboratory Animal Resources, 1996) and were approved by our institutional animal care and use committee.
Chemistry
General Experimental
Unless otherwise noted, chemicals were commercially available and used as received without further purification. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous material, unless otherwise stated. Reactions were monitored by thin layer chromatography (TLC) carried out on precoated silica gel PE SIL G/UV plates (Whatman), using UV light as the visualizing agent. Flash silica gel chromatography was performed by hand using silica gel 60A (Fischer, 230–400 Mesh) or using prepacked silica columns on a Teledyne Isco Combiflash 200 eluting with ethyl acetate:hexane or dichloromethane:methanol, as stated. When necessary, reverse phase chromatography was performed using prepacked C18 columns and a Teledyne Isco Combiflash 200 eluting with water:acetonitrile + 0.1% TFA. High resolution ion trap mass spectra were obtained using a Finnigan LCQ Advantage Max mass spectrometer. The purity of tested compounds was determined using an Agilent 100 HPLC system. All the assayed compounds possess >95% purity. All the NMR spectra were recorded on a Bruker 400 model spectrometer in either DMSO-d6 or CDCL3. Chemical shifts (δ) for 1H NMR spectra are reported in parts per million to residual solvent protons. Chemical shifts (δ) for 13C NMR spectra are reported in parts per million to residual solvent carbons. NMR data were analyzed using ACD Labs NMR Processor Academic Edition, and the following abbreviations were used to explain multliplicities: s = singlet, d = doublet, t = triplet, q = quartet, m =multiplet, br = broad.
Synthesis of 2-amino derivatives: Standard Procedure
To a solution of 2-bromo-1,4-naphthoquinone in abs EtOH was added an excess of the corresponding amine (2 equiv, unless otherwise stated), and the reaction was stirred at room temperature and monitored by TLC. Most reactions were complete within 10 minutes. While some reactions precipitated pure product which was collected by vacuum filtration, others required chromatographic purification.
2-amino-1,4-naphthoquinone (1d)
6.25 g NaN3 was dissolved in 15 mL H2O and acidified with 5 mL glacial acetic acid. The NaN3 solution was added to a solution of 1,4-naphthoquinone (5 g, 29 mmol) dissolved in 100 mL of THF/H2O (4:1) and stirred at room temperature. After 6 hrs, the reaction was concentrated in vacuo and redissolved in ethyl acetate. The resulting solution was washed with 1 M NaOH and saturated NaCl. Multiple extractions were required. The extracts were combined, dried with MgSO4, and concentrated in vacuo. The reddish brown residue was purified by column chromatography (silica gel, 50% v/v ethyl acetate/hexane) to yield 4.8 g fluffy bright orange crystals 1d (96% yield). MS m/z calcd (M+) 173.05, found 173.04. 1H NMR (400 MHz, DMSO-d6) Shift 7.94 (dd, J = 7.15, 19.70 Hz, 2H), 7.82 (dt, J = 1.00, 7.53 Hz, 1H), 7.69 – 7.76 (m, 1H), 5.82 (s, 1H).
2-amino-3-methyl-1,4-naphthoquinone (1e)
Dissolved 2-methyl-1,4-naphthoquinone (560 mg, 3.3 mmol) in methanol (30 mL) and placed under inert atmosphere. Dissolved 1.37 g NaN3 in 10 mL water and acidified to pH 4 (83 drops of 12 M HCl). Added sodium azide solution to reaction flask and slowly heated reaction to 50°C and then stirred for 5 hrs. Reaction was quenched with water and extracted with ethyl acetate (2x) The organic layers were combined and washed with saturated NaCl solution, dried with Mg2SO4, and concentrated in vacuo. The reaction was purified on silica gel eluting with ethyl acetate and hexane (3:7) to yield 423 mg orange powder 1e (69% yield). MS m/z calcd (M+) 188.07, found 188.04. 1H NMR (400 MHz, DMSO-d6) Shift 7.96 – 8.03 (m, 2H), 7.83 (dt, J = 1.25, 7.53 Hz, 1H), 7.71 – 7.78 (m, 1H), 6.86 (s, 2H), 1.97 (s, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift 0.53, 28.28, 9.45, 39.58, 132.22, 125.73, 134.80, 125.94, 125.93, 134.81.
2-(methylamino)naphthalene-1,4-dione (2a)
To a solution of 2-bromo-1,4-naphthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added and excess of aqueous methylamine solution (40%, 207 uL, 2.3 mmol). The reaction was stirred for 10 min at rt, then concentrated in vacuo and purified on silica gel eluting with ethyl acetate and hexane to yield 116 mg reddish orange powder (52% yield). MS m/z calcd (M+) 188.2, found 188.2. 1H NMR (400 MHz, DMSO-d6) Shift 7.97 (dd, J = 7.40, 13.93 Hz, 2H), 7.84 (dt, J = 1.13, 7.47 Hz, 1H), 7.64 – 7.78 (m, 2H), 5.61 (s, 1H), 2.80 (d, J = 5.02 Hz, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift 38.88, 27.43, 99.25, 131.78, 134.75, 125.53.
2-(ethylamino)naphthalene-1,4-dione (2b)
Ethylamine HCl (196 mg, 2.4 mmol, 2 equiv) and K2CO3 (331 mg, 2.4 mmol, 2 equiv) were dissolved in water (3 mL) and added to a solution of 2-bromo-1,4-naphthoquinone (283 mg, 1.2 mmol, 1 equiv) dissolved in abs EtOH (40 mL). Reaction stirred at rt for 10 min, then concentrated in vacuo and purified on silica gel eluting with ethyl acetate and hexane to yield 142 mg brown powder (59% yield). MS m/z calcd (M+) 202.02, found 202.1. 1H NMR (400 MHz, DMSO-d6) Shift 7.96 – 8.07 (m, 2H), 7.88 (dt, J = 1.13, 7.47 Hz, 2H), 7.74 – 7.82 (m, 1H), 7.61 (br. s., 1H), 5.73 (s, 1H), 3.27 (quin, J = 6.90 Hz, 2H), 1.23 (t, J = 7.28 Hz, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift 12.33, 39.17, 36.12, 98.94, 132.18, 134.92, 125.17.
2-(isopropylamino)naphthalene-1,4-dione (2d)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of isopropylamine (206 uL, 2.4 mmol). The reaction was stirred for 10 min at rt, then concentrated in vacuo and purified on silica gel eluting with ethyl acetate, followed by reverse phase purification on C18 silica to yield 240 mg bright orange powder (47% yield). 1H NMR (400 MHz, DMSO-d6) d 7.92 – 8.04 (m, 2H), 7.80 – 7.90 (m, 1H), 7.70 – 7.76 (m, 1H), 7.18 (d, J = 8.28 Hz, 1H), 5.71 (s, 1H), 1.22 (d, J = 6.40 Hz, 6H). C13-HSQC (400 MHz, DMSO-d6) (ppm) 21.52, 39.70, 43.86, 99.93, 132.64, 135.19, 125.63, 126.32.
2-(propylamino)naphthalene-1,4-dione (2e)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of propylamine (197 uL, 2.4 mmol). The reaction was stirred at rt for 10 min, then concentrated in vacuo and purified on silica gel eluting with ethyl acetate and hexane to yield 178 mg bright orange powder (69% yield). 1H NMR (400 MHz, DMSO-d6) Shift 7.92 – 8.03 (m, 2H), 7.84 (dt, J = 1.00, 7.53 Hz, 1H), 7.70 – 7.77 (m, 1H), 7.60 (br. s., 1H), 5.69 (s, 1H), 3.15 (q, J = 6.53 Hz, 2H), 2.51 (s, 7H), 1.60 (sxt, J = 7.28 Hz, 2H), 0.91 (t, J = 7.40 Hz, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 11.13, 11.12, 20.15, 39.58, 43.39, 98.90, 131.88, 134.94, 131.87, 134.93, 131.87, 125.26.
2-(cyclopentylamino)naphthalene-1,4-dione (2f)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of cyclopentylamine (237 uL, 2.4 mmol). The reaction was stirred at rt for 10 min, then concentrated in vacuo and purified on silica gel eluting with ethyl acetate and hexane to yield 225 mg red powder (39% yield). MS m/z calcd (M+) 242.29, found 242.1. 1H NMR (400 MHz, DMSO-d6) Shift 7.97 (dd, J = 7.40, 15.43 Hz, 2H), 7.84 (t, J = 7.03 Hz, 1H), 7.69 – 7.78 (m, 1H), 7.26 (d, J = 7.03 Hz, 1H), 5.70 (s, 1H), 3.71 – 3.94 (m, 1H), 1.86 – 2.08 (m, 2H), 1.61 – 1.78 (m, 4H), 1.49 – 1.61 (m, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 10.97, 20.36, 39.48, 43.30, 98.96, 131.81, 125.61, 134.87, 131.89, 125.03, 125.03, 134.86, 125.86, 131.80.
2-((cyclohexylmethyl)amino)naphthalene-1,4-dione (2g)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of cyclohexanemethylamine (312 uL, 2.4 mmol). Progress of the reaction was monitored with TLC. Compound was purified using chromatography on silica gel to yield 124 mg orange powder (38% yield).). MS m/z calcd (M+) 270.34, found 270.1. 1H NMR (400 MHz, DMSO-d6) Shift 7.97 (dd, J = 7.03, 17.57 Hz, 2H), 7.83 (dt, J = 1.00, 7.53 Hz, 1H), 7.70 – 7.77 (m, 1H), 7.64 (t, J = 6.15 Hz, 1H), 5.68 (s, 1H), 3.04 (t, J = 6.53 Hz, 2H), 1.50 – 1.81 (m, 6H), 1.05 – 1.32 (m, 3H), 0.81 – 1.03 (m, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 30.82, 25.66, 26.58, 25.66, 30.82, 6.40, 40.14, 48.34, 99.47, 132.52, 135.19, 125.62, 126.34.
2-(prop-2-yn-1-ylamino)naphthalene-1,4-dione (2h)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of propargylamine (154 uL, 2.4 mmol). Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane to yield 103 mg fuzzy golden brown crystals (20% yield). MS m/z calcd (M+) 212.22, found 212.1. 1H NMR (400 MHz, DMSO-d6) Shift 7.94 – 8.03 (m, 2H), 7.81 – 7.88 (m, 2H), 7.73 – 7.79 (m, 1H), 5.79 (s, 1H), 4.05 (dd, J = 2.51, 6.02 Hz, 2H), 3.29 (t, J = 2.45 Hz, 1H). C13-HSQC (400 MHz, DMSO-d6) Shift F1 (ppm) 31.03, 39.73, 75.10, 79.28, 101.64, 125.76, 125.91, 126.12, 126.13, 132.79, 135.37, 135.37, 148.45, 153.90.
2-(phenylamino)naphthalene-1,4-dione (2i)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of propargylamine (154 uL, 2.4 mmol). Compound was purified using chromatography on silica gel to yield 103 mg fuzzy golden brown crystals (20% yield).). MS m/z calcd (M+) 212.22, found 212.1. 1H NMR (400 MHz, DMSO-d6) Shift 7.94 – 8.03 (m, 2H), 7.81 – 7.88 (m, 2H), 7.73 – 7.79 (m, 1H), 5.79 (s, 1H), 4.05 (dd, J = 2.51, 6.02 Hz, 2H), 3.29 (t, J = 2.45 Hz, 1H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 21.52, 39.70, 43.86, 99.93, 132.64, 135.19, 125.63, 126.32.
2-(naphthalen-1-ylamino)naphthalene-1,4-dione (2j)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 1-naphthylamine (343 mg, 2.4 mmol). Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane, followed by reverse phase purification on C18 silica to yield 6.5 mg greyish purple powder (1% yield). MS m/z calcd (M+) 300.32, found 300.2. 1H NMR (400 MHz, DMSO-d6) Shift 7.96 – 8.18 (m, 4H), 7.31 – 7.55 (m, 6H), 7.26 (d, J = 7.53 Hz, 1H), 7.03 – 7.15 (m, 1H), 6.57 (s, 1H). C13-HSQC (400 MHz, DMSO-d6) Shift F1 (ppm) 39.58, 111.14, 107.15, 136.19, 112.78, 129.34, 128.11, 126.58, 124.96, 126.58, 126.58, 133.99, 126.58, 125.32, 121.82.
2-(benzylamino)naphthalene-1,4-dione (2k)
To a solution of 2-bromo-1,4-naphthoquinone (5 g, 21.1 mmol) in minimum amount of abs EtOH was added an excess benzylamine (4.6 mL, 42.2 mmol, 2 equiv). The reaction was stirred for 10 min at rt, then briefly concentrated in vacuo until copious amounts of orange precipitate were visible. the solution was then cooled to 4°C and vacuum filtered to yield 3.5 g fluffy bright orange crystals (59% yield). MS m/z calcd (M+) 264.1, found 264.1. 1H NMR (400 MHz, DMSO-d6) Shift 8.22 (t, J = 6.40 Hz, 1H), 8.01 (d, J = 7.03 Hz, 1H), 7.87 – 7.94 (m, 1H), 7.82 (dt, J = 1.13, 7.47 Hz, 1H), 7.70 – 7.77 (m, 1H), 7.31 – 7.40 (m, 4H), 7.22 – 7.30 (m, 1H), 5.57 (s, 1H), 4.46 (d, J = 6.53 Hz, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 39.62, 45.48, 100.68, 127.64, 129.05, 127.63, 132.36, 135.20, 125.67, 126.37.
2-(benzyl(methyl)amino)naphthalene-1,4-dione (2l)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of N-benzylmethylamine (310 uL, 2.4 mmol). Compound was taken up onto Celite and purified using chromatography on silica gel eluting with ethyl acetate and hexane to yield 230 mg bright orange powder (69% yield). MS m/z calcd (M+) 278.11, found 278.1. 1H NMR (400 MHz, DMSO-d6) Shift 7.87 – 7.97 (m, 2H), 7.81 (dt, J = 1.28, 7.43 Hz, 1H), 7.70 – 7.78 (m, 1H), 7.33 – 7.41 (m, 2H), 7.30 (d, J = 6.97 Hz, 3H), 5.85 (s, 1H), 4.85 (s, 2H), 3.08 (s, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 39.73, 40.38, 56.81, 106.52, 127.31, 128.99, 129.08, 127.31, 133.02, 134.48, 124.91, 126.76, 134.48.
2-((3-phenethylpropyl)amino)naphthalene-1,4-dione (2m)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of phenethylamine (302 uL, 2.4 mmol). The reaction was stirred for 30 min at rt and the precipitate filtered to yield 90 mg bright orange powder (27% yield). MS m/z calcd (M+) 278.32, found 278.4. 1H NMR (400 MHz, DMSO-d6) Shift 7.96 (dd, J = 7.78, 14.31 Hz, 1H), 7.69 – 7.88 (m, 2H), 7.56 (t, J = 5.77 Hz, 1H), 7.15 – 7.41 (m, 5H), 5.75 (s, 1H), 3.26 – 3.54 (m, 2H), 2.99 – 3.14 (m, 1H), 2.81 – 2.98 (m, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 33.55, 33.55, 39.77, 43.57, 43.57, 100.01, 125.71, 126.27, 126.77, 129.01. 132.64, 135.24.
2-((pyridin-4-ylmethyl)amino)naphthalene-1,4-dione (2n)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 4-picolylamine (243 uL, 2.4 mmol). Compound was purified using chromatography on silica gel, followed by C18 silica gel, to yield 87.8 mg yellow powder (28% yield). MS m/z calcd (M+) 264.1, found 264.1. 1H NMR (400 MHz, DMSO-d6) Shift 8.74 (d, J = 5.27 Hz, 2H), 8.27 (t, J = 6.53 Hz, 1H), 8.04 (d, J = 6.78 Hz, 1H), 7.89 – 7.95 (m, 1H), 7.84 (dt, J = 1.13, 7.47 Hz, 1H), 7.73 – 7.80 (m, 3H), 5.56 (s, 1H), 4.69 (d, J = 6.53 Hz, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm)39.83, 43.80, 101.07, 123.72, 123.72, 125.35, 125.93, 132.32, 134.90, 145.16.
2-((pyridin-3-ylmethyl)amino)naphthalene-1,4-dione (2o)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 3-(aminomethyl)pyridine (244 uL, 2.4 mmol). Compound was purified using chromatography on silica gel to yield 50 mg orange powder (15% yield). 1H NMR (400 MHz, DMSO-d6) Shift 8.61 (d, J = 1.76 Hz, 1H), 8.47 (dd, J = 1.51, 4.77 Hz, 1H), 8.23 (t, J = 6.40 Hz, 1H), 8.01 (d, J = 7.03 Hz, 1H), 7.88 – 7.95 (m, 1H), 7.69 – 7.86 (m, 3H), 7.37 (dd, J = 4.77, 7.78 Hz, 1H), 5.65 (s, 1H), 4.50 (d, J = 6.53 Hz, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 39.23, 42.70, 100.63, 123.21, 132.10, 134.85, 134.84, 125.22, 125.87, 148.52, 148.52.
2-((pyridin-2-ylmethyl)amino)naphthalene-1,4-dione (2p)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 2-picolylamine (247 uL, 2.4 mmol). The reaction was stirred for 10 minutes and then concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane, followed by reverse phase purification on C18 silica to yield 90 mg yellowish powder (29% yield). MS m/z calcd (M+) 265.09, found 265.1. 1H NMR (400 MHz, DMSO-d6) Shift 8.59 (br. s., 1H), 8.10 (t, J = 6.05 Hz, 1H), 8.02 (d, J = 7.70 Hz, 1H), 7.92 (d, J = 7.70 Hz, 1H), 7.80 – 7.87 (m, 2H), 7.71 – 7.78 (m, 1H), 7.31 – 7.52 (m, 2H), 5.61 (s, 1H), 4.56 (br. s., 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 39.72, 46.52, 101.13, 122.49, 123.49, 125.86, 125.86, 132.62, 135.29, 138.79, 148.41.
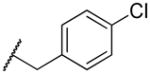
2-((4-chlorobenzyl)amino)naphthalene-1,4-dione (2q)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 4-chlorobenzylamine (292 uL, 2.4 mmol). Progress of the reaction was monitored with TLC. Precipitate was vacuum filtered after 20 min to give 267 mg sparkly orange powder (74% yield). MS m/z calcd (M+) 298.74, found 298.8. 1H NMR (400 MHz, DMSO-d6) Shift 8.15 (t, J = 6.40 Hz, 1H), 7.93 (d, J = 7.03 Hz, 1H), 7.83 (d, J = 6.78 Hz, 1H), 7.72 – 7.78 (m, 1H), 7.63 – 7.70 (m, 1H), 7.20 – 7.42 (m, 4H), 5.49 (s, 1H), 4.37 (d, J = 6.53 Hz, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm)39.48, 44.11, 46.15, 100.42, 128.86, 132.53, 134.84, 125.11, 125.98.
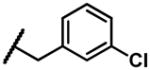
2-((3-chlorobenzyl)amino)naphthalene-1,4-dione (2r)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 3-chlorobenzylamine (293 uL, 2.4 mmol). Reaction was stirred for 30 min after which the precipitated solid was filtered to yield 199 mg bright orange powder (56% yield). MS m/z calcd (M+) 298.06, found 298.1. 1H NMR (400 MHz, DMSO-d6) Shift 8.23 (t, J = 6.53 Hz, 1H), 7.95 – 8.05 (m, 1H), 7.87 – 7.94 (m, 1H), 7.79 – 7.87 (m, 1H), 7.70 – 7.79 (m, 1H), 7.45 (s, 1H), 7.25 – 7.42 (m, 3H), 5.60 (s, 1H), 4.47 (d, J = 6.53 Hz, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 40.08, 44.86, 100.96, 125.82, 126.28, 126.48, 127.51, 127.51, 130.72, 132.58, 135.31.
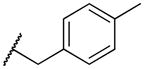
2-((4-methylbenzyl)amino)naphthalene-1,4-dione (2s)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 4-methylbenzylamine (306 uL, 2.4 mmol). Reaction was stirred for 40 min after which the precipitated solid was filtered to yield 187 mg (56% yield) reddish orange powder. MS m/z calcd (M+) 278.11, found 278.1. 1H NMR (400 MHz, DMSO-d6) Shift 8.25 (t, J = 6.40 Hz, 1H), 8.00 – 8.12 (m, 1H), 7.93 – 8.00 (m, 1H), 7.89 (dt, J = 1.00, 7.53 Hz, 1H), 7.75 – 7.85 (m, 1H), 7.27 – 7.36 (m, 2H), 7.17 – 7.27 (m, 2H), 5.62 (s, 1H), 4.47 (d, J = 6.27 Hz, 2H), 2.34 (s, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 20.78, 40.11, 45.39, 100.80, 129.73, 127.84, 132.56, 135.44, 125.74, 126.52.
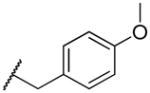
2-((4-methoxybenzyl)amino)naphthalene-1,4-dione (2t)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 4-methoxybenzylamine (314 uL, 2.4 mmol). Reaction was stirred for 10 min after which the precipitated solid was filtered to yield 192 mg (55% yield) slightly orangish yellow powder. MS m/z calcd (M+) 294.11, found 294.0. 1H NMR (400 MHz, DMSO-d6) Shift 8.17 (t, J = 6.40 Hz, 1H), 7.95 – 8.05 (m, 1H), 7.87 – 7.94 (m, 1H), 7.82 (dt, J = 1.13, 7.47 Hz, 1H), 7.68 – 7.77 (m, 1H), 7.29 (d, J = 8.53 Hz, 2H), 6.91 (d, J = 8.53 Hz, 2H), 5.59 (s, 1H), 4.37 (d, J = 6.53 Hz, 2H), 3.73 (s, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm)39.99, 45.03, 55.57, 100.90, 114.25, 125.77, 126.47, 129.05, 132.58, 135.43, 146.84, 152.29.
methyl 4-(((1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)methyl)benzoate (2u)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of Methyl 4-(aminomethyl)benzoate hydrochloride (569 mg, 2.4 mmol) and K2CO3 dissolved in water. Reaction was stirred for 10 min after which the precipitated solid was filtered to yield 189 mg (49% yield) yellow powder. MS m/z calcd (M+) 322.1, found 322.1. 1H NMR (400 MHz, DMSO-d6) Shift 8.27 (t, J = 6.40 Hz, 1H), 8.01 (d, J = 7.53 Hz, 1H), 7.87 – 7.98 (m, 3H), 7.82 (dt, J = 1.00, 7.53 Hz, 1H), 7.70 – 7.78 (m, 1H), 7.49 (d, J = 8.03 Hz, 2H), 5.53 (s, 1H), 4.54 (d, J = 6.53 Hz, 2H), 3.84 (s, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 40.03, 45.25, 52.64, 100.97, 125.56, 126.49, 127.84, 129.87, 132.61, 135.33, 146.82, 152.19.
2-((3-(trifluoromethyl)benzyl)amino)naphthalene-1,4-dione (2v)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 3-(trifluoromethyl)benzylamine (420 mg, 2.4 mmol). The reaction was stirred for 10 minutes and then concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane to yield 397 mg yellowish powder (48% yield). MS m/z calcd (M+) 332.09, found 332.1. 1H NMR (400 MHz, DMSO-d6) Shift 8.28 (t, J = 6.53 Hz, 1H), 8.01 (d, J = 6.78 Hz, 1H), 7.87 – 7.94 (m, 1H), 7.83 (dt, J = 1.13, 7.47 Hz, 1H), 7.72 – 7.79 (m, 2H), 7.69 (d, J = 7.53 Hz, 1H), 7.54 – 7.67 (m, 2H), 5.64 (s, 1H), 4.56 (d, J = 6.53 Hz, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 40.22, 45.02, 100.83, 124.20, 124.21, 126.33, 126.33, 130.13, 131.76, 135.16.
2-((3,4-dichlorobenzyl)amino)naphthalene-1,4-dione (2w)
To a solution of 2-bromo-1,4-napthoquinone (283 mg, 1.2 mmol) in abs EtOH (40 mL) was added an excess of 3,4-dichlorobenzylamine (422 mg, 2.4 mmol). MS m/z calcd (M+) 331.02, found 331.1. 1H NMR (400 MHz, DMSO-d6) Shift 8.22 (t, J = 6.53 Hz, 1H), 7.96 – 8.05 (m, 1H), 7.88 – 7.94 (m, 1H), 7.83 (dt, J = 1.00, 7.53 Hz, 1H), 7.71 – 7.78 (m, 1H), 7.67 (d, J = 1.76 Hz, 1H), 7.58 – 7.64 (m, 1H), 7.38 (dd, J = 1.76, 8.28 Hz, 1H), 5.61 (s, 1H), 4.46 (d, J = 6.53 Hz, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 40.10, 44.37, 101.12, 125.77, 126.44, 128.13, 129.75, 130.97, 132.60, 135.26.
Synthesis of 2-amido derivatives: Standard Procedure
Reactions were carried out under argon. To a solution of Compound 1 and NaH (3 equiv, 60% dispersion) dissolved in tetrahydofuran was slowly added an excess of the corresponding acyl chloride (1.5 equiv). Products were purified using chromatography followed by crystallization.
N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)acetamide (3a)
0.15 g (0.88 mmol) was dissolved in 2 mL acetic anhydride along with 0.2 mL glacial acetic acid, and the reaction was refluxed overnight. The reaction was allowed to cool to room temperature and the precipitated product was filtered and crystallized from ethyl acetate and hexane to yield 156 mg of fine yellow crystals (82% yield). MS m/z calcd (M+) 216.06, found 216.1. 1H NMR (400 MHz, DMSO-d6) Shift 9.95 (s, 1H), 8.02 – 8.11 (m, 1H), 7.94 – 8.01 (m, 1H), 7.80 – 7.93 (m, 2H), 7.70 (s, 1H), 2.25 (s, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift 25.27, 39.91,115.96, 134.11, 125.83, 134.10, 125.82.
N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)propionamide (3b)
To a solution of Compound 1d (0.3 g, 1.7 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in tetrahydofuran (THF, 20 mL) was slowly added an excess of propionyl chloride chloride (267 uL, 2.5 mmol, 1.5 equiv). The reaction was stirred at room temperature for 10 minutes. The reaction was quenched with water and extracted with dichloromethane (2x), washed with 1 M NaOH, 1 M HCl, and saturated NaCl. The organic layer was dried over Mg2SO4 and concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane to yield 54 mg mg yellow powder (% yield). MS m/z calcd (M+)229.07, found 229.9. 1H NMR (400 MHz, DMSO-d6) d ppm 1.07 (t, J=7.58 Hz, 3 H) 2.61 (q, J=7.34 Hz, 2 H) 7.72 (s, 1 H) 7.83 – 7.94 (m, 2 H) 7.95 – 8.01 (m, 1 H) 8.04 – 8.10 (m, 1 H) 9.84 (s, 1 H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 9.23, 40.18, 30.21, 116.32, 133.92, 135.23, 125.66, 126.74.
methyl (1,4-dioxo-1,4-dihydronaphthalen-2-yl)carbamate (3c)
To a solution of Compound 1d (0.3 g, 1.7 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in dry DMF and placed under argon. Methylchloroformate (197 uL, 2.5 mmol, 1.5 equiv) was added slowly. Reaction stirred at rt until starting material consumed as determined by TLC. Quenched reaction with excess water and extracted with DCM (3x). Combined organic layers, dried with Mg2SO4, and concentrated in vacuo. An excess of water was added to the remaining dark solution from which a brown solid precipitated. This brown solid was filtered and purified using silica gel (50/50 ethyl acetate/hexane with 0.1% Et3N). Crystallized with hot ethyl acetate/hexane to yield 133 mg crumbly greenish brown crystals (34% yield). 1H NMR (400 MHz, DMSO-d6) Shift 9.41 (s, 1H), 8.05 (dd, J = 1.25, 7.28 Hz, 1H), 7.95 – 8.01 (m, 1H), 7.81 – 7.93 (m, 2H), 7.34 (s, 1H), 3.76 (s, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm)31.13, 40.57, 53.82, 114.98, 126.21, 127.02, 134.40.
N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)isobutyramide (3d)
To a solution of Compound 1d (0.3 g, 1.7 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in tetrahydofuran (THF, 20 mL) was slowly added an excess of the corresponding isobutyryl chloride (267 uL, 2.5 mmol, 1.5 equiv). The reaction was stirred at room temperature for 10 minutes. The reaction was quenched with water and extracted with dichloromethane (2x), washed with 1 M NaOH, 1 M HCl, and saturated NaCl. The organic layer was dried over Mg2SO4 and concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane and crystallized with hot ethyl acetate and hexane to yield 54 mg yellow crystals (13% yield). MS m/z calcd (M+) 244.26, found 244.0. 1H NMR (400 MHz, DMSO-d6) Shift 9.77 (s, 1H), 8.03 – 8.09 (m, 1H), 7.94 – 7.99 (m, 1H), 7.82 – 7.92 (m, 2H), 7.71 (s, 1H), 1.09 (d, J = 6.97 Hz, 7H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm)
N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)cyclopentanecarboxamide (3e)
To a solution of Compound 1d (0.3 g, 1.7 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in tetrahydofuran (THF, 20 mL) was slowly added an excess of the corresponding cyclopentanecarbonyl chloride (300 uL, 2.5 mmol, 1.5 equiv). The reaction was stirred at room temperature for 5 minutes. The reaction was quenched with water and extracted with dichloromethane (2x), washed with 1 M NaOH, 1 M HCl, and saturated NaCl. The organic layer was dried over Mg2SO4 and concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane and crystallized with hot ethyl acetate and hexane to yield 83 mg yellow crystals (18% yield). MS m/z calcd (M+) 270.3, found 270.3. 1H NMR (400 MHz, DMSO-d6) Shift 9.75 (s, 1H), 8.06 (d, J = 7.34 Hz, 1H), 7.93 – 8.00 (m, 1H), 7.81 – 7.92 (m, 2H), 7.70 (s, 1H), 3.24 (quin, J = 7.61 Hz, 1H), 1.79 – 1.93 (m, 2H), 1.61 – 1.76 (m, 4H), 1.48 – 1.61 (m, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 26.16, 26.17, 30.30, 30.30, 40.31, 45.48, 116.47, 125.86, 126.66, 133.66, 135.27.
N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)cyclohexanecarboxamide (3f)
To a solution of Compound 1d (0.3 g, 1.7 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in tetrahydofuran (THF, 20 mL) was slowly added an excess of the corresponding cyclohexanecarbonyl chloride (337 uL, 2.5 mmol, 1.5 equiv). Products were purified using chromatography followed by crystallization. The reaction was stirred at room temperature for 5 minutes. The reaction was quenched with water and extracted with dichloromethane (2x), washed with 1 M NaOH, 1 M HCl, and saturated NaCl. The organic layer was dried over Mg2SO4 and concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane to yield 140 mg yellowish powder (29% yield). MS m/z calcd (M+) 284.3, found 384.3. 1H NMR (400 MHz, DMSO-d6) Shift 9.69 (s, 1H), 7.99 (dd, J = 1.13, 7.40 Hz, 1H), 7.87 – 7.92 (m, 1H), 7.73 – 7.85 (m, 2H), 7.63 (s, 1H), 1.51 – 1.81 (m, 5H), 1.00 – 1.38 (m, 6H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 25.33, 25.34, 29.44, 29.44, 40.28, 44.81, 116.33, 125.82, 126.87, 134.03, 135.40.
N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)benzamide (3g)
0.3 g (1.73 mmol) Compound 1d and 3 equiv NaH (60% dispersion, 0.2 g) were dissolved in 20 mL dry THF. To this was added 1.5 equiv. benzoyl chloride (301 μL). The reaction was quenched with water and extracted twice with DCM. The organic extracts were combined and washed sequentially with 1 M NaOH, 1 M HCl, and a saturated solution of NaCl. The extract was then dried over Mg2SO4 and concentrated in vacuo. The resulting powder was further purified by column chromatography (silica gel, 30/70% v/v ethyl acetate/hexane with 1% Et3N) and crystallized from ethyl acetate/hexane to yield 183 mg small, fine, bright yellow crystals (39% yield). MS m/z calcd (M+) 277.07, found 277.1. 1H NMR (400 MHz, DMSO-d6) Shift 9.74 (s, 1H), 8.10 – 8.15 (m, 1H), 7.87 – 8.07 (m, 5H), 7.79 (s, 1H), 7.67 – 7.74 (m, 1H), 7.57 – 7.65 (m, 2H).). C13-HSQC (400 MHz, DMSO-d6) Shift 126.1, 129.5, 117.0, 128.4, 40.1, 126.9, 126.1, 129.5, 117.1, 128.4, 40.1, 133.3, 129.4, 134.3, 135.3.
N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)-4-methylbenzamide (3h)
To a solution of Compound 1d (0.3 g, 1.7 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in tetrahydofuran (THF, 20 mL) was slowly added an excess of the corresponding isobutyryl chloride (267 uL, 2.5 mmol, 1.5 equiv). The reaction was stirred at room temperature for 10 minutes. The reaction was quenched with water and extracted with dichloromethane (2x), washed with 1 M NaOH, 1 M HCl, and saturated NaCl. The organic layer was dried over Mg2SO4 and concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane and crystallized with hot ethyl acetate and hexane to yield 103 mg yellow crystals (21% yield). MS m/z calcd (M+) 292.3, found 291.9. 1H NMR (400 MHz, DMSO-d6) Shift 9.65 (s, 1H), 8.08 – 8.16 (m, 1H), 7.99 – 8.07 (m, 1H), 7.85 – 7.99 (m, 4H), 7.77 (s, 1H), 7.42 (d, J = 8.03 Hz, 2H), 2.42 (s, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 21.56, 40.40, 129.90, 116.76, 128.16, 134.25, 135.51, 126.08, 126.73.
4-chloro-N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)benzamide (3i)
To a solution of Compound 1d (0.3 g, 1.7 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in tetrahydofuran (THF, 20 mL) was slowly added an excess of the corresponding 4-chlorobenzoyl chloride (327 uL, 2.5 mmol, 1.5 equiv). The reaction was stirred at room temperature for 5 minutes. The reaction was quenched with water and extracted with dichloromethane (2x), washed with 1 M NaOH, 1 M HCl, and saturated NaCl. The organic layer was dried over Mg2SO4 and concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane and crystallized with hot dichloromethane to yield 67 mg fluffy greenish yellow crystals (13% yield). MS m/z calcd (M+) 312.72, found 312.7. 1H NMR (400 MHz, DMSO-d6) Shift 9.80 (s, 1H), 8.00 – 8.09 (m, 1H), 7.89 – 7.98 (m, 3H), 7.79 – 7.89 (m, 2H), 7.70 (s, 1H), 7.60 (d, J = 8.53 Hz, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 40.15, 117.25, 126.08, 126.88, 129.22, 130.29, 134.25, 135.42.
3-chloro-N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)benzamide (3j)
To a solution of Compound 1d (0.3 g, 1.7 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in tetrahydofuran (THF, 20 mL) was slowly added an excess of the corresponding 3-chlorobenzoyl chloride (326 uL, 2.5 mmol, 1.5 equiv). The reaction was stirred at room temperature for 5 minutes. The reaction was quenched with water and extracted with dichloromethane (2x), washed with 1 M NaOH, 1 M HCl, and saturated NaCl. The organic layer was dried over Mg2SO4 and concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane, followed by C18 silica gel, to yield 380 mg greenish yellow powder (72% yield). MS m/z calcd (M+) 312.72, found 312.7. 1H NMR (400 MHz, CHLOROFORM-d) Shift 9.18 (br. s., 1H), 8.18 (t, J = 6.78 Hz, 1H), 8.04 (s, 1H), 7.96 (s, 1H), 7.75 – 7.89 (m, 2H), 7.63 (d, J = 8.53 Hz, 1H), 7.46 – 7.56 (m, 1H), 7.29 (s, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 40.08, 117.38, 126.02, 127.13, 127.13, 128.27, 131.08, 132.89, 134.31, 135.41.
N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)-2-phenylacetamide (3k)
To a solution of Compound 1d (0.5 g, 2.89 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in tetrahydofuran (THF, 20 mL) was slowly added an excess of the corresponding 2-phenacetyl chloride (573 uL, 4.34 mmol, 1.5 equiv). The reaction was stirred at room temperature for 1 hr. The reaction was quenched with water and extracted with dichloromethane (2x), washed with 1 M NaOH, 1 M HCl, and saturated NaCl. The organic layer was dried over Mg2SO4 and concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane to yield 33 m yellow powder (4% yield). MS m/z calcd (M+) 292.3, found 293.1. 1H NMR (400 MHz, CHLOROFORM-d) Shift 8.43 (br. s., 1H), 8.01 – 8.15 (m, 2H), 7.88 (s, 1H), 7.79 (dt, J = 1.25, 7.53 Hz, 1H), 7.67 – 7.74 (m, 1H), 7.34 – 7.52 (m, 5H), 3.85 (s, 2H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 44.98, 76.87, 129.43, 128.10, 129.42, 133.16, 135.00, 117.16, 126.62, 126.62.
2-([1,1′-biphenyl]-4-yl)-N-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)acetamide (3l)
To a solution of Compound 1d (0.3 g, 1.7 mmol, 1 equiv) and NaH (0.2 g, 5 mmol, 60% dispersion) dissolved in tetrahydofuran (THF, 20 mL) was slowly added an excess of the corresponding 3-chlorobenzoyl chloride (326 uL, 2.5 mmol, 1.5 equiv). Products were purified using chromatography followed by crystallization. The reaction was stirred at room temperature for 5 minutes. The reaction was quenched with water and extracted with dichloromethane (2x), washed with 1 M NaOH, 1 M HCl, and saturated NaCl. The organic layer was dried over Mg2SO4 and concentrated in vacuo. Compound was purified using chromatography on silica gel eluting with ethyl acetate and hexane, followed by C18 silica gel, to yield 380 mg greenish yellow powder (72% yield). MS m/z calcd (M+) 354.11, found 354.0. 1H NMR (400 MHz, DMSO-d6) d 9.79 (s, 1H), 8.24 (d, J = 8.07 Hz, 2H), 8.00 – 8.15 (m, 2H), 7.87 – 7.99 (m, 3H), 7.70 – 7.84 (m, 3H), 7.41 – 7.61 (m, 4H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 39.59, 129.68, 117.02, 127.49, 134.24, 127.49, 135.47, 126.20, 128.82, 126.90,131.36.
Synthesis of 2-ureyl derivatives: Standard Procedure
Reactions were carried out under argon. To a stirred solution of Compound 1 (0.1 g, 0.577 mmol, 1 equiv) dissolved in dimethylformamide (DMF, 10 mL) was added the corresponding isocyanate (0.577 mmol, 1 equiv) followed by 3 drops of triethylamine. The reaction was then slowly heated to 80°C and monitored using TLC. Upon completion, the reaction was allowed to cool to room temperature and quenched with water. Unless otherwise stated, the precipitated product was filtered and crystallized using hot ethyl acetate.
1-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)-3-ethylurea (4a)
To a stirred solution of Compound 1 (0.1 g, 0.577 mmol, 1 equiv) dissolved in dimethylformamide (DMF, 10 mL) was added ethyl isocyanate (100 μL, 0.577 mmol, 1 equiv) followed by 3 drops of triethylamine isocyanate. The reaction was heated to 80° C and stirred for 3 hrs. After allowing the flask to cool to rt, the reaction was quenched with water and the filtered precipitate was crystallized with hot ethyl acetate to yield 92 mg fine dark yellow crystals (65% yield). MS m/z calcd (M+) 245.08, found 245.5. 1H NMR (400 MHz, DMSO-d6) Shift 8.90 (s, 1H), 8.04 (dd, J = 0.88, 7.65 Hz, 1H), 7.96 (dd, J = 0.88, 7.40 Hz, 1H), 7.76 – 7.91 (m, 2H), 7.50 (t, J = 5.27 Hz, 1H), 7.47 (s, 1H), 3.07 – 3.22 (m, 2H), 1.07 (t, J = 7.15 Hz, 3H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 15.42, 40.05, 34.51, 152.38, 146.65, 112.42, 133.77, 135.40, 125.57, 135.40, 133.62, 126.97.
1-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)-3-isopropylurea (4b)
To a stirred solution of Compound 1 (0.1 g, 0.577 mmol, 1 equiv) dissolved in dimethylformamide (DMF, 10 mL) was added isopropyl isocyanate (57 μL, 0.577 mmol, 1 equiv) followed by 3 drops of triethylamine isocyanate. The reaction was heated to 80° C and stirred for 3 hrs. After allowing the flask to cool to rt, the reaction was quenched with water and the filtered precipitate was crystallized with hot ethyl acetate to give 40 mg crumbly tan crystals (27% yield). MS m/z calcd (M+) 259.1, found 259.0. 1H NMR (400 MHz, DMSO-d6) Shift 8.83 (s, 1H), 8.01 – 8.07 (m, 1H), 7.96 (dd, J = 0.75, 7.53 Hz, 1H), 7.77 – 7.92 (m, 2H), 7.42 – 7.53 (m, 2H), 3.76 (qd, J = 6.55, 13.24 Hz, 1H), 1.12 (d, J = 6.53 Hz, 6H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 23.13, 40.08, 147.26, 152.18, 41.71, 112.04, 133.48, 135.31, 125.92, 126.92.
1-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)-3-phenylurea (4c)
To a stirred solution of Compound 1 (0.1 g, 0.577 mmol, 1 equiv) dissolved in dimethylformamide (DMF, 10 mL) was added phenyl isocyanate (63 uL, 0.577 mmol, 1 equiv) followed by 3 drops of triethylamine. The reaction was heated to 100° C and stirred for 3 hrs. After allowing the flask to cool to rt, the reaction was quenched with water and extracted with DCM. The organic extract was concentrated in vacuo, and purified with HPLC. An M+ of 293 came off very slowly at 100% MeCN. The appropriate tubes were combined, and the product was crystallized from ethyl acetate to yield 27 mg powdery neon orange crystals (16% yield). MS m/z calcd (M+) 293.09, found 292.8. 1H NMR (400 MHz, DMSO-d6) Shift 9.88 (s, 1H), 9.19 (s, 1H), 8.09 (d, J = 7.28 Hz, 1H), 7.99 (d, J = 7.53 Hz, 1H), 7.81 – 7.95 (m, 2H), 7.52 (s, 1H), 7.49 (d, J = 7.78 Hz, 2H), 7.34 (t, J = 7.78 Hz, 2H), 7.02 – 7.11 (m, 1H). C13-HSQC (400 MHz, DMSO-d6) Shift 129.5, 135.3, 133.9, 123.4, 126.9, 126.0, 1289.5, 113.2, 119.0, 40.1.
1-benzyl-3-(1,4-dioxo-1,4-dihydronaphthalen-2-yl)urea (4d)
To a stirred solution of Compound 1 (0.1 g, 0.577 mmol, 1 equiv) dissolved in dimethylformamide (DMF, 10 mL) was added benzyl isocyanate (106 uL, 0.577 mmol, 1 equiv) followed by 3 drops of triethylamine. The reaction was then slowly heated to 80°C and monitored using TLC. After 4 hrs, the reaction was allowed to cool to room temperature and quenched with water. The precipitated product was filtered and crystallized using hot ethyl acetate to yield 139 mg bright yellow crystals (78% yield). MS m/z calcd (M+) 307.1, found 307.0. 1H NMR (400 MHz, DMSO-d6) Shift 9.05 (s, 1H), 8.05 (dd, J = 1.00, 7.53 Hz, 1H), 7.94 – 8.00 (m, 1H), 7.79 – 7.91 (m, 1H), 7.49 (s, 1H), 7.16 – 7.43 (m, 6H), 6.44 (t, J = 5.90 Hz, 1H), 4.35 (d, J = 5.77 Hz, 1H), 4.24 (d, J = 6.02 Hz, 1H). C13-HSQC (400 MHz, DMSO-d6) Shift (ppm) 39.98, 43.07, 126.81, 128.99, 112.23, 133.55, 135.17, 125.72, 126.81.
4-oxo-N-phenyl-4H-chromene-2-carboxamide (5c)
4-oxo-4H-1-benzopyran-2-carboxylic acid (0.5 g, 2.63 mmol) was dissolved in 20 mL DMF and placed under argon. The solution was cooled to 0°C and thionyl chloride was added slowly and the mixture stirred for 30 min on ice. After, aniline was added and the reaction was stirred at rt. overnight. The reaction was quenched with sodium bicarbonate solution, and the precipitate was filtered to yield 84 mg yellow powder (12% yield). MS m/z calcd (M+) 266.02, found 266.1. 1H NMR (400 MHz, DMSO-d6) Shift 10.77 (br. s., 1H), 8.09 (dd, J = 1.38, 7.91 Hz, 1H), 7.90 – 7.99 (m, 1H), 7.83 – 7.88 (m, 1H), 7.78 (d, J = 8.28 Hz, 2H), 7.57 (t, J = 7.15 Hz, 1H), 7.41 (t, J = 7.91 Hz, 2H), 7.17 (t, J = 7.40 Hz, 1H), 6.99 (s, 1H). C13-HSQC (400 MHz, DMSO-d6) (ppm) 40.30, 111.46, 124.75, 129.20, 126.40, 121.85, 119.21, 135.51, 125.28.
4-oxo-N-phenyl-4H-chromene-3-carboxamide (5d)
Chromone-3-carboxylic acid (0.5 g, 2.63 mmol) was dissolved in DCM and placed under argon. The solution was cooled to 0°C and thionyl chloride was added slowly and the mixture stirred for 30 min on ice. After, aniline was added and the reaction was stirred at rt. and filtered the white precipitate. Washed with 1 M NaOH overnight. Reaction quenched with 1 M NaHCO3 (x2), 1 M HCl, and saturated NaCl. The organic layer was dried with Mg2SO4, filtered, and concentrated in vacuo. The resulting powder was crystallized with ethyl acetate to yield 38 mg of fine, very pale yellow crystals (5% yield). MS m/z calcd (M+) 265.07, found 266.2. 1H NMR (400 MHz, DMSO-d6) Shift 11.34 (s, 1H), 9.21 (s, 1H), 8.27 (dd, J = 1.51, 8.03 Hz, 1H), 7.91 – 8.01 (m, 1H), 7.85 (d, J = 8.28 Hz, 1H), 7.74 (d, J = 7.78 Hz, 2H), 7.61 – 7.70 (m, 1H), 7.41 (t, J = 7.91 Hz, 2H), 7.12 – 7.20 (m, 1H). C13-HSQC (400 MHz, DMSO-d6) Shift 39.91, 125.82, 129.97, 127.10, 118.84, 118.83, 136.01, 125.83, 127.1.
Supplementary Material
Acknowledgments
We are grateful to Dr. Chris Lindsey and Dr. John Lemasters for their invaluable guidance and many insightful discussions. HT22 cells were a generous gift from Dr. Dave Schubert at Salk Institute for Biological Studies. This research was supported by a NIH/NHLBI pre-doctoral training fellowship (T32-HL007260-36) and grants from the National Center for Research Resources (5P20RR024485-02) and the National Institute of General Medical Sciences (8P20GM103542-02) from the National Institutes of Health, South Carolina Clinical and Translational Research Institute UL1RR029882, and in part by pilot research funding from an American Cancer Society Institutional Research Grant awarded to the Hollings Cancer Center, Medical University of South Carolina.
ABBREVIATIONS USED
- VK
Vitamin K
- VK1
Vitamin K1, phylloquinone
- VK2
Vitamin K2, menaquinone
- VK3
Vitamin K3, menadione
- GSH
glutathione
- CNS
central nervous system
- RIP1
Receptor-Interacting Protein 1
- RIP3
receptor-Interacting Protein 3
- MLKL
Mixed lineage kinase domain-like
- PGAM5
phosphoglycerate mutase family member 5
- Drp-1
Dynamin-related protein 1
- BSO
L-buthionine sulfoximine
- UBIAD1
UbiA prenyltransferase domain-containing protein 1
- PINK1
PTEN-induced putative kinase 1
- ClogP
calculated logP
- tPSA
total polar surface area
- TLC
thin layer chromatography
- THF
tetrahydrofuran
- PC50
concentration producing 50% protection
- TC50
concentration producing 50% toxicity
- HDAC
histone deacetylase
- Nec-1
Necrostatin-1
- Ideb
Idebenone
- ROS
reactive oxygen species
- Rho123
dihydrorhodamine 123
- CM-H2DCFDA
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester
- DPPH
2,2-diphenyl-1-picrylhydrazyl
- HO-1
Heme oxygenase 1
- NQO-1 NAD(P)H
quinone oxioreductase 1
- ALS
amytropic lateral sclerosis
- ATP
adenosine triphosphate
- ETC
electron transport chain
- t-BOOH
tert-butylhydroperoxide
- i.p
intraperitoneal
- PARL
presenilins-associated rhomboid-like protease
- DMEM
Dulbecco’s Modified Eagles’s medium
- MTS
3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)2-H-tetrazolium
- DMSO
dimethylsulfoxide
- HBSS
Hank’s Balanced Salt Solution
- DTNB
5,5′-dithiobis-2-nitrobenzoic acid
- TNB
5-thio-2-nitrobenzoic acid
- FITC
Fluorescein isothiocyanate
- qRT-PCR
quantitative real time-polymerase chain reaction
- LDS
lithium dodecyl sulfate
- IgG
immunoglobulin G
- HRP
horseradish peroxidase
Footnotes
Supporting Information Available. Supplementary figures and tables, and detailed experimental procedures; copies of HPLC and all spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 2.Simonian NA, Coyle JT. Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 1996;36:83–106. doi: 10.1146/annurev.pa.36.040196.000503. [DOI] [PubMed] [Google Scholar]
- 3.Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- 4.Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K, Youdim MB. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- 5.Sofic E, Lange KW, Jellinger K, Riederer P. Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci Lett. 1992;142:128–130. doi: 10.1016/0304-3940(92)90355-b. [DOI] [PubMed] [Google Scholar]
- 6.Sagara Y, Schubert D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J Neurosci. 1998;18:6662–6671. doi: 10.1523/JNEUROSCI.18-17-06662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsumoto K, Lo EH, Pierce AR, Halpern EF, Newcomb R. Secondary elevation of extracellular neurotransmitter amino acids in the reperfusion phase following focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:114–124. doi: 10.1097/00004647-199601000-00014. [DOI] [PubMed] [Google Scholar]
- 8.Albrecht P, Lewerenz J, Dittmer S, Noack R, Maher P, Methner A. Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc- as a neuroprotective drug target. CNS Neurol Disord Drug Targets. 2010;9:373–382. doi: 10.2174/187152710791292567. [DOI] [PubMed] [Google Scholar]
- 9.Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 10.Suttie JW. Mechanism of action of vitamin K: synthesis of gamma-carboxyglutamic acid. CRC Crit Rev Biochem. 1980;8:191–223. doi: 10.3109/10409238009105469. [DOI] [PubMed] [Google Scholar]
- 11.Price PA. Role of vitamin-K-dependent proteins in bone metabolism. Annu Rev Nutr. 1988;8:565–583. doi: 10.1146/annurev.nu.08.070188.003025. [DOI] [PubMed] [Google Scholar]
- 12.Shearer MJ, Bach A, Kohlmeier M. Chemistry, nutritional sources, tissue distribution and metabolism of vitamin K with special reference to bone health. J Nutr. 1996;126:1181S–1186S. doi: 10.1093/jn/126.suppl_4.1181S. [DOI] [PubMed] [Google Scholar]
- 13.Thijssen HH, Drittij-Reijnders MJ. Vitamin K status in human tissues: tissue-specific accumulation of phylloquinone and menaquinone-4. Br J Nutr. 1996;75:121–127. doi: 10.1079/bjn19960115. [DOI] [PubMed] [Google Scholar]
- 14.Nickerson ML, Kostiha BN, Brandt W, Fredericks W, Xu KP, Yu FS, Gold B, Chodosh J, Goldberg M, Lu da W, Yamada M, Tervo TM, Grutzmacher R, Croasdale C, Hoeltzenbein M, Sutphin J, Malkowicz SB, Wessjohann L, Kruth HS, Dean M, Weiss JS. UBIAD1 mutation alters a mitochondrial prenyltransferase to cause Schnyder corneal dystrophy. PLoS One. 2010;5:e10760. doi: 10.1371/journal.pone.0010760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakagawa K, Hirota Y, Sawada N, Yuge N, Watanabe M, Uchino Y, Okuda N, Shimomura Y, Suhara Y, Okano T. Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature. 2010;468:117–121. doi: 10.1038/nature09464. [DOI] [PubMed] [Google Scholar]
- 16.Suhara YHN, Okitsu T, Sakai M, Watanabe M, Nakagawa K, Wada A, Takeda K, Takahashi K, Tokiwa H, Okano T. Structure–Activity Relationship of Novel Menaquinone-4 Analogues: Modification of the Side Chain Affects their Biological Activities. J Med Chem. 2012;55:1553–1558. doi: 10.1021/jm2013166. [DOI] [PubMed] [Google Scholar]
- 17.Okano T, Shimomura Y, Yamane M, Suhara Y, Kamao M, Sugiura M, Nakagawa K. Conversion of phylloquinone (Vitamin K1) into menaquinone-4 (Vitamin K2) in mice: two possible routes for menaquinone-4 accumulation in cerebra of mice. J Biol Chem. 2008;283:11270–11279. doi: 10.1074/jbc.M702971200. [DOI] [PubMed] [Google Scholar]
- 18.Tsaioun KI. Vitamin K-dependent proteins in the developing and aging nervous system. Nutr Rev. 1999;57:231–240. doi: 10.1111/j.1753-4887.1999.tb06950.x. [DOI] [PubMed] [Google Scholar]
- 19.Sundaram KS, Lev M. Regulation of sulfotransferase activity by vitamin K in mouse brain. Arch Biochem Biophys. 1990;277:109–113. doi: 10.1016/0003-9861(90)90557-f. [DOI] [PubMed] [Google Scholar]
- 20.Vos M, Esposito G, Edirisinghe JN, Vilain S, Haddad DM, Slabbaert JR, Van Meensel S, Schaap O, De Strooper B, Meganathan R, Morais VA, Verstreken P. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science. 2012;336:1306–1310. doi: 10.1126/science.1218632. [DOI] [PubMed] [Google Scholar]
- 21.Allison AC. The possible role of vitamin K deficiency in the pathogenesis of Alzheimer’s disease and in augmenting brain damage associated with cardiovascular disease. Med Hypotheses. 2001;57:151–155. doi: 10.1054/mehy.2001.1307. [DOI] [PubMed] [Google Scholar]
- 22.Sakaue M, Mori N, Okazaki M, Kadowaki E, Kaneko T, Hemmi N, Sekiguchi H, Maki T, Ozawa A, Hara S, Arishima K, Yamamoto M. Vitamin K has the potential to protect neurons from methylmercury-induced cell death in vitro. J Neurosci Res. 2011;89:1052–1058. doi: 10.1002/jnr.22630. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Lin JC, Wang H, Peterson JW, Furie BC, Furie B, Booth SL, Volpe JJ, Rosenberg PA. Novel role of vitamin k in preventing oxidative injury to developing oligodendrocytes and neurons. J Neurosci. 2003;23:5816–5826. doi: 10.1523/JNEUROSCI.23-13-05816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2:541–553. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fieser LFaHJL. Reaction of hydrazoic acid with naphthoquinones. Journal of the American Chemical Society. 1935;57:1482–1484. [Google Scholar]
- 26.Valente C, Moreira R, Guedes RC, Iley J, Jaffar M, Douglas KT. The 1,4-naphthoquinone scaffold in the design of cysteine protease inhibitors. Bioorg Med Chem. 2007;15:5340–5350. doi: 10.1016/j.bmc.2007.04.068. [DOI] [PubMed] [Google Scholar]
- 27.Tandon VK, Singh RV, Yadav DB. Synthesis and evaluation of novel 1,4-naphthoquinone derivatives as antiviral, antifungal and anticancer agents. Bioorg Med Chem Lett. 2004;14:2901–2904. doi: 10.1016/j.bmcl.2004.03.047. [DOI] [PubMed] [Google Scholar]
- 28.Nagai S. 2-(Ureido or alkoxycarbonylamino)-1,4-naphthoquinones as agricultural fungicides. Jpn Kokai Tokkyo Koho. 1979 [Google Scholar]
- 29.Ha JS, Park SS. Glutamate-induced oxidative stress, but not cell death, is largely dependent upon extracellular calcium in mouse neuronal HT22 cells. Neurosci Lett. 2006;393:165–169. doi: 10.1016/j.neulet.2005.09.056. [DOI] [PubMed] [Google Scholar]
- 30.van Leyen K, Siddiq A, Ratan RR, Lo EH. Proteasome inhibition protects HT22 neuronal cells from oxidative glutamate toxicity. J Neurochem. 2005;92:824–830. doi: 10.1111/j.1471-4159.2004.02915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tobaben S, Grohm J, Seiler A, Conrad M, Plesnila N, Culmsee C. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ. 2011;18:282–292. doi: 10.1038/cdd.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Topliss JG. Utilization of operational schemes for analog synthesis in drug design. J Med Chem. 1972;15:1006–1011. doi: 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- 33.Inks ES, Josey BJ, Jesinkey SR, Chou CJ. A novel class of small molecule inhibitors of HDAC6. ACS Chem Biol. 2012;7:331–339. doi: 10.1021/cb200134p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan S, Schubert D, Maher P. Oxytosis: A novel form of programmed cell death. Curr Top Med Chem. 2001;1:497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- 35.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 36.Blois MS. Antioxidant Determinations by the Use of a Stable Free Radical. Nature. 1958;181:1199–1200. [Google Scholar]
- 37.Molyneux P. The use of the stable free radical diphenylpicrylhydrazyl (DPPH) for estimating antioxidant activity. Songklanakarin J Sci Technol. 2003;26:211–219. [Google Scholar]
- 38.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007;35:7074–7086. doi: 10.1093/nar/gkm638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukui M, Song JH, Choi J, Choi HJ, Zhu BT. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur J Pharmacol. 2009;617:1–11. doi: 10.1016/j.ejphar.2009.06.059. [DOI] [PubMed] [Google Scholar]
- 41.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 42.Ten VS, Starkov A. Hypoxic-ischemic injury in the developing brain: the role of reactive oxygen species originating in mitochondria. Neurol Res Int. 2012;2012:542976. doi: 10.1155/2012/542976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta. 2010;1802:80–91. doi: 10.1016/j.bbadis.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 44.Lifshitz J, Friberg H, Neumar RW, Raghupathi R, Welsh FA, Janmey P, Saatman KE, Wieloch T, Grady MS, McIntosh TK. Structural and functional damage sustained by mitochondria after traumatic brain injury in the rat: evidence for differentially sensitive populations in the cortex and hippocampus. J Cereb Blood Flow Metab. 2003;23:219–231. doi: 10.1097/01.WCB.0000040581.43808.03. [DOI] [PubMed] [Google Scholar]
- 45.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 46.Lemasters JJ, Nieminen AL. Mitochondrial oxygen radical formation during reductive and oxidative stress to intact hepatocytes. Biosci Rep. 1997;17:281–291. doi: 10.1023/a:1027332611839. [DOI] [PubMed] [Google Scholar]
- 47.Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 48.Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 49.Breckenridge DG, Kang BH, Kokel D, Mitani S, Staehelin LA, Xue D. Caenorhabditis elegans drp-1 and fis-2 regulate distinct cell-death execution pathways downstream of ced-3 and independent of ced-9. Mol Cell. 2008;31:586–597. doi: 10.1016/j.molcel.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- 51.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583–21587. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 54.Grohm J, Kim SW, Mamrak U, Tobaben S, Cassidy-Stone A, Nunnari J, Plesnila N, Culmsee C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012;19:1446–1458. doi: 10.1038/cdd.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 56.Morimoto BH, Koshland DE., Jr Induction and expression of long- short-term neurosecretory potentiation in a neural cell line. Neuron. 1990;5:875–880. doi: 10.1016/0896-6273(90)90347-i. [DOI] [PubMed] [Google Scholar]
- 57.Maher P, Davis JB. The role of monoamine metabolism in oxidative glutamate toxicity. Journal of Neuroscience. 1996;16:6394–6401. doi: 10.1523/JNEUROSCI.16-20-06394.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 59.Davis JB, Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652:169–173. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 60.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 61.Hippalgaonkar K, Majumdar S, Kansara V. Injectable lipid emulsions-advancements, opportunities and challenges. AAPS Pharm Sci Tech. 2010;11:1526–1540. doi: 10.1208/s12249-010-9526-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.