Abstract
Adenoviruses (Ads) have shown promise as vectors for gene delivery in clinical trials. Efficient viral targeting to a tissue of choice requires both ablation of the virus’ original tropism and engineering of an efficient receptor-mediated uptake by a specific cell population. We have developed a series of adapters binding to the virus with such high affinity that they remain fully bound for >10 d, block its natural receptor binding site and mediate interaction with a surface receptor of choice. The adapter contains two fused modules, both consisting of designed ankyrin repeat proteins (DARPins), one binding to the fiber knob of adenovirus serotype 5 and the other binding to various tumor markers. By solving the crystal structure of the complex of the trimeric knob with three bound DARPins at 1.95-Å resolution, we could use computer modeling to design a link to a trimeric protein of extraordinary kinetic stability, the capsid protein SHP from the lambdoid phage 21. We arrived at a module which binds the knob like a trimeric clamp. When this clamp was fused with DARPins of varying specificities, it enabled adenovirus serotype 5-mediated delivery of a transgene in a human epidermal growth factor receptor 2-, epidermal growth factor receptor-, or epithelial cell adhesion molecule-dependent manner with transduction efficiencies comparable to or even exceeding those of Ad itself. With these adapters, efficiently produced in Escherichia coli, Ad can be converted rapidly to new receptor specificities using any ligand as the receptor-binding moiety. Prefabricated Ads with different payloads thus can be retargeted readily to many cell types of choice.
Keywords: protein design, tumor targeting, viral retargeting, X-ray crystallography, protein engineering
Adenoviruses (Ads) are actively being developed as vectors for in vivo gene delivery to diagnose and treat human disease. They can incorporate up to 35 kb of foreign DNA, are safe because they do not integrate into the host cell genome, and can infect dividing and nondividing cells (1–3). However, achieving efficient and specific gene transfer to disease-affected tissues by Ads constitutes an enormous technological challenge, because Ads—like any other virus—lack specificity for diseased tissues. Furthermore, regardless of the delivery route, Ads undergo numerous interactions with nontarget, normal tissues, including cellular and molecular components of the immune system.
The challenge in efficient and highly specific retargeting of Ads thus lies in ablating the Ads’ natural interactions and facilitating uptake of the virus by the cell population or tissue of choice in a highly specific manner.
Ad capsids are composed of three major proteins, the hexon that forms the shell of the capsid, the penton base, and the fiber that associates with the penton base to form the penton capsomers. The entrance of Ads into the cell first requires the interaction of the knob domain of the fiber with a primary receptor on the target cell. The best-studied and most frequently used serotype, Ad5, binds to the Coxsackievirus-and-Ad receptor (CAR) (4, 5), but other serotypes use different primary receptors, such as sialic acid, CD46, desmoglein 2, CD80, or CD86 (6–9). Independent of the type of primary receptor, in a second step the penton base of the Ad makes contact with integrin receptors on the cell surface, initiating the subsequent uptake of the virus by receptor-mediated endocytosis (10; for review see refs. 11 and 12).
For efficient targeting of Ad5, a first major task is to ablate the virion’s natural tropism, and this ablation has been achieved by introducing mutations into the knob, hexon, and/or penton base within motifs mediating interactions with cell-surface molecules or blood components, leading to reduced transduction efficiency (13–19). The second challenge is to retarget the Ad5 specifically to the diseased cells/tissue. Because the natural receptor usually is expressed at low levels in the desired cell populations or might not be accessible in the tissue because of the polarity of CAR expression and the localization to difficult-to-reach tight junctions (20), the virus must be tagged with a new specificity for receptors that are expressed on cells in the diseased tissue.
To retarget Ad5, two types of strategies have been followed: the genetic fusion to Ad coat proteins, mainly established for fiber fusions (16, 21–25), and the development of bispecific adapters (for review see refs. 14 and 26).
A strategy of creating a generic, universal bispecific adapter without the need to produce new virus for every new receptor target seems compelling. Previously we and others have reported the use of bispecific adapters binding to a virus capsid protein for specific adenovirus-mediated targeting (14, 26–37). The successful execution of the adapter strategy depends critically on our ability to design protein adapters for Ad that allow the targeting of any desired disease-associated receptor and whose association with the Ad virion is highly stable and can be fine-tuned to be compatible with the virion’s structural integrity and its step-wise disintegration during cell entry (38).
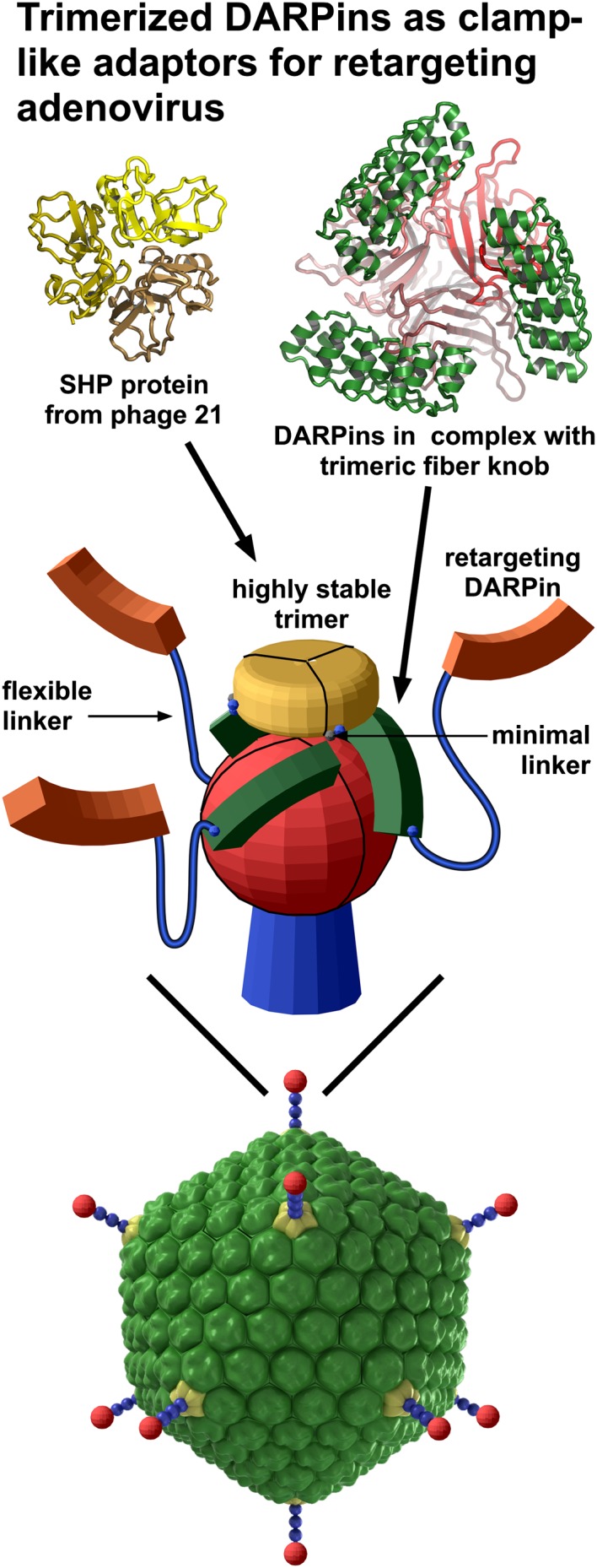
As an important step in this technology development, we have designed adapters derived from a trimeric clamp that associates virtually irreversibly with Ad virions. To do so, we exploited unique properties of designed ankyrin repeat proteins (DARPins) that had been selected to bind the mutant Ad5 knob∆TAYT (37). We solved the crystal structure of the DARPin/knob complex at 1.95-Å resolution. We designed a molecule based on this result in which the knob-binding DARPins were trimerized using an unusually stable trimerization domain, SHP of the lambdoid phage 21. As a result, these trimeric fusion proteins clamp the knob so tightly that they become an integral structural component of the virus. Importantly, however, as our transduction studies showed, this highly stable association is fully compatible with virus disintegration during targeted infection. By fusing this clamp genetically to DARPins specific for biomarkers of human tumors, we could achieve specific transduction of cells expressing the tumor cell markers human epidermal growth factor receptor 2 (HER2), epithelial cell adhesion molecule (EpCAM), or epidermal growth factor receptor (EGFR).
Results
For efficient retargeting of Ad using an adapter-based strategy, it is crucial to develop adapters that have such a high affinity to the virion that they essentially do not dissociate at all from the virus particle for several days. Previously, we described an approach to develop an adapter based on DARPins: One type of DARPin that was selected to bind the Ad5 knob (37) was arranged as a linear multimer to wrap around the Ad5 knob and fused to another retargeting DARPin specific for HER2. This adapter was shown to enable transduction of HEK293 cells overexpressing HER2 with the Ad–adapter complex. However, the system showed increased efficiency only at higher than stoichiometric ratios, with an excess of adapter over knob, for mono- and divalent adapters, thus suggesting some dissociation. Also, a trivalent DARPin offered no further improvement over a divalent DARPin, probably because not all knob subunits could be occupied in the linear arrangement of knob-binding DARPins and therefore affinity was not increased further. To create even more tightly binding adapters, we solved the crystal structure of a knob-binding DARPin in complex with the Ad5 knob and used this structure as the basis for designing a tightly binding trimer clamping the trimeric structure of the knob.
Structure Determination of Ad5 Knob/Darpin 1D3 Complex.
To aid the design of improved adapters, the exact positioning of the DARPin 1D3 on the knob∆TAYT was determined by X-ray crystallography of the complex. The complex crystallized in space group P212121 with six polypeptide chains in the asymmetric unit (Table S1). The 1.95-Å resolution crystal structure revealed an exact 3:1 stoichiometry with three DARPin 1D3 molecules binding to one Ad5 knob trimer (Fig. 1 A and B).
Fig. 1.

Crystal structure of Ad5 knob/1D3 complex. Overview of the complex showing the Ad5 knob and DARPin 1D3 molecules in two perpendicular orientations as red and green ribbons, respectively. (A) Top view. (B) Side view. The orientation of the fiber knob is shown schematically in red. (C) Superposition of the Ad5 knob/1D3 complex with the CAR D1 domain of Ad37 (PDB ID: 2J12; light blue ribbon) or CAR D1 from Ad12 (PDB ID: 1KAC; violet ribbon). The Ad5 knob trimer is shown as red and gray surfaces, and the Ad37 and Ad12 knobs have been omitted. Only one chain of the 1D3 DARPin is shown in green. The boxed area is magnified on the right. Here green and light blue sticks indicate residues from 1D3 and CAR D1, respectively. Labels in roman and italic characters refer to 1D3 and CAR D1, respectively.
Although all three 1D3 DARPins were well defined in the final electron density maps (Fig. S1), the analysis of their B-factors, a measure of thermal stability, revealed pronounced differences. The average B-factors for the Ad5 knob trimer with chain labels A, B, and C are 32.6, 34.0, and 41.2 Å2; for the cognate DARPins with chain labels D, E, and F these values are 45.2, 51.1, and 69.3 Å2, respectively. Thus, the average B-factor is significantly higher for DARPin F than for the others. This observation could be explained by either a lower occupancy, which would indicate that the Ad5 knob trimer cannot bind three DARPins simultaneously, or elevated thermal mobility caused by fewer contacts with neighboring molecules that are related by crystallographic symmetry. The analysis of the crystal lattice revealed that, in addition to the main interface contact with the Ad5 knob, DARPin chains D, E, and F also are involved in crystal contacts but with very different buried surface areas of 617, 900, and 42.4 Å2, respectively. Thus, DARPin chain F is fixed only loosely in the crystal lattice, explaining its increased average B-factor.
Previously, we observed that even at high concentrations DARPin 2E6 on average would bind with only 2.5 molecules to one Ad5 knob trimer in solution (37). DARPins 1D3 and 2E6 are closely related. They share identical interface residues and differ in just three framework positions outside the DARPin/Ad5 knob interface. Thus, it appears that binding to the trimeric knob has some inherent asymmetry.
The epitope to which 1D3 binds is located on the side opposite the Ad5 knob N terminus (where the fiber shaft is located) and fairly close to the threefold trimer axis (Fig. 1 and Fig. S2). The observed arrangement illustrates that 1D3 can recognize the full-length knob protein in the context of the Ad5 virion, as previously shown (37). Furthermore, the ΔTAYT deletion (residues 489–492), which has been reported to abolish CAR binding (39, 40), is located close to the Ad5 knob’s N terminus and shaft and hence remote from the 1D3 epitope. Therefore, 1D3 does not discriminate between WT and ΔTAYT isoforms of Ad5 knob (37). Furthermore, we predicted from the structure that binding of 1D3 to the knob would interfere directly with CAR binding to the WT knob. Mapping of the 1D3- and CAR-binding sites on the knob revealed that, even though the two epitopes are not fully overlapping, binding of CAR to the Ad5 knob/1D3 complex would create a clash between residues 13–19 and 43–46 from 1D3 and residues 62–65 and 84–86 from CAR (Fig. 1C). This inference was borne out by direct inhibition experiments (see below).
Most importantly, the structure of the Ad5 knobΔTAYT/1D3 complex aids in the design of improved recognition modules. In the complex, the C termini of the DARPins point away from Ad5 surface and are in an almost perfect triangular arrangement with a distance between them of ∼48 Å. Because of this configuration, we reasoned that the stability of the knob–DARPin complex could be improved by fusing three DARPins with a very stable trimerization module that supports exactly this spacing and orientation of bound DARPins on the knob without any additional flexibility. This fusion should result in a clamp with very high avidity.
Construction of Trimeric Adapters by Rational Design.
To test this reasoning, we chose to design a highly stable trimeric module to link three copies of the knob-binding DARPin 1D3 in an optimal arrangement to clamp the knob. First, the consensus C-capping repeat (C-cap) of the knob-binding DARPin was replaced by a C-cap of higher thermal stability and fewer structural fluctuations, as previously reported (41, 42). This approach seemed reasonable, because the C-cap was not involved in directly binding to the knob, and, indeed, with the new C-cap no differences in affinity were observed (Fig. S3).
Because the crystal structure showed that the C termini of the three knob-bound DARPins point away from the virus, we fused these termini to the small 11.8-kDa capsid protein SHP of the lambdoid phage 21, whose crystal structure we had previously determined (43). This protein forms a highly stable trimer that remains intact even in SDS gels and which dissociates and denatures with a half-life of 1 mo in solution (43). According to our design, this trimeric structure containing SHP was expected to be located above the knob, with the three DARPins grabbing the knob from three sides (Fig. 2 and Fig. S4). We reasoned that this clamp concept would work most efficiently when the linkers are as short as possible without leading to steric interference, to provide the highest local concentration of correctly oriented DARPins. To optimize the design, three variants of different lengths of the minimal linker between the knob-binding DARPin 1D3nc (“nc” for new cap) and SHP were constructed (Fig. 2 and Fig. S4). All these fusions were expressed in Escherichia coli and formed trimers even in the absence of a knob. This trimerization was quantitative, because no monomer peak could be detected using size-exclusion chromatography in combination with multiangle static light scattering (SEC-MALS). The trimers bound to the trimeric knob with a perfect 1:1 stoichiometry as determined by MALS (Fig. S5), in accordance with three DARPins binding to the three knob subunits. We did not observe cross-linking of knob proteins by the trimeric knob-binding modules, consistent with a clamp-like binding, thus eliminating the danger of virus cross-linking in later transduction experiments.
Fig. 2.

Construction of the trimeric clamp based on the crystal structure of the DARPin/knob complex. (A and B) Schematic representation of the two trimeric adapter complexes. The retargeting DARPins (orange) are fused via a long flexible linker (blue) to either the N terminus (A) or C terminus (B) of the knob-binding DARPin (green). The knob-binding DARPin is fused in turn by a very short minimal linker (blue dot) to the trimerization module (yellow), SHP from the lambdoid phage 21. The fusion of the knob-binding DARPin was always at the N terminus of SHP, which is on the same face and very close to the C terminus. Three variants of minimal linkers were used: SHP1 containing a single glycine (G), SHP2 containing the dipeptide glycine-alanine (GA), or SHP3 containing the 10 amino acids GLKAGADVNA. The knob and fiber shaft are shown in red. Below the model, the respective gene is shown schematically. (C) Detailed structural model with an indication of the experimental structures on which it is based (PDB ID: 4ATZ, this work).
Trimeric Adapters Conferred Ad-Mediated Gene Transfer in a HER2-Dependent Manner.
The potential of the 1D3nc_SHP knob-binding modules in Ad5-mediated gene transfer was investigated. As a model system, we chose to target HER2, a cell-surface receptor overexpressed on many tumor cells. Retargeting DARPins were fused either at the N or C terminus of the 1D3nc_SHP module (Fig. 2 and Fig. S4). Two different HER2-binding DARPins, G3 or 9.29 (44, 45), recognizing epitopes on different domains of the target HER2, or the control DARPin E2_5 (46) were tested. All adapters containing the knob-binding module and a retargeting DARPin could be expressed in high-yield, formed trimers (Fig. S6A) and bound both the knob and HER2 simultaneously, as determined by ELISA (Fig. S6B).
The purified adapters then were used to form a complex with the Ad5 virus encoding a luciferase reporter gene (Ad5luc), and the complex was used to transduce target cells. The use of Ad5 with an unmodified WT knob allowed evaluation of the CAR-dependent background transduction and comparison of the trimeric adapters to the previously described linear adapter (37). If the trimeric adapter was tightly bound, no CAR-mediated entry should be observed, because crystallographic data predicted that binding of DARPin 1D3 to the knob would block the knob from binding to CAR (Fig. 1C) (47). Indeed, this prediction was confirmed in competition experiments (Fig. S7) with the human CAR D1 domain, which interacts with the WT Ad5 knob domain.
The binding of adapter or CAR also was mutually exclusive in transduction of HEK293 Flp-In cells expressing high levels of CAR: At a ratio of one trimeric 1D3nc_SHP1 knob-binding clamp per trimeric knob, transduction with Ad5luc was blocked completely and could not be reduced further by a 10-fold excess of 1D3nc_SHP1 to knob (Fig. S8).
To investigate HER2-mediated transduction, Ad5luc was coated with the HER2-retargeting adapter at a ratio of one trimeric adapter to one trimeric knob. This complex was used to transduce HEK293 Flp-In cells stably overexpressing HER2 (293Flp/HER2) or, as control, the parental HEK293 Flp-In cells expressing only low amounts of HER2 (48). The activity of the luciferase was measured 14 h posttransduction (Fig. 3A). As evidenced by the difference in reporter expression in control and HER2-expressing cells, HER2-specific transduction was observed for both HER2 binders, G3 and 9.29, regardless of whether they were fused to the N or C terminus of the knob-binding module. However, the G3-based adapter resulted in a 30-fold increase in luciferase expression as compared with the non–CAR-mediated background transduction caused by Ad5 coated with the knob-binding module alone, whereas DARPin 9.29 was only 25- to 27-fold above background. The N-terminal fusion of the DARPin retargeting HER2 to the knob-binding unit seemed to yield higher luciferase units, 25- to 36-fold, compared with 20- to 24-fold of the corresponding C-terminal fusions. In this experimental set-up the different linker spacings between the knob-binding DARPin and SHP seemed to perform equally well.
Fig. 3.

Comparison of the transduction efficiency of WT Ad5 coated with various DARPin adapters. (A) HEK293 Flp-In cells (293Flp) or HEK293 Flp-In stably expressing HER2 (293Flp/HER2) were transduced with a multiplicity of 100 vp per cell coated with a 1:1 ratio of knob-binding DARPin to knob subunits. The trimeric adapter containing the trimeric knob-binding module 1D3nc_SHP1 (green bars), 1D3nc_SHP2 (red bars), or 1D3nc_SHP3 (blue bars) spaced by different linker lengths was fused to a DARPin binding to HER2, G3, or 9.29 or to a control DARPin, E2_5, respectively. These DARPins were fused to the N terminus (N) or C terminus (C) of the trimeric 1D3nc_SHP knob-binding module. (B) HEK293 Flp-In cells (293Flp) or HEK293 Flp-In cells stably expressing HER2 (293Flp/HER2) were transduced with 100 vp per cell coated with a 1:1 (green bars), 10:1 (red bars), or 100:1 (blue bars) ratio of knob-binding DARPin to knob subunits. The trimeric knob-binding module 1D3nc_SHP1 was fused to the HER2-binding DARPin G3 or to the control DARPin E2_5. The linear DARPins contained one (mono), two (bi), or three (tri) knob-binding modules in series and one HER2-binding module. Controls were cells alone (C); cells infected with virus alone (white bars; V); or cells infected with virus coated with the trimeric 1D3nc_SHP1 knob-binding module (without the HER2-binding module; V+A). Relative luciferase light units (RLU) were determined 14.5 h after infection. The assay was performed in duplicate; error bars show SD.
Next we verified the specificity of the HER2-mediated transduction using a competition assay (Fig. S9). The transduction was specific for HER2, because the luciferase expression could be reduced when the cells were preincubated with the DARPin G3 but not with a control, DARPin E2_5. We observed a high background signal in untransfected HEK293 Flp-In cells with all adapters containing a HER2-specific DARPin. To investigate whether this high background signal resulted from a low level of HER2 expression by the HEK293 Flp-In cells or from reinfection by newly produced virus via CAR, we investigated transduction efficiency in a HER2, CAR, and a system not expression E1 or E3, using the cell lines CHO Flp-In and CHO/HER2 (Fig. S10). Whereas the adapter containing the control DARPin E2_5 did not mediate transduction of any of the two cell lines, the adapter fused to the HER2-binding DARPin G3 mediated transduction of only the cell line overexpressing HER2 at a level 35-fold above background. As expected, when tested on CHO cells, luciferase expression was observed at the same low levels with either virus alone (i.e, Ad5luc coated with either of the two control adapters, 1D3nc_SHP1 alone or fused to the control DARPin E2_5, as well as with Ad5luc coated with the HER2-specific adapter containing the DARPin G3).
Next, the efficiency of the previously designed linear DARPin-based adapters (37) was compared with that of the trimeric adapters designed in this study (Fig. 3B). 293Flp/HER2 or the parental cell line as control was infected with adapter-coated virus at 100 virus particles (vp) per cell with different ratios of adapter to knob. When the trimeric adapters were used, most efficient transduction was achieved at the lowest tested ratio, 1:1, as is consistent with a perfect three DARPin to one knob stoichiometry and very tight binding. At this ratio, transduction of HER2-expressing cells was 15-fold more efficient than non–CAR-mediated background, i.e., Ad coated with the knob-binding module alone not containing an anti-HER2 DARPin. The decrease in luciferase activity with increasing adapter concentration can be explained by gradual saturation of HER2 with free adapters in excess.
The efficacy of transduction of 293Flp/HER2 cells enabled by the linear adapters was nearly equal to that achieved with the trimeric adapters (Fig. 3B). However, in the case of linear adapters and WT Ad5 knob proficient in CAR binding, the background signal of luciferase in HEK293 Flp-In cells was elevated and in some cases reached the intensities obtained from 293Flp/HER2 cells. This effect was most dramatic with the linear monovalent 2E6_G5 adapter, which had a much lower affinity than the linear bi- or trivalent 2E6_G5 adapters (37). When the ratio of adapter was increased 10- or 100-fold over knob subunit concentration, this background signal was reduced. Taken together, these two observations suggest that even at the highest ratio of these linear adapters tested (100:1) some CAR-binding sites within the knobs remained unblocked by adapters, allowing the virus to bind to CAR expressed equally on both cell lines. In conclusion, these experiments clearly show the gains in the specificity by increased affinity of the adapter to the virion that resulted from the rational design of trimeric adapters made possible by the structural data generated earlier in the present study.
Trimeric Adapters Mediate Transduction of Tumor Cell Lines in an HER2-Dependent Manner.
Because one possible application of the adapter strategy is its use in tumor targeting, it is essential to verify that various tumor cell lines can be transduced using a series of different tumor-associated surface receptors. As a first model system, we investigated the HER2-mediated transduction of adapter-coated Ad5luc using the HER2-overexpressing human breast tumor cell line BT474 (Fig. 4) (49). Transduction was achieved in an HER2-dependent manner when the DARPins G3 or 9.29 were fused to the knob-binding module, whereas only background luciferase activity was observed when the knob-binding module was fused to the control DARPin E2_5.
Fig. 4.

Transduction of adapter-coated Ad5 into BT474 tumor cells overexpressing HER2. BT474 cells were infected with a multiplicity of 100 vp per cell with a 1:1 ratio of trimeric adapter to knob. The trimerization module containing various minimal linker lengths [1D3nc_SHP1 with 1-aa spacing (white bars), 1D3nc_SHP2 with 2-aa spacing (hatched bars), or 1D3nc_SHP3 with 10-aa spacing (gray bars)] was fused to a DARPin binding to HER2, G3, or 9.29 or to a control DARPin E2_5. These DARPins were fused to either the N terminus or C terminus of the 1D3nc_SHP unit. Black bars indicate controls [C, cells without infection; V, cells infected with virus alone; A, cells infected with virus coated with the trimeric 1D3nc_SHP1 knob-binding module (without the HER2-binding module)]. RLU were determined 16 h after infection. The assay was performed in duplicate; error bars show the SD.
In contrast to the previous results using the HEK293 Flp-In cell line (Fig. 3A), which showed only moderate differences between the various adapter constructs, the differences were more dramatic using BT474 cells (Fig. 4). Whereas the C-terminal fusions of DARPin 9.29 showed transduction levels in the range of 40- to 57-fold above background (virus coated with the knob-binding module alone), G3 fusions were 49- to 102-fold over background, depending on the linker between the knob-binding DARPin 1D3nc and the SHP trimerization domain. For the N-terminal fusions the transduction levels of G3 were 107- to 231-fold over background, and the transduction levels of 9.29 were 164- to 293-fold over background. In conclusion, the N-terminal fusions showed much higher transduction levels than the C-terminal fusions. Furthermore, the most compact forms of the adapter (constructs containing SHP1 with a linker length of 1 aa or SHP2 with a linker length of 2 aa) as N-terminal fusions to the SHP trimerization domain seemed to be more efficient than SHP3 fusion proteins which contain an extended α-helix of 10 aa in the linker region between knob-binding DARPin 1D3nc and the SHP trimerization domain. Because the N-terminal fusion of the retargeting DARPin to the 1D3nc_SHP1 knob-binding module proved most efficient, it was used in all further experiments.
Trimeric Adapters Are Bound Stably to the Virus Particle with Very High Affinity.
Next the stability of interaction of the trimeric adapter with the virus was compared with that of the linear adapters reported earlier, measured in terms of functional affinity (37). First, we needed to test whether the virus itself was stable at room temperature for the 2-wk duration of the experiment. Ad5luc alone incubated without adapter showed the same level of luciferase activity at all time points investigated, indicating that the virus was stable for up to 2 wk at room temperature (Fig. 5A). Second, the stability of the adapters and competitor was investigated. Therefore, the proteins either were incubated at room temperature for 2 wk or were freshly thawed and compared by SDS/PAGE (Fig. S11). The proteins showed no degradation during the course of the experiment. Third, and most importantly, the dissociation of the adapters from the virions was tested. For this purpose, the trimeric adapter G3_1D3nc_SHP1 or the linear trivalent adapter (2E6)3_G5 was incubated with Ad5luc and subsequently incubated with a 100-fold excess of competitor (trimeric adapter without HER2-retargeting DARPin) for various time periods before transduction of BT474 cells (Fig. 5B). The purpose of the competitor was to prevent reassociation of the adapter, i.e., to make any dissociation event unidirectional and thus to elucidate the true off-rate. While the linear adapter (2E6)3_G5 showed a reduction to background level after only 1 d of incubation with competitor, the trimeric adapter still conferred the same high transduction of BT474 cells after 10 d as observed initially in a HER2-dependent manner. To show the importance of the competitor in this experiment, we found that in its absence, after transduction of BT474 cells, the same signal intensity was observed for both adapters for all time points tested (Fig. 5B). This control showed that the reduction of luciferase is not a result of the deterioration of the linear trivalent adapter but due to its faster dissociation, i.e., lower functional affinity.
Fig. 5.

Test of dissociation of DARPin-based adapters from the virus particle. (A) Integrity of infectivity of the virus itself and the bound knob-binding modules. Black bars represent infection with Ad5luc virus alone; gray bars represent infection with virus coated with 1D3nc_SHP1 (without the HER2-binding module) at an adapter:knob ratio of 1:1. Samples were incubated in PBS at room temperature for the time points indicated before infection of BT474 cells at 100 vp per cell. RLU were determined 16 h after infection. (B) Measurements of adapter dissociation from virus. Trimeric G3_1D3nc_SHP1 adapters (gray bars) or linear trivalent (2E6)3_G5 adapters (white bars), both containing three knob-binding DARPins and a HER2-binding DARPin (G3 or G5), were incubated with Ad5luc at a 1:1 ratio of adapter to knob at room temperature for 1.5 h in PBS before the addition of a 100-fold excess of competitor 1D3nc_SHP1 (with no HER2-binding module) (hatched bars) to prevent reassociation of dissociated adapter or no competitor (white bars) as control. Samples were incubated at room temperature for the time points indicated before infection of BT474 cells at 100 vp per cell. RLU were determined 16 h after infection. All assays were performed in duplicate; error bars show the SD.
Together these results show that the trimeric adapters do not dissociate measurably over 10 d and therefore display a much higher functional affinity than the linear trivalent adapter.
Trimeric Adapters Can Be Constructed Readily with Different Targeting Specificities.
To prove that the adapter strategy can be applied readily to other specificities, the HER2-binding module was replaced by modules recognizing other cell-surface receptors, such as EGFR and EpCAM. Adapters containing retargeting DARPins specific for EGFR or EpCAM were cloned, expressed, and purified. They were constructed in the adapter format 1D3nc_SHP as N-terminal fusions, and their efficiency in transducing tumor cell lines overexpressing either EGFR or EpCAM was tested.
For Ad retargeting to EGFR, the DARPins E01 or E69 that bind to two distinct epitopes on EGFR (44, 50) were used. The specificity and efficiency of EGFR-mediated transduction was investigated using the previously described human epidermal carcinoma tumor cell line A431 overexpressing EGFR (Fig. 6A) (50). Ad5luc was coated with the adapters at an adapter-to-knob ratio of 1:1, and cells were infected at a multiplicity of 100 vp per cell. Both E01 and E69 adapters mediated transduction of A431 cells with an efficiency of 200-fold over virus alone (which represented the background level) (Fig. 6A). No transduction was observed for the EGFR-negative cell line CHO (Fig. S12A), suggesting that the EGFR-mediated transduction is indeed receptor specific. In addition, E01 adapter-coated Ad5luc infected CHO cells transiently expressing EGFR but not when EpCAM was overexpressed or the control CHO cells (Fig. S13A).
Fig. 6.

Transduction of adapter-coated Ad5 using tumor cells overexpressing EGFR or EpCAM. (A) A431 cells were infected at a multiplicity of 100 vp per cell with a 1:1 ratio of trimeric adapter to knob. The trimeric 1D3nc_SHP1 knob-binding module was fused to either the EGFR-binding DARPin E01 (black bars) or E69 (hatched bars). Before infection the cells were incubated with 5 µM of the bispecific competitor E01_LZ3_E69 (LZ), a control DARPin E2_5, or no competitor (−). For controls (gray bars), cells were infected with virus alone (V) or with virus coated with 1D3nc_SHP1 (no EGFR-binding module) (V+A). (B) HT29 cells were infected at a multiplicity of 100 vp per cell with a 1:1 ratio of trimeric adapter to knob. The trimeric knob-binding module 1D3nc_SHP1 was fused to either the EpCAM-binding DARPin Ec4 (black bars) or Ac2 (striped bars). Before infection the cells were incubated with 5 µM of the bispecific competitor Ec1_LZ3_Ac2 (LZ), a control DARPin E2_5, or no competitor (−). For controls (gray bars), cells were infected with virus alone (V) or with virus coated with 1D3nc_SHP1 (with no EpCAM-binding module) (V+A). RLU were determined 16 h after infection. The assay was performed in duplicate; error bars show the SD.
To show specificity of the transduction, a competition experiment was performed in which the cells were preincubated with a DARPin alone. When a bivalent and bispecific E69_E01 competitor, dimerized by a leucine zipper (LZ3) (50), was used, the transduction efficiency was reduced almost to background level both for the E01 adapter (to 7.2%) and for the E69 adapter (to 13%). Using the control DARPin E2_5 for competition, no reduction of transduction efficiency was observed (Fig. 6A).
To investigate EpCAM-mediated gene-transfer, the DARPins Ec4 or Ac2 were used as retargeting DARPins. They previously had been reported to bind EpCAM specifically at different epitopes (51). Adapters containing either retargeting module were used to coat Ad5luc at an adapter-to-knob ratio of 1:1 and their transduction efficiency was investigated (Fig. 6B). Both adapters mediated infection of the previously described EpCAM-overexpressing human colorectal adenocarcinoma cell line HT29 (51) (for Ac2, 18-fold over background, for Ec4, 12-fold over background). The transduction was receptor specific, because no luciferase was detected using the control cell line HEK293T expressing no EpCAM (Fig. S12B). In addition, the Ac2 and Ec4 adapter-coated Ad5luc infected CHO cells transiently expressing EpCAM, but not when EGFR was overexpressed or when CHO cells were used as control (Fig. S13B).
Competition experiments were performed to investigate further the specificity of the EpCAM adapters. The signals obtained were reduced only slightly, with no statistical significance, when the cells were preincubated with the control DARPin E2_5. In contrast, when the bispecific competitor Ec1_ Ac2 was used [Ec1 binds to the same epitope as Ec4 (51)], dimerized by LZ3, transduction efficiency was reduced to 1.5% for the Ac2 adapter and to 33% for the Ec4 adapter, demonstrating the specificity of transduction (Fig. 6B).
In conclusion, the retargeting module of the adapter can be exchanged readily to target different cell-surface receptors, validating the generic application of the trimeric knob-binding adapter strategy.
Discussion
Here, we describe an approach using DARPins toward a generic, universal system for efficient retargeting of Ad5 to a cell population of interest using an adapter strategy. While a genetic fusion of retargeting proteins to Ad capsid proteins would require the new construction and production of virus for every new epitope targeted and every new payload, an adapter-based strategy allows the use of generic, premade Ads containing a certain payload. An adapter strategy also can be used with nonpeptidic ligands (52). To date such approaches have been hampered by the low affinity of the adapter to the virus, leading to loss of specific interaction over time; also, when the virus construct retained intrinsic binding to virus receptors (e.g., CAR), unspecific virus entry to any CAR-expressing cells would result (24, 27–35).
We previously reported the selection of knob-binding DARPins to the fiber knob domain which were able to bind to both the WT and the knob domain mutated in the TAYT motif ablating CAR binding (37, 39). These monovalent DARPins bound at an affinity in the low nanomolar range (1.35 nM for 2E6) as determined by surface plasmon resonance (SPR), already better than approaches using the soluble CAR D1 domain displaying an affinity of only 15 nM (53). Trimerization of CAR D1-based adapters had been achieved using either the phage T4 fibritin domain or an isoleucine GCN4 trimerization domain (54, 55), which increased the efficiency of retargeting, but to our knowledge affinities have not been reported. The greatest obstacle in using these adapters was that they could be used only with WT Ad5, thus maintaining its intrinsic binding to CAR. Our approach aimed at generating trimeric adapters that could bind to Ad5 ablated in CAR binding. By linear multimerization we had previously achieved a functional affinity of better than 100 pM, but biophysical measurements suggested that only two DARPins might bind tightly per knob, with the third binding more weakly (37).
The crystal structure of the knob-binding DARPin 1D3 in complex with the trimeric knob protein allowed us to determine that the stoichiometry was clearly defined as three DARPins binding to one trimeric knob (Fig. 1), even though the B-factor (a measure of thermal motion) was much higher for one of the DARPins than for the other two. In solution, all three DARPins of the complex probably would show equal thermal mobilities, similar to the B-factor of chain F. This finding was a clear indication that, if ideally arranged on the knob, three DARPins could be positioned with greatly increased functional affinity. Moreover, the determination of the arrangement of the DARPins around the knob by X-ray crystallography suggested a better strategy than wrapping the DARPins around the knob linearly. Because the C termini are exposed at the top, we could fuse a highly stable trimeric protein, the capsid-stabilizing protein SHP of lambdoid phage 21 (43), allowing the DARPins to “clamp” the knob (Fig. 2 and Fig. S4). This adapter arrangement binds to the knob virtually irreversibly over 10 d, whereas the linear trimeric versions show measurable dissociation (Figs. 3 and 5B). This design thus is a great advance over other existing adapter-based approaches (24, 27–35, 37, 56).
DARPins are the ideal scaffold for large-scale production of such adapters (46, 57), because the absence of disulfides allows efficient soluble expression of these adapters with their multiple domains in the E. coli cytoplasm at very high levels (50–100 mg/mL) of culture. They spontaneously form very clean trimers after expression (Figs. S5 and S6). These properties make a scale-up for therapeutic products very realistic.
We have shown here that the knob-binding module, consisting of a knob-binding DARPin and the trimerization domain SHP, can be fused readily to a cell-surface receptor-binding DARPin, which will retarget Ad5 to a cell population expressing the respective cell-surface receptor (Figs. 3, 4, and 6). Because we can generate such retargeting DARPin modules to essentially any target using ribosome or phage display (37, 44, 50, 51, 58–64), we can exploit these capabilities [e.g., in tumor therapy in animal models (63, 65–67)] to use Ad5 to deliver payloads with tumor-controlling potential.
Although the performance of our trimeric bispecific adapters presented here still must be evaluated in vivo, it is clear that they have greatly improved properties—such as very high functional affinity, convenient exchange of the retargeting module, and high-level production—that represent major advances over the previously described adapter approaches.
Future studies will elucidate the efficacy of the trimeric adapters in vivo in combination with Ads that are ablated in their interaction of cells mediated by the fiber, penton base, or hexon protein (14). Targeted Ads to date proved inefficient in vivo, especially when delivered systemically, as would be of the greatest clinical relevance, because the fiber-mediated transduction is undermined by clearance of Ad virions by Kupffer cells and robust fiber-independent transduction of hepatocytes (14, 68). In particular, it has been shown that the high efficacy of hepatic transduction by systemically injected Ad is caused by the extensive coverage of the virus surface by blood coagulation factor X. In essence, factor X functions as a bispecific targeting adapter linking the Ad hexon capsid protein hexon to heparin sulfate-containing proteoglycans present on hepatocytes (69, 70). Simply blocking the hexon-binding sites for factor X has not proven sufficient and was out-competed by the high excess of factor X in the blood (27). More promising might be modifications of the hexon by replacing all hypervariable regions (HVRs) with the HVRs of hexons of Ad serotypes that do not bind factor X (13, 71) or by replacing or deleting amino acids in HVR5 involved in factor X binding (13, 56). Undoubtedly, Ads will have to be modified in several surface features simultaneously, and the adapter strategy will be useful in rapidly determining the most effective ones.
Here we have demonstrated a versatile strategy for rapidly generating Ad adapters with various cell-surface receptor specificities that clamp the Ad knob and bind virtually irreversibly to the virion. This generic adapter-based approach represents a generically applicable method to redirect prefabricated Ads to any cell population of choice. Although Ad vectors still will need to be engineered to reduce nontarget cell uptake, we present in this study a generic contribution toward efficient retargeting in future clinical applications.
Materials and Methods
Cloning, Expression, and Purification of DARPins, Knob, and Adapters.
The knob-binding DARPins 1D3 and 2E6 were equipped with a stabilized C-cap (41, 42) and cloned into the expression vector pDST72 (44), a derivative of pQE30, by a PCR-based strategy. To construct adapters, the C terminus of the knob-binding DARPins (omitting the last three residues of the DARPin, which were disordered in the crystal structure) were fused to the N terminus of the phage capsid protein SHP, used as trimerization domain (43) (SHP, V13-P115), and was spaced by different linkers (SHP1, G; SHP2, GA; SHP3, GLKAGADVNA), introduced by a PCR-based strategy and cloned into pDST72. In a second step, the cDNA was inserted into pQIBi2_2 with either an N- or C-terminal fusion of the retargeting DARPins G3, 9.29, Ec4, Ac2, E01, or E69, reported earlier (44, 45, 50, 51), spaced by a (Gly4Ser)4 linker. These retargeting DARPins can be exchanged readily using either BamHI/HindIII (for N-terminal fusions) or BglII/BsaI (for C-terminal fusions). For competition, monomeric DARPins or bispecific DARPins with increased valency (LZ constructs) expressed and purified as previously described (37, 50, 51) were used. Expression of the MA(H)6 mutant knob∆TAYT was performed as previously described (37).
Purification of DARPins, knob, and adapters was performed using immobilized-metal affinity chromatography (Ni-NTA superflow; QIAGEN) as previously described (37, 50, 51). Additional purification using SEC was performed in some cases using a Superdex 200 16/60 column (GE Healthcare Biosciences) in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) at a flow rate of 1 mL/min. For cloning, expression, purification, and refolding of the human CAR D1 domain see SI Materials and Methods.
Crystallization of the Knob-DARPin Complex.
A first dataset was recorded in-house (MAR345 detector, Bruker Micro Star X-ray generator, Software GO 3.4.5.) at 2.7-Å resolution. A second dataset was recorded at the Swiss Light Source (Paul Scherrer Institute, Villagen, Switzerland) with a resolution of 1.95 Å, which was used for the refinement of the in-house–determined structure. The crystallization procedure is described in detail in SI Materials and Methods.
Cell Lines.
Cell lines stably overexpressing HER2 were established using the HEK293 Flp-In or CHO Flp-In cell line (Invitrogen Ltd). Both were generated using the plasmid pcDNA5/FRT (Invitrogen Ltd.) into which the cDNA for HER2 (Gene Service) was inserted by a PCR-based strategy. Cells were transfected with Lipofectamine 2000 (Invitrogen Ltd.), and stable integrants were selected using hygromycin at a concentration of 150 µg/mL for HEK cells and 800 µg/mL for CHO cells. The HER2-overexpressing human breast ductal carcinoma tumor cell line BT474 (HTB-20), the EGFR-overexpressing human epidermal carcinoma cell line A431 (CRL-1555), the EpCAM-overexpressing human colorectal adenocarcinoma line HT29 (HTB-38), and the human epithelial embryonic kidney cell line HEK293T (CRL-1573) were all obtained from American Type Culture Collection. Cells generally were grown in DMEM (Ham’s F-12 for CHO), 10% (vol/vol) FCS, penicillin/streptomycin, at 37 °C and 5% (vol/vol) CO2.
Virus Production.
A dual reporter based on an Ad5 expression system [kind gift of T. Trueb, University of Zurich (72, 73)] expressing both firefly luciferase and eGFP was cloned. After introduction of the bicistronic construct spaced by an internal ribosome entry site sequence into the transfer vector pCTA, Cre-mediated recombination was performed in the E. coli strain Bm28.5 for integration of the expression cassette into the viral genome (pAdlox). The recombinant adenoviral DNA was used to transfect the complementing cell line HEK293 WT using Lipofectamine 2000 (Invitrogen Ltd.). Recombinant virus was amplified over several rounds of infection. For purification of virus 30 × 15 cm plates were harvested and processed as previously described using a CsCl gradient (74). Virus concentration was determined using OD260 with a conversion factor of 9.09 × 1013 vp per absorbance unit at 260 nm (75).
Transduction Experiments.
If not otherwise stated, transduction experiments were performed in a 24-well format with 2 × 105 cells per well using a multiplicity of 100 vp per cell. Recombinant virus was incubated with a 1:1 ratio of adapter per knob for 1 h at room temperature. Cells were infected and incubated at 37 °C, 5% (vol/vol) CO2 for 1 h. The medium was replaced, and cells were incubated further for 14–18 h as indicated. For analysis of luciferase activity the medium was removed, cells were lysed in 300 μL cell lysis buffer, and activity was determined using luciferase reporter assay solution (Promega) and a Victor3 plate luminometer (PerkinElmer Inc.). For competition studies testing specificity, the cells were preincubated with competitor protein for 1–1.5 h before transduction. For competition studies investigating the stability of the adapter, adapter and virus were mixed and incubated for 1.5 h before a 100-fold excess of competitor was added and incubated for different lengths of time before infection.
Supplementary Material
Acknowledgments
We thank Beat Blattmann and Céline Stutz-Ducommun from the in-house National Centers of Competence in Research (NCCR) crystallization unit for their assistance during the crystallization process; Nikolas Stefan and Manuel Simon for sharing epithelial cell adhesion molecule-related reagents; Ykelien Boersma for epidermal growth factor receptor-related reagents; Philipp Schätzle for his introduction to adenovirus generation and production; and Simon Hansen for help in surface plasmon resonance evaluation. This work was funded by a grant from the NCCR Structural Biology to A.P., by European Research Council senior investigator Grant NEXTBINDERS 268621 (to A.P.), by the University of Zurich, and Grants R01 CA128807, R01 CA116621, and R21 EB012259 from the National Institutes of Health.
Footnotes
Conflict of interest statement: A.P. is a shareholder of Molecular Partners AG, which is commercializing the DARPin technology.
This article is a PNAS Direct Submission.
Data deposition: Crystallography, atomic coordinates, and structure factors reported in this paper have been deposited in the Protein Data Bank, www.pdb.org (ID code 4ATZ).
See Author Summary on page 3725 (volume 110, number 10).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213653110/-/DCSupplemental.
References
- 1.Amalfitano A, Parks RJ. Separating fact from fiction: Assessing the potential of modified adenovirus vectors for use in human gene therapy. Curr Gene Ther. 2002;2(2):111–133. doi: 10.2174/1566523024605618. [DOI] [PubMed] [Google Scholar]
- 2.Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2007—an update. J Gene Med. 2007;9(10):833–842. doi: 10.1002/jgm.1100. [DOI] [PubMed] [Google Scholar]
- 3.Verma IM, Weitzman MD. Gene therapy: Twenty-first century medicine. Annu Rev Biochem. 2005;74:711–738. doi: 10.1146/annurev.biochem.74.050304.091637. [DOI] [PubMed] [Google Scholar]
- 4.Bergelson JM, et al. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275(5304):1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 5.Tomko RP, Xu R, Philipson L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc Natl Acad Sci USA. 1997;94(7):3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnberg N, Edlund K, Kidd AH, Wadell G. Adenovirus type 37 uses sialic acid as a cellular receptor. J Virol. 2000;74(1):42–48. [PMC free article] [PubMed] [Google Scholar]
- 7.Gaggar A, Shayakhmetov DM, Lieber A. CD46 is a cellular receptor for group B adenoviruses. Nat Med. 2003;9(11):1408–1412. doi: 10.1038/nm952. [DOI] [PubMed] [Google Scholar]
- 8.Short JJ, et al. Adenovirus serotype 3 utilizes CD80 (B7.1) and CD86 (B7.2) as cellular attachment receptors. Virology. 2004;322(2):349–359. doi: 10.1016/j.virol.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 9.Wang H, et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat Med. 2011;17(1):96–104. doi: 10.1038/nm.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell. 1993;73(2):309–319. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- 11.Nemerow GR, Pache L, Reddy V, Stewart PL. Insights into adenovirus host cell interactions from structural studies. Virology. 2009;384(2):380–388. doi: 10.1016/j.virol.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell WC. Adenoviruses: Update on structure and function. J Gen Virol. 2009;90(Pt 1):1–20. doi: 10.1099/vir.0.003087-0. [DOI] [PubMed] [Google Scholar]
- 13.Alba R, et al. Identification of coagulation factor (F)X binding sites on the adenovirus serotype 5 hexon: Effect of mutagenesis on FX interactions and gene transfer. Blood. 2009;114(5):965–971. doi: 10.1182/blood-2009-03-208835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khare R, Chen CY, Weaver EA, Barry MA. Advances and future challenges in adenoviral vector pharmacology and targeting. Curr Gene Ther. 2011;11(4):241–258. doi: 10.2174/156652311796150363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koizumi N, et al. Modified adenoviral vectors ablated for coxsackievirus-adenovirus receptor, αv integrin, and heparan sulfate binding reduce in vivo tissue transduction and toxicity. Hum Gene Ther. 2006;17(3):264–279. doi: 10.1089/hum.2006.17.264. [DOI] [PubMed] [Google Scholar]
- 16.Krasnykh VN, Douglas JT, van Beusechem VW. Genetic targeting of adenoviral vectors. Mol Ther. 2000;1(5 Pt 1):391–405. doi: 10.1006/mthe.2000.0062. [DOI] [PubMed] [Google Scholar]
- 17.Nicklin SA, Wu E, Nemerow GR, Baker AH. The influence of adenovirus fiber structure and function on vector development for gene therapy. Mol Ther. 2005;12(3):384–393. doi: 10.1016/j.ymthe.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 18.Noureddini SC, Curiel DT. Genetic targeting strategies for adenovirus. Mol Pharm. 2005;2(5):341–347. doi: 10.1021/mp050045c. [DOI] [PubMed] [Google Scholar]
- 19.Coughlan L, et al. Tropism-modification strategies for targeted gene delivery using adenoviral vectors. Viruses. 2010;2(10):2290–2355. doi: 10.3390/v2102290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Philipson L, Pettersson RF. The coxsackie-adenovirus receptor—a new receptor in the immunoglobulin family involved in cell adhesion. Curr Top Microbiol Immunol. 2004;273:87–111. doi: 10.1007/978-3-662-05599-1_3. [DOI] [PubMed] [Google Scholar]
- 21.Belousova N, et al. Genetically targeted adenovirus vector directed to CD40-expressing cells. J Virol. 2003;77(21):11367–11377. doi: 10.1128/JVI.77.21.11367-11377.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belousova N, Mikheeva G, Gelovani J, Krasnykh V. Modification of adenovirus capsid with a designed protein ligand yields a gene vector targeted to a major molecular marker of cancer. J Virol. 2008;82(2):630–637. doi: 10.1128/JVI.01896-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krasnykh VN, Mikheeva GV, Douglas JT, Curiel DT. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J Virol. 1996;70(10):6839–6846. doi: 10.1128/jvi.70.10.6839-6846.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wickham TJ, et al. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins. J Virol. 1997;71(11):8221–8229. doi: 10.1128/jvi.71.11.8221-8229.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magnusson MK, et al. A transductionally retargeted adenoviral vector for virotherapy of Her2/neu-expressing prostate cancer. Hum Gene Ther. 2012;23(1):70–82. doi: 10.1089/hum.2011.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8(8):573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen CY, May SM, Barry MA. Targeting adenoviruses with factor x-single-chain antibody fusion proteins. Hum Gene Ther. 2010;21(6):739–749. doi: 10.1089/hum.2009.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dmitriev I, Kashentseva E, Rogers BE, Krasnykh V, Curiel DT. Ectodomain of coxsackievirus and adenovirus receptor genetically fused to epidermal growth factor mediates adenovirus targeting to epidermal growth factor receptor-positive cells. J Virol. 2000;74(15):6875–6884. doi: 10.1128/jvi.74.15.6875-6884.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebbinghaus C, et al. Functional and selective targeting of adenovirus to high-affinity Fcgamma receptor I-positive cells by using a bispecific hybrid adapter. J Virol. 2001;75(1):480–489. doi: 10.1128/JVI.75.1.480-489.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haisma HJ, et al. Selective targeting of adenovirus to αvβ3 integrins, VEGFR2 and Tie2 endothelial receptors by angio-adenobodies. Int J Pharm. 2010;391(1-2):155–161. doi: 10.1016/j.ijpharm.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 31.Hong SS, Galaup A, Peytavi R, Chazal N, Boulanger P. Enhancement of adenovirus-mediated gene delivery by use of an oligopeptide with dual binding specificity. Hum Gene Ther. 1999;10(16):2577–2586. doi: 10.1089/10430349950016627. [DOI] [PubMed] [Google Scholar]
- 32.Korokhov N, et al. Targeting of adenovirus via genetic modification of the viral capsid combined with a protein bridge. J Virol. 2003;77(24):12931–12940. doi: 10.1128/JVI.77.24.12931-12940.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pereboev AV, et al. Enhanced gene transfer to mouse dendritic cells using adenoviral vectors coated with a novel adapter molecule. Mol Ther. 2004;9(5):712–720. doi: 10.1016/j.ymthe.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 34.Reynolds PN, et al. A targetable, injectable adenoviral vector for selective gene delivery to pulmonary endothelium in vivo. Mol Ther. 2000;2(6):562–578. doi: 10.1006/mthe.2000.0205. [DOI] [PubMed] [Google Scholar]
- 35.Watkins SJ, Mesyanzhinov VV, Kurochkina LP, Hawkins RE. The ‘adenobody’ approach to viral targeting: Specific and enhanced adenoviral gene delivery. Gene Ther. 1997;4(10):1004–1012. doi: 10.1038/sj.gt.3300511. [DOI] [PubMed] [Google Scholar]
- 36.Wickham TJ, et al. Targeted adenovirus gene transfer to endothelial and smooth muscle cells by using bispecific antibodies. J Virol. 1996;70(10):6831–6838. doi: 10.1128/jvi.70.10.6831-6838.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dreier B, et al. Her2-specific multivalent adapters confer designed tropism to adenovirus for gene targeting. J Mol Biol. 2011;405(2):410–426. doi: 10.1016/j.jmb.2010.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zlotnick A, Stray SJ. How does your virus grow? Understanding and interfering with virus assembly. Trends Biotechnol. 2003;21(12):536–542. doi: 10.1016/j.tibtech.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Kirby I, et al. Mutations in the DG loop of adenovirus type 5 fiber knob protein abolish high-affinity binding to its cellular receptor CAR. J Virol. 1999;73(11):9508–9514. doi: 10.1128/jvi.73.11.9508-9514.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roelvink PW, Mi Lee G, Einfeld DA, Kovesdi I, Wickham TJ. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adenoviridae. Science. 1999;286(5444):1568–1571. doi: 10.1126/science.286.5444.1568. [DOI] [PubMed] [Google Scholar]
- 41.Wetzel SK, et al. Residue-resolved stability of full-consensus ankyrin repeat proteins probed by NMR. J Mol Biol. 2010;402(1):241–258. doi: 10.1016/j.jmb.2010.07.031. [DOI] [PubMed] [Google Scholar]
- 42.Wetzel SK, Settanni G, Kenig M, Binz HK, Plückthun A. Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J Mol Biol. 2008;376(1):241–257. doi: 10.1016/j.jmb.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 43.Forrer P, Chang C, Ott D, Wlodawer A, Plückthun A. Kinetic stability and crystal structure of the viral capsid protein SHP. J Mol Biol. 2004;344(1):179–193. doi: 10.1016/j.jmb.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 44.Steiner D, Forrer P, Plückthun A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J Mol Biol. 2008;382(5):1211–1227. doi: 10.1016/j.jmb.2008.07.085. [DOI] [PubMed] [Google Scholar]
- 45.Zahnd C, et al. A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J Mol Biol. 2007;369(4):1015–1028. doi: 10.1016/j.jmb.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 46.Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol. 2003;332(2):489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 47.Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science. 1999;286(5444):1579–1583. doi: 10.1126/science.286.5444.1579. [DOI] [PubMed] [Google Scholar]
- 48.Gensler M, Buschbeck M, Ullrich A. Negative regulation of HER2 signaling by the PEST-type protein-tyrosine phosphatase BDP1. J Biol Chem. 2004;279(13):12110–12116. doi: 10.1074/jbc.M309527200. [DOI] [PubMed] [Google Scholar]
- 49.Belousova N, et al. Development of a targeted gene vector platform based on simian adenovirus serotype 24. J Virol. 2010;84(19):10087–10101. doi: 10.1128/JVI.02425-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boersma YL, Chao G, Steiner D, Wittrup KD, Plückthun A. Bispecific designed ankyrin repeat proteins (DARPins) targeting epidermal growth factor receptor inhibit A431 cell proliferation and receptor recycling. J Biol Chem. 2011;286(48):41273–41285. doi: 10.1074/jbc.M111.293266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stefan N, et al. DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J Mol Biol. 2011;413(4):826–843. doi: 10.1016/j.jmb.2011.09.016. [DOI] [PubMed] [Google Scholar]
- 52.Simon M, Zangemeister-Wittke U, Plückthun A. Facile double-functionalization of designed ankyrin repeat proteins using click and thiol chemistries. Bioconjug Chem. 2012;23(2):279–286. doi: 10.1021/bc200591x. [DOI] [PubMed] [Google Scholar]
- 53.Kirby I, et al. Identification of contact residues and definition of the CAR-binding site of adenovirus type 5 fiber protein. J Virol. 2000;74(6):2804–2813. doi: 10.1128/jvi.74.6.2804-2813.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kashentseva EA, Seki T, Curiel DT, Dmitriev IP. Adenovirus targeting to c-erbB-2 oncoprotein by single-chain antibody fused to trimeric form of adenovirus receptor ectodomain. Cancer Res. 2002;62(2):609–616. [PubMed] [Google Scholar]
- 55.Kim J, et al. Targeting adenoviral vectors by using the extracellular domain of the coxsackie-adenovirus receptor: Improved potency via trimerization. J Virol. 2002;76(4):1892–1903. doi: 10.1128/JVI.76.4.1892-1903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alba R, et al. Biodistribution and retargeting of FX-binding ablated adenovirus serotype 5 vectors. Blood. 2010;116(15):2656–2664. doi: 10.1182/blood-2009-12-260026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Binz HK, et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22(5):575–582. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- 58.Boersma YL, Plückthun A. DARPins and other repeat protein scaffolds: Advances in engineering and applications. Curr Opin Biotechnol. 2011;22(6):849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 59.Dreier B, Plückthun A. Rapid selection of high-affinity binders using ribosome display. Methods Mol Biol. 2012;805:261–286. doi: 10.1007/978-1-61779-379-0_15. [DOI] [PubMed] [Google Scholar]
- 60.Hanes J, Jermutus L, Weber-Bornhauser S, Bosshard HR, Plückthun A. Ribosome display efficiently selects and evolves high-affinity antibodies in vitro from immune libraries. Proc Natl Acad Sci USA. 1998;95(24):14130–14135. doi: 10.1073/pnas.95.24.14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanes J, Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci USA. 1997;94(10):4937–4942. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Plückthun A. Ribosome display: A perspective. Methods Mol Biol. 2012;805:3–28. doi: 10.1007/978-1-61779-379-0_1. [DOI] [PubMed] [Google Scholar]
- 63.Tamaskovic R, Simon M, Stefan N, Schwill M, Plückthun A. Designed ankyrin repeat proteins (DARPins) from research to therapy. Methods Enzymol. 2012;503:101–134. doi: 10.1016/B978-0-12-396962-0.00005-7. [DOI] [PubMed] [Google Scholar]
- 64.Zahnd C, Amstutz P, Plückthun A. Ribosome display: Selecting and evolving proteins in vitro that specifically bind to a target. Nat Methods. 2007;4(3):269–279. doi: 10.1038/nmeth1003. [DOI] [PubMed] [Google Scholar]
- 65.Martin-Killias P, Stefan N, Rothschild S, Plückthun A, Zangemeister-Wittke U. A novel fusion toxin derived from an EpCAM-specific designed ankyrin repeat protein has potent antitumor activity. Clin Cancer Res. 2011;17(1):100–110. doi: 10.1158/1078-0432.CCR-10-1303. [DOI] [PubMed] [Google Scholar]
- 66.Theurillat JP, et al. Designed ankyrin repeat proteins: A novel tool for testing epidermal growth factor receptor 2 expression in breast cancer. Mod Pathol. 2010;23(9):1289–1297. doi: 10.1038/modpathol.2010.103. [DOI] [PubMed] [Google Scholar]
- 67.Zahnd C, et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: Effects of affinity and molecular size. Cancer Res. 2010;70(4):1595–1605. doi: 10.1158/0008-5472.CAN-09-2724. [DOI] [PubMed] [Google Scholar]
- 68.Xu Z, Tian J, Smith JS, Byrnes AP. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J Virol. 2008;82(23):11705–11713. doi: 10.1128/JVI.01320-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalyuzhniy O, et al. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc Natl Acad Sci USA. 2008;105(14):5483–5488. doi: 10.1073/pnas.0711757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Waddington SN, et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132(3):397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 71.Roberts DM, et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature. 2006;441(7090):239–243. doi: 10.1038/nature04721. [DOI] [PubMed] [Google Scholar]
- 72.Chartier C, et al. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J Virol. 1996;70(7):4805–4810. doi: 10.1128/jvi.70.7.4805-4810.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mohanty S, et al. Overexpression of IRS2 in isolated pancreatic islets causes proliferation and protects human beta-cells from hyperglycemia-induced apoptosis. Exp Cell Res. 2005;303(1):68–78. doi: 10.1016/j.yexcr.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 74.Becker TC, et al. Use of recombinant adenovirus for metabolic engineering of mammalian cells. Methods Cell Biol. 1994;43(Pt A):161–189. doi: 10.1016/s0091-679x(08)60603-2. [DOI] [PubMed] [Google Scholar]
- 75.Mittereder N, March KL, Trapnell BC. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol. 1996;70(11):7498–7509. doi: 10.1128/jvi.70.11.7498-7509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]