

Abstract
The origin and biological role of dynamic motions of folded enzymes is not yet fully understood. In this study, we examine the molecular determinants for the dynamic motions within the β-barrel of superoxide dismutase 1 (SOD1), which previously were implicated in allosteric regulation of protein maturation and also pathological misfolding in the neurodegenerative disease amyotrophic lateral sclerosis. Relaxation-dispersion NMR, hydrogen/deuterium exchange, and crystallographic data show that the dynamic motions are induced by the buried H43 side chain, which connects the backbones of the Cu ligand H120 and T39 by a hydrogen-bond linkage through the hydrophobic core. The functional role of this highly conserved H120–H43–T39 linkage is to strain H120 into the correct geometry for Cu binding. Upon elimination of the strain by mutation H43F, the apo protein relaxes through hydrogen-bond swapping into a more stable structure and the dynamic motions freeze out completely. At the same time, the holo protein becomes energetically penalized because the twisting back of H120 into Cu-bound geometry leads to burial of an unmatched backbone carbonyl group. The question then is whether this coupling between metal binding and global structural motions in the SOD1 molecule is an adverse side effect of evolving viable Cu coordination or plays a key role in allosteric regulation of biological function, or both?
Keywords: allostery, local unfolding, metal binding, protein aggregation, structural frustration
Dynamic motions of folded proteins in several cases are found to be essential for allosteric control of ligand binding, adaptive orientation of active-site moieties, and communication between distant sites in protein structures and complexes (1–4). Despite this fundamental role of dynamic motions in biological function, the molecular factors that control structural fluctuations and conformational heterogeneity within folded proteins remain largely unexplored. Also, it is not yet known if there is any relation, or principal difference, between the functional motions of proteins and the supposedly adverse structural fluctuations implicated in protein misfolding and aggregation (5, 6). An interesting system for shedding more light on these questions is the enzyme superoxide dismutase 1 (SOD1) associated with pathological misfolding and aggregation in the neurodegenerative disease amyotrophic lateral sclerosis (ALS). Native SOD1 is a symmetric homodimer of two Ig-like β-barrels, each of which coordinates one catalytic Cu+/2+ ion and one structural Zn2+ ion. The redox active Cu+/2+ ion is ligated directly to the side of the monomer β-barrel, whereas the Zn2+ ties together the long loops IV and VII to a densely packed dome over the active site. The role of this dome is twofold. It composes a selectivity filter for substrates to the Cu+/2+ site (7) and also provides a critical part of the dimer interface (8). As a consequence, the dimerization of SOD1 becomes tightly controlled by metal binding via the structure of loops IV and VII, and the conserved C57–C146 disulfide linkage (9): if the metals and disulfide bond are lost, the protein dissociates into free monomers. Metal loss also unleashes characteristic dynamic motions of the central barrel scaffolds (10–13) that are most pronounced in the β-sheets facing the metal-binding sites. Together, these features form an intricate network of structural communication within the SOD1 molecule, spanning from the dynamic motions of the monomeric scaffold, via metal binding and the active-site loops, to the dimer interface. As a clue to the functional role of the communication, the binding of a single Zn2+ ion to one subunit of the dynamic apoSOD1 dimer is found to alter the structure and stability of the entire molecule (14). Based on the general view that globular proteins with compromised structural rigidity are at increased risk of misfolding (5, 6), the dynamic apoSOD1 molecule also has drawn attention as a possible precursor for pathological aggregation in ALS (12, 13, 15). In this study, we show by a combination of NMR, X-ray crystallography, and protein engineering that the origin of these dynamic motions is the built-in strain of a conserved hydrogen-bond linkage through the hydrophobic core. Upon truncation of this linkage by the mutation H43F, the SOD1 structure relaxes through H-bond swapping to a more ideal sheet architecture and the dynamics disappear. At a dynamic level, the results unravel the mechanism of an intricate allosteric crosstalk and show how a single side chain in the core can indirectly control a broad repertoire of functional traits. As a side effect of this structural crosstalk, however, the properties of the SOD1 molecule become globally sensitive to mutational perturbation, shedding light on its high sequence conservation and gain of neurotoxic function upon structurally distant mutations.
Results
Dynamic Motions of the apoSOD1 Barrel.
The dynamic motions of the apoSOD1 barrel are localized mainly to the β-strands facing the active site, and are indicated experimentally by diminished NMR NOE couplings, relaxation-dispersion measurements (11), and backbone hydrogen-exchange [hydrogen/deuterium (H/D)] kinetics (16). To enhance the structural resolution of these dynamic motions as much as possible, we focus here on a SOD1 variant with truncated loops IV and VII (apoSODΔIVΔVII) (16). An advantage of this variant is that it eliminates interference from the active-site loops, which otherwise leads to NMR line broadening at low temperatures, at which the barrel motions are most pronounced. In most other respects, apoSODΔIVΔVII behaves as the wild-type monomer (apoSOD1pwt) (16). Measurements of 15N Carr–Purcell–Meiboom–Gill (CPMG) relaxation profiles show here that 34 of the 110 apoSODΔIVΔVII residues are involved in dynamic exchange (Fig. 1 and Table S1). Global data analysis (SI Text) shows further that this exchange is well described by a two-state relaxation (kex = k1 + k−1) between a highly populated ground state (GS) and a high free-energy state (HS) with occupancies (pGS) and (pHS), respectively.
Fig. 1.

Dynamic motions of apoSOD1ΔIVΔVII measured by CPMG NMR. (A) The dynamic motions (red) are centered in the active-site sheet (β4 and β5) and the adjacent strand β6. (B) Representative data showing the CPMG amplitude decrease at high temperature. (C) Calculated chemical shift differences between the species undergoing dynamic exchange. (D) The population of the high-energy state decreases by protein destabilization by temperature, pH, and urea. (E) Sequence outline of apoSOD1ΔIVΔVII showing the positions with dynamic exchange (red), the corresponding dynamics of apoSOD1pwt (stars), and the GAG replacements of loops IV and VII.
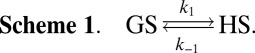 |
The highest density of dynamic residues is found in β4 to β6 (Fig. 1), in good agreement with previous CPMG measurements of apoSOD1pwt (11) and H/D exchange analysis (16). Besides some loss of signal at positions that previously interfaced the dynamic loop-IV structure, the main effect of loop removal overall is lower CPMG amplitudes. The reason for this slight amplitude loss seems to be decreased occupancy of the high-energy state, consistent with the slight gain in global protein stability of apoSODΔIVΔVII (Table 1 and SI Text). As a sharp outlier in this trend, the CPMG amplitude of I99 in β6 displays a nearly eightfold increase upon loop removal, rendering this position the strongest signal in apoSODΔIVΔVII. Judging by the unaffected H/D exchange rates of β6 and the neighboring β3, this signal increase likely is not the result of the increased dynamics of β6 itself (16). Rather it seems induced by altered fluctuations of β5 against which the I99 side chain packs (Fig. 1).
Table 1.
Kinetic and thermodynamic parameters determined from CPMG NMR relaxation-dispersion experiments
| Parameter | apoSOD1pwt | SOD1ΔIVΔVII | SOD1ΔIVΔVII in 1.5 M urea |
 (%)* (%)*
|
1.80 (0.07) | 0.63 (0.15) | |
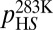 (%)* (%)*
|
0.70 (0.03) | 0.14 (0.02) | |
 (%)* (%)*
|
0.80 (0.10) | 0.34 (0.02) | 0.06 (0.02) |
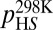 (%)* (%)*
|
0.16 (0.02) | 0.03 (0.01) | |
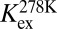 (kHz)* (kHz)*
|
5.85 (0.4) | 2.80 (0.23) | |
 (kHz)* (kHz)*
|
3.68 (0.3) | 2.39 (0.34) | |
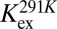 (kHz)* (kHz)*
|
2.59 (0.2) | 2.95 (0.4) | 3.75 (0.6) |
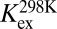 (kHz)* (kHz)*
|
2.00 (0.6) | 3.18 (1.0) | |
| ΔGGS-HS (kcal/mol)† | 2.79 (1.15)‡ | 3.72 (2.69) | 4.94 (6.25) |
| ΔHGS-HS (kcal/mol)† | −13.6 (1.2)‡ | −19.23 (1.89) | −23.76 (4.33) |
| ΔSGS-HS (cal/molK)† | −55 (2)‡ | −80 (10) | −96 (20) |
Dynamic Exchange with a High-Energy State That Is More Compact than the Ground State.
To shed further light on the molecular nature of the GS-to-HS transition (Scheme 1), we examined how the relaxation-dispersion behavior of apoSODΔIVΔVII responds to changes in temperature, pH, and [urea]. The results show that the population of the high-energy state decreases with increasing temperature, from pHS = 1.8 ± 0.1% at 278 K to pHS = 0.16 ± 0.02% at 298K (Fig. 1 and Table 1), consistent with previous data for apoSOD1pwt (11). Correspondingly, the exchange rate constant decreases from kex = 5,800 s−1 to kex = 2,000 s−1 (Table 1). Such reversed temperature dependence of transition from ground state to high-energy state is not expected for a local unfolding event unless the system is in the cold-unfolding regime (17). Rather, elevated temperature is predicted to favor disorder. As a complement to the thermal scans, we monitored how the transition from ground state to high-energy state responds to lowered pH. The CPMG profiles obtained at pH 7.3, 6.3, and 5.3 yield pHS values of 3.20 ± 0.08%, 1.8 ± 0.1%, and 0.98 ± 0.08%, respectively (Fig. 1, Table 1, SI Text, and Fig. S1A). Accordingly, the occupancy of high-energy state displays a consistent decrease with protein stability, and not the other way around, as expected for high-energy states that are partly ruptured intermediates. As a final test of this peculiarity we destabilized apoSODΔIVΔVII with urea, which is the general method for quantifying changes in solvent-accessible surface area in protein-folding equilibria (18). The results show that the occupancy of the high-energy state decreases from pHS = 1.8 ± 0.1% to pHS = 0.6 ± 0.2% upon addition of 1.5 M urea (Table 1, SI Text, and Fig. S1B), that is, the transition between ground state and high-energy state involves burial of solvent-accessible surface area. From the value of  , it is further evident that the extent of burial is sizable and corresponds to 29% of that in the global folding transition (SI Text and Fig. S1C). Because the CPMG data show no response to protein concentration (SI Text and Fig. S1D), we conclude that the burial is not an effect of intermolecular association but stems from consolidation of the monomer itself. To allow for such consolidation, the solution state of apoSOD1 needs to be expanded more than indicated from X-ray data, consistent with earlier findings by Banci et al. (13). It also follows that the transition from ground state to high-energy state is not a local unfolding event but, quite the opposite, a folding transition into a more compact state. An intriguing possibility would be that this compact state actually is analogous to the X-ray structure of the apoSOD1 molecule, selected by crystallization because of its high structural order.
, it is further evident that the extent of burial is sizable and corresponds to 29% of that in the global folding transition (SI Text and Fig. S1C). Because the CPMG data show no response to protein concentration (SI Text and Fig. S1D), we conclude that the burial is not an effect of intermolecular association but stems from consolidation of the monomer itself. To allow for such consolidation, the solution state of apoSOD1 needs to be expanded more than indicated from X-ray data, consistent with earlier findings by Banci et al. (13). It also follows that the transition from ground state to high-energy state is not a local unfolding event but, quite the opposite, a folding transition into a more compact state. An intriguing possibility would be that this compact state actually is analogous to the X-ray structure of the apoSOD1 molecule, selected by crystallization because of its high structural order.
Truncation of Conserved Hydrogen-Bond Linkage Eliminates β-Sheet Dynamics.
Characteristic of Ig-like structures is the conserved “tyrosine corner motif,” comprising a buried tyrosyl moiety that hydrogen bonds to a backbone carbonyl group at position n – 4 or 5 (19). In SOD1, this feature is replaced with a conserved “histidine corner motif”: the buried imidazole of H43 hydrogen bonds to the backbone of T39 in loop III (Fig. 2). Of particular interest is that H43 also binds to the backbone of the Cu+/2+ ligand H120, thereby forming the hub for a buried hydrogen-bond chain that extends through the SOD1 core (Fig. 2). Via the backbone of L38, the chain connects further to the backbone of G93 in loop V, a hotspot for ALS-provoking mutations (15). To examine the impact of this conserved linkage on the observed barrel dynamics, we replaced H43 with a phenylalanine. The apo mutant maintains a well-dispersed 1H-15N-HSQC (heternonuclear single quantum coherence) spectrum characteristic of a structurally ordered protein (Fig. 2, SI Text, and Fig. S2A). Even so, the chemical shift differences induced by the mutation are significant and reveal long-range effects (Fig. 2 and Table S2), which matches well with the regions found to undergo dynamic exchange in apoSOD1pwt and apoSODΔIVΔVII (SI Text). The H43F mutation also seems to increase the protein’s content of β structure as measured by secondary structure propensity (SSP) analysis, particularly in the regions of β2, β4−β6, and β7 (Fig. S2A and SI Text). This indicates that the mutation H43F imposes order on the SOD1 structure. Consistently, the mutation H43F also abolishes the relaxation-dispersion dynamics of the SOD1 barrel: the CPMG decays flatten completely, save for a single residual signal from I99 in β6 at low temperatures (Fig. 2 and SI Text). The same loss of barrel dynamics follows mutation H43F in apoSOD1pwt (SI Text and Fig. S3). On this basis, we conclude that the conserved hydrogen-bond linkage mediated by the side chain of H43 induces the dynamic motions of the SOD1 barrel.
mutant maintains a well-dispersed 1H-15N-HSQC (heternonuclear single quantum coherence) spectrum characteristic of a structurally ordered protein (Fig. 2, SI Text, and Fig. S2A). Even so, the chemical shift differences induced by the mutation are significant and reveal long-range effects (Fig. 2 and Table S2), which matches well with the regions found to undergo dynamic exchange in apoSOD1pwt and apoSODΔIVΔVII (SI Text). The H43F mutation also seems to increase the protein’s content of β structure as measured by secondary structure propensity (SSP) analysis, particularly in the regions of β2, β4−β6, and β7 (Fig. S2A and SI Text). This indicates that the mutation H43F imposes order on the SOD1 structure. Consistently, the mutation H43F also abolishes the relaxation-dispersion dynamics of the SOD1 barrel: the CPMG decays flatten completely, save for a single residual signal from I99 in β6 at low temperatures (Fig. 2 and SI Text). The same loss of barrel dynamics follows mutation H43F in apoSOD1pwt (SI Text and Fig. S3). On this basis, we conclude that the conserved hydrogen-bond linkage mediated by the side chain of H43 induces the dynamic motions of the SOD1 barrel.
Fig. 2.

Mutation H43F freezes out the dynamics. (A) Conserved H120–H43–T39 linkage. (B) The mutant H43F has an altered but highly dispersed HSQC spectrum. (C) CPMG profiles showing diminished dynamics for apo .
.
Mutation H43F Leads to Consolidation of the Active-Site Sheet.
The impact of the H43F mutation on the barrel breathing motions was examined by H/D exchange analysis, which measures the time it takes for each individual amide proton to exchange with solvent deuterons through local or global unfolding. In essence, apoSOD1pwt and apoSODΔIVΔVII display three levels of H/D exchange rates (16): (i) fast exchange, in which the amide protons are replaced within the experimental dead time (∼5 min); (ii) intermediate exchange in the EX2 regime, in which the exchange rate constant  is proportional to the population of locally “open” species; and (iii) slow exchange in the EX1 regime, in which
is proportional to the population of locally “open” species; and (iii) slow exchange in the EX1 regime, in which  follows the rate constant for global unfolding. For apoSODΔIVΔVII, the fast and intermediate exchanges are localized mainly to the dynamic sheet below the active site, whereas the back sheet exchanges slowly through global unfolding, save for the first few residues of β2 (SI Text and Table S3). The pattern, which fits well with CPMG data (Fig. 1), indicates that the motions of the active-site sheet are complex and energetically disperse, describing openings of less than 2.2 kcal/mol for dead-time exchange and excitations of >3.5 kcal/mol for intermediate exchange (SI Text), as well as transitions into the compact HS (Scheme 1). As observed with CPMG analysis, the dynamic motions undergo radical changes upon truncation of the H120–H43–T39 linkage. The H/D exchange rates of the active-site sheet show a marked decrease where, most notably, the amide of G44 in β4 changes from dead-time exchange in apoSODΔIVΔVII to full protection in apo
follows the rate constant for global unfolding. For apoSODΔIVΔVII, the fast and intermediate exchanges are localized mainly to the dynamic sheet below the active site, whereas the back sheet exchanges slowly through global unfolding, save for the first few residues of β2 (SI Text and Table S3). The pattern, which fits well with CPMG data (Fig. 1), indicates that the motions of the active-site sheet are complex and energetically disperse, describing openings of less than 2.2 kcal/mol for dead-time exchange and excitations of >3.5 kcal/mol for intermediate exchange (SI Text), as well as transitions into the compact HS (Scheme 1). As observed with CPMG analysis, the dynamic motions undergo radical changes upon truncation of the H120–H43–T39 linkage. The H/D exchange rates of the active-site sheet show a marked decrease where, most notably, the amide of G44 in β4 changes from dead-time exchange in apoSODΔIVΔVII to full protection in apo (Table S3). At the same time, H43F increases the exchange rates in β2 and β3 at the opposite side of the protein, and in the beta cap connecting the two sheets (Fig. 3). Accordingly, the ordering of the active-site sheet occurs at the expense of the structural rigidity of β2, β3, and the beta cap: one frustration seems to replace another.
(Table S3). At the same time, H43F increases the exchange rates in β2 and β3 at the opposite side of the protein, and in the beta cap connecting the two sheets (Fig. 3). Accordingly, the ordering of the active-site sheet occurs at the expense of the structural rigidity of β2, β3, and the beta cap: one frustration seems to replace another.
Fig. 3.

Mutation H43F decreases the H/D exchange rates ( ) in the active-site sheet β4, β5, and β7 and increases
) in the active-site sheet β4, β5, and β7 and increases 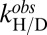 in the opposing part of the structure.
in the opposing part of the structure.
X-Ray Crystallography.
To pinpoint the structural rearrangement underlying the diminished dynamics upon mutation H43F, we solved the X-ray structures of apoSOD1ΔIVΔVII, apo , and holo
, and holo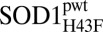 to resolutions of 1.9 Å, 2.8 Å, and 1.3 Å, respectively (SI Text and Table S4). The most striking feature of the apo
to resolutions of 1.9 Å, 2.8 Å, and 1.3 Å, respectively (SI Text and Table S4). The most striking feature of the apo structure is that the twisted backbone carbonyl group of H120 (β7), after being released from H43, relaxes into the plane of the sheet and H-bonds to the amide of G44 (β4) (Fig. 4). Following this relaxation, there is a tightening of the H-bond network between β7 and β4, where the average H-bond length decreases from 2.9 ± 0.13 Å to 2.8 ± 0.21 Å. The tightening also extends to β5 and is accompanied by a new surface interaction between the side chains of H120 and D53. The loss of dynamic motions upon mutation H43F thus seem to be the result of ordering of the active-site sheet, fully consistent with the HSQC and solution-state SSP analysis in Fig. S2A. Because of this extensive structural rearrangement, apoSOD1ΔIVΔVII and apo
structure is that the twisted backbone carbonyl group of H120 (β7), after being released from H43, relaxes into the plane of the sheet and H-bonds to the amide of G44 (β4) (Fig. 4). Following this relaxation, there is a tightening of the H-bond network between β7 and β4, where the average H-bond length decreases from 2.9 ± 0.13 Å to 2.8 ± 0.21 Å. The tightening also extends to β5 and is accompanied by a new surface interaction between the side chains of H120 and D53. The loss of dynamic motions upon mutation H43F thus seem to be the result of ordering of the active-site sheet, fully consistent with the HSQC and solution-state SSP analysis in Fig. S2A. Because of this extensive structural rearrangement, apoSOD1ΔIVΔVII and apo cannot strictly be seen as the same state. Equally revealing, we find in holo
cannot strictly be seen as the same state. Equally revealing, we find in holo that metal coordination to the H120 side chain wrenches back its backbone carbonyl group into the core (Fig. 4). This shows that the strained geometry of H120 in the wild-type protein is indeed required for metal binding and even establishes, at the cost of desolvating an unmatched polar moiety in the H43F mutant, that there seem to be no alternative metal-binding geometries available.
that metal coordination to the H120 side chain wrenches back its backbone carbonyl group into the core (Fig. 4). This shows that the strained geometry of H120 in the wild-type protein is indeed required for metal binding and even establishes, at the cost of desolvating an unmatched polar moiety in the H43F mutant, that there seem to be no alternative metal-binding geometries available.
Fig. 4.

Removal of the conserved H120–H43–T39 linkage by mutation H43F. (A) X-ray structures of apoSOD1ΔIVΔVII (PDB entry 4BCZ) and apo (PDB entry 4BD4) show that H43F leads to relaxation of the twisted backbone carbonyl of H120 into the sheet where it H-bonds to the amide of G44. (B) Cu2+ coordination twists back the carbonyl of H120 into the core, forcing the protein into a native-like metal-binding geometry. Holo
(PDB entry 4BD4) show that H43F leads to relaxation of the twisted backbone carbonyl of H120 into the sheet where it H-bonds to the amide of G44. (B) Cu2+ coordination twists back the carbonyl of H120 into the core, forcing the protein into a native-like metal-binding geometry. Holo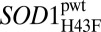 (PDB entry 4BCZ).
(PDB entry 4BCZ).
Effects on Folding, Metallation, and Enzymatic Activity.
The global folding reaction of the apoSOD1pwt and apoSOD1ΔIVΔVII monomers is described to a good approximation by a cooperative two-state transition (9, 16):
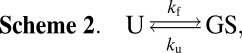 |
where U is the globally unfolded species, GS is the folded ground state in Scheme 1, and kf and ku are the folding and unfolding rate constants, respectively. The resulting chevron plots of logkf and logku vs. [GdmCl] are shown in Fig. 5, and the kinetic parameters derived from these plots are listed in Table 1. The curvatures of the unfolding limbs are linked to changes of rate-limiting step at high [GdmCl] and are discussed in SI Text. From the very small effects of the H43F mutation on the global stabilities of apoSOD1pwt and apoSOD1ΔIVΔVII, it is apparent that the penalty of removing the H120–H43–T39 linkage is compensated for by new favorable interactions in the folded ground state, in full accordance with the structural rearrangement of the apo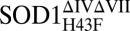 structure in Fig. 4. In contrast, the H43F mutation yields a pronounced destabilization of the fully metallated species holo
structure in Fig. 4. In contrast, the H43F mutation yields a pronounced destabilization of the fully metallated species holo , reflected by a selective increase of ku (Fig. 5 and Table 2). Again, the result is in full accordance with the X-ray data, in which the maintained geometry of the metal-bound species leaves the wrenched H120 backbone amide unsatisfied upon H43 removal (Fig. 4). Removal of the H120–H43 bond also is expected to decrease metal affinity. To examine this possibility, we measured the effect of EDTA on the unfolding kinetics of the holo proteins, which specifically targets the metal-off rates (20) (Fig. 5, Folding Analysis, and Table S5). Consistent with previous studies (20), holoSOD1pwt displays elevated ku values in the presence of EDTA, exposing the apparent rate constant of metal loss, koff (Fig. 5). This rate constant of the fully metallated protein most likely describes the simultaneous loss of both metal ions, because metal loss from the Zn site in the absence of a loaded Cu site is considerably faster (20), i.e., koff is strongly controlled by the Cu site. Upon H43F mutation, however, ku of holo
, reflected by a selective increase of ku (Fig. 5 and Table 2). Again, the result is in full accordance with the X-ray data, in which the maintained geometry of the metal-bound species leaves the wrenched H120 backbone amide unsatisfied upon H43 removal (Fig. 4). Removal of the H120–H43 bond also is expected to decrease metal affinity. To examine this possibility, we measured the effect of EDTA on the unfolding kinetics of the holo proteins, which specifically targets the metal-off rates (20) (Fig. 5, Folding Analysis, and Table S5). Consistent with previous studies (20), holoSOD1pwt displays elevated ku values in the presence of EDTA, exposing the apparent rate constant of metal loss, koff (Fig. 5). This rate constant of the fully metallated protein most likely describes the simultaneous loss of both metal ions, because metal loss from the Zn site in the absence of a loaded Cu site is considerably faster (20), i.e., koff is strongly controlled by the Cu site. Upon H43F mutation, however, ku of holo converges with that of the apo protein. The mutation thus seems to increase koff to a point at which metal dissociation no longer limits the unfolding kinetics. Taken together, this shows that the H120–H43–T39 linkage not only induces structural malleability of the apo protein, but also maintains high metal affinity. The obvious question, then, is how does this conserved feature affect enzyme activity? To find out, we compared the activity of holoSOD1pwt and holo
converges with that of the apo protein. The mutation thus seems to increase koff to a point at which metal dissociation no longer limits the unfolding kinetics. Taken together, this shows that the H120–H43–T39 linkage not only induces structural malleability of the apo protein, but also maintains high metal affinity. The obvious question, then, is how does this conserved feature affect enzyme activity? To find out, we compared the activity of holoSOD1pwt and holo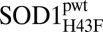 at two different pHs: at pH 9.5 with the direct KO2 assay and at pH 7.4 with a modified pyrogallol assay (21) (SI Text). The results show that the impact of the H43F mutation on the enzymatic turnover is modest: the activity per Cu+/2+-loaded monomer goes down by a factor of ∼5 (SI Text). As a control, we also determined the effect of H43F mutation on the activity of wild-type holo dimer and yielded a correspondingly modest reduction factor of ∼3 (SI Text). Notably, this reduction in activity upon H43F mutation is smaller than that observed for splitting the dimer interface (SI Text).
at two different pHs: at pH 9.5 with the direct KO2 assay and at pH 7.4 with a modified pyrogallol assay (21) (SI Text). The results show that the impact of the H43F mutation on the enzymatic turnover is modest: the activity per Cu+/2+-loaded monomer goes down by a factor of ∼5 (SI Text). As a control, we also determined the effect of H43F mutation on the activity of wild-type holo dimer and yielded a correspondingly modest reduction factor of ∼3 (SI Text). Notably, this reduction in activity upon H43F mutation is smaller than that observed for splitting the dimer interface (SI Text).
Fig. 5.

Folding chevron plots (logkf and logku) and the apparent metal-off rate constants (logkoff) for the proteins analyzed in this study. Parameters from fits are listed in Table 2. logku in the presence of EDTA, corresponding to logkoff, is shown as red ● and red ▲ for 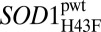 and SOD1pwt, respectively. logkoff at 0 M GdmCl was determined from fluorescence of the Zn2+-specific chelator Zinpyr.
and SOD1pwt, respectively. logkoff at 0 M GdmCl was determined from fluorescence of the Zn2+-specific chelator Zinpyr.
Table 2.
Kinetic parameters determined from folding kinetics
| Parameter | apoSOD1pwt | holoSOD1pwt | 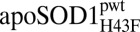 |
 |
apoSOD1ΔIVΔVII | 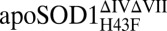 |
log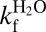 * *
|
−1.75 (0.20) | −0.25 (0.05) | −1.42 (0.09) | −0.20 (0.05) | −1.06 (0.04) | −0.74 (0.07) |
| mf (M−1)* | −2.85 (0.78) | −2.21 (0.04) | −3.49 (0.39) | −1.98 (0.08) | −2.29 (0.05) | −2.21 (0.10) |
log * *
|
−3.36 (0.09) | −8.29 (0.22) | −3.29 (0.04) | −5.45 (0.14) | −4.35 (0.03) | −4.77 (0.10) |
| mu (M−1)* | 1.16 (0.06) | 1.64 (0.08) | 1.14 (0.02) | 1.06 (0.05) | 0.77 (0.01) | 0.85 (0.03) |
| log Kpart* | −3.14 (0.15) | −3.87 (0.24) | −3.56 (0.10) | −3.64 (0.29) | −4.09 (0.23) | −3.93 (0.91) |
| m’’ (M−1)* | 1.01 (0.05) | 0.96 (0.07) | 0.93 (0.02) | 0.76 (0.04) | 0.74 (0.03) | 0.66 (0.11) |
| ΔGGS-U (kcal/mol)† | 2.19 (0.39) | 10.93 (0.37) | 2.54 (0.18) | 7.14 (0.26) | 4.47 (0.10) | 5.48 (0.23) |
| MP (M)‡ | 0.40 (0.10) | 2.09 (0.07) | 0.40 (0.04) | 1.73 (0.01) | 1.07 (0.02) | 1.32 (0.02) |


Discussion
The integrity and structural stability of natural proteins rely generally on funnel-like folding energy landscapes leading to unique final states (22). To comply with biological function, however, this basic funnel sometimes must be perturbed to allow several competing low-energy states and, thus, local motions of the native structure (3, 23–26). In this study, we demonstrate that the molecular basis for such functional complexity may be surprisingly simple: the global dynamic motions of apoSOD1 are induced by the strain of a single conserved hydrogen-bond linkage and may be switched on and off by a single point mutation (Fig. 6).
Fig. 6.

Switching off the dynamic motions by mutation H43F. (A) Dynamic positions of apoSOD1ΔIVΔVII (red) with intact H120–H43–T39 linkage. (B) H-bond relaxation and loss of dynamics in the mutant apo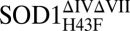 . (C) Metallation of apo
. (C) Metallation of apo drives the protein back to wild-type–like distortion of the β sheet.
drives the protein back to wild-type–like distortion of the β sheet.
Dynamic Motions from Evolutionary Engineered Frustration.
Mechanistically, the H120–H43–T39 linkage across the SOD1 core seems to promote dynamic motions by frustration (24), i.e., the protein is strained into a geometry in which it cracks up and starts to rattle between multiple low-energy minima. Judging from CPMG data (Fig. 1), the epicenter of this cracking is in the active-site sheet (Fig. 6). From the lack of exchange at H120 and G41, it also is evident that the straining bond between H120 and H43 does not transiently break during the dynamic motions but remains intact (Fig. 6). The frustration nevertheless is relieved upon H43F mutation, which relaxes the protein into a fixed ground state with continuous hydrogen bonding between β4 and β7 (Fig. 6). Even so, the influence of the H43F mutation is not limited to the folded ground state but also is seen by H/D exchange at higher energy levels, at which the diminished breathing in the β4–β5 region is accompanied by loosening of the beta cap and strands β2 and β3. This shows that the structural interplay indeed is long range and extends across the entire SOD1 monomer (Fig. 3). Despite the ordered appearance of the crystal structures, the apoSOD1 scaffold thus is equipped with a rich repertoire of correlated movements, consistent with earlier predictions from simulations (27, 28) and NMR studies (13). The apoSOD1 barrel also controls the motions of the functional loops: the CPMG profiles of the barrel are correlated precisely with those of loops IV and VII, and mutation H43F freezes the motions in both regions (SI Text); vice versa, metallation of the protein’s Zn2+ site freezes the motions of the barrel via consolidation of loops IV and VII (11). Taken together, this points at a two-way coupling between the barrel and loop motions, with interesting implications for the function of the SOD1 dynamics.
Functional Role of the SOD1 Dynamics.
The coupled structural and dynamic effects of the H43F mutations raise the question, do all these features relate to biological function, or are some merely side effects of conflicting evolutionary design? From X-ray data, it is clear that the H120–H43–T39 linkage is critical for distorting the Cu+/2+ ligand H120 into native coordination geometry, thus priming the apo structure for metal binding (Fig. 4). Consistently, the H43F mutant selectively destabilizes the holo state and shows radically increased metal-off rates (Fig. 5): elimination of the apoSOD1 distortion weakens the protein’s ability to hang on to its metals. It then is possible that the plasticity arising from the apo-state distortion presents an additional benefit by allowing adaptive orientation of metal-binding ligands. During catalytic turnover, the Cu+/2+ ion is redox active, which might induce cyclic structural changes between different coordination geometries. Such changes conceivably may lead to adverse stability differences between oxidized and reduced species (29) unless accommodated for by the protein structure. A related system might be the electron shuttle azurin, in which CPMG analysis identifies local structural motions around the coordinated Cu+ ion (30). The suggested role of this azurin flexibility, or lack of rigid protein matrix, is to facilitate electron transfer (30). From this perspective, and from the growing evidence that active-site dynamics are critical for enzyme catalysis (31, 32), it is notable that the H43F mutation has only a modest impact on SOD1 activity. One explanation might be that the activity of SOD1 is near diffusion control (7) and not limited by dynamics at the observed timescales. The task of the protein matrix then would be to select the substrate electrostatically and to protect the Cu+/2+ ion from reacting with other molecules (7). Instead, the most apparent role of the SOD1 dynamics is in structural crosstalk: the coupling between barrel and loop motions explains mechanistically how fractional metallation of one monomer influences not only the binding of subsequent metal ions, but also the structure and stability of the entire homodimer (14, 27, 33, 34). Where there are tuneable dynamics there also is allostery (4), even if it happens to pass undetected in conformationally biased X-ray structures (13). As an indication that the SOD1 crosstalk indeed is functional, it comprises 37% of the protein’s residues, complying well with the benchmark for other allosteric proteins (26). The precise biological function of this structural interplay, however, is not yet clear. One possibility is that it regulates molecular assembly, perhaps by contributing to more concerted enzymatic activation in the maturation pathway (35).
Aggregation and Disease.
Finally, we ask whether the partly ruptured apoSOD monomer is also the mysterious precursor for pathologic aggregation in ALS. After all, its structural plasticity need not be solely a functional advantage but also might carry aberrant side effects arising from conflicting evolutionary optimization (36). Of particular interest here is the high-energy state HS (Scheme 1), as seemingly analogous species are implicated as precursors in β2-microglobulin and lysozyme amyloidosis (5, 6). In contrast to the high-energy intermediates of β2-microglobulin and lysozyme, however, we observe here that the high-energy state HS is not on the pathway to the unfolded state, but is more compact than the solution ground state (Table 1). Together with the finding that destabilization of the apo monomer with urea accelerates the fibrillation kinetics in vitro (37), this puts precise constraints on the aggregation mechanism: the fibrillation precursor needs to be more expanded than the folded ground state, which excludes the involvement of the high-energy state HS. Instead, the precursor for apoSOD fibrillation appears to be the globally denatured state (37), consistent with an observed maximum of the fibrillation kinetics above 5 M urea, at which the occupancy of D is nearly 1 and the occupancy of the high-energy state has decreased from 0.01 to less than 10−4.7 (Table 1, SI Text, and Fig. S4). On this basis, we conclude that the dynamic motions of the apoSOD1 barrel do not compromise the protein’s solubility. The most imminent explanation as to why the SOD1 motions in this respect are different from other amyloidogenic proteins is that they are functionally evolved. A predicted consequence of the SOD1 crosstalk, nevertheless, is that the protein becomes globally sensitive to mutations. Peripheral mutations need not only perturb locally but also may modulate the structure allosterically over long distances. An example of such long-range modulation is the observation that the common ALS-associated SOD1 mutation G93A affects the metal-binding ligands more than 18 Å away (38). For an enzyme whose stability, maturation, and cellular lifetimes are intimately coupled to the metal-binding equilibria (20), such global effects might be critical, shedding light not only on why the ALS-provoking mutations are so broadly distributed in the SOD1 structure, but also on why the SOD1 sequence is so highly conserved across divergent species (39). To this end, our data show that the evolutionary pathway to such complex functional dynamics may be very short: in this case, the change of a ubiquitous tyrosine corner into a cross-core histidine link is just one mutation away.
Materials
Protein Preparation.
Mutagenesis, expression, and purification were performed as in refs. 9 and 16, and experiments were in Bis-Tris HCl buffer at pH 6.3 unless otherwise stated.
NMR.
CPMG relaxation rates were measured on a Bruker Avance 700-MHz spectrometer. Assignment of apo was by a standard set of experiments (SI Text) on a Bruker 900-MHz spectrometer. Relaxation rates R1, R2, and NOE were determined as described in ref. 16 on a Bruker 600-MHz spectrometer. H/D exchange analysis was by 1:1 sample dilution in D2O, and detection was on a Bruker 700-MHz spectrometer, as described in ref. 16. Detection dead time was 3 min, and HSQC spectra were recorded every 10 min.
X-ray Crystallography.
Crystallization was by hanging drop as described in (SI Text) and data were collected at 100 K at the MAX Laboratory synchrotron in Lund, Sweden. Data collection statistics are listed in (SI Text). All structures were solved by molecular replacement using Phaser with monomeric 1MFM as a starting model. Final refinement, validation, and geometrical statistics are described in (SI Text). Structures are deposited in www.pdb.org with accession numbers 4BCY for holo , 4BCZ for apoSOD1ΔIVΔVII, and 4BD4 for apo
, 4BCZ for apoSOD1ΔIVΔVII, and 4BD4 for apo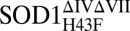 .
.
Folding Kinetics.
Protein concentration was 4 μM, and mixing was by a PiStar-180 stopped-flow apparatus (Applied Photophysics) or done manually on a Varian Cary Eclipse, with excitation at 280 nm and emission above 305 nm or at 360 nm. Data were analyzed with Kaleidagraph (Synergy Software) (SI Text).
Metal-Off Rates.
Metal-off rates (koff) were determined by GdmCl-induced unfolding in 2 mM EDTA. At 0 M GdmCl, koff was determined by mixing holo protein with the Zn2+-specific fluorescent chelator Zinpyr (Sigma-Aldrich).
Supplementary Material
Acknowledgments
Funding was from the Swedish Research Council (VR 2009–5580), the Knut and Alice Wallenberg Foundation, the Bertil Hållsten Foundation, and Hjärnfonden. Access to research infrastructure activity in the Seventh Framework Programme of the European Council (Project 261863, Bio-NMR) is acknowledged.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Data Bank, www.pdb.org (PDB ID codes 4BCY, 4BCZ, and 4BD4).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1217306110/-/DCSupplemental.
References
- 1.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13(9):831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Popovych N, Tzeng SR, Tonelli M, Ebright RH, Kalodimos CG. Structural basis for cAMP-mediated allosteric control of the catabolite activator protein. Proc Natl Acad Sci USA. 2009;106(17):6927–6932. doi: 10.1073/pnas.0900595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olsson U, Wolf-Watz M. Overlap between folding and functional energy landscapes for adenylate kinase conformational change. Nat Commun. 2010;1:111. doi: 10.1038/ncomms1106. [DOI] [PubMed] [Google Scholar]
- 4.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57(3):433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 5.Canet D, et al. Local cooperativity in the unfolding of an amyloidogenic variant of human lysozyme. Nat Struct Biol. 2002;9(4):308–315. doi: 10.1038/nsb768. [DOI] [PubMed] [Google Scholar]
- 6.Eichner T, Kalverda AP, Thompson GS, Homans SW, Radford SE. Conformational conversion during amyloid formation at atomic resolution. Mol Cell. 2011;41(2):161–172. doi: 10.1016/j.molcel.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Getzoff ED, et al. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature. 1992;358(6384):347–351. doi: 10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- 8.Hörnberg A, Logan DT, Marklund SL, Oliveberg M. The coupling between disulphide status, metallation and dimer interface strength in Cu/Zn superoxide dismutase. J Mol Biol. 2007;365(2):333–342. doi: 10.1016/j.jmb.2006.09.048. [DOI] [PubMed] [Google Scholar]
- 9.Lindberg MJ, Normark J, Holmgren A, Oliveberg M. Folding of human superoxide dismutase: Disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci USA. 2004;101(45):15893–15898. doi: 10.1073/pnas.0403979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banci L, Bertini I, Cramaro F, Del Conte R, Viezzoli MS. Solution structure of Apo Cu,Zn superoxide dismutase: role of metal ions in protein folding. Biochemistry. 2003;42(32):9543–9553. doi: 10.1021/bi034324m. [DOI] [PubMed] [Google Scholar]
- 11.Teilum K, et al. Transient structural distortion of metal-free Cu/Zn superoxide dismutase triggers aberrant oligomerization. Proc Natl Acad Sci USA. 2009;106(43):18273–18278. doi: 10.1073/pnas.0907387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banci L, et al. Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc Natl Acad Sci USA. 2009;106(17):6980–6985. doi: 10.1073/pnas.0809845106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banci L, et al. NMR characterization of a “fibril-ready” state of demetalated wild-type superoxide dismutase. J Am Chem Soc. 2011;133(2):345–349. doi: 10.1021/ja1069689. [DOI] [PubMed] [Google Scholar]
- 14.Potter SZ, et al. Binding of a single zinc ion to one subunit of copper-zinc superoxide dismutase apoprotein substantially influences the structure and stability of the entire homodimeric protein. J Am Chem Soc. 2007;129(15):4575–4583. doi: 10.1021/ja066690+. [DOI] [PubMed] [Google Scholar]
- 15.Nordlund A, Oliveberg M. Folding of Cu/Zn superoxide dismutase suggests structural hotspots for gain of neurotoxic function in ALS: Parallels to precursors in amyloid disease. Proc Natl Acad Sci USA. 2006;103(27):10218–10223. doi: 10.1073/pnas.0601696103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danielsson J, Kurnik M, Lang L, Oliveberg M. Cutting off functional loops from homodimeric enzyme superoxide dismutase 1 (SOD1) leaves monomeric β-barrels. J Biol Chem. 2011;286(38):33070–33083. doi: 10.1074/jbc.M111.251223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliveberg M, Tan YJ, Fersht AR. Negative activation enthalpies in the kinetics of protein folding. Proc Natl Acad Sci USA. 1995;92(19):8926–8929. doi: 10.1073/pnas.92.19.8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fersht AR. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman; 1999. [Google Scholar]
- 19.Hamill SJ, Cota E, Chothia C, Clarke J. Conservation of folding and stability within a protein family: The tyrosine corner as an evolutionary cul-de-sac. J Mol Biol. 2000;295(3):641–649. doi: 10.1006/jmbi.1999.3360. [DOI] [PubMed] [Google Scholar]
- 20.Leinartaite L, Saraboji K, Nordlund A, Logan DT, Oliveberg M. Folding catalysis by transient coordination of Zn2+ to the Cu ligands of the ALS-associated enzyme Cu/Zn superoxide dismutase 1. J Am Chem Soc. 2010;132(38):13495–13504. doi: 10.1021/ja1057136. [DOI] [PubMed] [Google Scholar]
- 21.Marklund S. Spectrophotometric study of spontaneous disproportionation of superoxide anion radical and sensitive direct assay for superoxide dismutase. J Biol Chem. 1976;251(23):7504–7507. [PubMed] [Google Scholar]
- 22.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys. 2005;38(3):245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 23.Miyashita O, Onuchic JN, Wolynes PG. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc Natl Acad Sci USA. 2003;100(22):12570–12575. doi: 10.1073/pnas.2135471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreiro DU, Hegler JA, Komives EA, Wolynes PG. On the role of frustration in the energy landscapes of allosteric proteins. Proc Natl Acad Sci USA. 2011;108(9):3499–3503. doi: 10.1073/pnas.1018980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, Wolynes PG, Takada S. Frustration, specific sequence dependence, and nonlinearity in large-amplitude fluctuations of allosteric proteins. Proc Natl Acad Sci USA. 2011;108(9):3504–3509. doi: 10.1073/pnas.1018983108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daily MD, Gray JJ. Local motions in a benchmark of allosteric proteins. Proteins. 2007;67(2):385–399. doi: 10.1002/prot.21300. [DOI] [PubMed] [Google Scholar]
- 27.Khare SD, Dokholyan NV. Common dynamical signatures of familial amyotrophic lateral sclerosis-associated structurally diverse Cu, Zn superoxide dismutase mutants. Proc Natl Acad Sci USA. 2006;103(9):3147–3152. doi: 10.1073/pnas.0511266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding F, Dokholyan NV. Dynamical roles of metal ions and the disulfide bond in Cu, Zn superoxide dismutase folding and aggregation. Proc Natl Acad Sci USA. 2008;105(50):19696–19701. doi: 10.1073/pnas.0803266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winkler JR, Wittung-Stafshede P, Leckner J, Malmström BG, Gray HB. Effects of folding on metalloprotein active sites. Proc Natl Acad Sci USA. 1997;94(9):4246–4249. doi: 10.1073/pnas.94.9.4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korzhnev DM, Karlsson BG, Orekhov VY, Billeter M. NMR detection of multiple transitions to low-populated states in azurin. Protein Sci. 2003;12(1):56–65. doi: 10.1110/ps.0225403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295(5559):1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 32.Watt ED, Shimada H, Kovrigin EL, Loria JP. The mechanism of rate-limiting motions in enzyme function. Proc Natl Acad Sci USA. 2007;104(29):11981–11986. doi: 10.1073/pnas.0702551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banci L, et al. Backbone dynamics of human Cu,Zn superoxide dismutase and of its monomeric F50E/G51E/E133Q mutant: The influence of dimerization on mobility and function. Biochemistry. 2000;39(31):9108–9118. doi: 10.1021/bi000067z. [DOI] [PubMed] [Google Scholar]
- 34.Schuyler AD, Carlson HA, Feldman EL. Computational methods for identifying a layered allosteric regulatory mechanism for ALS-causing mutations of Cu-Zn superoxide dismutase 1. Proteins. 2011;79(2):417–427. doi: 10.1002/prot.22892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rae TD, Torres AS, Pufahl RA, O’Halloran TV. Mechanism of Cu,Zn-superoxide dismutase activation by the human metallochaperone hCCS. J Biol Chem. 2001;276(7):5166–5176. doi: 10.1074/jbc.M008005200. [DOI] [PubMed] [Google Scholar]
- 36.Nordlund A, et al. Functional features cause misfolding of the ALS-provoking enzyme SOD1. Proc Natl Acad Sci USA. 2009;106(24):9667–9672. doi: 10.1073/pnas.0812046106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lang L, Kurnik M, Danielsson J, Oliveberg M. Fibrillation precursor of superoxide dismutase 1 revealed by gradual tuning of the protein-folding equilibrium. Proc Natl Acad Sci USA. 2012;109(44):17868–17873. doi: 10.1073/pnas.1201795109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Museth AK, Brorsson AC, Lundqvist M, Tibell LA, Jonsson BH. The ALS-associated mutation G93A in human copper-zinc superoxide dismutase selectively destabilizes the remote metal binding region. Biochemistry. 2009;48(37):8817–8829. doi: 10.1021/bi900703v. [DOI] [PubMed] [Google Scholar]
- 39.Sandelin E, Nordlund A, Andersen PM, Marklund SS, Oliveberg M. Amyotrophic lateral sclerosis-associated copper/zinc superoxide dismutase mutations preferentially reduce the repulsive charge of the proteins. J Biol Chem. 2007;282(29):21230–21236. doi: 10.1074/jbc.M700765200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.