

Abstract
Carbonic anhydrases (CAs, EC 4.2.1.1) are a group of metalloenzymes that play important roles in carbon metabolism, pH regulation, CO2 fixation in plants, ion transport etc., and are found in all eukaryotic and many microbial organisms. This family of enzymes catalyzes the interconversion of CO2 and HCO3−. There are at least 16 different CA isoforms in the alpha structural class (α-CAs) that have been isolated in higher vertebrates, with CA isoform II (CA II) being ubiquitously abundant in all human cell types. CA inhibition has been exploited clinically for decades for various classes of diuretics and anti-glaucoma treatment. The characterization of the overexpression of CA isoform IX (CA IX) in certain tumors has raised interest in CA IX as a diagnostic marker and drug target for aggressive cancers and therefore the development of CA IX specific inhibitors. An important goal in the field of CA is to identify, rationalize, and design potential compounds that will preferentially inhibit CA IX over all other isoforms of CA. The variations in the active sites between isoforms of CA are subtle and this causes non-specific CA inhibition which leads to various side effects. In the case of CA IX inhibition, CA II along with other isoforms of CA provide off-target binding sites which is undesirable for cancer treatment. The focus of this article is on CA IX inhibition and two different structural approaches to CA isoform specific drug designing: tail approach and fragment addition approach.
Introduction
Carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous metalloenzymes that catalyze the reversible hydration/dehydration of CO2/HCO3−1,2. All CAs identified to date use a metalhydroxide mechanism to mediate the two-step ping-pong catalytic mechanism.
Classification
CA has been divided into five classes: α (found in mammals, prokaryotes, algae and fungi), β (found mainly in plants and some prokaryotes), γ (present only in some forms of bacteria), and two other sub-classes: δ and ζ (similar to class β, found in diatoms)3–8. Human class α, the most studied of all classes, consists of at least 15 isoforms6,9. Of these 15 isoforms, three are non-catalytic because they lack the Zn-metal in the active site which is essential for the enzyme function. These non-catalytic isoforms (CA VIII, X and XI) are referred to as CA related proteins (CARPs). The remaining 12 catalytic isoforms differ from each other in catalytic activity, tissue localization and cellular distribution (cytosolic (CA I-III, VII, XIII), membrane bound (CA IV, IX, XII, XIV), secretory (CA VI) or mitochondrial (CA VA, VB))4,10–25. Table 1 provides a list of one PDB ID for each solved crystal structures of catalytic isoforms of α-CA (mostly human); Table 2 and 3 summarize primary sequence identity and structural similarity among these isoforms, respectively.
Table 1.
List of isoforms of α-CA crystal structures along with their respective PDB IDs, and source organism.
Table 2.
Primary sequence identity % (upper right) and number of conserved residues (lower left) among various α-CA isoforms.
| I | II | III | IV | V | VI | VII | IX | XII | XIV | |
|---|---|---|---|---|---|---|---|---|---|---|
| I | - | 60.5 | 54.4 | 28.8 | 43.7 | 30.9 | 50.6 | 31.3 | 29.8 | 32.6 |
| II | 158 | - | 58.9 | 32.4 | 49.4 | 32.5 | 55.9 | 31.4 | 28.4 | 36.0 |
| III | 142 | 153 | - | 30.2 | 43.3 | 32.8 | 52.9 | 29.5 | 27.3 | 33.0 |
| IV | 78 | 88 | 82 | - | 27.9 | 31.3 | 33.1 | 29.4 | 26.0 | 34.2 |
| V | 114 | 129 | 113 | 76 | - | 28.6 | 47.9 | 29.0 | 24.3 | 32.6 |
| VI | 83 | 87 | 88 | 85 | 77 | - | 35.3 | 41.0 | 35.4 | 41.4 |
| VII | 132 | 146 | 138 | 90 | 125 | 95 | - | 34.6 | 31.6 | 36.3 |
| IX | 85 | 85 | 80 | 80 | 79 | 110 | 94 | - | 31.2 | 41.9 |
| XII | 79 | 75 | 72 | 70 | 64 | 93 | 84 | 83 | - | 37.8 |
| XIV | 86 | 82 | 88 | 93 | 87 | 110 | 97 | 113 | 99 | - |
Table 3.
Structural main chain RMSDs (Å) among various isoforms of human CAs.
| I | II | III | IV | V* | VI | VII | IX | XII | XIII | XIV | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| I | - | 0.98 | 0.87 | 2.48 | 1.02 | 1.55 | 0.99 | 2.02 | 2.07 | 0.85 | 1.52 |
| II | - | 0.96 | 2.29 | 0.98 | 1.48 | 0.77 | 2.05 | 2.17 | 0.76 | 1.50 | |
| III | - | 2.37 | 1.01 | 1.69 | 0.86 | 1.79 | 1.99 | 0.85 | 1.41 | ||
| IV | - | 1.65 | 1.52 | 2.36 | 2.81 | 2.78 | 1.39 | 2.48 | |||
| V* | - | 1.47 | 0.83 | 1.82 | 1.89 | 1.30 | 1.42 | ||||
| VI | - | 1.52 | 1.35 | 1.58 | 1.58 | 1.49 | |||||
| VII | - | 1.79 | 1.97 | 0.77 | 1.42 | ||||||
| IX | - | 1.87 | 2.06 | 1.88 | |||||||
| XII | - | 1.17 | 1.86 | ||||||||
| XIII | - | 1.18 | |||||||||
| XIV | - |
Murine CA
Structure and function
CA isoform II (CA II) is a monomeric 29 kDa protein composed of 260 amino acids with a mixed α/β globular protein fold. It is the best studied and most abundant isoform of CA present in humans, comprising a 10 β-strand sheet flanked by 7 α-helices. The active site zinc present at the base of the 15 Å deep conical cleft is tetrahedrally coordinated by three histidine residues (H94, H96 and H119) and a hydroxyl ion. Clustering of various amino acid residues in and around the rim of the active site of CA, forms various amino acid selective pockets. The active site cavity is partitioned into two very different but conserved environments comprising a cluster of hydrophobic amino acids (V121, V143, L198, V207 and W209) on one side, and hydrophilic amino acids (Y7, N62, H64, N67, T199 and T200) lining the surface on the other side. While the rim of the active site exhibits surface pockets that are characteristic to the various isoforms (Fig. 1A).
Figure 1.

(A) Various pockets (encircled mesh) within and around the active site of CA II78. (B): An inhibitor, PDB ID: 3OYS48, extending out of the active site of CA II (white surface), stabilized by hydrophobic (red) and hydrophilic (blue) regions. Active site Zn is shown as a blue sphere.
The contrasting environments within the active site aid in enhancing the catalytic efficiency of the enzyme. The first step, hydration of CO2 that occurs in the hydrophobic pocket (Fig. 1B), is the nucleophilic attack on incoming CO2 by a Zn-bound OH− to produce HCO3−. The binding of HCO3− at the metal is weak and accordingly is displaced by a water molecule. In the second step, the Zn-H2O loses a proton (H+) leaving the Zn in its original Zn-OH− conformation through a network of well-ordered H-bonded waters to several hydrophilic residues (Y7, N62 and N67 in CA II) that line the active site (Fig. 1B). These residues are found to be highly conserved among various isoforms of α-CA. This network stretches from the Zn-bound solvent to the proton shuttling residue H64.
| step1: |
| step2: |
Extensive research has shown that this proton transfer step (step 2) is rate limiting in CA catalysis and His64 to be the proton donor/acceptor in the process 26–31. The enzyme displays very strong pH dependence for both kcat and kcat/KM that are defined by a single ionization corresponding to a pKa of ~7 30,32–35.
Physiological role
CA is involved in several physiological roles including rapid access to bicarbonate in red blood corpuscles (RBCs), fluid secretion, acid/base balance and thus pH regulation, gluconeogenesis, ureagenesis, gastric acid production, tumorigenesis, lipogenesis, electrolyte secretion, and transport of CO2 from tissues4,10,11,13–18,36. CO2 released as a part of respiration by tissues is weakly soluble in blood and thus, in order to be transported, is converted to HCO3− by CA in the RBCs. Furthermore, the role of CA in diseases such as glaucoma has long been known. Over secretion of aqueous humor in the eye causes increased intra-ocular pressure. Reduction in CA activity decreases the secretion of HCO3− and aqueous humor, thereby reducing the pressure. Recent research on the role of CA in epilepsy, obesity, pain, and cancers of breast and intestine has drawn attention3–6,8,37,38.
CA Inhibition
As CAs are involved in many physiological reactions as mentioned above, CA isoforms are therapeutic targets with the potential to be inhibited to treat diseases like glaucoma, edema, epilepsy and obesity. Broadly, CA inhibitors (CAIs) that bind to the metal center can be divided into two main classes, by forming: (1) trigonal-bipyramidal species (e.g. formates, cyanates) by adding an extra functional group to the already hydroxyl bound zinc or, (2) tetrahedral adducts (e.g. sulfonamides, bisulfites) with zinc, by displacing the zinc-bound hydroxyl. However, inhibitors like coumarins (that bind away from Zn), nitrates (that bind near the Zn but do not coordinate with the metal), and trithiocarbonates (that bind to the Zn in a distorted tetrahedron coordination) have also been reported39–44.
Extensive research has been performed on various classes of CAIs45–49. The sulfonamide derived compounds are buried deep in the active site, displacing solvent molecules, with the sulfonamide amine nitrogen binding directly to the active site zinc atom (distance ~ 2.0 Å) and accepting a hydrogen bond from OG1 of T199 and with the O2 of the sulfonamide accepting a hydrogen bond from the main-chain nitrogen atom of T199 at a distance ~ 3.0 Å. The overall Zn(N)4 coordination can be described as a distorted tetrahedron. The compound moieties extend out of the active site and are stabilized by both hydrophilic and hydrophobic residues (Fig. 1B).
CA IX and Cancer
Cancer continues to be the second most leading cause of death in the United States, after cardiovascular diseases. Accounting for 1.6 million cases in 2011, cancer was estimated to kill 571,000 people in the U.S.50.
It has now been well documented that tumor hypoxia is associated with poor prognosis and increased tumor aggressiveness, and CA IX (an endogenous marker for tumor hypoxia) upregulation that regulates the pH of tumor cells. In contrast to the other CA isoforms, CA IX has been implicated to play a role in regulation of cell proliferation, adhesion, and malignant cell invasion. Interestingly, CA IX is over expressed in human epithelial tumors derived from tissues that normally do not express these isoforms, including carcinomas of the cervix, lungs, kidneys, prostate, and breast. There is also evidence that CA IX allows tumors to acclimate to a hypoxic microenvironment, promoting tumor cell proliferation, and that CA IX expression is related to poor survival in patients51.
Most tumors experience a structurally and functionally disturbed microcirculation of oxygen which pathophysiologically causes an inadequate supply of oxygen (or hypoxia)4,37,50,52–55. In tumors such as gliomas/ependymomas4, mesotheliomas4, tumors of kidney56, carcinomas of bladder57, uterus/cervix58,59, pharynx60, head/neck61, breast62–64, esophagus, follicles, brain, vulva, squamous/basal cells4 and lungs65, hypoxia or a mutation in von Hippel-Lindau (VHL) factor leads to an overexpression of CA IX (up to 150 fold)4,37,62 through hypoxia inducible factor (HIF) cascade4,37,50,52–55,64.
CA IX is a transmembrane protein37. Based on sequence similarity, CA IX has been recognized as a multidomain protein consisting of an N-terminal proteoglycan like (PG) domain, a CA catalytic domain, a transmembrane segment (TM), and an intracytoplasmic (IC) portion66. The CA catalytic domain presents a significant sequence identity (30-40%) to other catalytic CA isozymes. Both CA and PG domains are glycosylated67. A cDNA coding for the CA IX protein was first cloned by Pastorek et al.68, and the CA9 gene was further characterized66. According to recent biochemical and structure reports, the CA IX is a dimer67 and shows similarity with CA II not only in terms of catalytic efficiency, but also affinity for inhibitors3,4,15,37 like sulfonamides69–75 and sulfamates74–76.
So far, only one structure of CA IX77 crystallized in complex with acetazolamide (a classic inhibitor of CA) is available in the PDB. The structures of CA II (PDB ID: 3KS3)78 and CA IX (PDB ID: 3IAI)77 are very similar and have a Cα RMSD of 1.5 Å (Fig. 2). However, there are several amino acid residues differences in and around the rim of the active site of the two isoforms. Ala65, Phe131, Asn67, Gln69, Ile91 and Leu204 in CA II are structurally equivalent to Ser65, Val131, Gln67, Thr69, Leu91 and Ala204 in CA IX, respectively. It is interesting to note that 3 of these differences lie in a region that forms a hydrophobic pocket on the rim of the active site where some of the already known inhibitors bind CA II49,73,79–89. It is these differences which could be exploited to design inhibitors which prefer binding to one CA compared to another. Differences in the active site residues of CA II and CA IX also account for a variation in the size of their active site cavities. With a volume of 290.4 Å3 and surface area of 245.3 Å2, the active site of CA II is larger than that of CA IX (volume = 270.7 Å3, surface area = 215.3 Å2). These calculations were performed using the online web server of CASTp90.
Figure 2.
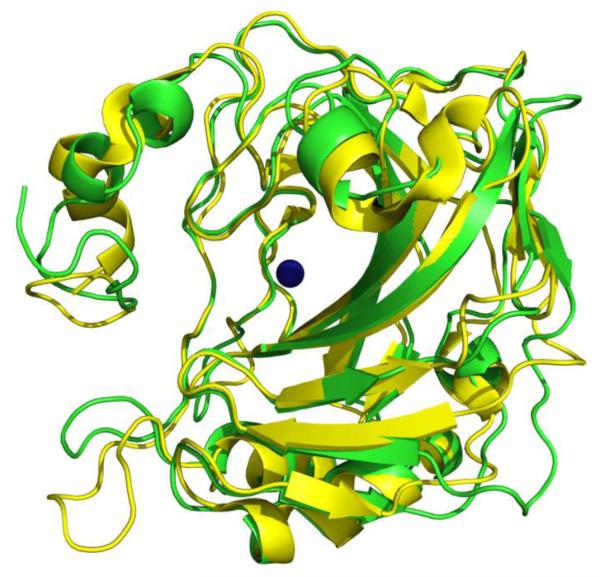
Superposition of CA II78 (yellow) and CA IX77 (green), showing high conservation in secondary structures. Active site Zn is shown as a blue sphere.
Isoform Specific Drug Design
One of the major issues in targeting CA IX for the treatment of tumors is its structural similarity with CA II. In fact, most isoforms of CA bear only few differences in the active site which makes it difficult for designing inhibitors that would specifically bind to one isoform over the others. This non-specific binding (or off-target inhibition) to other isoforms of CA by clinically used (U.S. Food and Drug Administration approved) CA II inhibitory drugs is what causes various side effects during the course of treatment of glaucoma, such as eye irritation, watering, blurred vision, taste changes, constipation and diarrhea4. This has given rise to the need to get greater insights into the fine structural differences within the isoforms and exploit these to design novel and more specific inhibitors in order to circumvent the problem of off-target binding. Although, not many isoform specific inhibitors are known yet, but new sulfonamides and other non-sulfur based compounds are continuously being reported to find derivatives with better inhibition profiles as compared to the promiscuous, first generation inhibitors 14,91,92. Various docking and kinetic studies to test and characterize novel and already existing inhibitors for isoform specificity are going on51,93.
Rational drug design or ligand design is the process of finding new compounds based on the knowledge of a biological target. However, once a ligand has been designed and proven to be effective in inhibiting or enhancing its appropriate target, there are certain issues that need to be resolved before the ligand can be used as a drug. These issues or rules were first defined as the Lipinski’s rule of five94. The rule describes pharmacokinetic properties like absorption, distribution, metabolism, excretion (ADME) of an ideal orally administered drug. According to this rule, for a drug (ligand) to be orally administered, it should not violate more than one of the following criteria: the ligand should have no more than 5 H-bond donors, 10 H-bond acceptors, 500 Dalton of molecular weight, and an octanol-water partition coefficient (log P) of 5. However, over the years there have been various changes to this rule, like the ones suggested by Ghose et al.95 and thus it is no longer followed verbatim. Some of the most successful results yielded by rational, structure based computer aided drug design have produced drugs like Saquinavir (HIV protease inhibitor)96, Dorzolamide (CA inhibitor)97,98 and Iminatib (tyrosine kinase inhibitor)99.
One of the most common approaches that have been used for designing CAIs is referred to as the ring approach where an aromatic ring is attached directly to the sulfonamide group to enhance binding affinities. This approach has been calibrated and exploited over the last few decades8. There is already enough information available about high affinity inhibitors against CA and more compounds are added to an ever growing database. However, it is not the binding affinity but isoform-specificity that is lacking and hence, the ring approach has become less important. Combinatorial chemistry is another widely used, although not a very successful approach to rapidly synthesize or computationally simulate a large number of structurally related molecules. The failure to take molecular flexibility of drugs into account could well be considered as the biggest shortcoming of this approach100.
As mentioned earlier, CA IX has been observed to be a key player in tumorigenesis and tumor progression101. One of the major issues with cancer treatment is the inability of most drugs to distinguish cancerous cells from normal cells. Overexpression of CA IX specifically in cancerous cells makes it a good drug target.
Protein Data Bank (PDB) Mining Approach
As shown in Fig. 1A earlier, clustering of various amino acid residues in and around the active site of CA, forms various pockets. Different sulfonamide inhibitors’ “tails” prefer different orientations and active site rim pockets as they extend out of from the Zn. Their orientation within a pocket depends largely on the interactions between the terminal tail atoms of the inhibitor and the residues that form the pocket. A list of all the inhibitors deposited in the PDB in complex with CA II, has been compiled. The inhibitors were then categorized based on their structural similarity and only one coordinate file for each inhibitor was included, shortening the original list of 400 structures to 145 non-redundant structures. Each of these structures were visualized in COOT102 and analyzed based on which pocket the inhibitors bind in. As shown in the flowchart (Fig. 3), 115 inhibitors were found to bind in one general pocket, while 14 inhibitors (Fig. 4) occupied a “selective” pocket (Fig. 5). Based on the observed active site binding interactions of these inhibitors in the crystal structures and their respective inhibition constants, the goal is to structurally characterize aspects of these interactions that would lead to more potent, selective CA IX sulfonamide inhibitors.
Figure 3.

Stepwise findings from Protein Data Bank mining. 14 inhibitors deposited in the PDB were found to be bound in the “selective” pocket which has different hydrophobicity in CA II and CA IX.
Figure 4.


Structures of the 14 inhibitors that occupy the “selective” pocket in CA II.
Figure 5.

(A) The 14 inhibitors49,73,79–89 occupying the “selective” pocket in CA II (white surface), where 3 residues differ from CA IX (shown in green). Active site Zn is shown as a blue sphere. (B) Active site zoomed.
The PDB deposited structure of CA IX, solved to a resolution of 2.2 Å, crystallized in P61 space group as a dimer. Coordinates of the aforementioned 129 inhibitors from the non-redundant list were used to model the respective inhibitors into the crystal structure of CA IX (PDB ID: 3IAI)77. The structures of CA II (PDB ID: 3KS3)78 and CA IX (PDB ID: 3IAI)77 are very similar although, there is a difference in three residues (N67Q, E69T and I91L) that form part of this “selective” pocket in the active site. It is this pocket where 14 inhibitors from the non-redundant list of PDBs were found to be bound. This raises opportunities for designing and altering the tails of these 14 inhibitors in a manner that might preferentially enhance their KIs for CA IX over CA II.
With the objective of enhancing the CA II inhibitor database to enable studies using the tail approach, more and more crystal structures of CA II in complex with various novel inhibitors belonging to different structural classes are being solved45–49.
To attain selective inhibition of CA IX and to relate its catalytic activity to extracellular acidification, the hypothesis is that the inhibition characteristics for CA IX will vary from CA II (based on crystal structures). The specific differences in amino acids projecting into the active site (namely, residues at positions 65, 67, 69. 91 and 131 (CA II numbering)) will have a direct effect on the specificity of inhibitor binding.
Among various other approaches that are currently under study, tail approach (a word coined by Supuran and co-workers)8 can be used to enhance the aqueous solubility of inhibitors in order to decrease the side effects caused by drugs which are only soluble in highly acidic solvents. The approach (Fig. 6) could also involve altering the terminal regions of an already known inhibitor such that the tail interacts specifically with residues on the surface of one isoform (CA IX) and not others.
Figure 6.
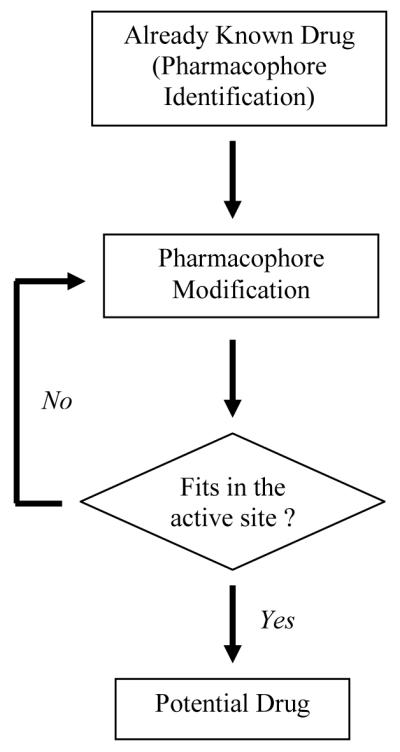
Approach to isoform specific drug design and enhancement of inhibition profiles for currently known drugs.
Fragment Addition Approach
This is an entirely different approach to drug design as it uses ab initio methodology to find random fragments that may bind to a protein. Once such fragments are identified, the forces and interactions that stabilize this non Zn-bound molecule within the active site can be mapped using structure activity relationships (SARs) among different isoforms of CA and exploited to design an isoform specific inhibitor. Thereafter, several of these binding regions could be used in fragment addition based drug design, as explained by SAR by NMR by Shuker et al.103.
There are various fragment libraries available that can be used for binding studies and drug design. The choice of which fragment library to choose depends largely on the experiment that is going to be performed. One of such libraries is the “Zenobia fragment library” that comprises of 352 compounds (soluble up to 200 mM in DMSO). Moreover, all these “fragment” compounds have molecular weights, polar surface area, hydrogen bond donors/acceptors in the range defined by the Lipinski’s rule of five.
Fragment addition approach could also be combined with tail approach by exploiting the zinc binding property of sulfonamide groups. Compounds (fragments) which bind at unique regions in CA IX could be covalently linked to the zinc binding sulfonamide group in classical CAIs.
Conclusion and Discussion
Carbonic anhydrase IX (CA IX) has been shown to play a critical role in cancer proliferation. CA IX overexpression in certain types of cancers has led to the use of CA IX as a biomarker for cancer diagnosis and prognosis. In addition, much knowledge has been acquired on CA II inhibition for various classes of diuretics and systemically acting antiglaucoma agents. The differences in the active sites of different isoforms of CA are subtle and this causes non-specific CA inhibition which leads to side effects. The PDB was mined for previously solved α-CA inhibitor complex structures (mainly CA II), and a sub-set of selected structures were superposed with the structure of CA IX. For example, the inhibitor from a CA II complex (PDB ID: 3N2P)49 when modeled in CA IX (PDB ID 3IAI)77 appears to form one extra hydrogen bond with the protein (Fig. 7). This is because Q97 in CA IX is one C-C bond longer than the N67 in CA II and thus extends further towards the inhibitor to be close enough to form a H-bond. Surface area (SA) buried by an inhibitor in a crystal structure with CA II, has been compared with that of the same inhibitor modeled into CA IX. A reduction of buried SA by 10 Å2 corresponds to a decrease in Gibbs free energy by 0.3 kcal/mol104. Although the buried SA in almost all the 14 inhibitors with CA IX is smaller (larger ΔG) than that with CA II, the presence of extra hydrogen bonds in the former compensates for the gain in ΔG by a larger magnitude (H-bond energy values obtained from Wendler et al.105). These findings can be hereafter correlated to the inhibition of CA II and CA IX to provide SARs that may help in understanding the varied binding affinities of the inhibitors toward different α-CA isoforms (Table 4). This data can further be used to guide in silico design of new CA inhibitors (CAIs) that would preferentially bind to CA IX (on-target) over others CAs (off-target).
Figure 7.

Stick representation of (A) PDB ID 3N2P49, and (B) the same inhibitor modeled into the active site of CA IX, PDB ID 3IAI77. When modeled into CA IX, this inhibitor forms an extra H-bond (shown with dotted black line). Active site Zn is sown as a blue sphere. Residues are as labeled.
Table 4.
Differences in buried surface area, hydrogen bonds, and difference in Gibbs free energy (ΔG) associated with H-bonds among published CA II-inhibitor crystal complexes and modeled CA IX-inhibitor complexes.
| PDB IDs | Buried Surface Area (Å2) | Gain in ΔG in CA IX (kcal/mol) |
# of Hydrogen Bonds | ||
|---|---|---|---|---|---|
| CA II | CA IX | CA II | CA IX | ||
| 3N2P49 | 369.10 | 362.83 | 0.19 | 4 | 5 |
| 1I8Z79 | 443.37 | 423.51 | 0.60 | 5 | 5 |
| 1I9179 | 434.65 | 426.42 | 0.25 | 5 | 5 |
| 1IF980 | 373.02 | 354.52 | 0.55 | 4 | 4 |
| 3BL181 | 406.18 | 375.43 | 0.92 | 4 | 4 |
| 3FFP82 | 335.25 | 313.03 | 0.67 | 4 | 4 |
| 3HLJ83 | 339.61 | 316.01 | 0.71 | 3 | 3 |
| 1ZE873 | 362.12 | 336.70 | 0.76 | 3 | 4 |
| 2FOU84 | 358.57 | 345.67 | 0.39 | 4 | 5 |
| 2HL485 | 358.58 | 342.58 | 0.48 | 5 | 6 |
| 2WD386 | 400.05 | 414.40 | −0.43 | 5 | 5 |
| 3K3487 | 398.95 | 367.60 | 0.94 | 4 | 4 |
| 3MNU88 | 341.60 | 322.31 | 0.58 | 5 | 5 |
| 3HKT89 | 376.31 | 356.40 | 0.60 | 5 | 2 |
Acknowledgement
The work has been supported by Alumni Fellowship Award and Grinter Award from University of Florida (MA), HHMI Science for Life (BK) and National Institutes of Health grant GM25154 (RM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Messerchmidt A, Bode W, Cygler M. Handbook of Metalloproteins. John Wiley and Sons Ltd.; 2004. [Google Scholar]
- 2.Domsic JF, Avvaru BS, Kim CU, Gruner SM, Agbandje-McKenna M, Silverman DN, McKenna R. J. Biol. Chem. 2008;283:30766–30771. doi: 10.1074/jbc.M805353200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scozzafava A, Mastrolorenzo A, Supuran CT. Expert Opinion on Therapeutic Patents. 2006;16:1627–1664. doi: 10.1517/13543776.16.1.27. [DOI] [PubMed] [Google Scholar]
- 4(a).Supuran CT. Nat. Rev. Drug Discov. 2008;7(2):168–81. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]; (b) Supuran CT, Scozzafava A, Conway J. Carbonic anhydrase: its inhibitors and activators. CRC Press; 2004. [Google Scholar]
- 5.Pastorekova S, Parkkila S, Pastorek J, Supuran CT. J Enzyme Inhib Med Chem. 2004;19:199–229. doi: 10.1080/14756360410001689540. [DOI] [PubMed] [Google Scholar]
- 6.Chegwidden WR, Carter ND. The carbonic anhydrases: new horizons. Birkhäuser; 2000. [PubMed] [Google Scholar]
- 7.Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, Weibel DB, Whitesides GM. Chem. Rev. 2008;108:946–1051. doi: 10.1021/cr050262p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Supuran CT, Scozzafava A, Casini A. Med Res Rev. 2003;23:146–189. doi: 10.1002/med.10025. [DOI] [PubMed] [Google Scholar]
- 9.Ulmasov B, Waheed A, Shah GN, Grubb JH, Sly WS, Tu C, Silverman DN. Proc. Natl. Acad. Sci. U.S.A. 2000;97:14212–14217. doi: 10.1073/pnas.97.26.14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimori I, Minakuchi T, Onishi S, Vullo D, Cecchi A, Scozzafava A, Supuran CT. Bioorg. Med. Chem. 2007;15:7229–7236. doi: 10.1016/j.bmc.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 11.Vullo D, Franchi M, Gallori E, Antel J, Scozzafava A, Supuran CT. J. Med. Chem. 2004;47:1272–1279. doi: 10.1021/jm031057+. [DOI] [PubMed] [Google Scholar]
- 12.Nishimori I, Vullo D, Innocenti A, Scozzafava A, Mastrolorenzo A, Supuran CT. J. Med. Chem. 2005;48:7860–7866. doi: 10.1021/jm050483n. [DOI] [PubMed] [Google Scholar]
- 13.Nishimori I, Minakuchi T, Onishi S, Vullo D, Scozzafava A, Supuran CT. J. Med. Chem. 2007;50:381–388. doi: 10.1021/jm0612057. [DOI] [PubMed] [Google Scholar]
- 14.Vullo D, Voipio J, Innocenti A, Rivera C, Ranki H, Scozzafava A, Kaila K, Supuran CT. Bioorg. Med. Chem. Lett. 2005;15:971–976. doi: 10.1016/j.bmcl.2004.12.052. [DOI] [PubMed] [Google Scholar]
- 15.Vullo D, Franchi M, Gallori E, Pastorek J, Scozzafava A, Pastorekova S, Supuran CT. Bioorg. Med. Chem. Lett. 2003;13:1005–1009. doi: 10.1016/s0960-894x(03)00091-x. [DOI] [PubMed] [Google Scholar]
- 16.Vullo D, Innocenti A, Nishimori I, Pastorek J, Scozzafava A, Pastoreková S, Supuran CT. Bioorg. Med. Chem. Lett. 2005;15:963–969. doi: 10.1016/j.bmcl.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 17.Nishimori I, Vullo D, Innocenti A, Scozzafava A, Mastrolorenzo A, Supuran CT. Bioorg. Med. Chem. Lett. 2005;15:3828–3833. doi: 10.1016/j.bmcl.2005.06.055. [DOI] [PubMed] [Google Scholar]
- 18.Lehtonen J, Shen B, Vihinen M, Casini A, Scozzafava A, Supuran CT, Parkkila A-K, Saarnio J, Kivelä AJ, Waheed A, Sly WS, Parkkila SJ. Biol. Chem. 2004;279:2719–2727. doi: 10.1074/jbc.M308984200. [DOI] [PubMed] [Google Scholar]
- 19.Supuran CT, Ilies MA, Scozzafava A. European Journal of Medicinal Chemistry. 1998;33:739–751. [Google Scholar]
- 20.Supuran CT, Scozzafava A, Ilies MA, Briganti F. J. Enzym. Inhib. 2000;15:381–401. doi: 10.1080/14756360009040695. [DOI] [PubMed] [Google Scholar]
- 21.Scozzafava A, Briganti F, Ilies MA, Supuran CT. J. Med. Chem. 2000;43:292–300. doi: 10.1021/jm990479+. [DOI] [PubMed] [Google Scholar]
- 22.Winum J-Y, Temperini C, El Cheikh K, Innocenti A, Vullo D, Ciattini S, Montero J-L, Scozzafava A, Supuran CT. J. Med. Chem. 2006;49:7024–7031. doi: 10.1021/jm060807n. [DOI] [PubMed] [Google Scholar]
- 23.Saczewski F, Sławiński J, Kornicka A, Brzozowski Z, Pomarnacka E, Innocenti A, Scozzafava A, Supuran CT. Bioorg. Med. Chem. Lett. 2006;16:4846–4851. doi: 10.1016/j.bmcl.2006.06.064. [DOI] [PubMed] [Google Scholar]
- 24.De Simone G, Vitale RM, Di Fiore A, Pedone C, Scozzafava A, Montero J-L, Winum J-Y, Supuran CT. J. Med. Chem. 2006;49:5544–5551. doi: 10.1021/jm060531j. [DOI] [PubMed] [Google Scholar]
- 25.Köhler K, Hillebrecht A, Schulze Wischeler J, Innocenti A, Heine A, Supuran CT, Klebe G. Angew. Chem. Int. Ed. Engl. 2007;46:7697–7699. doi: 10.1002/anie.200701189. [DOI] [PubMed] [Google Scholar]
- 26.Liljas A, Kannan KK, Bergstén PC, Waara I, Fridborg K, Strandberg B, Carlbom U, Järup L, Lövgren S, Petef M. Nature New Biol. 1972;235:131–137. doi: 10.1038/newbio235131a0. [DOI] [PubMed] [Google Scholar]
- 27.Nair SK, Christianson DW. J. Am. Chem. Soc. 1991;113:9455–9458. [Google Scholar]
- 28.Fisher Z, Hernandez Prada JA, Tu C, Duda D, Yoshioka C, An H, Govindasamy L, Silverman DN, McKenna R. Biochemistry. 2005;44:1097–1105. doi: 10.1021/bi0480279. [DOI] [PubMed] [Google Scholar]
- 29.Steiner H, Jonsson B-H, Lindskog S. European Journal of Biochemistry. 1975;59:253–259. doi: 10.1111/j.1432-1033.1975.tb02449.x. [DOI] [PubMed] [Google Scholar]
- 30.Tu CK, Silverman DN, Forsman C, Jonsson BH, Lindskog S. Biochemistry. 1989;28:7913–7918. doi: 10.1021/bi00445a054. [DOI] [PubMed] [Google Scholar]
- 31.Duda D, Tu C, Qian M, Laipis P, Agbandje-McKenna M, Silverman DN, McKenna R. Biochemistry. 2001;40:1741–1748. doi: 10.1021/bi002295z. [DOI] [PubMed] [Google Scholar]
- 32.Lindskog S. Pharmacol. Ther. 1997;74:1–20. doi: 10.1016/s0163-7258(96)00198-2. [DOI] [PubMed] [Google Scholar]
- 33.Silverman DN, McKenna R. Acc. Chem. Res. 2007;40:669–675. doi: 10.1021/ar7000588. [DOI] [PubMed] [Google Scholar]
- 34.Roy A, Taraphder S. J Phys Chem B. 2007;111:10563–10576. doi: 10.1021/jp073499t. [DOI] [PubMed] [Google Scholar]
- 35.Fisher SZ, Maupin CM, Budayova-Spano M, Govindasamy L, Tu C, Agbandje-McKenna M, Silverman DN, Voth GA, McKenna R. Biochemistry. 2007;46:2930–2937. doi: 10.1021/bi062066y. [DOI] [PubMed] [Google Scholar]
- 36.Alterio V, Vitale RM, Monti SM, Pedone C, Scozzafava A, Cecchi A, De Simone G, Supuran CT. J. Am. Chem. Soc. 2006;128:8329–8335. doi: 10.1021/ja061574s. [DOI] [PubMed] [Google Scholar]
- 37.Thiry A, Dogné J-M, Masereel B, Supuran CT. Trends Pharmacol. Sci. 2006;27:566–573. doi: 10.1016/j.tips.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 38.Stams T, Nair SK, Okuyama T, Waheed A, Sly WS, Christianson DW. Proc. Natl. Acad. Sci. U.S.A. 1996;93:13589–13594. doi: 10.1073/pnas.93.24.13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mangani S, Håkansson K. Eur. J. Biochem. 1992;210:867–871. doi: 10.1111/j.1432-1033.1992.tb17490.x. [DOI] [PubMed] [Google Scholar]
- 40.Håkansson K, Carlsson M, Svensson LA, Liljas A. J. Mol. Biol. 1992;227:1192–1204. doi: 10.1016/0022-2836(92)90531-n. [DOI] [PubMed] [Google Scholar]
- 41.Jönsson BM, Håkansson K, Liljas A. FEBS Lett. 1993;322:186–190. doi: 10.1016/0014-5793(93)81565-h. [DOI] [PubMed] [Google Scholar]
- 42.Liljas A, Håkansson K, Jonsson BH, Xue Y. Eur. J. Biochem. 1994;219:1–10. doi: 10.1007/978-3-642-79502-2_1. [DOI] [PubMed] [Google Scholar]
- 43.Eriksson AE, Kylsten PM, Jones TA, Liljas A. Proteins. 1988;4:283–293. doi: 10.1002/prot.340040407. [DOI] [PubMed] [Google Scholar]
- 44.Temperini C, Scozzafava A, Supuran CT. Bioorg. Med. Chem. Lett. 2010;20:474–478. doi: 10.1016/j.bmcl.2009.11.124. [DOI] [PubMed] [Google Scholar]
- 45.Biswas S, Aggarwal M, Güzel Ö, Scozzafava A, McKenna R, Supuran CT. Bioorg. Med. Chem. 2011;19:3732–3738. doi: 10.1016/j.bmc.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Carta F, Aggarwal M, Maresca A, Scozzafava A, McKenna R, Masini E, Supuran CT. J. Med. Chem. 2012;55:1721–1730. doi: 10.1021/jm300031j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carta F, Aggarwal M, Maresca A, Scozzafava A, McKenna R, Supuran CT. Chem. Commun. (Camb.) 2012;48:1868–1870. doi: 10.1039/c2cc16395k. [DOI] [PubMed] [Google Scholar]
- 48.Hen N, Bialer M, Yagen B, Maresca A, Aggarwal M, Robbins AH, McKenna R, Scozzafava A, Supuran CT. J. Med. Chem. 2011;54:3977–3981. doi: 10.1021/jm200209n. [DOI] [PubMed] [Google Scholar]
- 49.Pacchiano F, Aggarwal M, Avvaru BS, Robbins AH, Scozzafava A, McKenna R, Supuran CT. Chem. Commun. (Camb.) 2010;46:8371–8373. doi: 10.1039/c0cc02707c. [DOI] [PubMed] [Google Scholar]
- 50.Brahimi-Horn MC, Pouysségur J. FEBS Lett. 2007;581:3582–3591. doi: 10.1016/j.febslet.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 51.Cianchi F, Vinci MC, Supuran CT, Peruzzi B, De Giuli P, Fasolis G, Perigli G, Pastorekova S, Papucci L, Pini A, Masini E, Puccetti LJ. Pharmacol. Exp. Ther. 2010;334:710–719. doi: 10.1124/jpet.110.167270. [DOI] [PubMed] [Google Scholar]
- 52.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 53.Semenza GL. Cancer Metastasis Rev. 2007;26:223–224. doi: 10.1007/s10555-007-9058-y. [DOI] [PubMed] [Google Scholar]
- 54.Hon W-C, Wilson MI, Harlos K, Claridge TDW, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY. Nature. 2002;417:975–978. doi: 10.1038/nature00767. [DOI] [PubMed] [Google Scholar]
- 55.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim Av, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 56.Dorai T, Sawczuk I, Pastorek J, Wiernik PH, Dutcher JP. Cancer Invest. 2006;24:754–779. doi: 10.1080/07357900601062321. [DOI] [PubMed] [Google Scholar]
- 57.Ord JJ, Agrawal S, Thamboo TP, Roberts I, Campo L, Turley H, Han C, Fawcett DW, Kulkarni RP, Cranston D, Harris AL. J. Urol. 2007;178:677–682. doi: 10.1016/j.juro.2007.03.112. [DOI] [PubMed] [Google Scholar]
- 58.Swietach P, Vaughan-Jones RD, Harris AL. Cancer Metastasis Rev. 2007;26:299–310. doi: 10.1007/s10555-007-9064-0. [DOI] [PubMed] [Google Scholar]
- 59.Hutchison GJ, Valentine HR, Loncaster JA, Davidson SE, Hunter RD, Roberts SA, Harris AL, Stratford IJ, Price PM, West CML. Clin. Cancer Res. 2004;10:8405–8412. doi: 10.1158/1078-0432.CCR-03-0135. [DOI] [PubMed] [Google Scholar]
- 60.Sung FL, Hui EP, Tao Q, Li H, Tsui NBY, Dennis Lo YM, Ma BBY, To KF, Harris AL, Chan ATC. Cancer Lett. 2007;253:74–88. doi: 10.1016/j.canlet.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 61.Koukourakis MI, Giatromanolaki A, Sivridis E, Pastorek J, Karapantzos I, Gatter KC, Harris AL. Int. J. Radiat. Oncol. Biol. Phys. 2004;59:67–71. doi: 10.1016/j.ijrobp.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 62.Potter CPS, Harris AL. Br. J. Cancer. 2003;89:2–7. doi: 10.1038/sj.bjc.6600936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hussain SA, Ganesan R, Reynolds G, Gross L, Stevens A, Pastorek J, Murray PG, Perunovic B, Anwar MS, Billingham L, James ND, Spooner D, Poole CJ, Rea DW, Palmer DH. Br. J. Cancer. 2007;96:104–109. doi: 10.1038/sj.bjc.6603530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Trastour C, Benizri E, Ettore F, Ramaioli A, Chamorey E, Pouysségur J, Berra E. Int. J. Cancer. 2007;120:1451–1458. doi: 10.1002/ijc.22436. [DOI] [PubMed] [Google Scholar]
- 65.Swinson DEB, Jones JL, Richardson D, Wykoff C, Turley H, Pastorek J, Taub N, Harris AL, O’Byrne KJ. J. Clin. Oncol. 2003;21:473–482. doi: 10.1200/JCO.2003.11.132. [DOI] [PubMed] [Google Scholar]
- 66.Opavský R, Pastoreková S, Zelník V, Gibadulinová A, Stanbridge EJ, Závada J, Kettmann R, Pastorek J. Genomics. 1996;33:480–487. doi: 10.1006/geno.1996.0223. [DOI] [PubMed] [Google Scholar]
- 67.Hilvo M, Baranauskiene L, Salzano AM, Scaloni A, Matulis D, Innocenti A, Scozzafava A, Monti SM, Di Fiore A, De Simone G, Lindfors M, Jänis J, Valjakka J, Pastoreková S, Pastorek J, Kulomaa MS, Nordlund HR, Supuran CT, Parkkila S. J. Biol. Chem. 2008;283:27799–27809. doi: 10.1074/jbc.M800938200. [DOI] [PubMed] [Google Scholar]
- 68.Pastorek J, Pastoreková S, Callebaut I, Mornon JP, Zelník V, Opavský R, Zat’ovicová M, Liao S, Portetelle D, Stanbridge EJ. Oncogene. 1994;9:2877–2888. [PubMed] [Google Scholar]
- 69.Di Fiore A, Pedone C, D’Ambrosio K, Scozzafava A, De Simone G, Supuran CT. Bioorg. Med. Chem. Lett. 2006;16:437–442. doi: 10.1016/j.bmcl.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 70.Abbate F, Coetzee A, Casini A, Ciattini S, Scozzafava A, Supuran CT. Bioorg. Med. Chem. Lett. 2004;14:337–341. doi: 10.1016/j.bmcl.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 71.Svastová E, Hulíková A, Rafajová M, Zat’ovicová M, Gibadulinová A, Casini A, Cecchi A, Scozzafava A, Supuran CT, Pastorek J, Pastoreková S. FEBS Lett. 2004;577:439–445. doi: 10.1016/j.febslet.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 72.Cecchi A, Hulikova A, Pastorek J, Pastoreková S, Scozzafava A, Winum J-Y, Montero J-L, Supuran CT. J. Med. Chem. 2005;48:4834–4841. doi: 10.1021/jm0501073. [DOI] [PubMed] [Google Scholar]
- 73.Menchise V, De Simone G, Alterio V, Di Fiore A, Pedone C, Scozzafava A, Supuran CT. J. Med. Chem. 2005;48:5721–5727. doi: 10.1021/jm050333c. [DOI] [PubMed] [Google Scholar]
- 74.Abbate F, Winum J-Y, Potter BVL, Casini A, Montero J-L, Scozzafava A, Supuran CT. Bioorg. Med. Chem. Lett. 2004;14:231–234. doi: 10.1016/j.bmcl.2003.09.064. [DOI] [PubMed] [Google Scholar]
- 75.Winum J-Y, Vullo D, Casini A, Montero J-L, Scozzafava A, Supuran CT. J. Med. Chem. 2003;46:2197–2204. doi: 10.1021/jm021124k. [DOI] [PubMed] [Google Scholar]
- 76.Winum J-Y, Vullo D, Casini A, Montero J-L, Scozzafava A, Supuran CT. J. Med. Chem. 2003;46:5471–5477. doi: 10.1021/jm030911u. [DOI] [PubMed] [Google Scholar]
- 77.Alterio V, Hilvo M, Di Fiore A, Supuran CT, Pan P, Parkkila S, Scaloni A, Pastorek J, Pastorekova S, Pedone C, Scozzafava A, Monti SM, De Simone G. Proc. Natl. Acad. Sci. U.S.A. 2009;106:16233–16238. doi: 10.1073/pnas.0908301106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Avvaru BS, Kim CU, Sippel KH, Gruner SM, Agbandje-McKenna M, Silverman DN, McKenna R. Biochemistry. 2010;49:249–251. doi: 10.1021/bi902007b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim C-Y, Whittington DA, Chang JS, Liao J, May JA, Christianson DW. J. Med. Chem. 2002;45:888–893. doi: 10.1021/jm010163d. [DOI] [PubMed] [Google Scholar]
- 80.Grzybowski BA, Ishchenko AV, Kim C-Y, Topalov G, Chapman R, Christianson DW, Whitesides GM, Shakhnovich EI. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1270–1273. doi: 10.1073/pnas.032673399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Temperini C, Cecchi A, Scozzafava A, Supuran CT. Bioorg. Med. Chem. Lett. 2008;18:2567–2573. doi: 10.1016/j.bmcl.2008.03.051. [DOI] [PubMed] [Google Scholar]
- 82.Crocetti L, Maresca A, Temperini C, Hall RA, Scozzafava A, Mühlschlegel FA, Supuran CT. Bioorg. Med. Chem. Lett. 2009;19:1371–1375. doi: 10.1016/j.bmcl.2009.01.038. [DOI] [PubMed] [Google Scholar]
- 83.Baranauskienė L, Hilvo M, Matulienė J, Golovenko D, Manakova E, Dudutienė V, Michailovienė V, Torresan J, Jachno J, Parkkila S, Maresca A, Supuran CT, Gražulis S, Matulis D. J Enzyme Inhib Med Chem. 2010;25:863–870. doi: 10.3109/14756360903571685. [DOI] [PubMed] [Google Scholar]
- 84.Jude KM, Banerjee AL, Haldar MK, Manokaran S, Roy B, Mallik S, Srivastava DK, Christianson DW. J. Am. Chem. Soc. 2006;128:3011–3018. doi: 10.1021/ja057257n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Di Fiore A, Scozzafava A, Winum J-Y, Montero J-L, Pedone C, Supuran CT, De Simone G. Bioorg. Med. Chem. Lett. 2007;17:1726–1731. doi: 10.1016/j.bmcl.2006.12.099. [DOI] [PubMed] [Google Scholar]
- 86.Woo LWL, Jackson T, Putey A, Cozier G, Leonard P, Acharya KR, Chander SK, Purohit A, Reed MJ, Potter BVL. J. Med. Chem. 2010;53:2155–2170. doi: 10.1021/jm901705h. [DOI] [PubMed] [Google Scholar]
- 87.Behnke CA, Le Trong I, Godden JW, Merritt EA, Teller DC, Bajorath J, Stenkamp RE. Acta Crystallogr. D Biol. Crystallogr. 2010;66:616–627. doi: 10.1107/S0907444910006554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Di Fiore A, Monti SM, Innocenti A, Winum J-Y, De Simone G, Supuran CT. Bioorg. Med. Chem. Lett. 2010;20:3601–3605. doi: 10.1016/j.bmcl.2010.04.114. [DOI] [PubMed] [Google Scholar]
- 89.Lopez M, Paul B, Hofmann A, Morizzi J, Wu QK, Charman SA, Innocenti A, Vullo D, Supuran CT, Poulsen S-A. J. Med. Chem. 2009;52:6421–6432. doi: 10.1021/jm900914e. [DOI] [PubMed] [Google Scholar]
- 90.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. Nucleic Acids Res. 2006;34:W116–118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gieling RG, Babur M, Mamnani L, Burrows N, Telfer BA, Carta F, Winum J-Y, Scozzafava A, Supuran CT, Williams KJ. J. Med. Chem. 2012;55:5591–5600. doi: 10.1021/jm300529u. [DOI] [PubMed] [Google Scholar]
- 92.McDonald PC, Winum J-Y, Supuran CT, Dedhar S. Oncotarget. 2012;3:84–97. doi: 10.18632/oncotarget.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dubois L, Peeters S, Lieuwes NG, Geusens N, Thiry A, Wigfield S, Carta F, McIntyre A, Scozzafava A, Dogné J-M, Supuran CT, Harris AL, Masereel B, Lambin P. Radiother Oncol. 2011;99:424–431. doi: 10.1016/j.radonc.2011.05.045. [DOI] [PubMed] [Google Scholar]
- 94.Oprea TI, Davis AM, Teague SJ, Leeson PD. J Chem Inf Comput Sci. 2001;41:1308–1315. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- 95.Ghose AK, Viswanadhan VN, Wendoloski JJ. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 96.Ghosh AK, Thompson WJ, Fitzgerald PM, Culberson JC, Axel MG, McKee SP, Huff JR, Anderson PS. J. Med. Chem. 1994;37:2506–2508. doi: 10.1021/jm00042a002. [DOI] [PubMed] [Google Scholar]
- 97.Amari S, Aizawa M, Zhang J, Fukuzawa K, Mochizuki Y, Iwasawa Y, Nakata K, Chuman H, Nakano T. J Chem Inf Model. 2006;46:221–230. doi: 10.1021/ci050262q. [DOI] [PubMed] [Google Scholar]
- 98.Greer J, Erickson JW, Baldwin JJ, Varney MD. J. Med. Chem. 1994;37:1035–1054. doi: 10.1021/jm00034a001. [DOI] [PubMed] [Google Scholar]
- 99.Craig JC, Duncan IB, Hockley D, Grief C, Roberts NA, Mills JS. Antiviral Res. 1991;16:295–305. doi: 10.1016/0166-3542(91)90045-s. [DOI] [PubMed] [Google Scholar]
- 100.Feher M, Schmidt JM. J Chem Inf Comput Sci. 2003;43:218–227. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- 101.Li Y, Wang H, Oosterwijk E, Tu C, Shiverick KT, Silverman DN, Frost SC. Cancer Invest. 2009;27:613–623. doi: 10.1080/07357900802653464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Emsley P, Cowtan K. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 103.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 104.The Practice of Medicinal Chemistry.
- 105.Wendler K, Thar J, Zahn S, Kirchner B. J Phys Chem A. 2010;114:9529–9536. doi: 10.1021/jp103470e. [DOI] [PubMed] [Google Scholar]
- 106.Alterio V, Monti SM, Truppo E, Pedone C, Supuran CT, De Simone G. Org. Biomol. Chem. 2010;8:3528–3533. doi: 10.1039/b926832d. [DOI] [PubMed] [Google Scholar]
- 107.Duda DM, Tu C, Fisher SZ, An H, Yoshioka C, Govindasamy L, Laipis PJ, Agbandje-McKenna M, Silverman DN, McKenna R. Biochemistry. 2005;44:10046–10053. doi: 10.1021/bi050610h. [DOI] [PubMed] [Google Scholar]
- 108.Boriack-Sjodin PA, Heck RW, Laipis PJ, Silverman DN, Christianson DW. Proc. Natl. Acad. Sci. U.S.A. 1995;92:10949–10953. doi: 10.1073/pnas.92.24.10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pilka ES, Kochan G, Oppermann U, Yue WW. Biochem. Biophys. Res. Commun. 2012;419:485–489. doi: 10.1016/j.bbrc.2012.02.038. [DOI] [PubMed] [Google Scholar]
- 110.Di Fiore A, Truppo E, Supuran CT, Alterio V, Dathan N, Bootorabi F, Parkkila S, Monti SM, De Simone G. Bioorg. Med. Chem. Lett. 2010;20:5023–5026. doi: 10.1016/j.bmcl.2010.07.051. [DOI] [PubMed] [Google Scholar]
- 111.Whittington DA, Waheed A, Ulmasov B, Shah GN, Grubb JH, Sly WS, Christianson DW. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9545–9550. doi: 10.1073/pnas.161301298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Di Fiore A, Monti SM, Hilvo M, Parkkila S, Romano V, Scaloni A, Pedone C, Scozzafava A, Supuran CT, De Simone G. Proteins. 2009;74:164–175. doi: 10.1002/prot.22144. [DOI] [PubMed] [Google Scholar]
- 113.Whittington DA, Grubb JH, Waheed A, Shah GN, Sly WS, Christianson DW. J. Biol. Chem. 2004;279:7223–7228. doi: 10.1074/jbc.M310809200. [DOI] [PubMed] [Google Scholar]